

Abstract
The Escherichia coli sequence type 131 (ST131)-O25b:H4 clone has spread worldwide and become responsible for a significant proportion of multidrug-resistant extraintestinal infections. We generated humanized monoclonal antibodies (MAbs) that target the lipopolysaccharide O25b antigen conserved within this lineage. These MAbs bound to the surface of live bacterial cells irrespective of the capsular type expressed. In a serum bactericidal assay in vitro, MAbs induced >95% bacterial killing in the presence of human serum as the complement source. Protective efficacy at low antibody doses was observed in a murine model of bacteremia. The mode of action in vivo was investigated by using aglycosylated derivatives of the protective MAbs. The significant binding to live E. coli cells and the in vitro and in vivo efficacy were corroborated in assays using bacteria grown in human serum to mimic relevant clinical conditions. Given the dry pipeline of novel antibiotics against multidrug-resistant Gram-negative pathogens, passive immunization with bactericidal antibodies offers a therapeutic alternative to control infections caused by E. coli ST131-O25b:H4.
INTRODUCTION
Escherichia coli is a member of the intestinal commensal flora. Certain variants (pathotypes) of the species, however, can cause either intestinal or extraintestinal infections, such as urinary tract infection, meningitis, or bacteremia (1). Extraintestinal pathogenic E. coli (ExPEC) strains harbor a large array of virulence traits that enable them to cause disease outside the intestinal tract. ExPEC strains have been evolving antibiotic resistance, often a combined resistance against most of the clinically relevant antibiotics, such as fluoroquinolones, aminoglycosides, and β-lactam antibiotics. Typically, multidrug-resistant (MDR) strains are compromised in their fitness and virulence, which restricts their prevalence to a nosocomial setting and conversely limits their spread in the community. Some successful MDR clonal lineages do, however, retain high virulence potential (2, 3). The E. coli clonal lineage sequence type 131 (ST131)-O25b:H4, first described in 2008 (4, 5), has spread globally not only in hospitals (as do most other MDR clones) but also in the community (6–9). This clone is responsible for ∼15% (up to 25% [10, 11]) of all extraintestinal E. coli infections and represents the majority of fluoroquinolone-resistant isolates (12) and about half of the extended-spectrum β-lactamase (ESBL)-producing isolates (13). The progressive acquisition of additional resistance phenotypes in ST131-O25b:H4 strains leaves very few effective antibiotics for treatment of patients infected by members of this lineage (14). Even more alarming is the recent appearance of carbapenem-resistant ST131 isolates (15–17). Recently, ST131-O25b:H4 strains were shown to predominate among carbapenem-resistant E. coli isolates (18). A major clinical concern is the lack of development of novel antibiotics against Gram-negative pathogens, again leaving very limited treatment options (19). The potential emergence and subsequent spread of pan-resistant E. coli strains emphasizes the urgent need to develop alternative therapeutic approaches, such as monoclonal antibodies (MAbs).
Lipopolysaccharide (LPS) of Gram-negative bacteria has long been considered an attractive target for active and passive immunization approaches (20, 21). Antibodies against the lipid A (endotoxin) or core oligosaccharide portions of the LPS molecule are expected to have primarily an antiendotoxin function by neutralizing or sequestering endotoxin in the circulation (20). Their antibacterial effect is restricted because of the low accessibility of these epitopes on live bacteria, as they are masked by the abundant O side chains and/or the capsular polysaccharide (22). Conversely, it has been shown that antibodies specific to the O antigens of LPS can trigger bacterial killing by the complement system alone or, alternatively, through opsonophagocytic killing. In models of bacteremia using different animal species, antibacterial O-specific MAbs afford higher protection than those that target the core oligosaccharide portions of the LPS (23, 24). Bactericidal antibodies directed against the O antigens of LPS may therefore offer an effective therapeutic alternative to antibiotics in the fight against MDR clones. In this article, we describe humanized IgG1 MAbs specific to the conserved O antigen of the E. coli ST131-O25b:H4 clone that induce complement-mediated killing in vitro and give high protective efficacy in a murine model of bacteremia.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Two previously described ST131-O25b clinical isolates (81009 and 3O) (25, 26) that were confirmed genotypically (MLST typed by the Achtman scheme [27] and O25b-specific PCR) and phenotypically (serotyped by O25 rabbit serum and with O25b-specific MAbs) were used in this study. Strain 81009 expresses a K5-type capsular polysaccharide, while strain 3O expresses a non-K5 capsule, confirmed by the use of a K5-specific lytic phage (Statens Serum Institute). A collection of ST131 strains representing different pulsotypes was kindly provided by G. Peirano and J. Pitout (University of Calgary, Canada) (28).
Bacteria were routinely grown in Luria-Bertani (LB) broth (Fisher Scientific) or on Trypticase soy agar (TSA) plates (bioMérieux).
When bacteria were cultured in the presence of human serum, the serum samples obtained from healthy volunteers were pooled (from a minimum of 3 donors) and depleted of E. coli-specific antibodies according to a previously published method (29). Complement was heat inactivated at 56°C for 40 min, and human serum samples were diluted to 50% with 3% human albumin (Albiomin, Biotest), 1.67 diluted RPMI 1640 (Life Technologies), and 12 μM l-glutamine (Sigma-Aldrich) in Dulbecco's phosphate-buffered saline (DPBS) with calcium and magnesium (Life Technologies).
For in vivo experiments, bacteria were grown in LB broth or in pooled human serum (PAA) that was heat inactivated and diluted in RPMI 1640 without phenol red or l-glutamine (Life Technologies) to 50% final concentration.
Generation and selection of humanized monoclonal antibodies targeting the LPS O25b antigen.
BALB/cJRj mice were immunized three times with ∼1 × 107 CFU of either E. coli 81009 or E. coli 81009Δkps (30). Mice with the highest titers against O25b antigen were boosted, and their splenocytes were subjected to hybridoma fusion as previously described (30). From five independent fusions, a total of 23 hybridoma clones were selected based on specific binding to purified O25b LPS, assessed by immunoblots and ELISA, and surface staining of live E. coli O25b cells by flow cytometry. Sequencing of murine MAbs was performed by cloning cDNA specific to the VL and VH regions into commercial cloning vectors. Selected hybridoma clones were expressed as chimeric MAbs (i.e., the mouse variable regions were fused to human IgG1 constant domains and to kappa light chains). Three selected chimeric MAbs were subjected to humanization by CDR grafting technology (Fusion Antibodies Ltd., Belfast, United Kingdom). Briefly, the hypervariable (CDR) mouse antibody sequences were inserted into human framework sequences that were predicted in silico to be the most closely related to the original mouse frameworks. The best humanized MAb from each lineage was selected for further studies based on antigen binding affinity, surface staining, bactericidal activity, and in vivo protective efficacy. Antibodies were routinely expressed by CHO cells (Evitria AG, Schlieren, Switzerland) and purified through MabSelect or MabSelect SuRe resins. Aglycosylated MAb variants were generated by introducing N297Q mutations in the heavy chain.
Immunoblots.
Immunoblotting was performed as described previously (30). Purified and separated LPS blotted onto polyvinylidene difluoride (PVDF) was reacted with 1 μg/ml of O25b-specific monoclonal antibodies or with LPS core-specific murine MAb WN1 222-5 (Hycult Biotech). Binding of MAbs was detected by horseradish peroxidase (HRP)-conjugated goat F(ab′)2 anti-human IgG (Southern Biotech) or goat F(ab′)2 anti-mouse IgG (Southern Biotech) at 1:40,000 dilution.
Biolayer interferometry (BLI).
Antibody binding was measured by immobilizing biotinylated O25b polysaccharide antigen prepared as described previously (30) on streptavidin sensors (ForteBio, Pall Life Sciences) and monitoring the association of the MAbs (10 μg/ml) to the preloaded sensors for 10 min in DPBS containing 1% bovine serum albumin (BSA), followed by dissociation in the same buffer. The Kd (dissociation constant), kon (association rate), and koff (dissociation rate) values were determined using Data Analysis 7 software (ForteBio, Pall Life Sciences). Polyreactivity against nonrelated antigens was measured by immobilizing the MAbs on anti-human capture sensors (ForteBio, Pall Life Sciences) and monitoring the response for the association of 30 μg/ml antigen in solution for 10 min in PBS containing 1% BSA. Response values of <0.05 nm were considered negative. All polyreactivity antigens were purchased from Sigma-Aldrich.
Flow cytometry.
Surface staining was performed as previously described (30). Overnight cultures of bacteria were diluted 1:100 in LB broth or in a 50% depleted heat-inactivated human serum pool and grown at 37°C to mid-log phase. Bacteria (106 CFU) were reacted with MAbs in the concentration range of 0.01 to 160 μg/ml, followed by staining with 4 μg/ml of Alexa Fluor 488-conjugated goat anti-human IgG secondary antibody (Life Technologies) and 5 μM SYTO-62 nucleic acid stain (Life Technologies). Samples were quantified in a BD Accuri C6 flow cytometer (BD Biosciences), and data were analyzed using FCS Express software version 4 (De Novo Software).
Serum bactericidal assay.
Serum bactericidal assay (SBA) was performed in a 50% depleted human serum pool diluted with DPBS supplemented with calcium and magnesium. The reaction mixture contained ∼5 × 103 CFU from LB broth- or serum-grown mid-log-phase bacterial suspension and 10 or 20 μg/ml MAb, respectively. Mixtures without any antibody and with isotype-matched irrelevant MAb were included as controls. Bacteria were enumerated by plating appropriate dilutions following 3 h (LB broth-grown bacteria) or 5 h (serum-grown bacteria) incubation at 37°C with shaking at 410 rpm. Killing mediated by specific MAbs was expressed as killing (%) = 100 − [(CFUMAb/CFUcontrol antibody) × 100].
Animal experiments.
All animal experiments were performed according to Austrian law (BGBl. I Nr. 114/2012, approved by MA58, Vienna). Female 6- to 8-week-old BALB/cJRj mice (Janvier) were used in all experiments.
The protective efficacy of MAbs was assessed by intraperitoneal injection of MAbs diluted in DPBS in a total volume of 500 μl 24 h prior to challenge with a >90% lethal dose (determined in pilot studies) of E. coli 81009 or 3O strains. Control groups received isotype-matched (human IgG1) irrelevant MAb at the same dose. Challenge was performed intravenously with 100 μl of bacterial suspension. Bacteria were grown to mid-log phase (optical density at 600 nm [OD600] of ∼0.5) in LB broth, washed with DPBS, and diluted to the target inoculum (1 × 109 CFU/ml for 81009 and 1.5 × 109 CFU/ml for 3O). Alternatively, bacteria were grown in 50% depleted and heat-inactivated human serum until mid-log phase (OD600 of ∼0.2). Mice were challenged with serum-grown bacteria from frozen glycerol stocks washed and diluted in DPBS to 3 × 109 CFU/ml. In all cases, survival was monitored daily for 2 weeks. Statistical analysis was performed by the log rank (Mantel-Cox) test using GraphPad Prism 5.04 software. Differences were considered statistically significant when the P value was <0.05.
RESULTS
Binding characteristics of three humanized O25b-specific MAbs.
First, O25b-reactive MAbs were generated with standard hybridoma technology by immunizing mice with E. coli ST131-O25b:H4 cells. Mouse MAbs displaying the best binding to purified LPS and live bacterial cells were selected for the generation of murine-human IgG1 chimeric antibodies, three of which were humanized by CDR grafting technology, as described in Materials and Methods.
The antigen specificities of humanized MAbs 3E9-11, 2A7-01, and 4D5-02 were demonstrated by immunoblotting using purified LPS. All selected MAbs bound to the O25b antigen, whereas no binding was observed to unrelated, i.e., non-O25 LPS, molecules (Fig. 1). MAb 4D5-02 cross-reacted with the O25a antigen, while the other two MAbs did not, suggesting that MAb 4D5-02 recognizes a different epitope that is shared by O25a and O25b.
FIG 1.
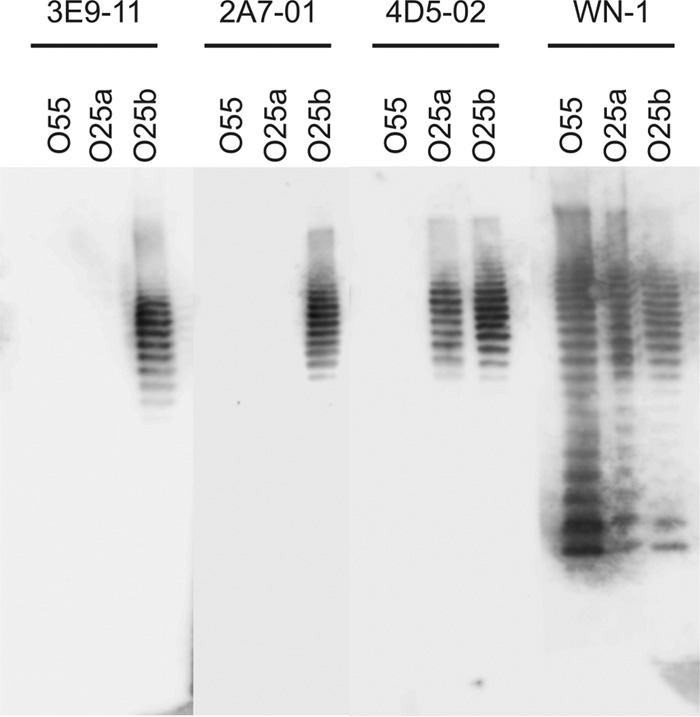
Immunoreactivity of O25b-specific humanized MAbs with purified LPS. Purified LPS samples from E. coli serotypes O25b, O25a, and O55 (control) were separated and blotted onto PVDF membranes that were reacted with the indicated MAbs. The inner-core-specific cross-reactive WN-1 222-5 MAb reacting with all LPS types was used as a positive control.
The binding characteristics were further investigated by biolayer interferometry (BLI; ForteBio) using biotinylated O25b polysaccharides. The affinity of all of the MAbs was in the range of 10−7 to 10−8 M (Table 1), which is in good agreement with values published for other anti-carbohydrate MAbs (31, 32). Importantly, no polyreactive characteristic for any of the MAbs was detected based on lack of binding to unrelated antigens, such as DNA, gelatin, fetuin, and dextran (Table 1).
TABLE 1.
Binding of O25b MAbs to the cognate antigen (O25b polysaccharide), expressed as affinity, association, and dissociation constants, and binding to unrelated antigens, expressed as response values
| MAb | Kd (M−1) | kon (M−1s−1) | koff (s−1) | Polyreactivity response (nm) |
|||
|---|---|---|---|---|---|---|---|
| Dextran | DNA | Fetuin | Gelatin | ||||
| 3E9 chimeric | 5.56E–08 | 2.83E+05 | 1.57E–02 | −0.0114 | −0.0114 | −0.0076 | −0.0159 |
| 3E9-11 | 6.94E–08 | 2.44E+05 | 1.69E–02 | −0.0124 | −0.0183 | −0.0145 | −0.016 |
| 2A7 chimeric | 1.12E–07 | 1.42E+05 | 1.59E–02 | −0.0178 | −0.0175 | −0.0146 | −0.0161 |
| 2A7-01 | 1.15E–07 | 1.62E+05 | 1.86E–02 | −0.0151 | −0.0157 | −0.0163 | −0.0165 |
| 4D5 chimeric | 1.49E–08 | 4.36E+04 | 6.51E–04 | −0.0107 | −0.0033 | −0.0034 | 0.0024 |
| 4D5-02 | 3.82E–08 | 3.86E+04 | 1.47E–03 | −0.0182 | −0.0143 | −0.0144 | −0.0167 |
| Irrelevant control MAb | 0.0056 | 0.0121 | −0.0224 | −0.0164 | |||
Binding to the native O25b antigens on the surface of live E. coli cells was assessed by flow cytometry. We observed comparably high levels of surface binding to two different ST131-O25b:H4 strains expressing different capsule types with all three antibodies at concentrations of ≥20 μg/ml (Fig. 2A). At lower antibody concentrations, the 3E9-11 and 2A7-01 MAbs displayed significantly higher surface binding intensity than that found for 4D5-02. In addition, surface binding was confirmed using a panel of clinical ST131 isolates representing different pulsotypes (28). All pulsotypes, with the exception of pulsotype O (previously shown to express the O16 antigen), were strongly stained by all three MAbs (see Table S1 in the supplemental material).
FIG 2.

Surface staining of live E. coli cells with O25b-specific MAbs. Two E. coli ST131-O25b:H4 clinical isolates, 81009 and 3O, were grown to mid-log phase in LB broth (A) or in 50% depleted heat-inactivated human serum (B) and stained with MAbs at a concentration range of 0.01 to 160 μg/ml. Fluorescent intensity of bacteria was determined by flow cytometry using labeled secondary IgG.
Encouraged by the observation that Gram-negative bacteria grown under in vivo-like conditions demonstrate changes in gene expression (33, 34), particularly those involved in the synthesis and export of surface molecules, we also measured antibody binding to live E. coli cells grown in the presence of human serum. We observed reduced surface staining of serum-grown bacteria; however, it was still considered intense based on the 10- to 100-fold increase in median fluorescent intensity relative to the negative-control antibody (Fig. 2B).
In vitro bactericidal activity of O25b MAbs.
To measure the bactericidal activity attributable to the three humanized MAbs, an SBA was performed to detect antibody-mediated complement-dependent killing of E. coli cells. We observed an antibody concentration-dependent bacterial killing with all three antibodies (Fig. 3A), which was completely abolished upon heat inactivation of human serum (see Fig. S1 in the supplemental material). Consistent with the surface staining data, 3E9-11 proved to be the most efficacious MAb at low antibody concentrations, resulting in >95% killing compared to that of control antibody during a 3-hour incubation. The other two MAbs required higher concentrations (>10 to 20 μg/ml) to reach the same level of bactericidal efficacy.
FIG 3.
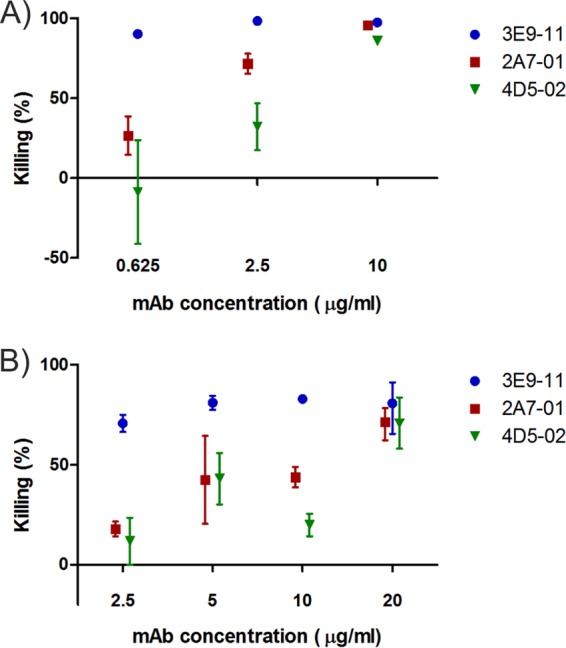
Serum bactericidal activity of O25b MAbs. Bactericidal effect was expressed as percent reduction of CFU relative to the bacterial numbers recovered from the control (irrelevant MAb) group. (A) Bacteria grown in LB broth incubated with humanized MAbs for 3 h. Combined results of 3 independent experiments shown as means ± standard errors. (B) Bacteria grown in 50% depleted heat-inactivated human serum incubated with humanized MAbs for 5 h. Combined results of 2 independent experiments shown as ranges (B).
Similarly, a highly efficacious bactericidal effect was observed when the assay was performed using bacteria grown in depleted human serum (Fig. 3B). Serum-grown bacteria required a higher antibody concentration and longer incubation (5 h versus 3 h) than LB broth-grown bacteria to give a comparable effect.
In vivo protection.
The protective efficacy of the selected humanized MAbs was tested in a murine bacteremia model using two unrelated ST131-O25b:H4 challenge strains. At an antibody dose of 100 μg/mouse (corresponding to ∼5 mg/kg dose), all three tested MAbs provided comparable high levels of protection against both strains (Fig. 4A and B). Considering the differences between the three MAbs observed in vitro, we were interested in comparing the protective efficacies as a function of antibody concentration. Passive immunization of mice with MAb doses between 1 and 150 μg (corresponding to ∼0.05 to 7.5 mg/kg and an estimated 1.3 to 200 μg/ml serum levels) revealed that MAb 3E9-11 was the most efficacious, while 4D5-02 was the least potent at lower antibody doses (Fig. 4B). This efficacy ranking was consistent with the in vitro bactericidal activities measured with the three MAbs (Fig. 3A).
FIG 4.
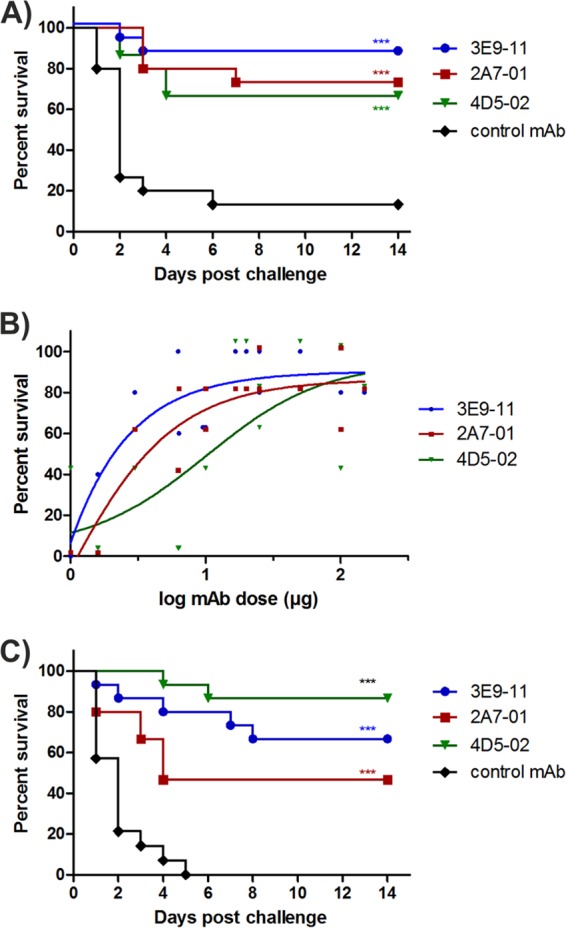
Protective efficacy of selected humanized MAbs in a murine lethal bacteremia model. Passive immunization was performed by intraperitoneal injections of MAbs 24 h prior to lethal intravenous challenge with strain 3O grown in LB broth (A), strain 81009 grown in LB broth (B), or strain 81009 grown in depleted human serum (C). Doses of 100 μg/mouse were used routinely, and survival curves for 14 days are depicted in panels A and C. Dose response curves with three selected MAbs within the range of 1 to 150 μg were performed, and survival rates over control at day 14 postinjection are plotted in panel B. Combined results of 2 (A) or 3 (C) independent experiments are shown. P values were calculated using the log rank test (***, P < 0.001).
In light of the lower surface-staining intensity and delayed bactericidal effect observed with serum-grown compared to LB broth-grown bacteria (Fig. 2B and 3B), we measured the protective efficacy of MAbs against bacteria preconditioned in human serum. Survival data from these experiments confirmed protection comparable to that observed for LB broth-grown bacteria at the same MAb dose (Fig. 4B and C).
In order to corroborate that the protection seen in the mouse model was mediated by the activation of complement, aglycosylated versions of the three MAbs were tested. These aglycosylated antibodies were generated by replacing the asparagine residue in the Fc domain of IgGs (N297Q) known to be essential for N-linked glycosylation and effective activation of the complement system via C1q binding. In these comparative studies, we used the minimal protective dose of each antibody, which was 5, 10, and 25 μg/mouse for 3E9-11, 2A7-01, and 4D5-02, respectively (based on data shown in Fig. 4B), or a 50-μg/mouse dose for all MAbs, which was considered to result in antibody excess. At the minimal protective MAb doses, the significant protection elicited by the glycosylated IgGs was lost upon use of the aglycosylated versions (Fig. 5A). Surprisingly, however, the protective capacity was found to be independent of the glycosylation status at the higher MAb dose (Fig. 5B).
FIG 5.
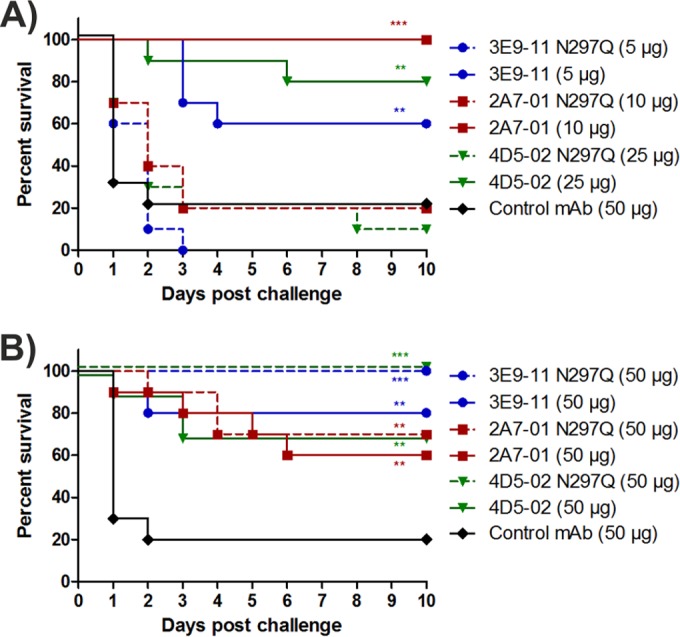
In vivo efficacy of aglycosylated MAbs. Mice were passively immunized with a minimal protective dose (A) or an excessive dose (B) of three different O25b-specific MAbs and their aglycosylated variants. Survival curves following a subsequent lethal challenge by strain 81009 are shown. Graphs show combined results of 2 independent experiments with groups of 5 mice each. P values (log rank test) are shown where survival was significantly different relative to the control group (**, P < 0.01; ***, P < 0.001).
To investigate this unexpected finding, we measured the in vitro bactericidal effect of aglycosylated MAbs side by side with the glycosylated (wild-type) counterparts. Surprisingly, at high doses, aglycosylated MAbs induced 70% to 92% of the killing, elicited by the glycosylated MAbs. Nevertheless, their bactericidal effect decreased rapidly upon dilution of the antibodies (Fig. 6).
FIG 6.
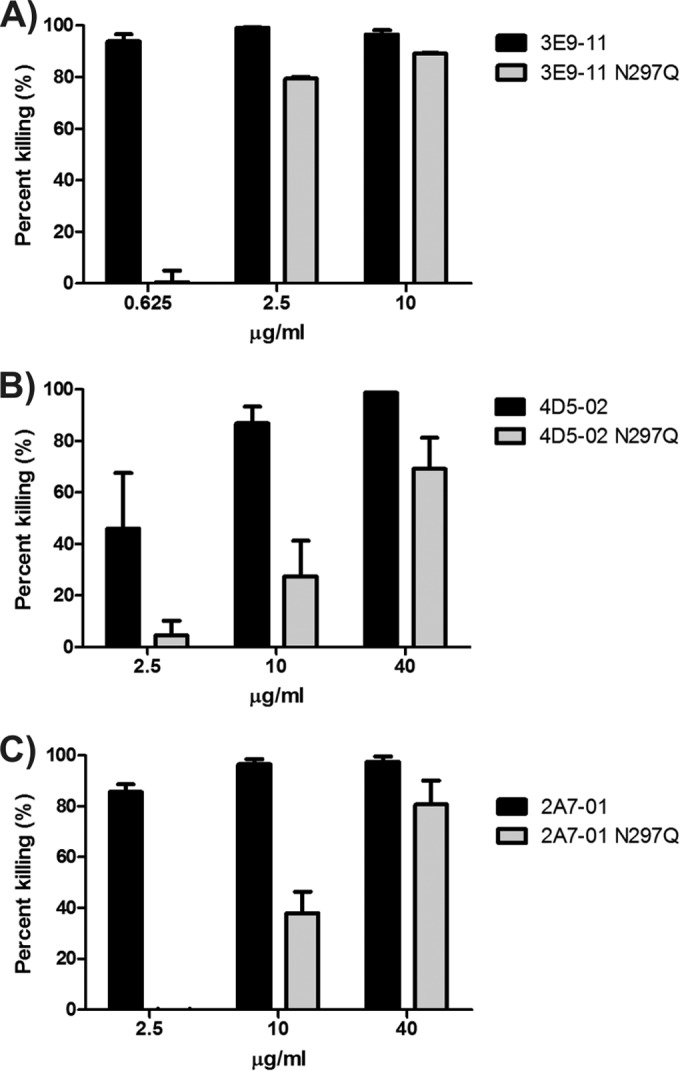
In vitro bactericidal efficacy of aglycosylated MAbs. Bactericidal activities of three aglycosylated MAbs were determined simultaneously with their glycosylated counterparts. Bactericidal activity is expressed as percent killing, i.e., CFU recovered in test groups relative to the control group (irrelevant MAb control) following 3 h of incubation in 50% depleted human serum. (A) MAb 3E9-11; (B) MAb 4D5-02; (C) MAb 2A7-01. Black bars, wild-type MAbs; gray bars, Fc mutated to remove glycosylation. Means with range for 2 independent assays are shown.
DISCUSSION
Passive immunization with hyperimmune sera (serum therapy) was a standard treatment option in the preantibiotic era. As we move toward a possible post-antibiotic era, it may be prudent to reconsider the merits of this general approach. Current state-of-the-art biomedical research and biopharmaceutical manufacturing capabilities make it possible to generate highly purified human/humanized MAbs against a range of pathogenic microorganisms. MAbs directed against nonhuman targets and as a class of therapeutics in general have an excellent safety record and are well tolerated in the clinic (unlike therapy with serum). To date, there are two licensed MAb products against infectious targets (35, 36) and many more in the clinical phase of testing or under preclinical development (37, 38).
The challenges associated with the development of antibacterial antibodies include finding molecular targets that are accessible on the surface of a given bacterium and simultaneously shared by clinically relevant strains. Since conserved bacterial surface molecules are masked by highly variable surface polysaccharides in E. coli (and other enterobacterial pathogens), it is difficult to find a target that can provide broad-spectrum protection. The concept of “magic bullets” that are able to treat a broad variety of pathogens might be considered unrealistic and should be replaced by rational designs of species/subspecies-specific therapeutic approaches. These novel antimicrobials with targeted coverage are not expected to induce widespread resistance or to have detrimental effects on normal microbiota.
In this paper, we described highly efficacious MAbs targeting the unique O antigen (30) of a clinically relevant MDR E. coli clonal lineage. The humanized MAbs binding to the O25b antigen were shown to confer high levels of protection in a murine model of bacteremia. This is in accordance with previous reports suggesting that the LPS O antigen is a protective antigen, i.e., that antibodies against these epitopes can elicit protection (23, 24, 39, 40).
The primary aim of the study was to identify MAbs capable of triggering bactericidal effects mediated solely by the complement system, i.e., without the involvement of phagocytes. Activation of the classic complement pathway is initiated by the binding of C1q to the antigen-bound antibody complex. C1q is a hexamer molecule that requires binding to several Fc regions for efficient activation of the complement cascade. This is supported by the greater complement-activating potential of pentameric IgM molecules compared to that of IgG subclasses. In order to be able to develop therapeutic MAbs of the IgG isotype with such an intended mode of action, we considered it important to select a target antigen that is highly abundant on the surface. We envisioned that adjacent binding of multiple IgG molecules may efficiently trigger the binding of C1q and hence activate the classic complement pathway. Indeed, the main mode of action of O25b-specific MAbs appears to be complement-mediated killing, as confirmed both in vitro and in vivo. In vivo, at minimal protective doses, aglycosylation of MAbs eliminated protection, confirming an Fc-mediated mode of action. Interestingly, however, at excessive MAb doses, even aglycosylated MAbs provided protection. One possible explanation for this protection is that aglycosylated MAbs retain some residual capacity of complement activation. This is supported by previous reports showing ∼30% residual binding of C1q and the consequent ability of aglycosylated MAbs to lyse sensitized erythrocytes at higher concentrations (41, 42). Our data showed an ∼10-fold reduction in bactericidal effect in vitro and a 2- to 10-fold higher MAb dose required for in vivo protection with aglycosylated MAbs, which showed good correlation with the previous results. Nevertheless, other studies showed a complete loss of C1q binding for aglycosylated MAbs (43). We hypothesize that the level of abundance of the different target antigens may at least partially explain these seemingly contradictory results. Nevertheless, we cannot rule out that the efficacy of a large amount of aglycosylated MAbs originates from involvement of other complement pathways or via other modes of action in vivo (e.g., agglutination or endotoxin neutralization). Further animal experiments (in murine and nonmurine models) and in vitro assays are needed to clarify these possibilities. Still, the principal mode of action for protection by the O25b-specific MAbs described in this study seemed to be the antibody-dependent complement-mediated bactericidal effect that was confirmed by in vitro experiments.
We considered it important to test O25b-specific MAbs against bacteria grown under in vivo-like conditions. Investigating bacterial pathogens cultured in common laboratory media may result in observations that are not directly relevant to the clinic because of the differences in the types of bacterial antigens expressed in situ under different environmental conditions. Recently, a transcriptome analysis of E. coli grown in serum versus that grown in LB broth revealed that the envelope substantially realigned when cultured in serum (33, 34). Importantly, while the accessibility of O25b antigens was lower when grown in serum (as shown by flow cytometry) due to the abundance of LPS, avid binding of MAbs to this target may still mediate a bactericidal effect. In the mouse protection studies, as little as 100 μg of the MAbs was shown to be highly protective against a challenge with 3 × 108 CFU of serum-grown mid-log-phase E. coli cells (and even lower doses against LB broth-grown bacteria). This MAb dose corresponded to 5 mg/kg, a relatively low concentration compared to other anti-infective MAbs tested in preclinical experiments (37, 38) or clinical trials (35, 36). Furthermore, in human bacteremia, the live bacterial numbers rarely exceeded 103 to 104 CFU/ml of blood (44).
Since there are ∼180 structurally and hence antigenically different O types of E. coli, the O25b-specific MAbs target only those E. coli cells that express this particular LPS antigen. However, the overrepresentation of isolates belonging to the ST131-O25b:H4 clonal group among MDR extraintestinal infections still justifies development of a MAb directed against this target. It was demonstrated that within the ST131 lineage, all strains that expressed the O25b antigen, irrespective of capsular and pulsotype, were bound by the tested MAbs.
Given the targeted specificity, a companion diagnostic tool that can rapidly identify infections by representatives of this clone would present a significant clinical advantage and cost benefit. Recently, we published a prototype of a bead-based agglutination assay that, with further development, may be appropriate for this purpose (30). Since this highly specific and sensitive diagnostic tool also utilizes O25b-specific MAbs, it would identify only isolates that in fact express the O25b antigen (in contrast to genotyping) and so would reliably identify patients who might benefit from passive immunotherapy with O25b-specific MAbs.
We envision that MAbs may provide a therapeutic alternative for strains resistant to available antibiotics. Although carbapenem-resistant E. coli strains are currently not prevalent, this may rapidly change, as exemplified by the related pathogen Klebsiella pneumoniae. On the other hand, LPS-specific MAbs may exert endotoxin-neutralizing potential and hence complement antibiotic therapy. Such a synergistic mode of action may be desirable given the concerns with LPS release associated with some antibiotics (45, 46). Experiments aiming to show additive and/or synergistic effects of antibiotics and MAbs are ongoing.
Supplementary Material
ACKNOWLEDGMENTS
We thank Katharina Havlicek and Manuel Zerbs for excellent technical assistance and Fraser Leslie for critical reading of the manuscript.
E. coli strains 81009 and 3O were kindly provided by Agnes Sonnevend and Aranzazu Valverde, respectively. ST131 strains covering different pulsotypes were kindly provided by Gisele Peirano and Johann Pitout (University of Calgary, Canada).
We are employees of Arsanis Biosciences GmbH and hold shares in the company.
The research work was substantially supported by the General Programme of the Austrian Research Promotion Agency (FFG).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04494-14.
REFERENCES
- 1.Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 2.Da Silva GJ, Mendonca N. 2012. Association between antimicrobial resistance and virulence in Escherichia coli. Virulence 3:18–28. doi: 10.4161/viru.3.1.18382. [DOI] [PubMed] [Google Scholar]
- 3.Pitout JD. 2012. Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front Microbiol 3:9. doi: 10.3389/fmicb.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Canica MM, Park YJ, Lavigne JP, Pitout J, Johnson JR. 2008. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J Antimicrob Chemother 61:273–281. doi: 10.1093/jac/dkm464. [DOI] [PubMed] [Google Scholar]
- 5.Coque TM, Novais A, Carattoli A, Poirel L, Pitout J, Peixe L, Baquero F, Canton R, Nordmann P. 2008. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum beta-lactamase CTX-M-15. Emerg Infect Dis 14:195–200. doi: 10.3201/eid1402.070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peirano G, Pitout JD. 2010. Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: the worldwide emergence of clone ST131 O25:H4. Int J Antimicrob Agents 35:316–321. doi: 10.1016/j.ijantimicag.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Rogers BA, Sidjabat HE, Paterson DL. 2011. Escherichia coli O25b-ST131: a pandemic, multiresistant, community-associated strain. J Antimicrob Chemother 66:1–14. doi: 10.1093/jac/dkq415. [DOI] [PubMed] [Google Scholar]
- 8.Woodford N, Turton JF, Livermore DM. 2011. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 9.Nicolas-Chanoine MH, Bertrand X, Madec JY. 2014. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev 27:543–574. doi: 10.1128/CMR.00125-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banerjee R, Strahilevitz J, Johnson JR, Nagwekar PP, Schora DM, Shevrin I, Du H, Peterson LR, Robicsek A. 2013. Predictors and molecular epidemiology of community-onset extended-spectrum beta-lactamase-producing Escherichia coli infection in a Midwestern community. Infect Control Hosp Epidemiol 34:947–953. doi: 10.1086/671725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams-Sapper S, Diep BA, Perdreau-Remington F, Riley LW. 2013. Clonal composition and community clustering of drug-susceptible and -resistant Escherichia coli isolates from bloodstream infections. Antimicrob Agents Chemother 57:490–497. doi: 10.1128/AAC.01025-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207:919–928. doi: 10.1093/infdis/jis933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. 2010. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin Infect Dis 51:286–294. doi: 10.1086/653932. [DOI] [PubMed] [Google Scholar]
- 14.Qureshi ZA, Doi Y. 2014. Escherichia coli sequence type 131: epidemiology and challenges in treatment. Expert Rev Anti Infect Ther 12:597–609. doi: 10.1586/14787210.2014.899901. [DOI] [PubMed] [Google Scholar]
- 15.Bonnin RA, Poirel L, Carattoli A, Nordmann P. 2012. Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLoS One 7:e34752. doi: 10.1371/journal.pone.0034752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimou V, Dhanji H, Pike R, Livermore DM, Woodford N. 2012. Characterization of Enterobacteriaceae producing OXA-48-like carbapenemases in the UK. J Antimicrob Chemother 67:1660–1665. doi: 10.1093/jac/dks124. [DOI] [PubMed] [Google Scholar]
- 17.Morris D, Boyle F, Ludden C, Condon I, Hale J, O'Connell N, Power L, Boo TW, Dhanji H, Lavallee C, Woodford N, Cormican M. 2011. Production of KPC-2 carbapenemase by an Escherichia coli clinical isolate belonging to the international ST131 clone. Antimicrob Agents Chemother 55:4935–4936. doi: 10.1128/AAC.05127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peirano G, Bradford PA, Kazmierczak KM, Badal RE, Hackel M, Hoban DJ, Pitout JD. 2014. Global incidence of carbapenemase-producing Escherichia coli ST131. Emerg Infect Dis 20:1928–1931. doi: 10.3201/eid2011.141388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coates AR, Halls G, Hu Y. 2011. Novel classes of antibiotics or more of the same? Br J Pharmacol 163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross AS. 2014. Anti-endotoxin vaccines: back to the future. Virulence 5:219–225. doi: 10.4161/viru.25965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagy G, Pal T. 2008. Lipopolysaccharide: a tool and target in enterobacterial vaccine development. Biol Chem 389:513–520. doi: 10.1515/BC.2008.056. [DOI] [PubMed] [Google Scholar]
- 22.Overbeek BP, Veringa EM. 1991. Role of antibodies and antibiotics in aerobic gram-negative septicemia: possible synergism between antimicrobial treatment and immunotherapy. Rev Infect Dis 13:751–760. doi: 10.1093/clinids/13.4.751. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman WD, Pollack M, Banks SM, Koev LA, Solomon MA, Danner RL, Koles N, Guelde G, Yatsiv I, Mouginis T. 1994. Distinct functional activities in canine septic shock of monoclonal antibodies specific for the O polysaccharide and core regions of Escherichia coli lipopolysaccharide. J Infect Dis 169:553–561. doi: 10.1093/infdis/169.3.553. [DOI] [PubMed] [Google Scholar]
- 24.Bailat S, Heumann D, Le RD, Baumgartner JD, Rietschel ET, Glauser MP, Di PF. 1997. Similarities and disparities between core-specific and O-side-chain-specific antilipopolysaccharide monoclonal antibodies in models of endotoxemia and bacteremia in mice. Infect Immun 65:811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szijarto V, Pal T, Nagy G, Nagy E, Ghazawi A, al-Haj M, El KS, Sonnevend A. 2012. The rapidly emerging ESBL-producing Escherichia coli O25-ST131 clone carries LPS core synthesis genes of the K-12 type. FEMS Microbiol Lett 332:131–136. doi: 10.1111/j.1574-6968.2012.02585.x. [DOI] [PubMed] [Google Scholar]
- 26.Novais A, Viana D, Baquero F, Martinez-Botas J, Canton R, Coque TM. 2012. Contribution of IncFII and broad-host IncA/C and IncN plasmids to the local expansion and diversification of phylogroup B2 Escherichia coli ST131 clones carrying blaCTX-M-15 and qnrS1 genes. Antimicrob Agents Chemother 56:2763–2766. doi: 10.1128/AAC.06001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peirano G, van der Bij AK, Freeman JL, Poirel L, Nordmann P, Costello M, Tchesnokova VL, Pitout JD. 2014. Characteristics of Escherichia coli sequence type 131 isolates that produce extended-spectrum beta-lactamases: global distribution of the H30-Rx sublineage. Antimicrob Agents Chemother 58:3762–3767. doi: 10.1128/AAC.02428-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerca N, Maira-Litran T, Jefferson KK, Grout M, Goldmann DA, Pier GB. 2007. Protection against Escherichia coli infection by antibody to the Staphylococcus aureus poly-N-acetylglucosamine surface polysaccharide. Proc Natl Acad Sci U S A 104:7528–7533. doi: 10.1073/pnas.0700630104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szijarto V, Lukasiewicz J, Gozdziewicz TK, Magyarics Z, Nagy E, Nagy G. 2014. Diagnostic potential of monoclonal antibodies specific to the unique O-antigen of multi-drug resistant epidemic E. coli clone ST131-O25b:H4. Clin Vaccine Immunol 21:930–939. doi: 10.1128/CVI.00685-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerstenbruch S, Brooks CL, Kosma P, Brade L, Mackenzie CR, Evans SV, Brade H, Muller-Loennies S. 2010. Analysis of cross-reactive and specific anti-carbohydrate antibodies against lipopolysaccharide from Chlamydophila psittaci. Glycobiology 20:461–472. doi: 10.1093/glycob/cwp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller-Loennies S, Brade L, Mackenzie CR, Di Padova FE, Brade H. 2003. Identification of a cross-reactive epitope widely present in lipopolysaccharide from enterobacteria and recognized by the cross-protective monoclonal antibody WN1 222-5. J Biol Chem 278:25618–25627. doi: 10.1074/jbc.M302904200. [DOI] [PubMed] [Google Scholar]
- 33.Huja S, Oren Y, Biran D, Meyer S, Dobrindt U, Bernhard J, Becher D, Hecker M, Sorek R, Ron EZ. 2014. Fur is the master regulator of the extraintestinal pathogenic Escherichia coli response to serum. mBio 5:e01460-14. doi: 10.1128/mBio.01460-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miajlovic H, Cooke NM, Moran GP, Rogers TR, Smith SG. 2014. Response of extraintestinal pathogenic Escherichia coli to human serum reveals a protective role for Rcs-regulated exopolysaccharide colanic acid. Infect Immun 82:298–305. doi: 10.1128/IAI.00800-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kummerfeldt CE. 2014. Raxibacumab: potential role in the treatment of inhalational anthrax. Infect Drug Resist 7:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott LJ, Lamb HM. 1999. Palivizumab. Drugs 58:305–311. doi: 10.2165/00003495-199958020-00009. [DOI] [PubMed] [Google Scholar]
- 37.Xu ZQ, Flavin MT, Flavin J. 2014. Combating multidrug-resistant Gram-negative bacterial infections. Expert Opin Investig Drugs 23:163–182. doi: 10.1517/13543784.2014.848853. [DOI] [PubMed] [Google Scholar]
- 38.Oleksiewicz MB, Nagy G, Nagy E. 2012. Anti-bacterial monoclonal antibodies: back to the future? Arch Biochem Biophys 526:124–131. doi: 10.1016/j.abb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Massironi SM, Arslanian C, Carneiro-Sampaio MM, Pontes GN. 2011. Minimal concentration of human IgM and IgG antibodies necessary to protect mice from challenges with live O6 Escherichia coli. FEMS Immunol Med Microbiol 63:193–201. doi: 10.1111/j.1574-695X.2011.00841.x. [DOI] [PubMed] [Google Scholar]
- 40.Dunn DL, Bogard WC Jr, Cerra FB. 1985. Efficacy of type-specific and cross-reactive murine monoclonal antibodies directed against endotoxin during experimental sepsis. Surgery 98:283–290. [PubMed] [Google Scholar]
- 41.Leatherbarrow RJ, Rademacher TW, Dwek RA, Woof JM, Clark A, Burton DR, Richardson N, Feinstein A. 1985. Effector functions of a monoclonal aglycosylated mouse IgG2a: binding and activation of complement component C1 and interaction with human monocyte Fc receptor. Mol Immunol 22:407–415. doi: 10.1016/0161-5890(85)90125-7. [DOI] [PubMed] [Google Scholar]
- 42.Lund J, Takahashi N, Pound JD, Goodall M, Jefferis R. 1996. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J Immunol 157:4963–4969. [PubMed] [Google Scholar]
- 43.Tao MH, Morrison SL. 1989. Studies of aglycosylated chimeric mouse-human IgG: role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol 143:2595–2601. [PubMed] [Google Scholar]
- 44.Cross AS, Opal SM, Sadoff JC, Gemski P. 1993. Choice of bacteria in animal models of sepsis. Infect Immun 61:2741–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrison DC, Bucklin SE. 1996. Evidence for antibiotic-mediated endotoxin release as a contributing factor to lethality in experimental gram-negative sepsis. Scand J Infect Dis Suppl 101:3–8. [PubMed] [Google Scholar]
- 46.Holzheimer RG. 2001. Antibiotic induced endotoxin release and clinical sepsis: a review. J Chemother 13(Spec no 1):159–172. doi: 10.1179/joc.2001.13.Supplement-2.159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.