

Abstract
CPAF (chlamydial protease-like activity factor), a Chlamydia serine protease, is activated via proximity-induced intermolecular dimerization that triggers processing and removal of an inhibitory peptide occupying the CPAF substrate-binding groove. An active CPAF is a homodimer of two identical intramolecular heterodimers, each consisting of 29-kDa N-terminal and 35-kDa C-terminal fragments. However, critical residues for CPAF intermolecular dimerization, catalytic activity, and processing were defined in cell-free systems. Complementation of a CPAF-deficient chlamydial organism with a plasmid-encoded CPAF has enabled us to characterize CPAF during infection. The transformants expressing CPAF mutated at intermolecular dimerization, catalytic, or cleavage residues still produced active CPAF, although at a lower efficiency, indicating that CPAF can tolerate more mutations inside Chlamydia-infected cells than in cell-free systems. Only by simultaneously mutating both intermolecular dimerization and catalytic residues was CPAF activation completely blocked during infection, both indicating the importance of the critical residues identified in the cell-free systems and exploring the limit of CPAF's tolerance for mutations in the intracellular environment. We further found that active CPAF was always detected in the host cell cytoplasm while nonactive CPAF was restricted to within the chlamydial inclusions, regardless of how the infected cell samples were treated. Thus, CPAF translocation into the host cell cytoplasm correlates with CPAF enzymatic activity and is not altered by sample treatment conditions. These observations have provided new evidence for CPAF activation and translocation, which should encourage continued investigation of CPAF in chlamydial pathogenesis.
INTRODUCTION
During the investigation of chlamydial interactions with host cells, a unique protease-like activity was detected in the cytoplasmic fraction of Chlamydia-infected cells (1, 2). The chlamydial protease-like activity factor, designated CPAF, was purified as N-terminal 29-kDa (CPAFn) and C-terminal 35-kDa (CPAFc) fragments (3). These two fragments formed intramolecular heterodimers, which were required for CPAF to acquire proteolytic activity (4, 5). Further crystal structure (6) and biochemical (6–8) analyses revealed that CPAF was activated via proximity-dependent intermolecular dimerization that triggered sequential processing of an internal inhibitory peptide covering the region between residues R243 and S283. The inhibitory peptide, occupying the substrate-binding groove of CPAF, is eventually removed by sequential processing, which generates intermediate fragments of CPAF (CPAFi). The first cleavage occurs between residues M242 and R243, which permits the subsequent cis-cleavage between M264-V265 and S283-G284. At the same time, the organization of the catalytic triad, consisting of residues E558, S499, and H105, is optimized. The first cleavage is triggered by intermolecular dimerization. The residues F45, V52, and P534 are critical for the dimerization. Substitutional mutations of the residues required for intermolecular dimerization, catalytic activity, or processing have resulted in significant reduction or complete blockade of CPAF activation in cell-free assays (5, 6). Despite the detailed structural and biochemical characterization of CPAF, how CPAF is activated and regulated in Chlamydia-infected cells remains unknown.
CPAF was predominantly detected in the cytosolic fraction/portion of the infected cells (3). However, how CPAF gets into the host cell cytoplasm remains unknown. Nevertheless, CPAF is predicted to possess a secretion leader sequence at its N terminus, and this leader sequence is both processed from mature CPAF isolated from Chlamydia-infected cells and functional in directing bacterial protein secretion crossing the inner membrane in a sec-dependent manner (9). A chlamydial mutant deficient in type II secretion also significantly restricted CPAF secretion (10), suggesting that CPAF may get out of the outer membrane via the type II secretion apparatus, which, however, only sends CPAF into the lumen of the inclusion. Due to lack of knowledge about how CPAF reaches the host cell cytoplasm, some have questioned the validity of the detection of CPAF in the host cell cytoplasm (11) despite the fact that CPAF has been localized to the host cell cytosol consistently (3, 12–17). Interestingly, the cytosolically localized CPAF is not colocalized with the secreted Pgp3 protein (18, 19), which may suggest that CPAF and Pgp3, although both are secreted into the host cell cytoplasm, may have different host targets. CPAF has been implicated functionally in the interaction of a number of host cell substrate targets important for chlamydial pathogenesis (20). However, it was later shown that these host substrates were the result of an imprecise method that failed to inactivate the protease during sample harvest (21), which triggered discussions on the authenticity of CPAF's function (22–24).
CPAF is a highly conserved protease common to all Chlamydiaceae (25) whose secretion and cytosolic location need further investigation to definitively elucidate its function in chlamydial pathogenesis. The recent generation of CPAF-deficient chlamydial organisms has provided a genetic basis for investigating the functionality of CPAF (10). We successfully complemented a CPAF-deficient mutant Chlamydia trachomatis serovar L2 organism, designated RST17 or L2-17, with a plasmid-encoded CPAF, which has allowed us to further characterize CPAF in the context of chlamydial infection. The transformed L2-17 organisms expressing a plasmid-encoded CPAF mutated at one or multiple intermolecular dimerization contact, catalytic, or cleavage site residues still produced significant levels of active CPAF, while the same mutations in CPAF resulted in loss of activity in cell-free systems (5, 6). Thus, CPAF tolerated more mutations inside Chlamydia-infected cells than in cell-free systems. Nevertheless, a CPAF molecule mutated at both intermolecular dimerization contact and catalytic residues was completely blocked from activation during infection. Furthermore, the active CPAF was always detected in the host cell cytoplasm while nonactive CPAF was restricted to within the chlamydial inclusion regardless of how the infected cell samples were treated. These observations together demonstrated that CPAF translocation into the host cell cytoplasm correlates well with CPAF enzymatic activity, and CPAF remains a promising target for understanding chlamydial pathogenesis.
MATERIALS AND METHODS
Cell culture and chlamydial infection.
HeLa cells (human cervical epithelial carcinoma cells; ATCC CCL2) were grown and maintained in medium consisting of Dulbecco modified Eagle medium (DMEM) (Sigma, St. Louis, MO) and 10% fetal calf serum (FCS) (Gemini Bio-Products, West Sacramento, CA) in a humidified incubator in the presence of 5% CO2. The wild-type C. trachomatis L2 (L2wt) organisms (L2/LGV-434/Bu) were purchased from the ATCC, while the CPAF-deficient L2 organisms, designated RST17 or L2-17, and the corresponding isogenic CPAF-competent control, RST5 or L2-5, were kindly provided by Raphael Valdivia from Duke University (10). The various L2-17 transformants were generated as described below. All chlamydial organisms were propagated, purified, aliquoted, and stored as described previously (26). For infection, cells grown in 24-well or 6-well plates with or without coverslips were inoculated with chlamydial organisms as described previously (27). The infected cultures were processed at different time points after infection for either Western blot or immunofluorescence assays as described below.
Generation of plasmids coding for wild-type (CPAF-wt) or mutant CPAF.
To construct a CPAF expression plasmid, we first generated a DNA fragment consisting of the pgp4 promoter sequence (97 nucleotides upstream of the pgp4 gene [28]) and the cpaF gene (29), both from C. trachomatis serovar L2, and the transcriptional termination signal sequence from ct579 of C. trachomatis serovar D. The ct579 terminator is a Rho-independent terminator (30) consisting of 43 nucleotides, starting at the 43rd nucleotide downstream of the ct579 stop codon (31). The three DNA fragments were linked together by using PCR (AccuPrime Pfx SuperMix; Life Technologies, Grand Island, NY). The cpaF-containing DNA fragment, functioning as an independent operon, was inserted between the Escherichia coli origin of replication and the gfp gene of the pGFP::SW2 vector (kindly provided by Ian Clarke, University of Southampton [32]) using an In-fusion HD cloning kit (Clontech Laboratories Inc., Mountain View, CA) as described previously (26, 33). The fusion products were rescued by transformation into Stellar competent bacterial cells. The bacterial transformants were selected for ampicillin resistance on LB agar plates. Bacterial colonies were screened for green fluorescence under a fluorescence microscope. Green colonies were picked up for extraction of plasmids. The purified plasmids were subjected to DNA sequencing of the predicted fusion junction regions to confirm correct insertion of the cpaF operon. Plasmids with the correct fusion junction sequences were transformed into E. coli K-12 ER2925 (Dam-Dcm strain; New England BioLabs, Ipswich, MA) for amplification. The amplified plasmids were subjected to DNA sequencing of the entire plasmid. The plasmid with correct sequence (designated pGFP-CPAF-wt) was used for transforming L2-17 chlamydial organisms as described below. The pGFP-CPAF-wt plasmid was used as a template for introducing the desired mutations into the codons of the cpaF gene by incorporating the mutations into the primers used for In-fusion HD cloning (see Table S1 in the supplemental material for all the primer sequences). In order to simultaneously introduce multiple mutations into a single cpaF gene, multiple fusion fragments were produced for a single fusion. The mutated codons include those coding for the intermolecular dimerization contact residues F45, V52, and P534; the catalytic residues H105, S499, and E558; and the inhibitory peptide cleavage site residues L259 and L281 (5, 6). The sequences of all the primers used in the current study are listed in Table S1 in the supplemental material.
Chlamydial transformation.
The L2-17 organisms (10) were transformed with the various plasmids created as described above by using the transformation protocol initially developed by Wang et al. (32), but with additional modifications (26, 33). Briefly, 10 μl L2-17 organisms (1 × 107 inclusion-forming units [IFU]) and 10 μl plasmid DNA (∼7 μg) were mixed in a total volume of 200 μl CaCl2 buffer for 45 min at room temperature (RT). Freshly trypsinized HeLa cells (6 × 106 cells) resuspended in 200 μl CaCl2 buffer were added to the elementary body (EB)-plasmid mixture and incubated for another 20 min at room temperature, with occasional mixing. Each 70 μl of the final mixture was plated onto a single well of a 6-well plate, together with 1.5 ml of prewarmed DMEM plus 10% FCS. The culture plates were incubated at 37°C in 5% CO2 without cycloheximide or ampicillin for 24 h. The cultures were replenished with fresh medium containing cycloheximide (2 μg/ml) and ampicillin (5 μg/ml) and incubated for an additional 24 h. Inclusions positive for green fluorescence were identified under a fluorescence microscope and picked up for passage on a fresh monolayer of HeLa cells in the presence of ampicillin (20 μg/ml). The resultant inclusions that remained positive for green fluorescence were defined as generation 2 and were passed for additional generations to enrich green-fluorescence-positive organisms, which were finally plaque cloned, as described previously (26, 33), for further experiments. The final L2-17 transformants were L2-17-CPAFwt (expressing plasmid-encoded wild-type CPAF), L2-17-CPAF-F45A (expressing CPAF with alanine substitution for the dimerization residue phenylalanine 45), L2-17-CPAF-E558A (CPAF with alanine substitution for catalytic residue glutamic acid 558), L2-17-CPAF-FVP to A (CPAF with simultaneous alanine substitutions for dimerization contact residues F45, V52, and P534), L2-17-CPAF-HSE to A (CPAF with simultaneous alanine substitutions for catalytic residues H105, S499, and E558), L2-17-CPAF-L259E (CPAF with replacement of the inhibitory peptide cleavage site residue L259 with glutamic acid), L2-17-CPAF-L281G (CPAF with replacement of the inhibitory peptide cleavage site residue L281 with glycine), and L2-17-CPAF-EF to A (CPAF with simultaneous alanine replacements of both the catalytic residue E558 and dimerization contact residue F45 with alanine).
Western blot assays.
Western blot assays were carried out as described previously (34). Briefly, HeLa cells with chlamydial infection were solubilized in 2% SDS sample buffer and loaded onto SDS polyacrylamide gel wells. After electrophoresis, the resolved protein bands were transferred to nitrocellulose membranes for antibody detection. The primary antibodies were monoclonal antibody (MAb) 100a, able to recognize both CPAFc and full-length CPAF (CPAFf); MAb MC22, recognizing the chlamydial major outer membrane protein (MOMP) (3); and MAb M20, detecting host cell cytokeratin 8 (Sigma, St. Louis, MO). The primary antibody binding was probed with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody (Jackson ImmunoResearch) and visualized with an enhanced chemiluminescence (ECL) kit (Santa Cruz Biotechnology).
Immunofluorescence assay.
The immunofluorescence assay was carried out as described previously (3). Briefly, HeLa cells with chlamydial infection were fixed at 40 h postinfection with 4% paraformaldehyde (PF) dissolved in phosphate-buffered saline (PBS) for 1 h at RT, followed by different permeabilization conditions, including 2% saponin for 30 min to 4 h, 1% SDS for 40 min, or cold methanol for 10 min, or fixed with 10% formalin for 1 h followed by permeabilization with 0.1% Triton for 15 min, as indicated for individual experiments. All samples were colabeled for CPAF, L2 organisms, and DNA. The mouse anti-CPAFc MAb 100a plus a goat anti-mouse IgG conjugated with Cy3 (red; Jackson ImmunoResearch) were used to visualize CPAF. A rabbit anti-chlamydial-organism antibody (polyclonal antibody [PAb] 1006 [unpublished data]) plus a goat anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 488 (green; Jackson ImmunoResearch, West Grove, PA) were used to visualize chlamydial organisms. The DNA dye Hoechst 33258 (blue; Sigma) was used to visualize DNA. The fluorescence images were acquired by using an Olympus AX-70 fluorescence microscope equipped with a charge-coupled-device (CCD) camera, and the images were processed using Adobe Photoshop. Some immunolabeled cell samples were also examined using a confocal microscope to validate intracellular localization, as described previously (35).
RESULTS
CPAF with mutations of critical residues is still active during chlamydial infection.
Although CPAF has been extensively characterized using structural and biochemical approaches in cell-free systems, its proteolytic property during live infection inside Chlamydia-infected cells has not been evaluated. A CPAF-deficient C. trachomatis L2 organism, designated RST17 or L2-17, no longer expressed CPAF (Fig. 1A) (10). CPAF expression was restored by transforming the L2-17 organisms with a plasmid-borne CPAF-wt gene. The plasmid-encoded CPAF-wt was not only expressed and processed, but also translocated into the host cell cytoplasm (Fig. 1B). We then used the CPAF transformation approach to investigate the critical residues required for CPAF intermolecular dimerization, catalytic activity, and processing in Chlamydia-infected cells (Fig. 2). Compared to CPAF-wt, CPAF with alanine substitution for the intermolecular dimerization contact residue phenylalanine 45 (F45A) or the catalytic residue glutamic acid 558 (E558A) displayed more immature intermediate CPAFi and full-length CPAFf, suggesting that these critical residues are required for optimal maturation of CPAF. However, significant amounts of the mutant CPAF molecules were still processed into the mature form, CPAFc. Significant processing was also detected in cultures expressing CPAF with simultaneous alanine substitutions for the intermolecular dimerization residues F45, V52, and P534 (FVP to A) or catalytic residues H105, S499, and E558 (HSE to A) or CPAF with replacement of the inhibitory peptide cleavage site residue L259 with glutamic acid (L259E) or of L281 with glycine (L281G). Careful analyses of the CPAF processing or fragmentation patterns led us to reveal some minor differences among the mutants. Maturation by the catalytic residue mutants was less efficient, especially the HSE to A triple mutant, compared to the FVP to A triple intermolecular dimerization mutant. In addition, the two cleavage site residue mutants (L259E and L281G) displayed the least efficiency in converting into the mature CPAFc species. Nevertheless, CPAF was still significantly processed in all cultures, regardless of the types of mutation. CPAF processing and maturation must occur during infection (prior to the lysis of the infected cells), since the cell samples were harvested in 8 M urea, conditions under which no CPAF processing can take place (21, 36). We further monitored the proteolytic activity of the processed CPAF molecules by taking advantage of a previous finding that active CPAF can cleave cytokeratin 8 during sample processing in RIPA buffer (37). All the mutant CPAF molecules cleaved cytokeratin 8. These observations demonstrated that CPAF can tolerate more mutation in Chlamydia-infected cells than in cell-free systems, which was not caused by contamination with wild-type CPAF-expressing L2 organisms. This is because all inclusions in cultures infected with L2-17 transformants were positive for green fluorescent protein (GFP) under an inverted microscope prior to processing for immunofluorescence staining. More importantly, all L2 transformants were resequenced to confirm the corresponding CPAF mutations. Simultaneous substitutions for three intermolecular dimerization residues or all three catalytic residues were still insufficient to completely block CPAF activation, suggesting that the intermolecular dimerization and catalytic residues may be able to compensate for each other inside Chlamydia-infected cells.
FIG 1.
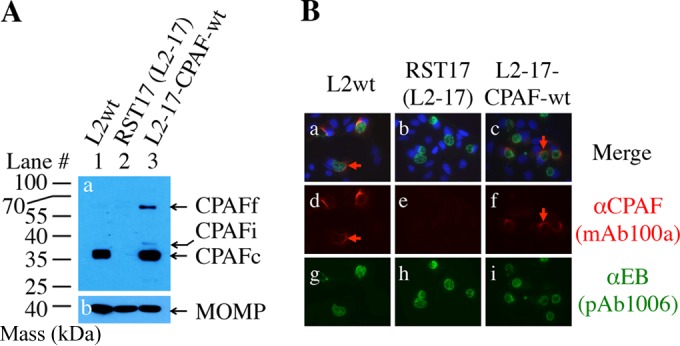
Complementation of a CPAF expression-deficient mutant with plasmid-encoded CPAF. (A) HeLa cells were infected with the following C. trachomatis L2 organisms: wild-type L2wt (lane 1), the CPAF-deficient mutant RST17 (L2-17) (lane 2), and L2-17 transformed with a plasmid that encodes wild-type CPAF (L2-17-CPAFwt) (lane 3). The cell samples were harvested at 40 h postinfection in 8 M urea to monitor CPAF processing by detecting the processed C-terminal fragment (CPAFc) with the anti-CPAF C terminus MAb 100a. Note that CPAFc was detected in cultures infected with either L2wt or L2-17-CPAFwt, but not in cultures infected with the CPAF-deficient L2-17 alone. Small amounts of both residual full-length CPAFf and intermediate CPAFi were also detected in the L2-17-CPAFwt culture. As a loading control, similar amounts of MOMP were detected in all samples (gel b; mouse anti-MOMP MAb MC22). (B) Parallel samples grown on coverslips in 24-well plates were processed for costaining of CPAF (mouse anti-CPAFc [αCPAF] MAb 100a) and chlamydial organisms (rabbit anti-chlamydial-organism PAb 1006), followed by visualization with a goat anti-mouse IgG conjugated with Cy3 (red) and goat anti-rabbit IgG conjugated with Cy2 (green), respectively. The DNA was labeled with Hoechst (blue). Note that CPAF expression and translocation were detected in cultures infected with either L2wt or L2-17-CPAFwt, but not in cultures infected with the CPAF-deficient L2-17. The red arrows indicate CPAF molecules localized outside the chlamydial inclusions.
FIG 2.
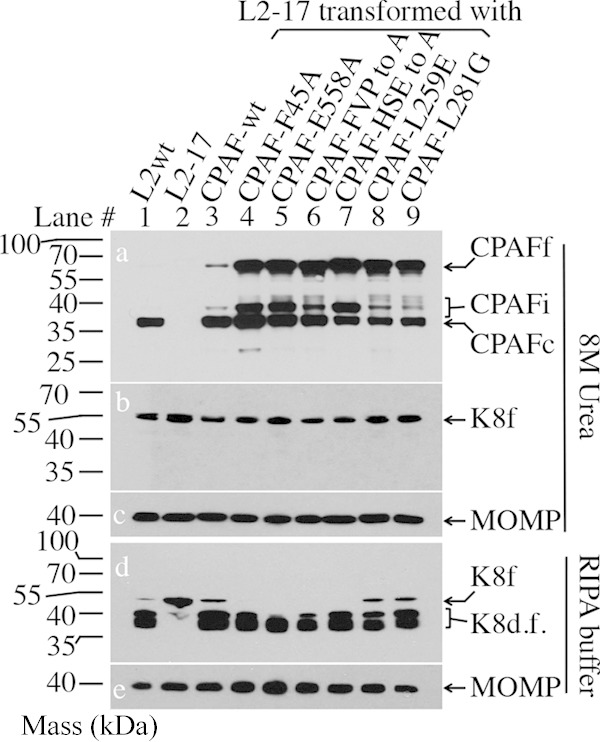
Effect of catalytic, dimerization, or cleavage site residue mutation on CPAF processing and activity during Chlamydia infection. HeLa cells were infected with the following L2 organisms: L2wt (lane 1); L2-17 (lane 2); L2-17 transformed with wild-type CPAF (CPAF-wt) (lane 3); CPAF with alanine substitution for the dimerization residue phenylalanine 45 (CPAF-F45A) (lane 4) or the catalytic residue glutamic acid 558 (CPAF-E558A) (lane 5); CPAF with simultaneous alanine substitutions for dimerization contact residues F45, V52, and P534 (CPAF-FVP to A) (lane 6) or catalytic residues H105, S499, and E558 (CPAF-HSE to A) (lane 7); or CPAF with replacement of the inhibitory peptide cleavage site residue L259 with glutamic acid (CPAF-L259E) (lane 8) or L281 with glycine (CPAF-L281G) (lane 9). The cell samples were harvested at 40 h postinfection in either 8 M urea (top) or RIPA buffer (bottom) to monitor CPAF processing (conversion of full-length CPAFf into CPAFi or CPAFc [a]) and cytokeratin 8 cleavage (conversion of full-length keratin 8 [K8f] into degradation fragments [K8d.f.] [b and d]), with MOMP as a loading control (c and e). Note that CPAF was still partially processed and that CPAF activity was detected in all cultures regardless of the CPAF mutation.
Simultaneous replacement of both catalytic and dimerization residues in the same molecule completely blocked CPAF processing and activation.
We next tested whether simultaneously replacing both a catalytic and a dimerization residue in the same CPAF molecule could prevent its activation (Fig. 3). CPAF with alanine substitutions for both the catalytic residue E588 and the dimerization contact residue F45 (CPAF-EF to A) was completely blocked from activation. No processed CPAF intermediates (CPAFi) or mature CPAFc, but only immature full-length CPAF, was detected in the culture infected with the L2-17-CPAF-EF to A organisms. The lack of CPAF processing was consistent with the lack of CPAF activity for degrading cytokeratin 8 by the double-mutant CPAF. However, triple substitutions for three catalytic or dimerization residues still allowed the CPAF mutants to maintain activity (Fig. 2). These observations together demonstrated that E558 and F45 are important in CPAF activation and suggested that catalytic activity and intermolecular dimerization may be complementary to each other for CPAF activation/activity during infection. Only when both catalytic activity and dimerization were disrupted at the same time could CPAF activation be completely blocked.
FIG 3.
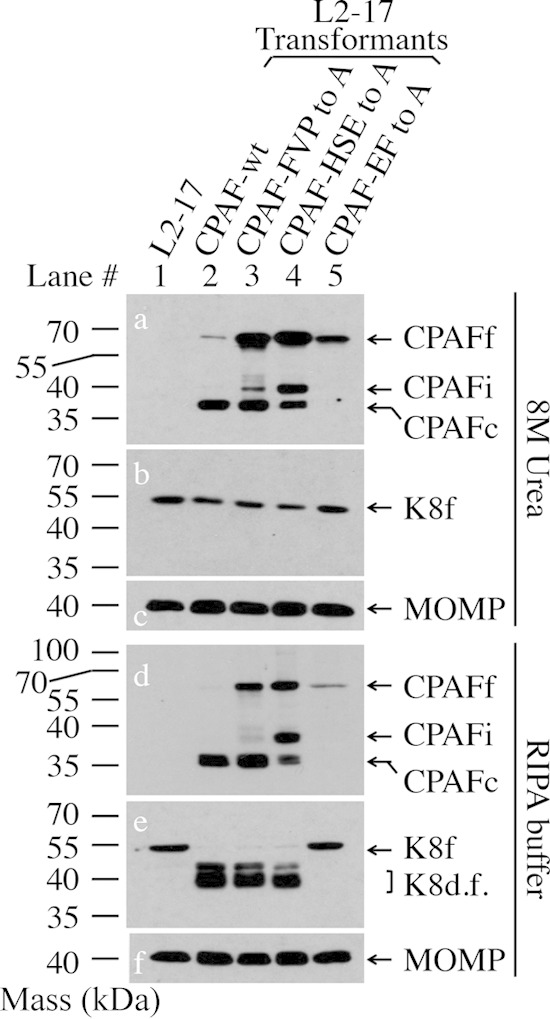
Effects of simultaneous substitutions for both catalytic and dimerization residues on CPAF processing and activity. HeLa cells were infected with L2-17 (lane 1) or L2-17 transformed with a plasmid encoding CPAF-wt (lane 2), CPAF-FVP to A (lane 3), CPAF-HSE to A (lane 4), or CPAF-EF to A (lane 5). The cell samples were harvested at 40 h postinfection in either 8 M urea (top) or RIPA buffer (bottom) for Western blotting detection of CPAF processing (a and d), keratin 8 cleavage (b and e), and MOMP (c and f). Note that CPAF processing was completely blocked and that no CPAF activity was detected in the culture infected with the L2-17-CPAF-EF to A organisms.
CPAF secretion is dependent on CPAF activity.
We also monitored the secretion patterns of the CPAF mutants under fluorescence microscopy (Fig. 4). CPAF secretion was detected in cultures infected with all but the organisms expressing the CPAF-EF to A mutants, correlating well with CPAF processing and activity. CPAF with mutations at either catalytic, intermolecular dimerization, or cleavage site residues still maintained significant processing and enzymatic activity (Fig. 2). Significant amounts of these mutant CPAF molecules were also detected in the host cytoplasm (Fig. 4). The catalytic and intermolecular dimerization CPAF-EF to A double mutant was no longer active (Fig. 3) and was not translocated into the host cell cytosol (Fig. 4). Nevertheless, a close look at the secretion patterns revealed some differences in the extents of translocation among the mutants that were detected in the host cell cytoplasm. Less complete translocation was found among the mutants with catalytic residue mutation, including the single-substitution mutant E558A and the HSE to A triple mutant, since more CPAF molecules from these mutants were retained within the inclusions. This distinct distribution pattern was further validated during the entire infection course (Fig. 5). Most of the CPAF-FVP to A intermolecular dimerization residue triple mutants were detected in the host cell cytoplasm, while a significant amount of the CPAF-HSE to A catalytic residue triple mutant was retained within the inclusions during the infection course. Despite the difference in host cell cytoplasm translocation, all CPAF molecules seemed to share similar expression time courses, with the first detection of CPAF at 12 h after infection. By 18 h after infection, CPAF translocation out of the inclusions into the host cell cytoplasm became detectable for both wild-type and mutant CPAFs, but not the CPAF-EF to A double mutant that totally lacked processing and activity (Fig. 3). This trend continued up to 48 h after infection (Fig. 5), and the distinct intracellular distribution of the CPAF-HSE to A versus the CPAF-EF to A mutants was confirmed under confocal microscopy (Fig. 6). These observations together demonstrated a strong correlation of CPAF translocation into the host cell cytoplasm with CPAF activity.
FIG 4.

Effect of CPAF alanine substitutional mutation on CPAF translocation. HeLa cells were infected with L2wt (a, k, and u); L2-17 (b, l, and v); L2-17-CPAFwt (c, m, and w); CPAF with alanine substitution for F45 (F45A) (d, n, and x) or E558 (E558A) (e, o, and y), F45, V52, and P534 (FVP to A) (f, p, and z), H105, S499, and E558 (HSE to A) (g, q, and aa), or E558 and F45 (EF to A) (j, t, and ad); or CPAF cleavage site residue substitution (L259E, h, r, and ab; L281G, i, s, and ac), as indicated. The infected cells were processed for immunofluorescence detection of CPAF (red), the L2 organism (green), and DNA (blue). Note that CPAF translocation was detected in cultures infected with all but the organisms expressing the CPAF-EF to A mutant. The red arrows indicate CPAF molecules localized outside the chlamydial inclusions.
FIG 5.
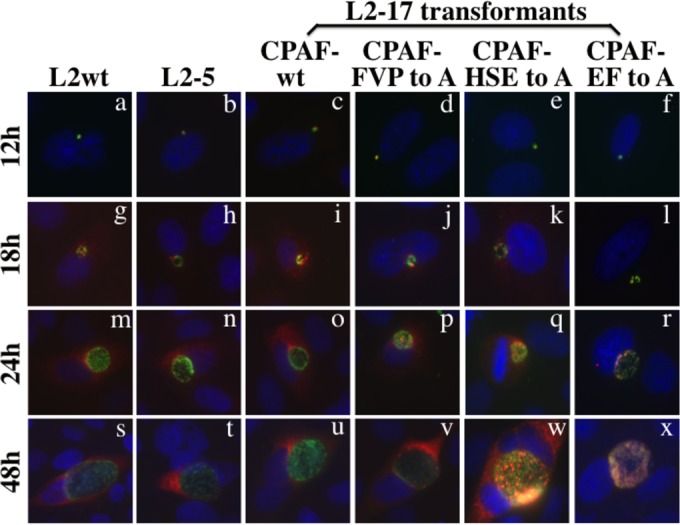
Monitoring CPAF expression and translocation during the infection course. HeLa cells were infected with L2wt (a, g, m, and s), the isogenic strain L2-5 (b, h, n, and t), L2-17-CPAFwt (c, i, o, and u), CPAF-FVP to A (d, j, p, and v), CPAF-HSE to A (e, k, q, and w), or CPAF-EF to A (f, l, r, and x), as indicated. The infected cells were processed for immunofluorescence detection of CPAF (red), the L2 organism (green), and DNA (blue) at various times after infection, as shown on the left. Note that both the chromosome- and plasmid-encoded CPAF molecules displayed similar expression time courses and that translocation of CPAF into the host cell cytoplasm was detected in cultures infected with all but the L2 organisms expressing the CPAF-EF to A mutant.
FIG 6.

Confocal microscopic imaging of CPAF expression and translocation. HeLa cells were infected with L2wt or L2-17 transformed with CPAF-wt or CPAF-EF to A, as indicated. The infected cells were processed for immunofluorescence detection of CPAF (red), the L2 organism (green), and DNA (blue) at 40 h after infection. Note that under confocal microscopy, both the chromosome- and plasmid-encoded wild-type CPAF molecules were translocated into the host cell cytoplasm, but the CPAF-EF to A mutant showed significant overlap with the intrainclusion chlamydial organisms without any significant translocation into the host cell cytoplasm (A), and these distinct intracellular distribution patterns were validated at various focal levels along the z axis (B).
CPAF secretion is not affected by the sample treatment conditions.
The availability of both secretable and nonsecretable CPAF allowed us to address concerns about the effects of sample treatment conditions on CPAF translocation (11, 24). The cultures infected with L2-17 organisms expressing either secretable or nonsecretable CPAF were treated under various conditions, and CPAF translocation was monitored under fluorescence microscopy (Fig. 7). The infected cell samples were fixed with 4% PF for 1 h at RT, followed by various permeabilization conditions, including 2% saponin for various times, 1% SDS for 10 min, and methanol for 10 min. Some samples were also fixed with formalin, followed by Triton permeabilization. We found that the secretable CPAF was always detected in the host cell cytosol without any significant overlap with the organisms, while the nonsecretable CPAF was restricted in the chlamydial inclusions regardless of the conditions used for treating the cell samples. These observations demonstrated that the cell sample processing conditions did not affect the secretability of CPAF.
FIG 7.
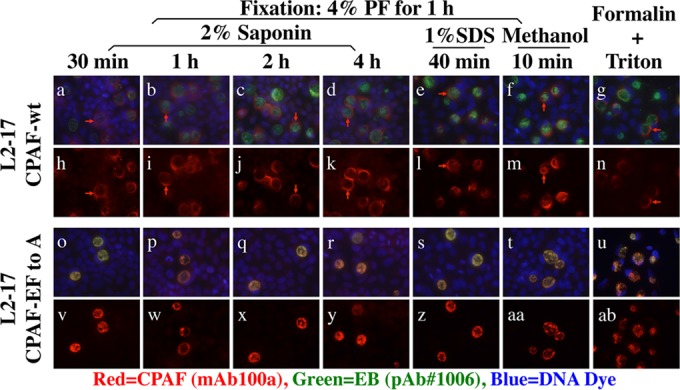
Effects of cell sample treatments on CPAF secretion. HeLa cells were infected with L2-17-CPAFwt (top two rows) or L2-17-CPAF-EF to A (bottom two rows). Forty hours after infection, the cell samples were fixed with 4% PF for 1 h at RT, followed by different permeabilization conditions, including 2% saponin for 30 min (a, h, o, and v), 1 h (b, i, p, and w), 2 h (c, j, q, and x), and 4 h (d, k, r, and y) or 1% SDS for 40 min (e, l, s, and z) or cold methanol for 10 min (f, m, t, and aa) or fixed with 10% formalin for 1 h, followed by permeabilization with 0.1% Triton for 15 min (g, n, u, and ab). All the samples were colabeled for CPAF (red), L2 organisms (green), and DNA (blue). Note that CPAF translocation into the host cell cytoplasm was always detected in cultures infected with L2-17-CPAF-wt but not in cultures infected with L2-17-CPAF-EF to A, regardless of the cell sample permeabilization conditions applied.
DISCUSSION
Since the chlamydial protease-like activity factor CPAF was discovered in 2001 (3), CPAF has attracted a great deal of interest in the chlamydial research field. Both biochemical and structural characterizations of CPAF have yielded a lot of new information, including the identification of critical residues important for intermolecular dimerization, catalytic activity, and inhibitory-peptide processing (6–8). However, how CPAF activity is regulated during chlamydial infection remains unknown. The availability of CPAF expression-deficient chlamydial organisms (10) and the successful transformation of the chlamydial organisms (26, 32, 33, 38, 39) allowed us to investigate CPAF inside Chlamydia-infected host cells. In the current study, we have presented experimental evidence to both validate the biochemical properties of CPAF previously characterized in cell-free systems and explore new features of CPAF activation and activity during chlamydial infection. First, a plasmid-encoded wild-type CPAF fully restored both CPAF activity and translocation in the CPAF-deficient L2-17 strain and displayed an expression and intracellular distribution pattern similar to that of the chromosomal CPAF from either L2-wt or the isogenic L2-5 strain during the infection course, suggesting that transformation of L2-17 with plasmid-encoded CPAF can be used to investigate CPAF properties. Second, CPAF mutated at critical residues required for intermolecular dimerization, catalytic activity, and peptide processing, as demonstrated in cell-free systems (5, 6), displayed only partial inhibition of activation. These mutants still maintained significant levels of CPAF activation/activity inside the Chlamydia-infected cells. This was true even when three important intermolecular dimerization residues or all three catalytic residues were simultaneously mutated. The dramatic differences in CPAF activation and activity between the cell-free systems and inside the infected cells suggest that there may be cofactors in the infected cells that aid in CPAF activation. Third, mutations in both intermolecular dimerization and catalytic residues in the same CPAF molecule completely blocked CPAF activation and activity during chlamydial infection. Together with the above observations, this result suggests that either the intermolecular dimerization or the catalytic residues may be sufficient for CPAF activation and activity, but simultaneous loss of both can no longer be rescued by the potential cofactors in the infected cells. Finally, as long as active CPAF was detected in the infected cells, CPAF was always localized in the cytoplasm of the infected cells. The CPAF with double mutations in both the intermolecular dimerization and catalytic residues (CPAF-FE to A) was no longer active and failed to translocate into the host cell cytoplasm. These observations demonstrated a correlation of CPAF translocation into the host cell cytoplasm with CPAF enzymatic activity. This correlation is further strengthened by the observation that the CPAF mutants with catalytic residue mutation seemed to show more severe deficiency in maturation/processing. These mutants were less efficient in translocating into the host cell cytoplasm.
How did CPAF tolerate more mutations inside the infected cells than in cell-free systems? It is difficult to imagine how a CPAF molecule with simultaneous alanine substitutions for all three catalytic residues, i.e., E558, S449, and H105, can carry out the processing of the internal inhibitory peptide and mature into active CPAF for cleaving host cytokeratin 8. The crystal structure of the wild-type CPAF revealed a water molecule sandwiched between the catalytic triad residues (6). This suggests that the catalytic triad is not tightly packed and the other adjacent residues may be able to replace the function of the catalytic triad residues, especially when CPAF is inside Chlamydia-infected cells. These alternative critical residues, which have not been identified in the cell-free system (based on the mutagenesis and crystal structure analyses), may be functional only inside cells, which explains why mutation of any of the 3 known critical residues resulted in significant reduction in CPAF activity in cell-free assays. Thus, the seemingly conflicting results from our in vivo study cannot disqualify the 3 known residues as catalytic residues in cell-free systems but suggest that there are additional critical residues that are required for CPAF activation in infected cells. The successful transformation of the CPAF-deficient chlamydial organisms presented in the current study has provided a platform to map these “in vivo critical residues.” We are consulting various structural biology experts about which other residues are the likely in vivo critical residues required for CPAF activation inside infected cells, although these extensive mapping experiments are beyond the scope of the current study.
A second possibility is that there are cofactors in the Chlamydia-infected cells that may compensate for the loss of function caused by replacement of catalytic residues identified in the cell-free systems. The cofactor may be a chlamydial protease or chlamydial-infection-activated protease that can activate CPAF in infected cells. If we can identify an inhibitor that cannot inhibit canonical CPAF activation (proximity-induced self-activation) but can block noncanonical activation pathways, we will be able to draw a more definitive conclusion about the existence of such cofactoral proteolytic activities for activating CPAF via a noncanonical pathway. However, extensive characterizations of the putative cofactors are beyond the scope of the current study and will be carried out in the future.
A third possibility for activation of the various “activation-deficient CPAF mutants” inside infected cells is that as the CPAF mutants accumulate in the inclusions to a threshold level in the late stage of infection, CPAF mutants may become activated in a concentration-dependent manner. The high concentration of CPAF inside infected cells may not be achievable in the cell-free system. The higher CPAF protein concentrations may overcome the deficiency in residues critical for either catalytic or intermolecular dimerization. This explanation is consistent with the proximity-induced activation model revealed in the cell-free system assay. Furthermore, the activated CPAF in the late stage of infection may contribute to the increased permeability of inclusion membranes and chlamydial exit.
The mutant CPAF with both catalytic and dimerization residues simultaneously replaced with alanine was no longer processed, lost all its enzymatic activity, and was strictly localized within the chlamydial inclusion. The generation of this mutant not only allowed us to confirm the critical roles of these two residues in CPAF activation, but also provided us with a tool to address concerns about the translocation of CPAF (11). We carefully compared the intracellular locations of the wild-type CPAF and the mutant CPAF under various cell sample treatment conditions. Regardless of how the cell samples were fixed and permeabilized, the wild-type CPAF was always detected in the host cell cytosol while the mutant CPAF was restricted to within the inclusions. Thus, we conclude that CPAF translocation into the host cytosol correlates with CPAF activity but is not altered by cell sample treatment conditions. Nevertheless, we are also aware that there are alternative explanations for the total lack of translocation of the CPAF-FE to A mutant with mutations at both catalytic and intermolecular dimerization residues. The amount of CPAF-FE to A was noted to be lower than those of the other CPAFs, which may be due to either a reduced expression level or decreased protein stability. The lower level of protein may result in less obvious processing and translocation. However, it is unlikely that a complete lack of processed CPAFc and translocated CPAF in CPAF-FE to A is due to the reduced level of the mutant, given the high detection sensitivity in both the Western blotting (Fig. 3) and immunofluorescence assay (Fig. 4 to 7) used in the current study.
The next question is how CPAF reaches the host cytosol. The sec-dependent pathway (9) and the type II secretion apparatus (10) may be able to transport CPAF into the lumen of chlamydial inclusions but cannot account for the CPAF in the host cell cytoplasm. An outer membrane vesicle budding mechanism may aid in CPAF's transportation into the host cell cytoplasm (20). This hypothesis is consistent with the following observations. The chlamydial reticulate body (RB) outer membrane was induced to undergo vesicularization (40, 41). Chlamydial-organism-free vesicles were detected both inside (42) and outside (43) inclusion membranes. The vesicularized CPAF may enter the host cell cytosol by the vesicle fusing with or passing through the inclusion membrane. Although outer membrane vesicles (OMVs) have been recognized as an essential means for Gram-negative bacteria to secrete virulence factors (44), the precise mechanisms by which OMVs are regulated remain unknown (45). C. trachomatis-infected cells may provide a unique opportunity for us to gain novel insights into the mechanisms of OMVs. As the tools for genetically manipulating chlamydial organisms are continuously developed and improved, we may be able to both address the role of OMVs in CPAF secretion and reveal novel mechanisms for regulating OMVs in general.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00275-15.
REFERENCES
- 1.Zhong G, Fan T, Liu L. 1999. Chlamydia inhibits interferon gamma-inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J Exp Med 189:1931–1938. doi: 10.1084/jem.189.12.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhong G, Liu L, Fan T, Fan P, Ji H. 2000. Degradation of transcription factor RFX5 during the inhibition of both constitutive and interferon gamma-inducible major histocompatibility complex class I expression in chlamydia-infected cells. J Exp Med 191:1525–1534. doi: 10.1084/jem.191.9.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med 193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong F, Sharma J, Xiao Y, Zhong Y, Zhong G. 2004. Intramolecular dimerization is required for the chlamydia-secreted protease CPAF to degrade host transcriptional factors. Infect Immun 72:3869–3875. doi: 10.1128/IAI.72.7.3869-3875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong F, Pirbhai M, Zhong Y, Zhong G. 2004. Cleavage-dependent activation of a chlamydia-secreted protease. Mol Microbiol 52:1487–1494. doi: 10.1111/j.1365-2958.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 6.Huang Z, Feng Y, Chen D, Wu X, Huang S, Wang X, Xiao X, Li W, Huang N, Gu L, Zhong G, Chai J. 2008. Structural basis for activation and inhibition of the secreted Chlamydia protease CPAF. Cell Host Microbe 4:529–542. doi: 10.1016/j.chom.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Chai J, Hart PJ, Zhong G. 2009. Identifying catalytic residues in CPAF, a Chlamydia-secreted protease. Arch Biochem Biophys 485:16–23. doi: 10.1016/j.abb.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D, Lei L, Flores R, Huang Z, Wu Z, Chai J, Zhong G. 2010. Autoprocessing and self-activation of the secreted protease CPAF in Chlamydia-infected cells. Microb Pathog 49:164–173. doi: 10.1016/j.micpath.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen D, Lei L, Lu C, Flores R, DeLisa MP, Roberts TC, Romesberg FE, Zhong G. 2010. Secretion of the chlamydial virulence factor CPAF requires the Sec-dependent pathway. Microbiology 156:3031–3040. doi: 10.1099/mic.0.040527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snavely EA, Kokes M, Dunn JD, Saka HA, Nguyen BD, Bastidas RJ, McCafferty DG, Valdivia RH. 2014. Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog Dis 71:336–351. doi: 10.1111/2049-632X.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bavoil PM, Byrne GI. 2014. Analysis of CPAF mutants: new functions, new questions (the ins and outs of a chlamydial protease). Pathog Dis 71:287–291. doi: 10.1111/2049-632X.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleba B, Stephens RS. 2008. Chlamydial effector proteins localized to the host cell cytoplasmic compartment. Infect Immun 76:4842–4850. doi: 10.1128/IAI.00715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Frohlich KM, Buckner L, Quayle AJ, Luo M, Feng X, Beatty W, Hua Z, Rao X, Lewis ME, Sorrells K, Santiago K, Zhong G, Shen L. 2011. Altered protein secretion of Chlamydia trachomatis in persistently infected human endocervical epithelial cells. Microbiology 157:2759–2771. doi: 10.1099/mic.0.044917-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heuer D, Brinkmann V, Meyer TF, Szczepek AJ. 2003. Expression and translocation of chlamydial protease during acute and persistent infection of the epithelial HEp-2 cells with Chlamydophila (Chlamydia) pneumoniae. Cell Microbiol 5:315–322. doi: 10.1046/j.1462-5822.2003.00278.x. [DOI] [PubMed] [Google Scholar]
- 15.Bauler LD, Hackstadt T. 2014. Expression and targeting of secreted proteins from Chlamydia trachomatis. J Bacteriol 196:1325–1334. doi: 10.1128/JB.01290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan P, Dong F, Huang Y, Zhong G. 2002. Chlamydia pneumoniae secretion of a protease-like activity factor for degrading host cell transcription factors required for [correction of factors is required for] major histocompatibility complex antigen expression. Infect Immun 70:345–349. doi: 10.1128/IAI.70.1.345-349.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dille S, Herbst K, Volceanov L, Nolke T, Kretz O, Hacker G. 2014. Golgi fragmentation and sphingomyelin transport to Chlamydia trachomatis during penicillin-induced persistence do not depend on the cytosolic presence of the chlamydial protease CPAF. PLoS One 9:e103220. doi: 10.1371/journal.pone.0103220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Chen D, Zhong Y, Wang S, Zhong G. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun 76:3415–3428. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen D, Lei L, Lu C, Galaleldeen A, Hart PJ, Zhong G. 2010. Characterization of Pgp3, a Chlamydia trachomatis plasmid-encoded immunodominant antigen. J Bacteriol 192:6017–6024. doi: 10.1128/JB.00847-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong G. 2011. Chlamydia trachomatis secretion of proteases for manipulating host signaling pathways. Front Microbiol 2:14–19. doi: 10.3389/fmicb.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen AL, Johnson KA, Lee JK, Sütterlin C, Tan M. 2012. CPAF: a chlamydial protease in search of an authentic substrate. PLoS Pathog 8:e1002842. doi: 10.1371/journal.ppat.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conrad TA, Yang Z, Ojcius D, Zhong G. 2013. A path forward for the chlamydial virulence factor CPAF. Microbes Infect 15:1026–1032. doi: 10.1016/j.micinf.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hacker G. 2014. The chlamydial protease CPAF: important or not, important for what? Microbes Infect 16:367–370. doi: 10.1016/j.micinf.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Zhong G. 2014. Question the questions on CPAF. Pathog Dis 72:3–4. doi: 10.1111/2049-632X.12205. [DOI] [PubMed] [Google Scholar]
- 25.Dong F, Zhong Y, Arulanandam B, Zhong G. 2005. Production of a proteolytically active protein, chlamydial protease/proteasome-like activity factor, by five different Chlamydia species. Infect Immun 73:1868–1872. doi: 10.1128/IAI.73.3.1868-1872.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong S, Yang Z, Lei L, Shen L, Zhong G. 2013. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J Bacteriol 195:3819–3826. doi: 10.1128/JB.00511-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricci S, Ratti G, Scarlato V. 1995. Transcriptional regulation in the Chlamydia trachomatis pCT plasmid. Gene 154:93–98. doi: 10.1016/0378-1119(94)00825-D. [DOI] [PubMed] [Google Scholar]
- 29.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 30.Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. 2000. Prediction of transcription terminators in bacterial genomes. J Mol Biol 301:27–33. doi: 10.1006/jmbi.2000.3836. [DOI] [PubMed] [Google Scholar]
- 31.Hefty PS, Stephens RS. 2007. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J Bacteriol 189:198–206. doi: 10.1128/JB.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Chen C, Gong S, Hou S, Qi M, Liu Q, Baseman J, Zhong G. 2014. Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J Bacteriol 196:989–998. doi: 10.1128/JB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong G, Reis e Sousa C, Germain RN. 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class II complexes formed by processing of exogenous protein. Proc Natl Acad Sci U S A 94:13856–13861. doi: 10.1073/pnas.94.25.13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Chen D, Sharma J, Cheng W, Zhong Y, Liu K, Jensen J, Shain R, Arulanandam B, Zhong G. 2006. The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect Immun 74:4826–4840. doi: 10.1128/IAI.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou S, Lei L, Yang Z, Qi M, Liu Q, Zhong G. 2013. Chlamydia trachomatis outer membrane complex protein B (OmcB) is processed by the protease CPAF. J Bacteriol 195:951–957. doi: 10.1128/JB.02087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong F, Su H, Huang Y, Zhong Y, Zhong G. 2004. Cleavage of host keratin 8 by a Chlamydia-secreted protease. Infect Immun 72:3863–3868. doi: 10.1128/IAI.72.7.3863-3868.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect Immun 81:636–644. doi: 10.1128/IAI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding H, Gong S, Tian Y, Yang Z, Brunham R, Zhong G. 2013. Transformation of sexually transmitted infection-causing serovars of Chlamydia trachomatis using blasticidin for selection. PLoS One 8:e80534. doi: 10.1371/journal.pone.0080534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsumoto A, Manire GP. 1970. Electron microscopic observations on the fine structure of cell walls of Chlamydia psittaci. J Bacteriol 104:1332–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsumoto A, Manire GP. 1970. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J Bacteriol 101:278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jorgensen I, Valdivia RH. 2008. Pmp-like proteins Pls1 and Pls2 are secreted into the lumen of the Chlamydia trachomatis inclusion. Infect Immun 76:3940–3950. doi: 10.1128/IAI.00632-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giles DK, Whittimore JD, LaRue RW, Raulston JE, Wyrick PB. 2006. Ultrastructural analysis of chlamydial antigen-containing vesicles everting from the Chlamydia trachomatis inclusion. Microbes Infect 8:1579–1591. doi: 10.1016/j.micinf.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 44.Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton BA. 2009. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog 5:e1000382. doi: 10.1371/journal.ppat.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haurat MF, Aduse-Opoku J, Rangarajan M, Dorobantu L, Gray MR, Curtis MA, Feldman MF. 2011. Selective sorting of cargo proteins into bacterial membrane vesicles. J Biol Chem 286:1269–1276. doi: 10.1074/jbc.M110.185744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.