

Abstract
Bacterial proteases are important virulence factors that inactivate host defense proteins and contribute to tissue destruction and bacterial dissemination. Outer membrane proteases of the omptin family, exemplified by Escherichia coli OmpT, are found in some Gram-negative bacteria. Omptins cleave a variety of substrates at the host-pathogen interface, including plasminogen and antimicrobial peptides. Multiple omptin substrates relevant to infection have been identified; nonetheless, an effective omptin inhibitor remains to be found. Here, we purified native CroP, the OmpT ortholog in the murine pathogen Citrobacter rodentium. Purified CroP was found to readily cleave both a synthetic fluorescence resonance energy transfer substrate and the murine cathelicidin-related antimicrobial peptide. In contrast, CroP was found to poorly activate plasminogen into active plasmin. Although classical protease inhibitors were ineffective against CroP activity, we found that the serine protease inhibitor aprotinin displays inhibitory potency in the micromolar range. Aprotinin was shown to act as a competitive inhibitor of CroP activity and to interfere with the cleavage of the murine cathelicidin-related antimicrobial peptide. Importantly, aprotinin was able to inhibit not only CroP but also Yersinia pestis Pla and, to a lesser extent, E. coli OmpT. We propose a structural model of the aprotinin-omptin complex in which Lys15 of aprotinin forms salt bridges with conserved negatively charged residues of the omptin active site.
INTRODUCTION
Bacterial proteases are critical virulence factors that play central roles at the host-pathogen interface. They contribute to bacterial virulence by degrading proteins of the host immune response, extracellular matrix proteins, and by interfering with host hemostasis. These proteases are secreted, attached to the cell surface, or embedded in the bacterial membrane. They usually belong to the main catalytic groups of proteases, namely, the serine, cysteine, and aspartate proteases and metalloproteases. Omptins constitute a unique group of integral outer membrane (OM) proteases implicated in pathogenicity and are present in a number of Gram-negative pathogens of the Enterobacteriaceae family, including Escherichia coli (OmpT), Yersinia pestis (Pla), Salmonella enterica (PgtE), Shigella flexneri (IcsP), and Citrobacter rodentium (CroP) (1–6). Omptin genes are most often part of mobile elements such as virulence plasmids or prophages, indicating that horizontal gene transfer likely played a role in the spread of these genes (7). For example, Y. pestis pla is part of the virulence plasmid pPCP1, whereas E. coli ompT is carried by cryptic prophages that inserted at various locations within the chromosome of different E. coli pathotypes (2, 8).
Members of the omptin family share 40 to 80% sequence identity at the amino acid level (7, 9). E. coli OmpT was the first omptin for which the structure was elucidated (10). OmpT adopts a β-barrel fold that consists of 10 antiparallel β-strands spanning the OM. The β-strands are linked by four short periplasmic loops and five surface-exposed loops, which surround the active-site groove and are responsible for substrate specificity (11). This overall structure is strictly conserved in other family members, including Y. pestis Pla (12). The interaction of omptins with the lipid A part of lipopolysaccharide (LPS) is essential for proteolytic activity (13, 14). Positively charged residues protruding from the barrel were shown to interact with the 4′ phosphate of lipid A, resulting in a locked conformation that is required for activity (10, 15).
Omptins were first classified as serine proteases, based on the presence of the Asp210-His212 dyad, which is reminiscent of the Asp-His-Ser triad of serine proteases (16). The OmpT crystal structure revealed the presence of the Asp83-Asp85 dyad on the opposite side of the active-site groove, and omptins were reclassified as aspartate proteases (10). The high-resolution crystal structure of Y. pestis Pla revealed the presence of a water molecule that is activated by the Asp210-His212 dyad and acts as a nucleophile to attack the substrate, while the Asp83-Asp85 dyad is proposed to participate in the stabilization of the catalytic intermediate (10, 12, 17). Together, these studies showed that omptins combine features of both serine and aspartate proteases and therefore constitute a unique family of proteases (12, 18). Previous studies on omptin inhibition reported that Zn2+, Cu2+, and benzamidine are able to inhibit OmpT activity (19–21). Classical inhibitors of the main classes of proteases are largely ineffective against omptins, most likely because of their unique catalytic mechanism (19, 20, 22). Promisingly, other studies indicated that the serine protease inhibitors aprotinin (bovine pancreatic trypsin inhibitor) and ulinastatin (urinary trypsin inhibitor) interfere with the activity of OmpT (23, 24).
Omptins were shown to preferentially cleave substrates at dibasic motifs (25, 26). This specificity is determined by the presence of the conserved Glu27 and Asp208 at the bottom of the deep S1 pocket and by Asp97 in the more shallow S1′ pocket (10). The physiological substrates of omptins consist of both host and bacterial proteins. The various omptins appear to have divergent substrate specificities, suggesting that each omptin evolved to fulfill specific functions necessary for successful colonization and infection. Most omptin substrates consist of proteins at the host-pathogen interface. For example, Pla (Plasminogen activator) of Y. pestis readily processes plasminogen into active plasmin, which promotes dissolution of fibrin clots and, in turn, bacterial dissemination (11). In contrast to Pla, E. coli OmpT poorly activates plasminogen (11, 12). Pla was proposed to contribute to Y. pestis survival and invasion by disrupting hemostasis through cleavage of the plasmin inhibitor α2-antiplasmin, plasminogen activator inhibitor 1, and the thrombin-activatable fibrinolysis inhibitor (11, 27, 28). Through this disruption of hemostasis, Pla has been shown to be essential for the progression of both bubonic and pneumonic plagues in murine models (29, 30). In addition, Caulfield et al. have recently uncovered the ability of Pla to degrade the apoptotic signaling protein Fas ligand (FasL) (31). By using Pla to degrade FasL, Y. pestis is capable of both disrupting the extrinsic apoptotic pathway and modifying the host's inflammatory response within murine lungs (31). Several omptins were reported to cleave host cationic antimicrobial peptides (AMPs), which play important roles in the innate immune defenses against pathogens (32–34). AMPs of the cathelicidin family, which often adopt an α-helical structure, are more susceptible to omptin degradation than defensins that are stabilized by three disulfide bonds (8). OmpT cleaves the human cathelicidin LL-37 at dibasic motifs, and the resulting cleavage products are devoid of bactericidal activity (35). Similarly, C. rodentium CroP degrades the murine cathelicidin-related antimicrobial peptide (CRAMP) (6). In addition to host protein degradation, omptins are involved in the processing and secretion of the passenger domains of some bacterial autotransporters. Secretion of the S. flexneri IcsA and Y. pestis YapA, YapE, and YapG autotransporters depends on the proteolytic activity of omptins IcsP and Pla, respectively (4, 5, 36, 37).
C. rodentium is an enteric pathogen that causes attaching and effacing lesions on epithelial cells in the mouse colon (38). These lesions are highly similar to those caused by enterohemorrhagic E. coli (EHEC) and enteropathogenic E. coli (EPEC) in the human gastrointestinal tract. Therefore, C. rodentium is used as a surrogate model to study the human-restricted pathogens EHEC and EPEC (39). Our previous work on the PhoPQ two-component system showed that the C. rodentium PhoQ sensor kinase does not respond to α-helical AMPs, unlike what is observed in S. enterica (6). This lack of PhoPQ activation by AMPs was attributed to the proteolytic activity of CroP, which degraded the α-helical AMPs before reaching the periplasmic space (6). In the present study, we confirm that the putative omptin catalytic dyad His212-Asp210 is required for CroP activity. We purified native CroP and characterized its substrate specificity against various known omptin substrates. Furthermore, we show that aprotinin inhibits CroP activity in a competitive manner. In addition to CroP, aprotinin was also found to inhibit OmpT and Pla. This is the first comprehensive study that examines the inhibitory activity of aprotinin against several omptins and describes its inhibitory mode of action.
MATERIALS AND METHODS
Bacterial growth conditions and reagents.
Bacteria were grown overnight with aeration at 37°C in Luria-Bertani (LB) broth and subcultured in N-minimal medium (40) supplemented with 0.2% glucose and 1 mM MgCl2 to an optical density at 600 nm (OD600) of 0.5. Chloramphenicol (30 μg/ml) or kanamycin (50 μg/ml) was added to the media when appropriate. The murine cathelicidin (CRAMP) was synthesized with purity greater than 90% (BioChemia). The fluorescence resonance energy transfer (FRET) substrate [2Abz-SLGRKIQIK(Dnp)-NH2] was purchased from AnaSpec. Glu-plasminogen and the plasmin substrate VLKpNA were from Sigma and Molecular Innovations, respectively. The protease inhibitors EDTA, leupeptin, pepstatin A, and aprotinin were from BioShop; phenylmethylsulfonyl fluoride (PMSF) was purchased from Sigma-Aldrich.
Construction of bacterial strains and plasmids.
Bacterial strains and plasmids used throughout the present study are listed in Table 1. DNA purification, cloning, and transformations were conducted according to standard procedures (41). All experiments were conducted in a BSL-2 laboratory in accordance with the McGill laboratory biosafety manual. The C. rodentium ΔwaaL deletion strain was made by sacB gene-based allelic exchange, as previously described (6). The double-deletion ΔwaaL ΔcroP strain was generated by conjugating the C. rodentium ΔwaaL strain with E. coli χ7213 transformed with plasmid pCR002 (6).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| C. rodentium | ||
| DBS100 | Wild type (ATCC 51459) | 38 |
| ΔcroP mutant | DBS100 (ΔcroP) | 6 |
| ΔcroP(pCRcroP) mutant | ΔcroP mutant transformed with pCRcroP | This study |
| ΔcroP(pYCcroP) mutant | ΔcroP mutant transformed with pYCcroP | This study |
| ΔcroP(pYCcroPMut) mutant | ΔcroP mutant transformed with pYCcroPMut | This study |
| ΔcroP(pYCpla) mutant | ΔcroP mutant transformed with pYCpla | This study |
| ΔwaaL mutant | DBS100 (ΔwaaL) | This study |
| ΔwaaLΔcroP mutant | DBS100 (ΔwaaL ΔcroP) | This study |
| ΔwaaLΔcroP(pYCpla) | ΔwaaL ΔcroP mutant transformed with pYCpla | This study |
| E. coli | ||
| χ7213 | thr-1 leuB6 fhuA21 lacY1 glnV44 recA1 asdA4 thi-1 RP4-2-Tc::Mu [-pir]; Kanr | 58 |
| EDL933 | EHEC O157:H7 | 59 |
| ΔompT mutant | EDL933 (ΔompT) | 35 |
| BL21 | F− ompT hsdS (rB− mB−) gal dcm | GE Healthcare |
| Plasmids | ||
| pCR002 | pRE112 containing ΔcroP mutation | 6 |
| pCRcroP | C. rodentium croP and its promoter cloned into pWSK129 | 6 |
| pWL214 | pUC18R6K-mini-Tn7T-Kan carrying PN25-tetR + Ptet-pla; Ampr Kanr | 30 |
| pACYC184 | Low-copy-number plasmid; Cmr Tetr | NEB |
| pYCcroP | pACYC184 containing croP under the control of its native promoter | This study |
| pYCcroPMut | pYCcroP containing croP with mutations H212A and D210A | This study |
| pYCpla | pACYC184 containing pla under the control of the croP promoter | This study |
Cmr, chloramphenicol resistance; Tetr, tetracycline resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance.
The croP gene under the control of its native promoter was isolated from plasmid pCRcroP by digestion with XbaI and SacI. This fragment was inserted into the pACYC184 plasmid in which the EcoRV site has been modified to a SacI site, generating plasmid pYCcroP. The plasmid expressing Pla under the control of the croP promoter region was generated by overlap extension PCR (42). The croP promoter fragments were amplified from C. rodentium wild-type genomic DNA using primers EXT1and EXT2 (Table 2). The extended pla open reading frame (ORF) fragment was amplified from plasmid pWL214 by using the primer pair EXT3/EXT4. The corresponding extended croP promoter and pla ORF fragments were combined, and overlap extension PCR was performed using the primer pair EXT1/EXT4. The resultant PCR products were digested with XbaI and SacI, gel purified, and ligated into pYCcroP previously digested with the same enzymes to generate plasmid pYCpla (Table 1). In a similar manner, point mutations H212A and D210A were introduced into croP by overlap extension PCR. The initial two fragments were amplified from pYCcroP using primer pairs EXT1/MUT2 and MUT3/MUT1. The primer pair EXT1/MUT1 was used for the final PCR resulting in the full-length croP mutant. The resultant PCR product was cloned into the XbaI and SacI restriction sites of plasmid pYCcroP, generating plasmid pYCcroPMut.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′–3′)a | Useb |
|---|---|---|
| EXT1 | CCCACTCTAGAGGATATTCAGCAGGATGGGC | croP promoter 5′F XbaI |
| EXT2 | GCCACAATAGAACTTTTCTTCATAGTGATGACTCCATTTTGTCAGGT | croP promoter 3′R pla extension |
| EXT3 | ACCTGACAAAATGGAGTCATCACTATGAAGAAAAGTTCTATTGTGGC | pla 5′F croP promoter extension |
| EXT4 | GCGAGCTCTCAGAAGCGATATTGCAGAC | pla 3′R SacI |
| MUT1 | GCGAGCTCTGCTCAGAAGGTATATTTCAC | croP 3′R SacI |
| MUT2 | GCGAGCGTAAGCTTCAGCATTATCCGATGCCCGTAC | croP MUT 3′R |
| MUT3 | GGATAATGCTGAAGCTTACGCTCGCGGGATCA | croP MUT 5′F |
| qEH810 | TCGGCTCCTTCCCGAATGGAG | qPCR EHEC ompT F |
| qEH811 | GATGCTTCCACCCAGCCGC | qPCR EHEC ompT R |
| qEH812 | GTGCTGCATGGCTGTCGTCA | qPCR EHEC 16S F |
| qEH813 | AGCACGTGTGTAGCCCTGGT | qPCR EHEC 16S R |
| qpla1009 | GGACTTGCAGGCCAGTATCGC | qPCR pla F |
| qpla1010 | AGCCGGATGTCTTCTCACGGA | qPCR pla R |
| qCR16F | TGTCTACTTGGAGGTTGTGCCCTT | qPCR C. rodentium 16S F |
| qCR16R | TGCAGTCTTCCGTGGATGTCAAGA | qPCR C. rodentium 16S R |
Restriction sites are underlined.
F, forward; R, reverse.
Purification of native CroP.
The C. rodentium ΔcroP strain transformed with pCRcroP was grown in LB broth to an approximate OD600 of 3. After centrifugation, bacterial pellets were resuspended in 50 mM Tris-HCl (pH 8.3) containing 5 mM MgCl2, and the cells were lysed by sonication. The lysate was centrifuged at 3,600 × g for 10 min at 4°C, and the resulting supernatant was further centrifuged at 100,000 × g for 1 h at 4°C to pellet bacterial membranes. OM and inner membrane fractions were separated using the Sarkosyl method (43), with modifications. The total membrane pellet was resuspended in 50 mM Tris-HCl (pH 8.3) supplemented with 5 mM MgCl2 and 0.1% Sarkosyl, incubated for 30 min at 10°C, and centrifuged at 100,000 × g for 1 h to pellet the Sarkosyl-insoluble fraction containing OM. Subsequently, the pellet was solubilized in 50 mM Tris-HCl (pH 8.3), 10 mM EDTA, and 1% Triton X-100. The OM suspension was incubated at 22°C for 30 min, followed by the removal of insoluble proteins by centrifugation at 12,000 × g for 10 min. The supernatant was applied to a HiTrap Q FF column (GE Healthcare) previously equilibrated with buffer A (25 mM Tris-HCl [pH 8.0], 50 mM NaCl, and 0.1% Triton X-100). Elution was performed using a linear gradient of buffer B (25 mM Tris-HCl [pH 8.0], 1 M NaCl and 0.1% Triton X-100). Fractions containing pure CroP were identified by SDS-PAGE and Coomassie blue staining, pooled, and dialyzed against phosphate-buffered saline (phosphate-buffered saline [PBS]; pH 7.4). Protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Scientific). Prior to use, purified CroP was incubated with a 3 M excess of rough LPS from E. coli EH100 (Sigma-Aldrich) overnight to ensure complete activation (16).
Western blotting.
Bacterial strains were grown to an OD600 of 0.5 in N-minimal medium and harvested by centrifugation. The bacterial pellets were then either used for Sarkosyl extraction to obtain OM fractions or resuspended in Laemmli sample buffer and boiled for 5 min to obtain whole-cell lysates. Protein samples were resolved by SDS-PAGE, and Western blotting with a rabbit polyclonal antibody raised against CroP was conducted as previously described (44). ImageJ software was used to measure the density of CroP bands on Western blot membranes.
Proteolytic cleavage of the FRET substrate.
The activity of purified CroP was assessed by incubating the enzyme with the FRET substrate (3 μM) in black 96-well microtiter plates (Costar). Fluorescence was monitored over 60 min with shaking between measurements at an excitation wavelength of 325 nm and emission wavelength of 430 nm in a BioTek FLx800 multidetection microplate reader equipped with an injector. Initial background fluorescence was subtracted from samples for normalization. For the determination of optimal pH, CroP (10 nM) activity was assessed in a mixed buffering system comprised of 50 mM citrate, phosphate, and Tris-HCl (pH 4 to 10). The relative activity was determined by calculating the area under the curve for a 30-min reaction relative to the average of triplicates at pH 7. Other assays using purified CroP were performed in PBS (pH 7.4).
Cleavage of the FRET substrate by bacterial cells expressing various omptins was conducted as previously described (35). Cells grown in N-minimal medium and normalized to an OD600 of 0.5 were centrifuged and resuspended in PBS. Bacteria (∼3 × 108 CFU/ml) were incubated with aprotinin for 15 min prior to injection of the FRET substrate (3 μM). The fluorescence was measured over 1 h, as described above. The area under the curve of all samples was determined using GraphPad Prism 5 software. Background fluorescence of the appropriate bacterial strain (C. rodentium ΔcroP or EHEC ΔompT) was subtracted from values. The relative activity was calculated with respect to the average of uninhibited triplicates.
Proteolytic cleavage of CRAMP.
CRAMP cleavage assays were carried out as previously described (35), with the following modifications. CRAMP (0.1 μM) was incubated at room temperature with either the indicated bacterial strains resuspended in PBS (∼3 × 108 CFU/ml) or purified CroP (0.4 μM). Samples were collected at the denoted time points, mixed with Tris-Tricine sample buffer (Bio-Rad), and boiled for 5 min. The influence of aprotinin on CRAMP cleavage by CroP was monitored at 5 min and carried out with the addition of aprotinin (200 and 400 μM). Peptide degradation products were resolved using 10 to 20% Tris-Tricine PAGE gels (Bio-Rad). Gels were fixed with 5% glutaraldehyde for 30 min, washed for 30 min with deionized water, and stained with Coomassie blue.
Plasminogen activation.
Activation of plasminogen by omptins was monitored as described elsewhere with modifications (11). Approximately 3 × 108 CFU/ml of bacterial cells, purified CroP (0.4 μM), or control buffer (PBS) was combined in wells with the plasmin substrate VLKpNA (4.5 mM) in triplicate. After 5 min, Glu-plasminogen (20.0 μg/ml) was added to each well. Cleavage of VLKpNA chromogenic substrate was monitored by measuring the absorbance at 405 nm every 10 min for 300 min. Absorbance was monitored with a Bio-Tek Power Wave ×340 spectrophotometer with shaking, followed by incubation at 37°C. For normalization of values, the initial background absorbance was subtracted from final measurements.
Mode of CroP inhibition by aprotinin.
Inhibition of CroP by aprotinin was further investigated using aprotinin (0 to 10 μM) and seven concentrations of the FRET substrate ranging from 0.66 to 16 μM. CroP (10 nM) incubated with aprotinin was added to a 400-μl cuvette containing the FRET substrate. Fluorescence (an excitation of 325 nm and an emission of 430 nm) was measured with a Varian Cary Eclipse fluorescence spectrophotometer over 7 min with measurements every 15 s. Initial background measurements were subtracted from the final reaction sample values. Initial velocity values (V0) were determined from the linear portion of the enzyme progression curves. Kinetics plots and constants for CroP were calculated from an average of four independent experiments. To determine the mode of inhibition, CroP kinetics data were fit to the Michaelis-Menten equation, and reciprocal values were calculated and fit to a best linear regression line in GraphPad Prism version 5. The Vmax and Km values were calculated from the Michaelis-Menten equation. The inhibition constant (Ki) was determined by fitting the substrate and velocity data to the competitive enzyme inhibition model with GraphPad Prism version 5.
qPCR.
Quantitative PCR (qPCR) analysis was conducted as previously described (35). Briefly, total RNA from bacteria grown in N-minimal medium was isolated using TRIzol reagents (Invitrogen) and treated with a DNA-free kit (Ambion) to remove residual DNA. Superscript III (Invitrogen) was used for the reverse transcription of RNA in triplicate. A reaction mixture void of Superscript III was also used as a negative control. qPCRs were performed in a Rotor-Gene 3000 thermal cycler (Corbett Research) using the Maxima SYBR green PCR kit (Fermentas) according to the manufacturer's instructions. Primers used are listed in Table 2. The levels of gene transcripts were normalized to 16S and analyzed using the 2−ΔΔCT method (45).
Molecular docking.
Docking of aprotinin (Protein Data Bank [PDB] code 4PTI) using OmpT (PDB code 1I78) or Pla (PDB code 2X55) was performed using the ClusPro 2.0 Web server (http://cluspro.bu.edu), taking into account electrostatic charge interactions (46). Molecular models of the omptin-aprotinin complex were generated using the program PIPER (47), as implemented in ClusPro 2.0 and clustered using a pairwise ligand root-mean-square-deviation criterion. The best scored models were subjected to energy minimization using the CHARMM potential to remove potential side chain clashes (48). The highest scoring models for OmpT and Pla were visualized using the PyMOL molecular graphics system (v1.5.0.4; Schrödinger, LLC).
RESULTS
CroP proteolytic activity depends on the Asp210-His212 catalytic dyad.
The C. rodentium CroP amino acid sequence is 74% identical to E. coli OmpT and contains all of the omptin catalytic residues (see Fig. S1 in the supplemental material) (17). To confirm that CroP and OmpT share similar catalytic mechanisms, we generated a CroP variant (CroPMut), in which alanine residues substitute the putative catalytic dyad residues Asp210 and His212. Western blot analysis of the OM fractions, using a polyclonal antibody against CroP, confirmed the presence of CroP or CroPMut at the OM of C. rodentium wild-type, ΔcroP(pYCcroP), and ΔcroP(pYCcroPMut) strains (Fig. 1A). The band corresponding to CroP was absent in the OM of the ΔcroP strain (Fig. 1A). These results indicate that both CroP and CroPMut are localized at the OM of C. rodentium. FRET substrate cleavage assays were performed to compare the activity of the various C. rodentium strains. C. rodentium wild-type and ΔcroP(pYCcroP) strains incubated with the FRET substrate resulted in a 2- to 3-fold increase in fluorescence compared to the C. rodentium ΔcroP and ΔcroP(pYCcroPMut) strains (Fig. 1B). These results clearly indicate that CroPMut present at the C. rodentium OM is inactive and unable to cleave the FRET substrate. To further confirm these results, AMP cleavage assays were performed by incubating the various C. rodentium strains with the murine cathelicidin CRAMP. Although the C. rodentium wild-type and ΔcroP(pYCcroP) strains generated two observable cleavage products and degraded the full-length peptide almost to completion, the ΔcroP and ΔcroP(pYCcroPMut) strains did not produce observable cleavage products (Fig. 1C). Together, these data show that the catalytic dyad Asp210-His212 is necessary for CroP proteolytic activity. They also confirm that CroP and OmpT have similar catalytic mechanisms.
FIG 1.
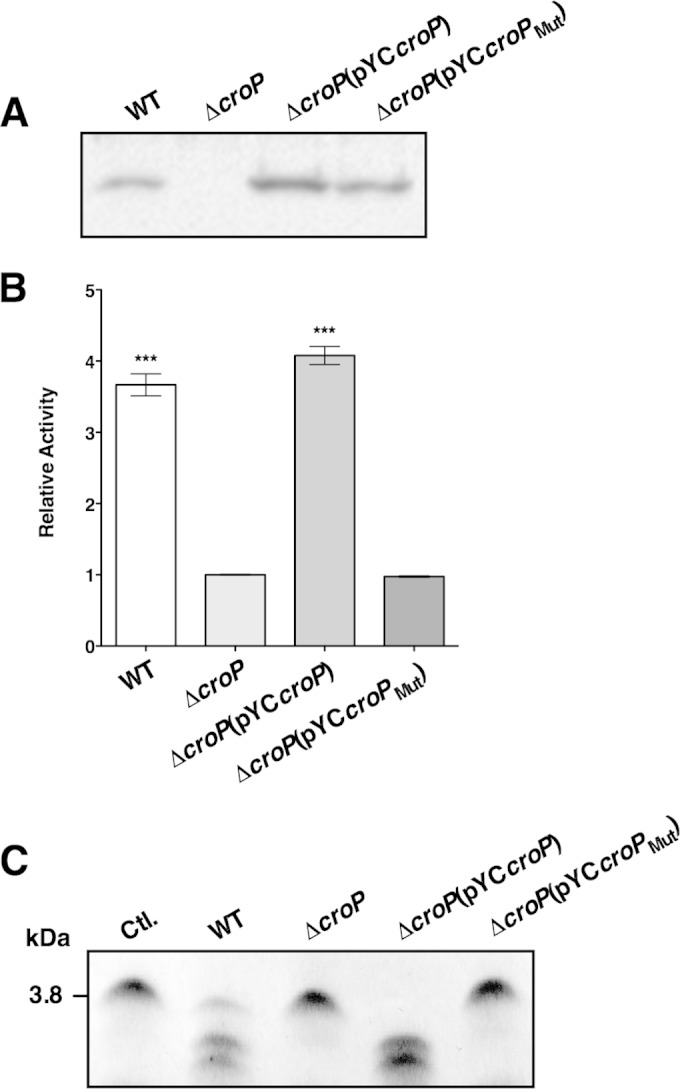
Expression and proteolytic activities of C. rodentium CroP and CroPMut. (A) Presence of CroP and CroPMut at the OM of C. rodentium. The OM fractions of C. rodentium wild-type, ΔcroP, ΔcroP(pYCcroP), and ΔcroP(pYCcroPMut) strains were resolved on an SDS–11% PAGE gel. Western blotting was done using a polyclonal rabbit antibody raised against purified CroP. (B) Proteolytic cleavage of the FRET substrate by CroP and CroPMut. The various C. rodentium strains were incubated with the FRET substrate for 60 min. The results are expressed as means ± the standard errors of the mean (SEM) of triplicate samples and are expressed relative to the activity of the C. rodentium ΔcroP strain. Statistical significance was assessed using a one-way analysis of variance, followed by Dunnett's post hoc comparison test (***, P < 0.001). (C) Proteolytic cleavage of CRAMP by CroP and CroPMut. CRAMP (0.1 μM) was incubated with the various C. rodentium strains for 15 min. After incubation, bacteria were pelleted by centrifugation, and supernatant aliquots were resolved by Tris-Tricine SDS-PAGE on 10 to 20% polyacrylamide gels and visualized by Coomassie blue staining. The data shown are representative of three independent experiments. Ctl., control; WT, wild type.
Purification of the native CroP protease.
Despite the high level of sequence identity between CroP and OmpT, CroP differs from OmpT by the deletion of two basic amino acids in the surface-exposed loop 4 (see Fig. S1 in the supplemental material). In OmpT, this dibasic motif (Lys217-Arg218) corresponds to the main autoproteolytic site that hampered early attempts to purify the native protease (16). The absence of this motif in CroP allowed us to purify the protein in its native state. The CroP protein was expressed in the C. rodentium ΔcroP strain transformed with the pWSK129-derived plasmid pCRcroP (Table 1) (6). Comparison of whole-cell lysates of ΔcroP and ΔcroP(pCRcroP) cells showed the appearance of a prominent band at ∼32 kDa in the ΔcroP(pCRcroP) lysate, which is close to the expected size of mature CroP (32.9 kDa) (see Fig. S2 in the supplemental material). CroP was purified from total membranes by Sarkosyl extraction of OMs, solubilization with Triton X-100 and anion-exchange chromatography. The purified product was visualized by SDS-PAGE, followed by Coomassie staining (see Fig. S2 in the supplemental material). After dialysis against PBS, purified CroP was incubated with a 3-fold molar excess of E. coli LPS, which is required for omptin activity (13, 15).
Cleavage of the FRET substrate by purified CroP.
The activity of purified CroP was examined by monitoring the increase in fluorescence emission indicative of FRET substrate cleavage at the RK dibasic motif [2Abz-SLGRKIQIK(Dnp)-NH2] (35). The pH activity profile of CroP was first examined using a mixed buffering system over a pH range of 4 to 10. CroP exhibited robust activity over a pH range of 6.0 to 8.0, with optimal activity at pH 7.0 (Fig. 2A). This CroP optimal pH value is consistent with those obtained for OmpT and Pla, and it differs from the low optimal pH value of aspartate proteases (12, 16). Cleavage of the FRET substrate using increasing amounts of CroP (0 to 10 nM) was then monitored over time. The results showed a dose-dependent increase in CroP activity against the FRET substrate (Fig. 2B). Purified CroP was used at a concentration of 10 nM for further experimentation.
FIG 2.
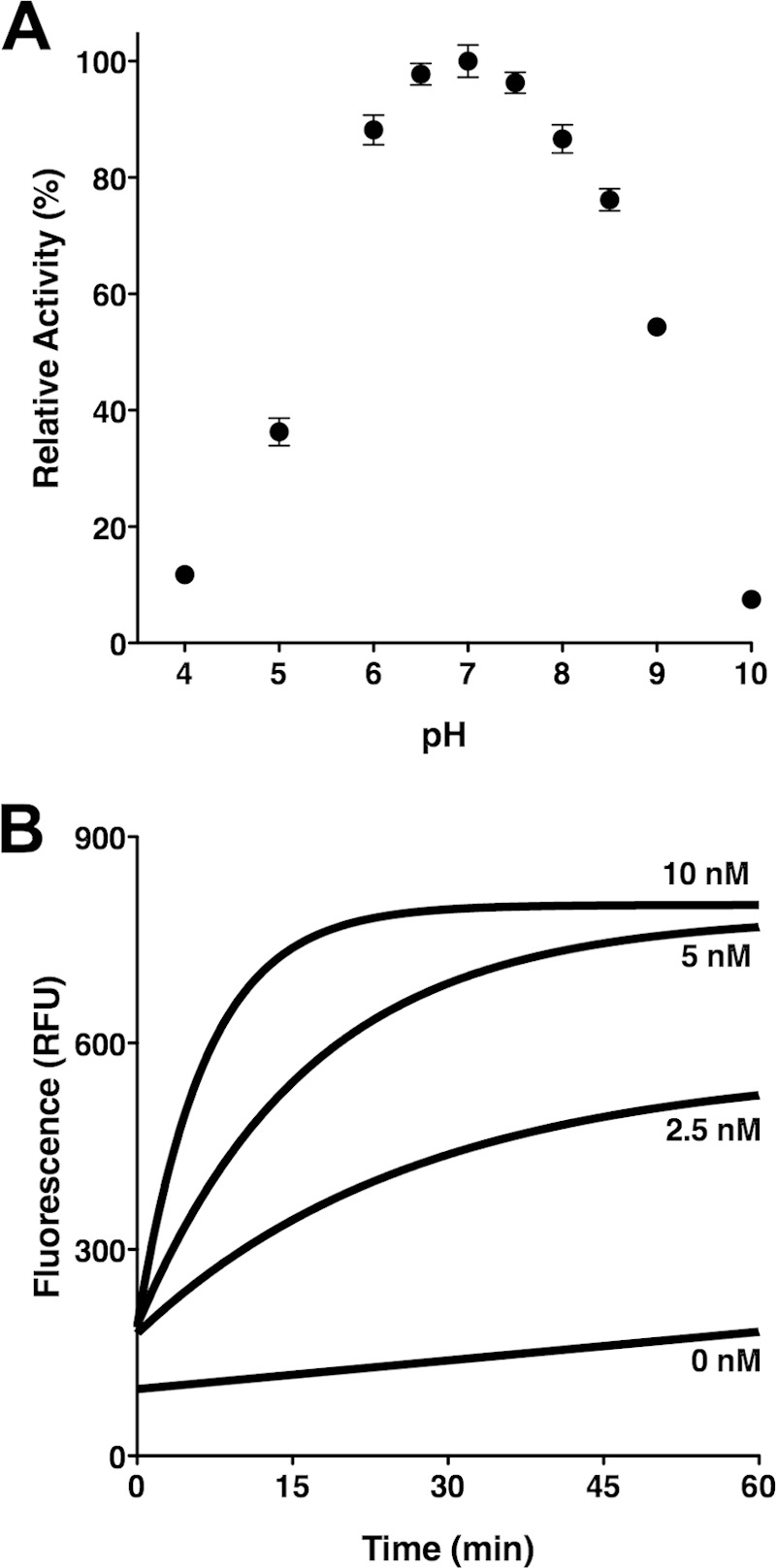
Proteolytic cleavage of the synthetic FRET substrate by purified CroP. (A) Effect of pH on CroP activity. Reactions were performed in a mixed buffering system (50 mM citrate, phosphate, and Tris-HCl) adjusted to the indicated pHs. The percentage of CroP activity is relative to the optimal pH. The results are expressed as means ± the SEM of triplicate samples and are representative of three independent experiments. (B) Dose-dependent increase of CroP activity. The FRET substrate was incubated with the indicated concentration of purified CroP in PBS. RFU, relative fluorescence units. The data shown are representative of three independent experiments performed in triplicate.
Purified CroP rapidly cleaves CRAMP but poorly activates plasminogen.
We have previously shown that CroP expressed by C. rodentium degrades the murine cathelicidin CRAMP, a 34-amino-acid AMP (6). A time course experiment monitoring the cleavage of CRAMP confirmed that purified CroP cleaves CRAMP. As shown in Fig. 3A, CroP rapidly cleaved CRAMP into two major cleavage products, with cleavage almost complete by 5 min. Previous studies showed that Y. pestis Pla readily cleaves plasminogen to generate plasmin, the serine protease that degrades fibrin clots (29). In contrast, E. coli OmpT is known to poorly activate plasminogen (11, 12). The ability of purified CroP to activate plasminogen was assessed by measuring plasmin activity using the chromogenic plasmin substrate VLKpNA. Incubation of purified CroP with Glu-plasminogen did not result in any notable increase in absorbance over a period of 300 min compared to the control sample (Fig. 3B), suggesting that CroP does not activate plasminogen well. To confirm this result, Glu-plasminogen was incubated with wild-type C. rodentium cells. Since the O-antigen of smooth LPS is known to interfere with the activation of plasminogen by omptins (49), these assays were performed using rough C. rodentium ΔwaaL strains that lack the O-antigen ligase WaaL. Compared to the control C. rodentium ΔwaaL ΔcroP strain, no substantial increase in absorbance was observed when C. rodentium ΔwaaL cells were incubated with Glu-plasminogen (Fig. 3B). As a positive control, incubation of Glu-plasminogen with the C. rodentium ΔwaaL ΔcroP(pYCpla) strain, which expresses Y. pestis Pla, resulted in a pronounced increase in absorbance (Fig. 3B). These data show that unlike Pla, CroP is a poor plasminogen activator. Taken together, these data suggest that CroP and OmpT may share similar substrate specificity.
FIG 3.

OmpT-like activity of purified CroP. (A) Proteolytic degradation of CRAMP by CroP. CRAMP was incubated with purified CroP with samples collected at the indicated times. Samples were resolved by Tris-Tricine SDS-PAGE on 10 to 20% polyacrylamide gels and visualized by Coomassie blue staining. The molecular mass of full-length CRAMP is indicated on the left. The data shown are representative of three independent experiments. (B) Cleavage of plasminogen into active plasmin by purified CroP and C. rodentium cells expressing various omptins. Glu-plasminogen and the plasmin substrate VLKpNA were incubated with purified CroP (□), control LPS buffer (○), and C. rodentium ΔwaaL (▲), ΔwaaL ΔcroP (⧫), and ΔwaaL ΔcroP(pYCpla) (●) strains. The absorbance at 405 nm was monitored over a 300-min period. For each data set, the initial absorbance was subtracted from all values. The data shown are expressed as means ± the SEM of triplicate samples and are representative of three independent experiments.
Aprotinin inhibits CroP activity.
Although the serine protease inhibitor aprotinin was reported to inhibit OmpT activity (23), it remains unclear whether it inhibits other omptins, including CroP. Having purified native CroP and a FRET assay to measure CroP activity allowed us to perform a small-scale screen for potential CroP inhibitors and confirm previous results obtained with OmpT. Inhibitors of the main classes of proteases, such as PMSF, leupeptin, pepstatin A, and EDTA, did not significantly affect CroP activity (Fig. 4A). In sharp contrast, addition of aprotinin at a concentration of 10 μM considerably decreased fluorescence emission, indicating inhibition of CroP proteolytic activity (Fig. 4A). Inhibition of CroP activity by aprotinin was further examined over a broad range of aprotinin concentrations (5 to 200 μM). Increasing concentrations of aprotinin resulted in a dose-dependent decrease in fluorescence emission, indicating that aprotinin inhibits CroP activity in a dose-dependent manner (Fig. 4B). CroP activity was reduced by ca. 93% at a concentration of aprotinin of 200 μM (Fig. 4B). To ensure that aprotinin or LPS was not interfering with fluorescence emission, we combined completely digested FRET substrate with either aprotinin (200 μM) or LPS (16 μM). No significant differences in fluorescence emission were observed between these samples compared to the PBS control (see Fig. S3 in the supplemental material). Together, these data show that aprotinin inhibits CroP activity at micromolar concentrations.
FIG 4.
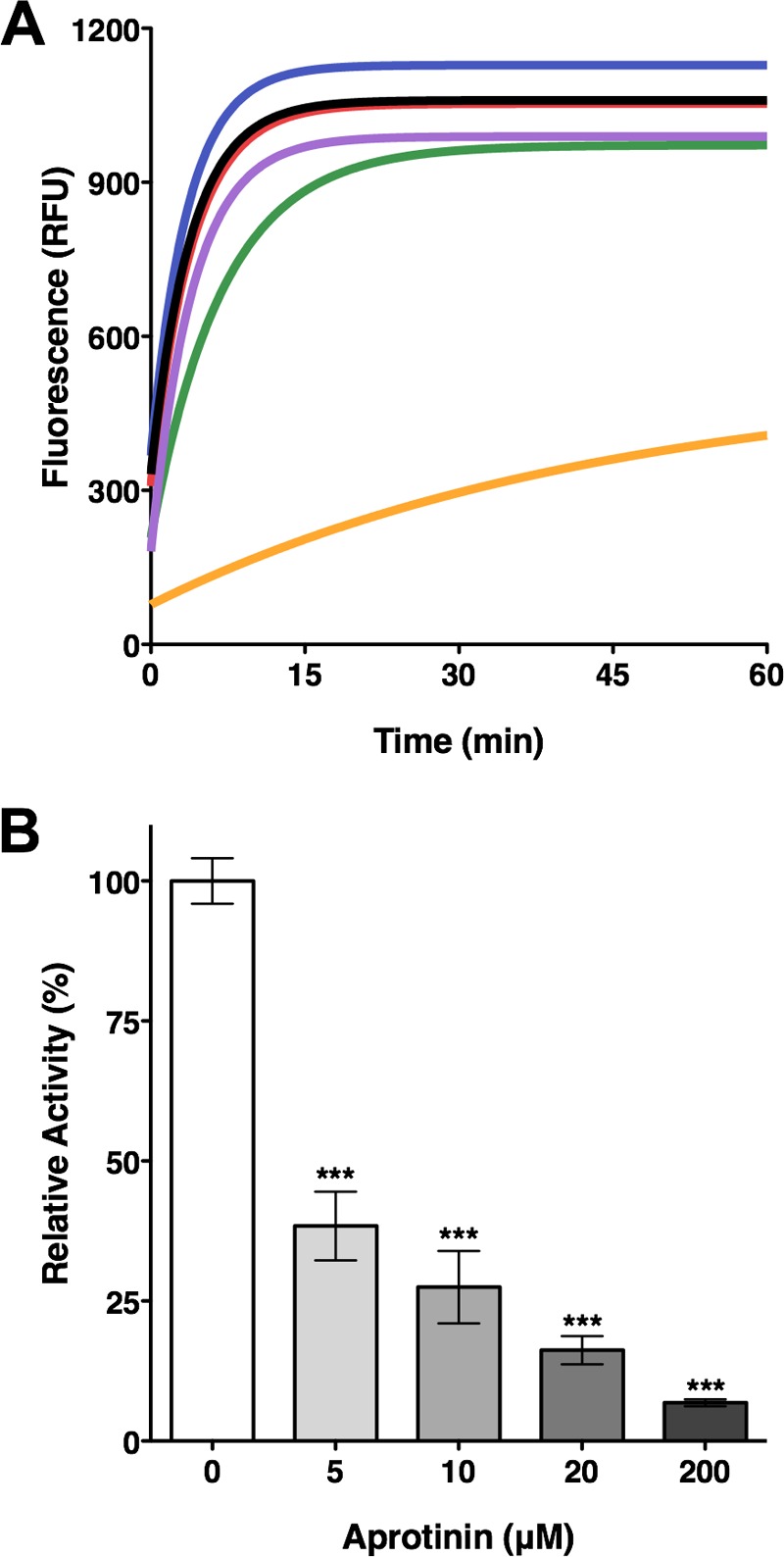
Inhibition of CroP activity by aprotinin. (A) Aprotinin inhibits cleavage of the FRET substrate by purified CroP. FRET assays were performed with 10 nM CroP in PBS (black) or in the presence of EDTA (2.5 mM, green), PMSF (2.5 mM, purple), leupeptin (10 μM, blue), pepstatin A (10 μM, red), or aprotinin (10 μM, orange). RFU, relative fluorescence units. The data shown are representative of three independent experiments performed in triplicate. (B) Inhibition of purified CroP by aprotinin is dose dependent. FRET assays were performed with 10 nM CroP in the presence of increasing concentrations of aprotinin, as indicated. The percentage of CroP activity is relative to the uninhibited reaction. The results are expressed as means ± the SEM of triplicate samples and are representative of three independent experiments. Statistical significance was assessed by one-way analysis of variance, followed by Dunnett's post hoc test (***, P < 0.001).
Aprotinin inhibits the cleavage of CRAMP by CroP.
To assess whether aprotinin inhibits the CroP-mediated cleavage of CRAMP, this AMP was incubated with purified CroP in the absence or presence of aprotinin and the reaction products were visualized on Tris-Tricine SDS-PAGE gels. Consistent with the results shown in Fig. 3A, CRAMP was completely cleaved by CroP into two smaller cleavage products in the absence of aprotinin (Fig. 5). When the cleavage assay was performed in the presence of aprotinin, the band corresponding to full-length CRAMP was still visible on gels, indicating inhibition of CRAMP cleavage (Fig. 5). In addition, inhibition of CRAMP cleavage appeared to be inversely proportional to the concentration of aprotinin present in the assay (Fig. 5). Taken together, these data show that aprotinin inhibits the cleavage of CRAMP by CroP.
FIG 5.

Aprotinin inhibits CRAMP proteolytic degradation by CroP. CRAMP was incubated with purified CroP in the presence of the indicated concentration of aprotinin for 5 min. Samples were resolved by Tris-Tricine SDS-PAGE on 10 to 20% polyacrylamide gels. Gels were stained with Coomassie blue. The molecular masses of aprotinin and CRAMP are indicated on the left. The results are representative of three independent experiments.
Aprotinin is a competitive inhibitor of CroP.
Aprotinin acts as a competitive inhibitor of trypsin and other serine proteases (50). Kinetic analyses were performed to determine the mode of aprotinin inhibition on CroP activity. Rates of the FRET substrate cleavage by purified CroP were measured in the presence of different concentrations of aprotinin. The Lineweaver-Burk plot analysis was indicative of competitive inhibition with increasing aprotinin concentrations resulting in an increase of the Km values, whereas Vmax values remained mostly unchanged (Fig. 6). The inhibition constant (Ki) of the cleavage of the FRET substrate by CroP was estimated to be 0.127 ± 0.035 μM. These data revealed that aprotinin inhibits CroP in a competitive manner and thus competes with the FRET substrate for binding to the CroP active site.
FIG 6.
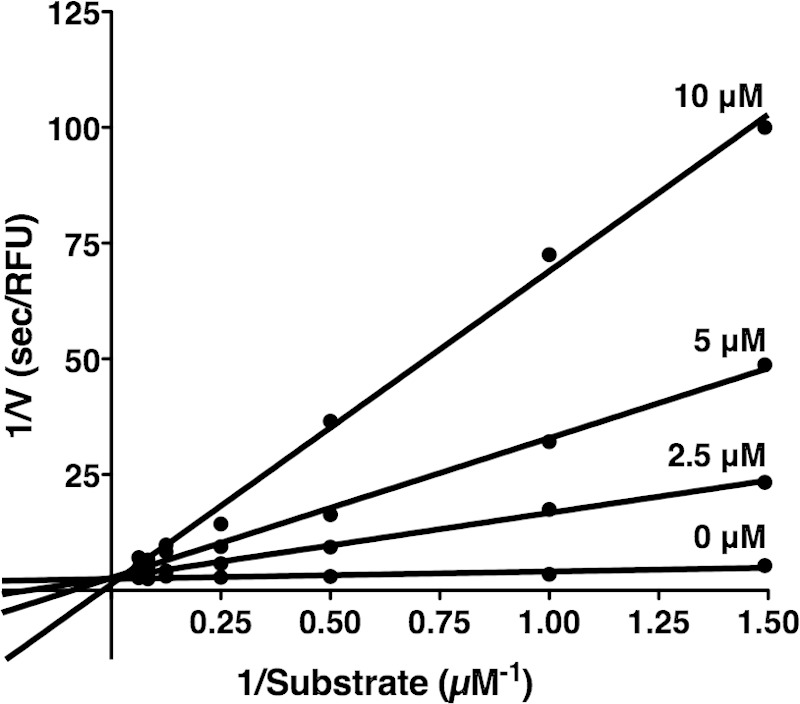
Aprotinin is a competitive inhibitor of CroP. A Lineweaver-Burk plot for inhibition of CroP by aprotinin is shown. The rates of the FRET substrate (0.66 to 16 μM) cleavage by CroP (10 nM) were measured in the presence of different concentrations of aprotinin, as indicated. The data shown are the mean of three independent experiments.
Aprotinin inhibits the activity of CroP present at the bacterial cell surface.
To determine whether aprotinin is able to inhibit the activity of CroP present at the OM of cells, we measured the ability of smooth wild-type C. rodentium cells to cleave the FRET substrate in the presence of increasing concentrations of aprotinin. The amount of CroP produced by wild-type C. rodentium was quantified by Western blotting, followed by density analysis of the Western blot signals (Fig. 7A). We estimated that our assays using C. rodentium whole cells contained approximately 24 ± 2 nM CroP, which is roughly twice the amount used for the purified CroP experiments shown in Fig. 4B. Increasing the concentration of aprotinin from 0 to 200 μM resulted in a steady decrease of CroP activity from 100% to ca. 17% (Fig. 7B). Compared to the experiment performed with 10 nM purified CroP (Fig. 4B), inhibition of CroP activity by aprotinin was less pronounced in the whole-cell experiment. This slight difference is likely due to nonspecific binding of aprotinin to bacterial cell surfaces, resulting in lowered concentrations of free inhibitor. These data indicate that aprotinin inhibits the activity of CroP in its native environment.
FIG 7.
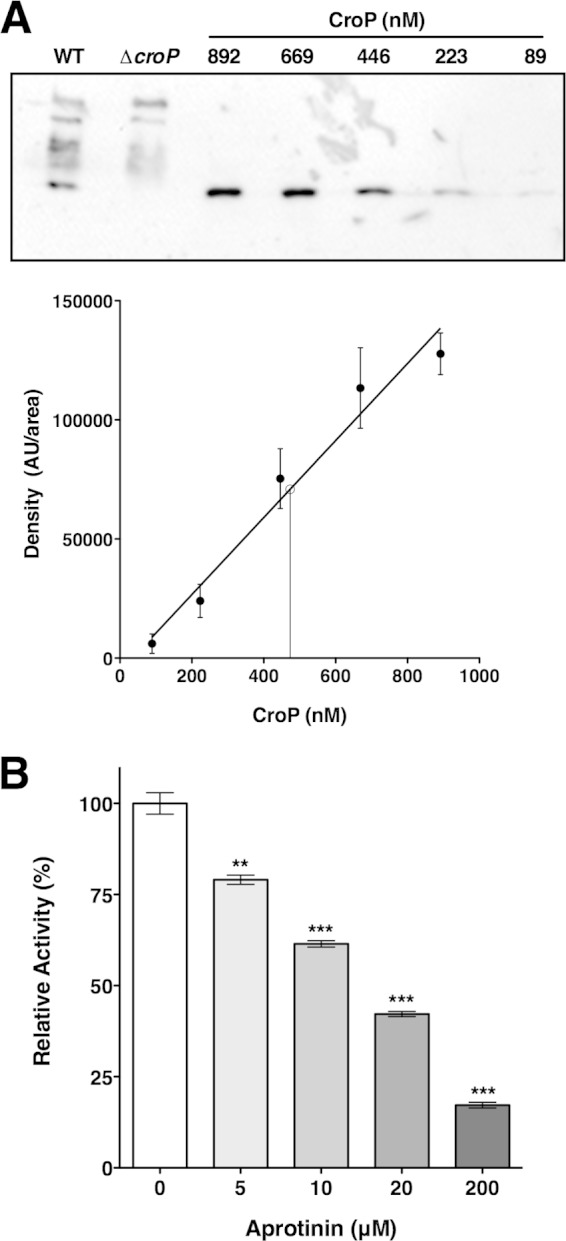
Aprotinin inhibits the activity of CroP present at the OM. (A) Production of CroP by C. rodentium wild type was quantified by densitometry analysis of Western blots by comparing to a standard curve of purified CroP. Western blots were performed in triplicate with the anti-CroP antibody. In the resulting standard curve of purified CroP (solid circles and line of best fit), data are expressed as means ± the SEM. The concentration of CroP in C. rodentium wild-type was interpolated from the standard curve (open circle and vertical line). AU, arbitrary units. (B) FRET assays were performed with wild-type C. rodentium cells in the presence of increasing concentrations of aprotinin, as indicated. The percentage of CroP activity is expressed relative to the uninhibited reaction. The results are expressed as means ± the SEM of triplicate samples and are representative of three independent experiments. Statistical significance was assessed by one-way analysis of variance followed by Dunnett's post hoc test (**, P < 0.01; ***, P < 0.001).
Aprotinin inhibits other omptins.
Since aprotinin was capable of inhibiting the activity of CroP present at the C. rodentium cell surface, we investigated the possibility that it inhibits omptins other than CroP. Inhibition of E. coli OmpT was examined by measuring the ability of EHEC EDL933 cells to cleave the FRET substrate in the presence of increasing concentrations of aprotinin. Although aprotinin appeared to be less potent at inhibiting OmpT compared to CroP, the proteolytic activity of OmpT was inhibited by ∼90% in the presence of 200 μM aprotinin (Fig. 8A). The Y. pestis pla gene was heterologously expressed in the C. rodentium ΔcroP strain. The proteolytic activity of Pla was gradually inhibited by incubating cells with increasing concentrations of aprotinin, and almost complete inhibition was observed at 200 μM aprotinin (Fig. 8B). A similar inhibition pattern was observed when pla was expressed in E. coli BL21 (see Fig. S4 in the supplemental material). As controls, expression of the EHEC ompT and Y. pestis pla genes were monitored by qPCR, and the presence of both omptins at the bacterial OM was assessed by measuring the proteolytic activity against the FRET substrate (see Fig. S5 in the supplemental material). Our data indicate that both omptin genes are expressed and suggest that Pla is present at the bacterial cell surface at higher levels than OmpT, which is consistent with the overexpression of Pla from a plasmid. Together, these data show that aprotinin inhibits not only the activity of CroP but also the activity of E. coli OmpT and Y. pestis Pla.
FIG 8.
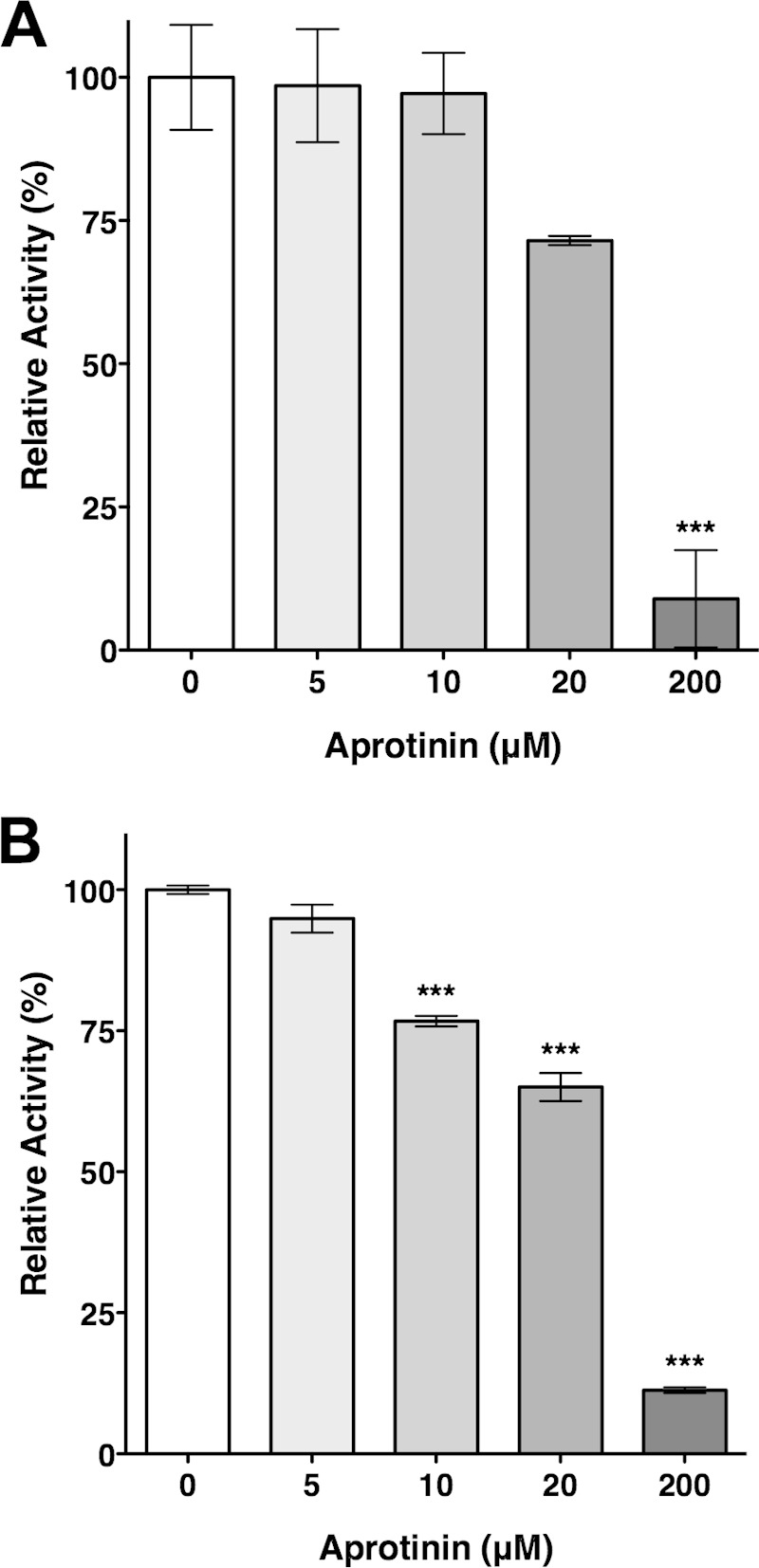
Inhibition of OmpT and Pla by aprotinin. FRET assays were performed with EHEC EDL933 cells containing wild-type levels of OmpT (A) and C. rodentium ΔcroP cells producing Pla from plasmid pYCpla (B). FRET assays were performed in triplicate in the presence of increasing concentrations of aprotinin, as indicated. The percentage of omptin activity is expressed relative to the uninhibited reaction. The results are expressed as means ± the SEM of triplicate samples and are representative of three independent experiments. Statistical significance was assessed by one-way analysis of variance followed by Dunnett's post hoc test (***, P < 0.001).
Docking model of the aprotinin-omptin complex.
Aprotinin forms highly stable complexes with trypsin. The P1 residue (Lys15) of aprotinin binds tightly to Asp189 of trypsin, which is located at the bottom of the S1 specificity pocket (50, 51). Based on the finding that aprotinin inhibits CroP in a competitive manner (Fig. 6), we hypothesized that Lys15 of aprotinin interacts with Glu27 and Asp208 (OmpT numbering), which are the two negatively charged residues that form the S1 specificity pocket of omptins (10, 12). A three-dimensional docking model of the aprotinin-omptin complex was generated as described in Materials and Methods. Essentially, similar models were obtained using the atomic coordinates of either OmpT or Pla. As shown in Fig. 9A, aprotinin fits within the omptin active-site groove and Lys15 of aprotinin protrudes into the S1 specificity pocket. Residue Lys15 of aprotinin is predicted to form a salt bridge with Glu27 but not with Asp208 of the S1 specificity pocket (Fig. 9B). In addition, Lys15 is predicted to interact with the catalytic residue Asp210 (Fig. 9B). Overall, this protein-docking model suggests that aprotinin inhibits trypsin and omptins through a similar mechanism.
FIG 9.
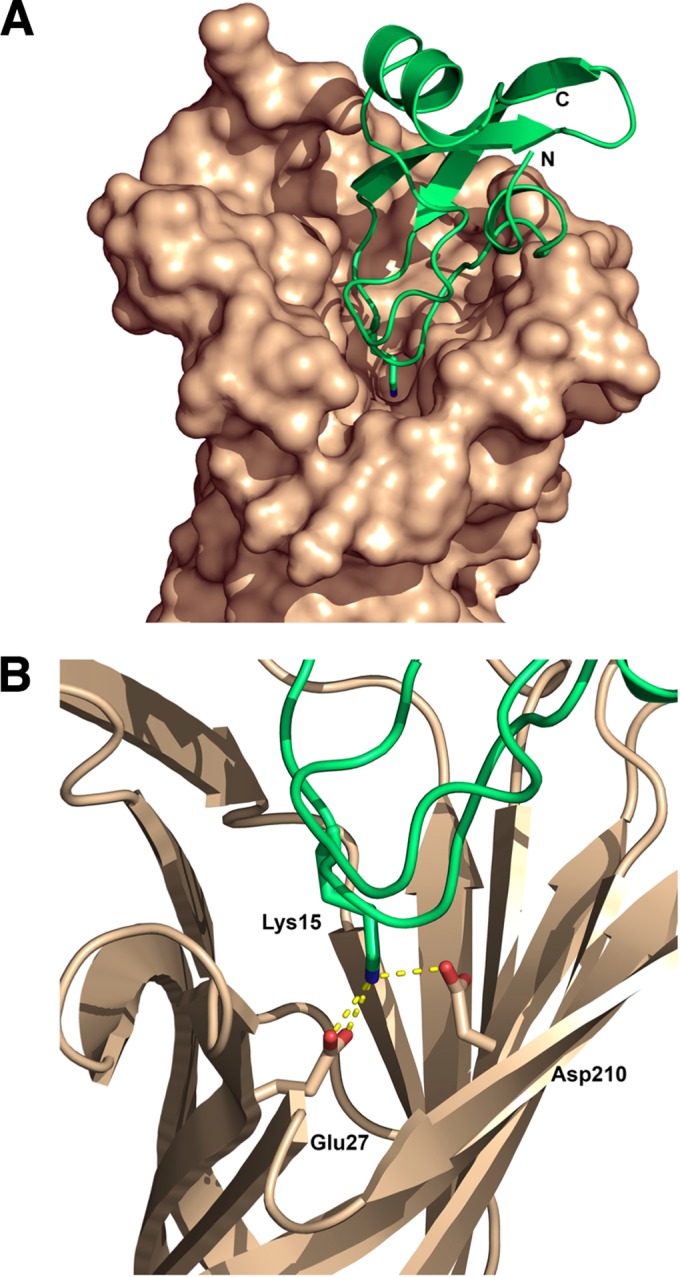
Model of interaction between aprotinin and OmpT. (A) Overview of aprotinin (green) docked within the OmpT active site (brown). The C and N termini of aprotinin are labeled accordingly. (B) Aprotinin residue Lys15 extending from the protease-binding loop. The predicted salt bridges of aprotinin Lys15 with the conserved omptins residues Glu27 and Asp210 are represented as yellow dashed lines. The diagrams were rendered using PyMOL.
DISCUSSION
Integral OM proteases of the omptin family, which are found exclusively in Gram-negative bacteria, play important roles in bacterial pathogenicity. Omptins have a unique mechanism of action combining features of both serine and aspartate proteases. Although this protease family constitutes a novel therapeutic target, specific omptin inhibitors are still unavailable. Having the C. rodentium omptin CroP purified in its native form, we analyzed the potential of purified CroP to cleave various relevant substrates. Purified CroP readily cleaved a synthetic FRET substrate and CRAMP, the sole murine cathelicidin AMP. However, unlike the Y. pestis omptin Pla, CroP did not significantly activate plasminogen into active plasmin. Most importantly, we found that the serine protease inhibitor aprotinin acts as a competitive inhibitor of CroP. In whole-cell assays, aprotinin was capable of inhibiting not only CroP activity but also the activity of E. coli OmpT and Y. pestis Pla. Therefore, aprotinin may represent a starting point for the development of specific omptin inhibitors with potential therapeutic applications.
Several proteins or peptides contributing to the host's innate defenses are either processed or inactivated through the proteolytic activity of omptins. However, whether these substrates are cleaved by all omptins remains unclear. The best-characterized omptin substrate, plasminogen, was shown to be readily activated by Pla, but poorly cleaved by OmpT (11, 12). Based on this finding and sequence identity, omptins have been divided into two subfamilies, namely, the Pla-like and OmpT-like omptin subfamilies (9). Our data showed that CroP poorly activates plasminogen into plasmin, as previously reported for OmpT (Fig. 3B). Previously, we reported that OmpT cleaves AMPs such as the human cathelicidin LL-37 (35, 44). Similarly, C. rodentium CroP rapidly cleaves the murine cathelicidin CRAMP (Fig. 3A) (6). These data are consistent with CroP and OmpT having similar substrate specificities. As suggested by the high sequence similarity between OmpT and CroP, our data indicate that CroP belongs to the OmpT subfamily of omptins.
Based on the involvement of the Asp83-Asp85 dyad in catalysis, omptins are mechanistically classified as aspartic proteases in the MEROPS database (52). However, several lines of evidence suggest that omptins are not aspartic proteases. For example, omptins deviate from the classical catalytic mechanism of aspartic proteases. An Asp210-His212 dyad is involved in the activation of the nucleophilic water molecule in omptins, whereas aspartic proteases use an Asp-Asp dyad to activate the water molecule. In contrast to aspartic proteases that have a low optimal pH of ∼4, both Pla and OmpT exhibit optimal pHs close to neutrality (12, 16). Our study provides additional evidences distinguishing omptins from aspartate proteases. First, CroP has a neutral optimal pH, like Pla and OmpT (Fig. 2A). Second, the activity of CroP is not inhibited by common protease inhibitors, including pepstatin A that is a potent inhibitor of aspartic proteases (53). Altogether, these lines of evidence support the reclassification of omptins apart from aspartic proteases, as proposed by others (12, 18).
Common protease inhibitors such as PMSF, leupeptin, pepstatin A, and EDTA have proven useful in the classification of proteases into serine, cysteine, and aspartic protease and metallo-protease families, respectively. In agreement with previous studies (19, 20), we found that these inhibitors do not affect CroP activity (Fig. 4A). In sharp contrast, we showed that the serine protease inhibitor aprotinin decreased the cleavage of the FRET substrate and CRAMP by purified CroP in a dose-dependent manner (Fig. 4 and 5). Aprotinin inhibited not only the activity of purified CroP but also the activity of CroP expressed by wild-type C. rodentium cells (Fig. 7). This suggests that aprotinin interacts with the extracellular domain of CroP, most likely with the CroP active site. Kinetic analyses revealed that aprotinin inhibits CroP in a competitive manner and thus confirms that aprotinin competes with the substrate for binding to the CroP active site (Fig. 6). In addition, we found that OmpT in wild-type EHEC EDL933 and Pla produced in the C. rodentium ΔcroP strain were also inhibited by aprotinin (Fig. 8). This is the first study to demonstrate that aprotinin inhibits multiple omptins.
Aprotinin is the most extensively studied Kunitz-type inhibitor of serine proteases. It is a competitive inhibitor that binds tightly to the S1 specificity pocket of trypsin mainly through the formation of a salt bridge between Lys15 of aprotinin and Asp189 of trypsin (51). Similar to Asp189 of trypsin, the S1 specificity pocket of omptins contains the negatively charged residues Glu27 and Asp208. Our computer-generated docking model of the omptin-aprotinin complex shown in Fig. 9 suggests that Lys15 of aprotinin makes two critical interactions with omptin residues. First, Lys15 interacts with Glu27 of the omptin S1 specificity pocket in a similar manner to that of the interaction with Asp189 of trypsin. Second, Lys15 forms a salt bridge with Asp210 that is part of the Asp210-His212 dyad and considered a catalytic residue, suggesting that residues involved in both catalysis and substrate specificity could be targeted by aprotinin variants. Surprisingly, no interaction was predicted with Asp208, the second negatively charged residue of the omptin S1 pocket. Although plausible, this structural model will require further experimental confirmation.
Although the main target of aprotinin is trypsin (Ki = 0.06 pM), it also potently inhibits other serine proteases such as plasmin (Ki = 1 nM), kallikrein (Ki = 0.09 nM), and chymotrypsin (Ki = 9 nM) (54). Although aprotinin inhibits CroP (Ki = 0.127 μM) to a lesser extent than the aforementioned proteases, CroP inhibition is greater than that of the serine proteases thrombin (Ki = 61 μM), elastase (Ki = 3.5 μM), and urokinase (Ki = 27.0 μM) (55–57). Therefore, the design of more potent (decreased Ki for omptins) and more specific (increased Ki for trypsin) omptin inhibitors appears to be feasible using a computational approach validated by experimental testing.
Because of their direct involvement in pathogenesis, omptins are prime candidates for therapeutic targeting. These proteases, which are found at the OM of important Gram-negative bacterial pathogens, have their active sites readily accessible to exogenous inhibitors. Since omptins are unique proteases with no homologs identified in mammalian hosts, specific omptin inhibitors are unlikely to have major off-target adverse drug effects. The finding that aprotinin inhibits CroP, OmpT, and Pla suggests that it is possible to develop inhibitors acting on multiple omptins that could be used for the treatment of various infections. Inhibition of the omptins expressed by various pathogens is expected to prevent invasion and dissemination of Y. pestis, to interfere with the cell-to-cell spread of S. flexneri in the intestinal epithelium, and to potentiate the effect of AMPs against E. coli pathotypes. Such antivirulence drugs targeting key virulence factors represent promising new therapeutic options in the fight against antibiotic-resistant bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Canadian Institutes of Health Research (MOP-15551) and the Natural Sciences and Engineering Research Council (RGPIN-217482). J.-L.T. was supported by a McGill Faculty of Medicine graduate fellowship.
We are grateful to William E. Goldman for providing plasmid pWL214. We thank Charles Viau for providing the C. rodentium ΔwaaL strain.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00136-15.
REFERENCES
- 1.Grodberg J, Dunn JJ. 1988. ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J Bacteriol 170:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sodeinde OA, Goguen JD. 1989. Nucleotide sequence of the plasminogen activator gene of Yersinia pestis: relationship to ompT of Escherichia coli and gene E of Salmonella typhimurium. Infect Immun 57:1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grodberg J, Dunn JJ. 1989. Comparison of Escherichia coli K-12 outer membrane protease OmpT and Salmonella typhimurium E protein. J Bacteriol 171:2903–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shere KD, Sallustio S, Manessis A, D'Aversa TG, Goldberg MB. 1997. Disruption of IcsP, the major Shigella protease that cleaves IcsA, accelerates actin-based motility. Mol Microbiol 25:451–462. doi: 10.1046/j.1365-2958.1997.4681827.x. [DOI] [PubMed] [Google Scholar]
- 5.Egile C, d'Hauteville H, Parsot C, Sansonetti PJ. 1997. SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol Microbiol 23:1063–1073. doi: 10.1046/j.1365-2958.1997.2871652.x. [DOI] [PubMed] [Google Scholar]
- 6.Le Sage V, Zhu L, Lepage C, Portt A, Viau C, Daigle F, Gruenheid S, Le Moual H. 2009. An outer membrane protease of the omptin family prevents activation of the Citrobacter rodentium PhoPQ two-component system by antimicrobial peptides. Mol Microbiol 74:98–111. doi: 10.1111/j.1365-2958.2009.06854.x. [DOI] [PubMed] [Google Scholar]
- 7.Hritonenko V, Stathopoulos C. 2007. Omptin proteins: an expanding family of outer membrane proteases in Gram-negative Enterobacteriaceae. Mol Membr Biol 24:395–406. doi: 10.1080/09687680701443822. [DOI] [PubMed] [Google Scholar]
- 8.Thomassin JL, Brannon JR, Kaiser J, Gruenheid S, Le Moual H. 2012. Enterohemorrhagic and enteropathogenic Escherichia coli evolved different strategies to resist antimicrobial peptides. Gut Microbes 3:556–561. doi: 10.4161/gmic.21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haiko J, Suomalainen M, Ojala T, Lahteenmaki K, Korhonen TK. 2009. Breaking barriers: attack on innate immune defenses by omptin surface proteases of enterobacterial pathogens. Innate Immun 15:67–80. doi: 10.1177/1753425909102559. [DOI] [PubMed] [Google Scholar]
- 10.Vandeputte-Rutten L, Kramer RA, Kroon J, Dekker N, Egmond MR, Gros P. 2001. Crystal structure of the outer membrane protease OmpT from Escherichia coli suggests a novel catalytic site. EMBO J 20:5033–5039. doi: 10.1093/emboj/20.18.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kukkonen M, Lahteenmaki K, Suomalainen M, Kalkkinen N, Emody L, Lang H, Korhonen TK. 2001. Protein regions important for plasminogen activation and inactivation of α2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol Microbiol 40:1097–1111. doi: 10.1046/j.1365-2958.2001.02451.x. [DOI] [PubMed] [Google Scholar]
- 12.Eren E, Murphy M, Goguen J, van den Berg B. 2010. An active site water network in the plasminogen activator pla from Yersinia pestis. Structure 18:809–818. doi: 10.1016/j.str.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 13.Kramer RA, Brandenburg K, Vandeputte-Rutten L, Werkhoven M, Gros P, Dekker N, Egmond MR. 2002. Lipopolysaccharide regions involved in the activation of Escherichia coli outer membrane protease OmpT. Eur J Biochem 269:1746–1752. doi: 10.1046/j.1432-1327.2002.02820.x. [DOI] [PubMed] [Google Scholar]
- 14.Kukkonen M, Korhonen TK. 2004. The omptin family of enterobacterial surface proteases/adhesins: from housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int J Med Microbiol 294:7–14. doi: 10.1016/j.ijmm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Eren E, van den Berg B. 2012. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J Biol Chem 287:23971–23976. doi: 10.1074/jbc.M112.376418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer RA, Zandwijken D, Egmond MR, Dekker N. 2000. In vitro folding, purification, and characterization of Escherichia coli outer membrane protease OmpT. Eur J Biochem 267:885–893. doi: 10.1046/j.1432-1327.2000.01073.x. [DOI] [PubMed] [Google Scholar]
- 17.Vandeputte-Rutten L, Gros P. 2002. Novel proteases: common themes and surprising features. Curr Opin Struct Biol 12:704–708. doi: 10.1016/S0959-440X(02)00393-7. [DOI] [PubMed] [Google Scholar]
- 18.Jarvinen HM, Laakkonen L, Haiko J, Johansson T, Juuti K, Suomalainen M, Buchrieser C, Kalkkinen N, Korhonen TK. 2013. Human single-chain urokinase is activated by the omptins PgtE of Salmonella enterica and Pla of Yersinia pestis despite mutations of active site residues. Mol Microbiol 89:507–517. doi: 10.1111/mmi.12293. [DOI] [PubMed] [Google Scholar]
- 19.Sugimura K, Higashi N. 1988. A novel outer-membrane-associated protease in Escherichia coli. J Bacteriol 170:3650–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimura K, Nishihara T. 1988. Purification, characterization, and primary structure of Escherichia coli protease VII with specificity for paired basic residues: identity of protease VII and OmpT. J Bacteriol 170:5625–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yam CH, Siu WY, Kaganovich D, Ruderman JV, Poon RY. 2001. Cleavage of cyclin A at R70/R71 by the bacterial protease OmpT. Proc Natl Acad Sci U S A 98:497–501. doi: 10.1073/pnas.98.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer RA, Vandeputte-Rutten L, de Roon GJ, Gros P, Dekker N, Egmond MR. 2001. Identification of essential acidic residues of outer membrane protease OmpT supports a novel active site. FEBS Lett 505:426–430. doi: 10.1016/S0014-5793(01)02863-0. [DOI] [PubMed] [Google Scholar]
- 23.Gill RT, DeLisa MP, Shiloach M, Holoman TR, Bentley WE. 2000. OmpT expression and activity increase in response to recombinant chloramphenicol acetyltransferase overexpression and heat shock in Escherichia coli. J Mol Microbiol Biotechnol 2:283–289. [PubMed] [Google Scholar]
- 24.Hui CY, Guo Y, He QS, Peng L, Wu SC, Cao H, Huang SH. 2010. Escherichia coli outer membrane protease OmpT confers resistance to urinary cationic peptides. Microbiol Immunol 54:452–459. doi: 10.1111/j.1348-0421.2010.00238.x. [DOI] [PubMed] [Google Scholar]
- 25.McCarter JD, Stephens D, Shoemaker K, Rosenberg S, Kirsch JF, Georgiou G. 2004. Substrate specificity of the Escherichia coli outer membrane protease OmpT. J Bacteriol 186:5919–5925. doi: 10.1128/JB.186.17.5919-5925.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dekker N, Cox RC, Kramer RA, Egmond MR. 2001. Substrate specificity of the integral membrane protease OmpT determined by spatially addressed peptide libraries. Biochemistry 40:1694–1701. doi: 10.1021/bi0014195. [DOI] [PubMed] [Google Scholar]
- 27.Haiko J, Laakkonen L, Juuti K, Kalkkinen N, Korhonen TK. 2010. The omptins of Yersinia pestis and Salmonella enterica cleave the reactive center loop of plasminogen activator inhibitor 1. J Bacteriol 192:4553–4561. doi: 10.1128/JB.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valls Serón M, Haiko J, De Groot PG, Korhonen TK, Meijers JCM. 2010. Thrombin-activatable fibrinolysis inhibitor is degraded by Salmonella enterica and Yersinia pestis. J Thromb Haemost 8:2232–2240. doi: 10.1111/j.1538-7836.2010.04014.x. [DOI] [PubMed] [Google Scholar]
- 29.Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. 1992. A surface protease and the invasive character of plague. Science 258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 30.Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 31.Caulfield AJ, Walker ME, Gielda LM, Lathem WW. 2014. The Pla protease of Yersinia pestis degrades Fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15:424–434. doi: 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stumpe S, Schmid R, Stephens DL, Georgiou G, Bakker EP. 1998. Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J Bacteriol 180:4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guina T, Yi EC, Wang H, Hackett M, Miller SI. 2000. A PhoP-regulated outer membrane protease of Salmonella enterica serovar Typhimurium promotes resistance to alpha-helical antimicrobial peptides. J Bacteriol 182:4077–4086. doi: 10.1128/JB.182.14.4077-4086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galvan EM, Lasaro MA, Schifferli DM. 2008. Capsular antigen fraction 1 and Pla modulate the susceptibility of Yersinia pestis to pulmonary antimicrobial peptides such as cathelicidin. Infect Immun 76:1456–1464. doi: 10.1128/IAI.01197-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomassin JL, Brannon JR, Gibbs BF, Gruenheid S, Le Moual H. 2012. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect Immun 80:483–492. doi: 10.1128/IAI.05674-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrenz MB, Pennington J, Miller VL. 2013. Acquisition of omptin reveals cryptic virulence function of autotransporter YapE in Yersinia pestis. Mol Microbiol 89:276–287. doi: 10.1111/mmi.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lane MC, Lenz JD, Miller VL. 2013. Proteolytic processing of the Yersinia pestis YapG autotransporter by the omptin protease Pla and the contribution of YapG to murine plague pathogenesis. J Med Microbiol 62:1124–1134. doi: 10.1099/jmm.0.056275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schauer DB, Falkow S. 1993. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect Immun 61:2486–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. 2005. Citrobacter rodentium of mice and man. Cell Microbiol 7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 40.Nelson DL, Kennedy EP. 1971. Magnesium transport in Escherichia coli. Inhibition by cobaltous ion. J Biol Chem 246:3042–3049. [PubMed] [Google Scholar]
- 41.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 42.Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 43.Filip C, Fletcher G, Wulff JL, Earhart CF. 1973. Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J Bacteriol 115:717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brannon JR, Thomassin JL, Desloges I, Gruenheid S, Le Moual H. 2013. Role of uropathogenic Escherichia coli OmpT in the resistance against human cathelicidin LL-37. FEMS Microbiol Lett 345:64–71. doi: 10.1111/1574-6968.12185. [DOI] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. 2004. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 47.Kozakov D, Brenke R, Comeau SR, Vajda S. 2006. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 48.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 49.Kukkonen M, Suomalainen M, Kyllonen P, Lahteenmaki K, Lang H, Virkola R, Helander IM, Holst O, Korhonen TK. 2004. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol Microbiol 51:215–225. doi: 10.1046/j.1365-2958.2003.03817.x. [DOI] [PubMed] [Google Scholar]
- 50.Vincent JP, Lazdunski M. 1972. Trypsin-pancreatic trypsin inhibitor association: dynamics of the interaction and role of disulfide bridges. Biochemistry 11:2967–2977. [DOI] [PubMed] [Google Scholar]
- 51.Huber R, Kukla D, Bode W, Schwager P, Bartels K, Deisenhofer J, Steigemann W. 1974. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. II. Crystallographic refinement at 1.9 Å resolution. J Mol Biol 89:73–101. [DOI] [PubMed] [Google Scholar]
- 52.Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marciniszyn J Jr, Hartsuck JA, Tang J. 1976. Mode of inhibition of acid proteases by pepstatin. J Biol Chem 251:7088–7094. [PubMed] [Google Scholar]
- 54.Fritz H, Wunderer G. 1983. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Drug Res 33:479–494. [PubMed] [Google Scholar]
- 55.Lestienne P, Bieth JG. 1978. The inhibition of human leukocyte elastase by basic pancreatic trypsin inhibitor. Arch Biochem Biophys 190:358–360. doi: 10.1016/0003-9861(78)90286-2. [DOI] [PubMed] [Google Scholar]
- 56.Lottenberg R, Sjak-Shie N, Fazleabas AT, Roberts RM. 1988. Aprotinin inhibits urokinase but not tissue-type plasminogen activator. Thromb Res 49:549–556. doi: 10.1016/0049-3848(88)90252-6. [DOI] [PubMed] [Google Scholar]
- 57.Pintigny D, Dachary-Prigent J. 1992. Aprotinin can inhibit the proteolytic activity of thrombin: a fluorescence and an enzymatic study. Eur J Biochem 207:89–95. [DOI] [PubMed] [Google Scholar]
- 58.Roland KCR, Sizemore D. 1999. Construction and evaluation of a Δcya Δcrp Salmonella typhimurium strain expressing avian pathogenic Escherichia coli O78 LPS as a vaccine to prevent airsacculitis in chickens. Avian Dis 43:429–441. doi: 10.2307/1592640. [DOI] [PubMed] [Google Scholar]
- 59.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med 308:681–685. doi: 10.1056/NEJM198303243081203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.