

Abstract
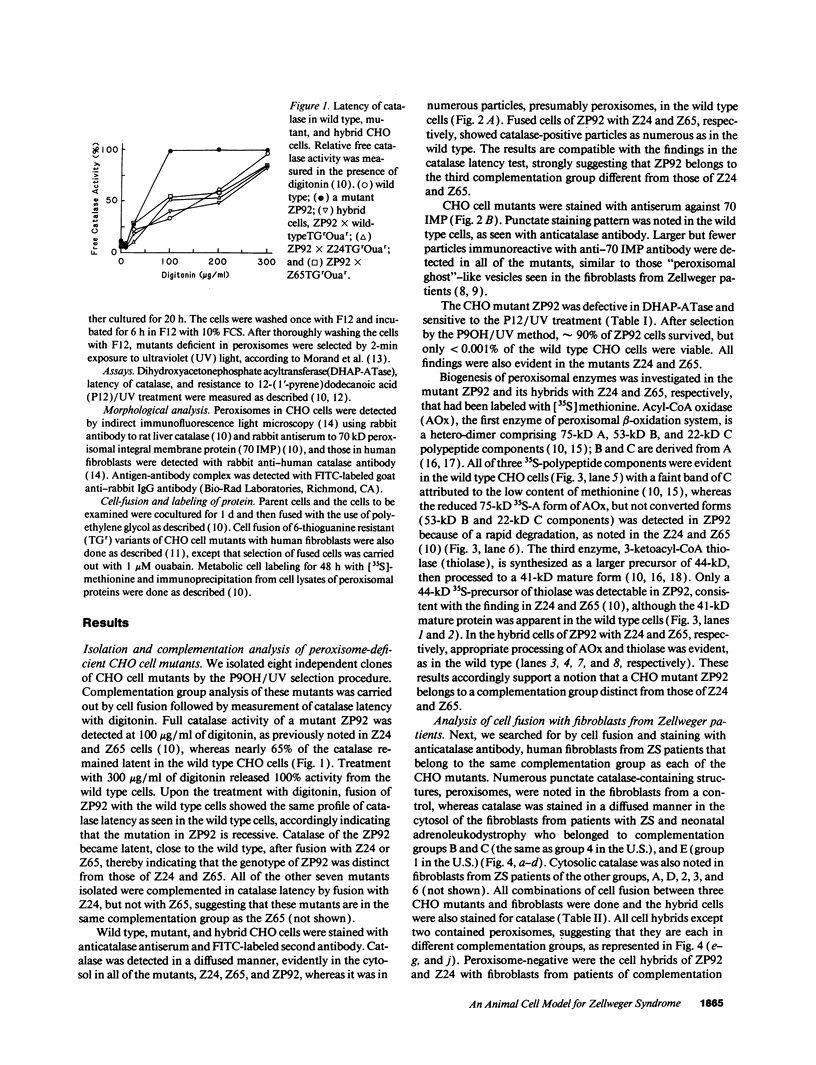
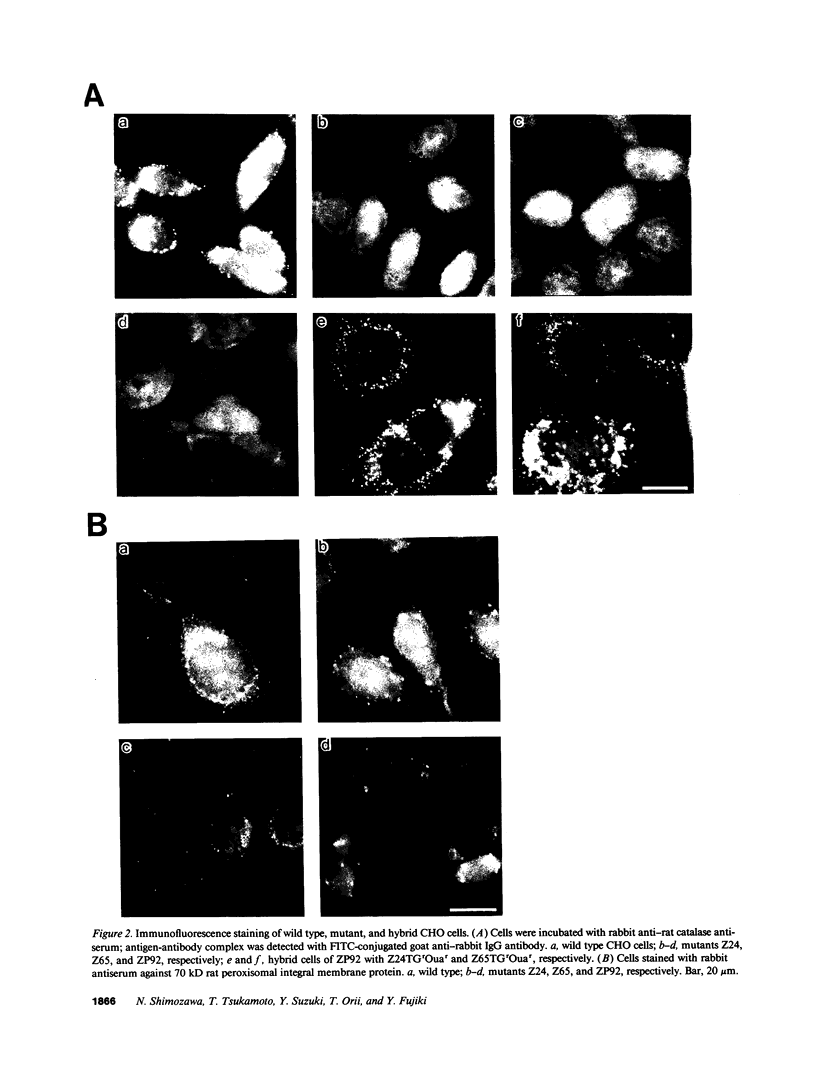
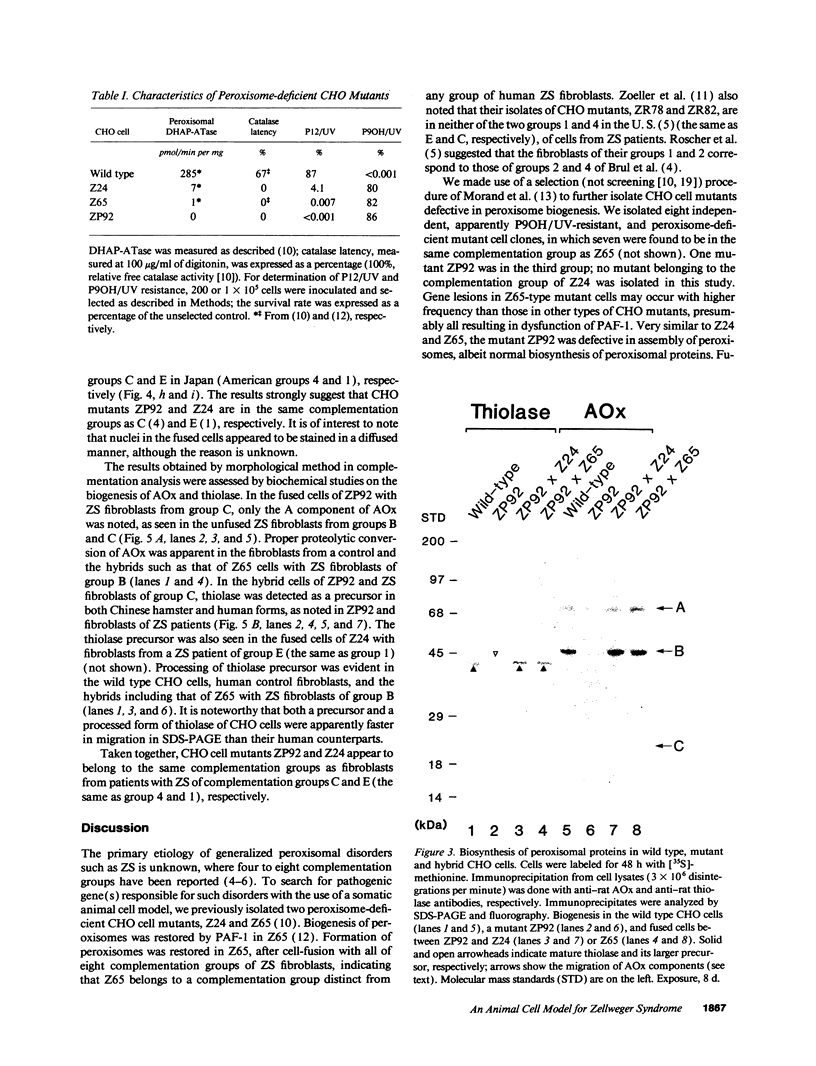
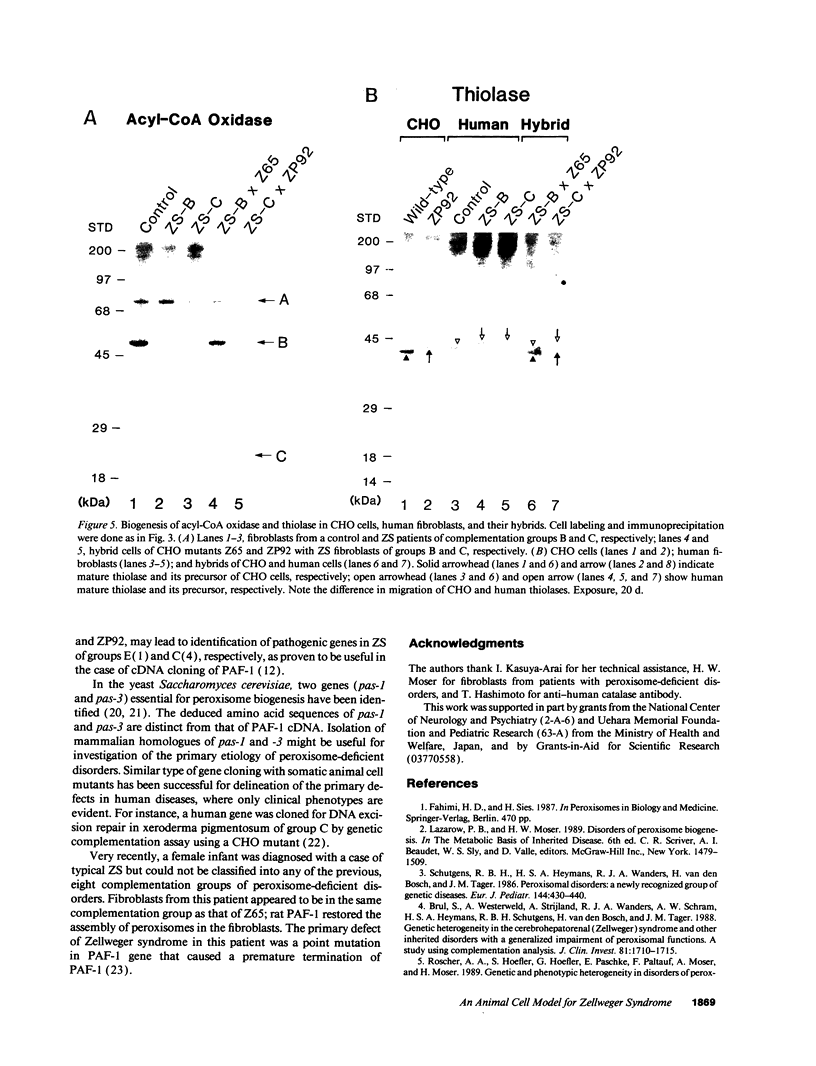
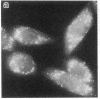
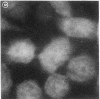
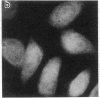
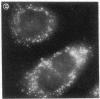
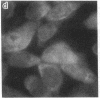
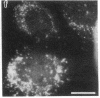
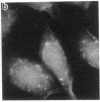
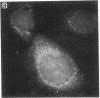
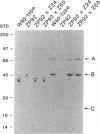
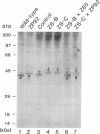
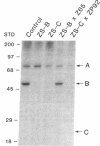
Generalized peroxisome-deficient disorders including cerebro-hepato-renal Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease are autosomal recessive diseases, where catalase-containing particles (peroxisomes) are morphologically absent. We previously isolated two Chinese hamster ovary (CHO) cell mutants (Z24 and Z65) that resemble the fibroblasts from patients with such diseases, in their defective peroxisome assembly (Tsukamoto, T., S. Yokota, and Y. Fujiki. 1990. J. Cell Biol. 110:651-660). Here we report isolation by the P9OH/UV method of a peroxisome-deficient CHO mutant, ZP92, of the third complementation group distinct from those of Z24 and Z65. Peroxisomal membrane ghosts were noted by immunochemical staining in all of the CHO mutants. Complementation analysis by cell fusion of the CHO mutants with cultured fibroblasts from patients with generalized peroxisomal disorders revealed that two CHO mutants (Z24 and ZP92) represent the human complementation groups, E (the same as group 1 in the U.S.) and C (the same as group 4), respectively. These CHO cell mutants are an apparently relevant animal cell model for studies on the molecular bases and primary defects of human peroxisome-deficient diseases.
Full text
PDF


Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Brul S., Westerveld A., Strijland A., Wanders R. J., Schram A. W., Heymans H. S., Schutgens R. B., van den Bosch H., Tager J. M. Genetic heterogeneity in the cerebrohepatorenal (Zellweger) syndrome and other inherited disorders with a generalized impairment of peroxisomal functions. A study using complementation analysis. J Clin Invest. 1988 Jun;81(6):1710–1715. doi: 10.1172/JCI113510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R., Wiebel F. F., Flessau A., Rytka J., Beyer A., Fröhlich K. U., Kunau W. H. PAS1, a yeast gene required for peroxisome biogenesis, encodes a member of a novel family of putative ATPases. Cell. 1991 Feb 8;64(3):499–510. doi: 10.1016/0092-8674(91)90234-p. [DOI] [PubMed] [Google Scholar]
- Fujiki Y., Rachubinski R. A., Mortensen R. M., Lazarow P. B. Synthesis of 3-ketoacyl-CoA thiolase of rat liver peroxisomes on free polyribosomes as a larger precursor. Induction of thiolase mRNA activity by clofibrate. Biochem J. 1985 Mar 15;226(3):697–704. doi: 10.1042/bj2260697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfischer S., Moore C. L., Johnson A. B., Spiro A. J., Valsamis M. P., Wisniewski H. K., Ritch R. H., Norton W. T., Rapin I., Gartner L. M. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science. 1973 Oct 5;182(4107):62–64. doi: 10.1126/science.182.4107.62. [DOI] [PubMed] [Google Scholar]
- Höhfeld J., Veenhuis M., Kunau W. H. PAS3, a Saccharomyces cerevisiae gene encoding a peroxisomal integral membrane protein essential for peroxisome biogenesis. J Cell Biol. 1991 Sep;114(6):1167–1178. doi: 10.1083/jcb.114.6.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S., Mori M., Takiguchi M., Tatibana M., Furuta S., Miyazawa S., Hashimoto T. Biosynthesis and intracellular transport of enzymes of peroxisomal beta-oxidation. J Biol Chem. 1984 May 25;259(10):6397–6402. [PubMed] [Google Scholar]
- Miyazawa S., Hayashi H., Hijikata M., Ishii N., Furuta S., Kagamiyama H., Osumi T., Hashimoto T. Complete nucleotide sequence of cDNA and predicted amino acid sequence of rat acyl-CoA oxidase. J Biol Chem. 1987 Jun 15;262(17):8131–8137. [PubMed] [Google Scholar]
- Miyazawa S., Osumi T., Hashimoto T., Ohno K., Miura S., Fujiki Y. Peroxisome targeting signal of rat liver acyl-coenzyme A oxidase resides at the carboxy terminus. Mol Cell Biol. 1989 Jan;9(1):83–91. doi: 10.1128/mcb.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand O. H., Allen L. A., Zoeller R. A., Raetz C. R. A rapid selection for animal cell mutants with defective peroxisomes. Biochim Biophys Acta. 1990 May 16;1034(2):132–141. doi: 10.1016/0304-4165(90)90066-6. [DOI] [PubMed] [Google Scholar]
- Roscher A. A., Hoefler S., Hoefler G., Paschke E., Paltauf F., Moser A., Moser H. Genetic and phenotypic heterogeneity in disorders of peroxisome biogenesis--a complementation study involving cell lines from 19 patients. Pediatr Res. 1989 Jul;26(1):67–72. doi: 10.1203/00006450-198907000-00019. [DOI] [PubMed] [Google Scholar]
- Santos M. J., Imanaka T., Shio H., Small G. M., Lazarow P. B. Peroxisomal membrane ghosts in Zellweger syndrome--aberrant organelle assembly. Science. 1988 Mar 25;239(4847):1536–1538. doi: 10.1126/science.3281254. [DOI] [PubMed] [Google Scholar]
- Schutgens R. B., Heymans H. S., Wanders R. J., van den Bosch H., Tager J. M. Peroxisomal disorders: a newly recognised group of genetic diseases. Eur J Pediatr. 1986 Feb;144(5):430–440. doi: 10.1007/BF00441734. [DOI] [PubMed] [Google Scholar]
- Shimozawa N., Tsukamoto T., Suzuki Y., Orii T., Shirayoshi Y., Mori T., Fujiki Y. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science. 1992 Feb 28;255(5048):1132–1134. doi: 10.1126/science.1546315. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Yamaguchi S., Orii T., Tsuneoka M., Tashiro Y. Nonspecific lipid transfer protein (sterol carrier protein-2) defective in patients with deficient peroxisomes. Cell Struct Funct. 1990 Oct;15(5):301–308. doi: 10.1247/csf.15.301. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T., Miura S., Fujiki Y. Restoration by a 35K membrane protein of peroxisome assembly in a peroxisome-deficient mammalian cell mutant. Nature. 1991 Mar 7;350(6313):77–81. doi: 10.1038/350077a0. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T., Yokota S., Fujiki Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in assembly of peroxisomes. J Cell Biol. 1990 Mar;110(3):651–660. doi: 10.1083/jcb.110.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G., van Ham R. C., Vermeulen W., Bootsma D., van der Eb A. J., Hoeijmakers J. H. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell. 1990 Aug 24;62(4):777–791. doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- Wiemer E. A., Brul S., Just W. W., Van Driel R., Brouwer-Kelder E., Van Den Berg M., Weijers P. J., Schutgens R. B., Van Den Bosch H., Schram A. Presence of peroxisomal membrane proteins in liver and fibroblasts from patients with the Zellweger syndrome and related disorders: evidence for the existence of peroxisomal ghosts. Eur J Cell Biol. 1989 Dec;50(2):407–417. [PubMed] [Google Scholar]
- Yajima S., Suzuki Y., Shimozawa N., Yamaguchi S., Orii T., Fujiki Y., Osumi T., Hashimoto T., Moser H. W. Complementation study of peroxisome-deficient disorders by immunofluorescence staining and characterization of fused cells. Hum Genet. 1992 Mar;88(5):491–499. doi: 10.1007/BF00219334. [DOI] [PubMed] [Google Scholar]
- Zoeller R. A., Allen L. A., Santos M. J., Lazarow P. B., Hashimoto T., Tartakoff A. M., Raetz C. R. Chinese hamster ovary cell mutants defective in peroxisome biogenesis. Comparison to Zellweger syndrome. J Biol Chem. 1989 Dec 25;264(36):21872–21878. [PubMed] [Google Scholar]
- Zoeller R. A., Raetz C. R. Isolation of animal cell mutants deficient in plasmalogen biosynthesis and peroxisome assembly. Proc Natl Acad Sci U S A. 1986 Jul;83(14):5170–5174. doi: 10.1073/pnas.83.14.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]