

Abstract
Mitochondrial dysfunction is known to be a contributing factor to a number of diseases including chronic alcohol induced liver injury. While there is a detailed understanding of the metabolic pathways and proteins of the liver mitochondrion, little is known regarding how changes in the mitochondrial proteome may contribute to the development of hepatic pathologies. Emerging evidence indicates that reactive oxygen and nitrogen species disrupt mitochondrial function through post-translational modifications to the mitochondrial proteome. Indeed, various new affinity labeling reagents are available to test the hypothesis that post-translational modification of proteins by reactive species contributes to mitochondrial dysfunction and alcoholic fatty liver disease. Specialized proteomic techniques are also now available, which allow for identification of defects in the assembly of multi-protein complexes in mitochondria and the resolution of the highly hydrophobic proteins of the inner membrane. In this review knowledge gained from the study of changes to the mitochondrial proteome in alcoholic hepatotoxicity will be described and placed into a mechanistic framework to increase understanding of the role of mitochondrial dysfunction in liver disease.
Keywords: Mitochondria, Alcohol, Liver, Oxidative stress, Nitric oxide, Proteomics, Post-translational modifications
INTRODUCTION
Long term heavy alcohol consumption is the most prevalent cause of liver-related morbidity and mortality in the United States. Excessive alcohol consumption is estimated to be the third leading cause of preventable death in the United States with up to 12 000 deaths each year attributed to alcoholic liver disease[1]. While it has long been held that the severity of alcoholic liver disease is dependent on the dose and duration of alcohol consumption, it has become increasingly clear that other factors may play a significant role in the development of liver disease[2]. Studies show that only 25% of heavy drinkers will develop alcoholic steatohepatitis and even less than 10% will progress to cirrhosis[3]. These observations have led to the general hypothesis that environmental, genetic, metabolic and/or viral factors, also referred to as "hits", may influence the progression from simple fatty liver (steatosis) to more serious liver diseases[2].
Evidence supports a "multi-hit" hypothesis in the pathophysiologic sequelae of alcoholic liver disease wherein the "first-hit" involves lipid accumulation in hepatocytes (steatosis), followed by "second-hits" that lead to more serious conditions such as alcoholic steatohepatitis, fibrosis, cirrhosis, and cancer. Examples of “second-hits” include metabolic stressors such as obesity, hypercholesterolemia, and hyperglycemia, which are components of the cardiometabolic syndrome, and environmental stressors, which may include dietary factors and pollutants such as tobacco smoke. Recent epidemiologic and clinical studies indicate that environmental tobacco smoke, a widespread toxicant, may accelerate fibrogenesis and increase the severity of a number of chronic liver diseases including hepatocellular carcinoma, hepatitis C-mediated liver injury, and primary biliary cirrhosis[4-8]. Moreover, alcohol, tobacco smoke, and obesity have been shown to be synergistic risk factors for hepatocellular carcinoma in patients[9]. Taken together, these studies clearly highlight the need to identify the molecular targets and mechanisms through which these potential stressors or "hits" interact to accelerate and worsen alcoholic liver disease.
Studies suggest a potential mechanistic link among chronic alcohol consumption and many of the proposed secondary stressors, which include increased oxidative damage, hypoxic stress, and disrupted redox cellular signaling. Interestingly, the mitochondrion is a primary target for many of these metabolic derangements, which can also differentially exacerbate hepatic pro-inflammatory responses. Furthermore, the mitochondrial proteome is exquisitely sensitive to modifications by reactive oxygen and nitrogen species (ROS/RNS) and thus offers a unique opportunity to investigate the molecular mechanisms underlying hepatic pathobiology from chronic alcohol consumption. Accordingly, this review article will present an overview of the emerging new roles of mitochondria and reactive species in the development of alcoholic liver disease.
MITOCHONDRIAL DYSFUNCTION IN ALCO-HOLIC LIVER DISEASE: BIOENERGETIC DEFECTS AND OXIDANT PRODUCTION
The effects of chronic alcohol consumption on liver have been extensively studied and have advanced our understanding of the molecular mechanisms responsible for alcohol hepatotoxicity. In general, chronic consumption of alcohol causes liver disease via oxidative stress, hypoxia, upregulation of pro-inflammatory cytokines, and bioenergetic defects involving the interactions of multiple liver cell types. Early in the disease process gut-derived endotoxin[10] activates Kupffer cells, which release a variety of potentially toxic substances including cytokines and ROS/RNS that negatively affect hepatic stellate cells and hepatocyte functions[11]. Consequently, a number of responses occur in hepatocytes including increased ROS/RNS production, mitochondrial damage, and altered nitric oxide (NO)-dependent control of respiration[12-14]. This profound disruption in mitochondrial metabolism contributes, in part, to liver disease by placing hepatocytes under bioenergetic stress. This is important because the inability to maintain hepatic ATP levels will predispose the liver to permanent damage due to a depression in the ATP requiring anabolic pathways responsible for replacing damaged and/or lost cellular macromolecules.
One critical change to hepatic mitochondria after chronic consumption of alcohol is a decrease in the rate of ATP synthesis. Conclusive evidence shows that chronic alcohol exposure depresses the activities of all the oxidative phosphorylation complexes by approximately 30%-50%, except Complex II[15,16]. Inhibition of mitochondrial protein synthesis due to alcohol-mediated damage to the mitochondrial DNA[13,17] and ribosomes[18,19] is proposed to contribute to decreased functioning of the oxidative phosphorylation system and depressed rates of ATP synthesis[20,21]. Recently, studies by Cunningham and colleagues have shown that both hepatic energy charge and NADH-linked respiration are decreased in mitochondria isolated from liver of non-human primates allowed to consume alcohol for 18 mo[22]; thus recapitulating the findings observed in rodent studies. This alcohol-dependent loss of mitochondrial function is also predicted to cause further increases in ROS/RNS production and oxidative damage to the organelle and liver.
During electron transport in the respiratory chain, electrons can "leak" from the respiratory complexes and be passed one at a time to molecular oxygen (O2) to form low amounts of superoxide anion (O2●–), which is proposed to be increased in hepatocytes due to alcohol-dependent alterations in mitochondria. Mitochondria contribute to the production of ROS in hepatocytes when alcohol is consumed through the metabolism of alcohol via distinct oxidative mechanisms and chronic alcohol-related alterations to the oxidative phosphorylation system[23,24]. First, ethanol metabolism increases the availability of reducing equivalents (i.e. NADH) to the mitochondrion, which results in the redox active semiquinone intermediates within Complexes I and III to be in a more "reduced" state, thereby facilitating the reduction of O2 to O2●–[25,26] (Figure 1). Moreover, in the chronic alcohol consumer, it is postulated that mitochondrial production of ROS is elevated not only due to increased NADH delivery to the respiratory chain, but also as a consequence of molecular defects to the respiratory complexes caused by chronic alcohol consumption[13,27-29].
Figure 1.
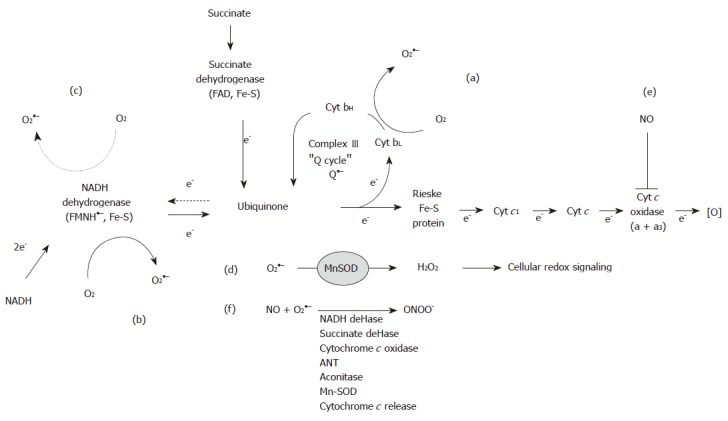
Superoxide production and reactive nitrogen species reactions in mitochondria. During electron transfer superoxide (O2●–) is generated within Complexes I (b, c) and III (a) due to the presence of stable semiquionine anion species. Solid arrows illustrate forward electron flow, whereas dashed arrows indicate reverse electron flow through Complex II to increase O2●– production in ComplexI(c). O2●– dismutation to hydrogen peroxide (H2O2) by manganese superoxide dismutase (MnSOD) affects cellular redox signaling pathways (d). Increased iNOS expression leads to diffusion of nitric oxide (NO) into mitochondria where it inhibits cytochrome c oxidase (e), which increases O2●– generation.
The mechanism of O2●– production by Complex III (ubiquinone cytochrome c reductase) is well understood, as it is linked to the reactions of the ubiquinone or "Q"-cycle[30]. As the levels of cytochrome b are decreased by chronic alcohol consumption[27,29] this defect is postulated to decrease the rate of re-oxidation of the ubisemiquinone anion (Q●–). This resulting increase in the steady-state levels of Q●– would subsequently lead to increased O2●– in mitochondria from alcohol exposed subjects (Figure 1A). In contrast, the mechanism responsible for increased O2●– production by Complex I (NADH dehydrogenase) in response to chronic alcohol consumption remains undefined because the sites responsible for generating O2●– are not known. For example, the semiquinone in the flavin mononucleotide and the various iron-sulfur centers of Complex I have all been implicated as potential sites of O2●– production (Figure 1B). Recent studies have demonstrated that the majority of O2●– generated in mitochondria respiring on the Complex II-linked substrate succinate occurs via reverse electron transport into Complex I[31-33]. This mechanism involves reverse electron flow from succinate to NAD+ providing reducing equivalents to the redox carriers of Complex I, which then function as sites of O2●– production (Figure 1C). The contribution of reverse electron flow through Complex I may be enhanced as a consequence of alcohol-dependent defects to the complex[27,28]. Similarly, mitochondrial levels of ROS may be increased by chronic alcohol consumption as a consequence of increased mitochondrial CYP450 2E1 levels[34] and as a by-product from matrix enzymes such as α-ketoglutarate dehydrogenase[35,36]. These higher rates of ROS production in the alcohol-damaged mitochondria are predicted to negatively affect mitochondrial function through oxidative damage to mitochondrial macro-molecules and cellular function through modulation of cellular redox signaling pathways (Figure 1D).
NITRIC OXIDE, REACTIVE NITROGEN SPECIES, AND MITOCHONDRIA PHYSIOLOGY: IMPACT ON ALCOHOL HEPATOTOXICITY
In this section some of the key characteristics by which nitric oxide (NO) and RNS affect mitochondrial function in the context of alcoholic liver injury will be presented. As discussed above, several studies have demonstrated that chronic alcohol consumption increases hepatocyte ROS production presumably from the mitochondrial respiratory chain[12,25,37]. As these increased levels of ROS exceed those required for signal transduction and detoxification via antioxidant systems, mitochondrial DNA, protein, and lipid damage is predicted to be enhanced as a consequence of chronic alcohol exposure. In addition, NO production is increased in response to chronic alcohol via induction of inducible nitric oxide synthase (iNOS)[12,38]. This has important ramifications for toxicity because NO and its metabolite peroxynitrite (ONOO-) have been implicated as key mediators of mitochondrial dysfunction[39,40]. Indeed, the detrimental effects of NO largely stem from excess NO diffusing into the mitochondrion and reacting with O2●– to produce ONOO-, a reactive metabolite that can directly or indirectly participate in reactions leading to inactivation of mitochondrial proteins via post-translational modifications[41]. Moreover, it is known that NO can regulate mitochondrial function through reversible binding at the heme site in cytochrome c oxidase, which inhibits oxygen consumption[42,43] and through cross-talk mechanisms with soluble guanylate cyclase[44]. Potential impacts of NO and ONOO- on mitochondria are illustrated in Figure 1E and F.
Exposure of mitochondria to low concentrations of NO results in the reversible inhibition of cytochrome c oxidase activity due to the competition of NO with O2 at the binuclear center of the enzyme[45-48]. Thus, NO has the effect to inhibit mitochondrial respiration. Studies from our laboratories have shown a novel role of NO in the pathogenesis of alcohol hepatotoxicity. We have demonstrated that the response of respiration to NO is altered by chronic alcohol consumption such that mitochondria from alcohol-fed animals are more sensitive to NO-dependent inhibition of respiration[14,38]. It is proposed that this loss of control of NO signaling per se, results in excessive inhibition of the respiratory chain leading to bioenergetic dysfunction (i.e. decreased ATP synthesis) and increased ROS production[49], which contribute to the development of alcoholic liver injury. The critical role of NO in alcohol hepatotoxicity is further supported because the increased sensitivity of mitochondrial respiration to inhibition by NO is absent in iNOS knockout mice fed alcohol chronically[38]. Therefore, this finding supports the hypothesis that the early induction of iNOS modifies the control of NO-dependent respiration, which contributes to the development of alcohol-dependent steatosis and inflammation[38,50].
It has been shown that NO-mediated inhibition of respiration alters activation of hypoxia responsive targets in cells such that increased NO interferes with the upregulation of molecules required by cells to adapt to hypoxic stress[51,52]. This may also have the effect of changing oxygen gradients in tissues. Indeed, this is precisely the series of events that we propose occurs with NO in alcohol hepatotoxicity[49], which has been shown to be associated with tissue hypoxia[53,54]. Several studies have clearly demonstrated that acute alcohol exposure "steepens" the hepatic oxygen gradient as a consequence of ethanol oxidation[55,56], thus rendering the pericentral regions of the liver lobule hypoxic[53]. Thus, the effect of acute hypoxia in an individual actively drinking is proposed to be exaggerated in the chronic alcohol consumer as a consequence of the alcohol dependent increase in inhibition of mitochondrial respiration by NO[14]. In fact, studies by Arteel and colleagues demonstrate that chronic alcohol consumption increases liver hypoxia[54]. This concept is important to consider because it suggests that the interaction of hypoxia and disrupted NO signaling in mitochondria may accelerate the progression from steatosis to more severe liver pathologies. Moreover, it is these conditions that will lead to the irreversible post-translational modification and inactivation of mitochondrial proteins, which are proposed to contribute to alcohol-mediated mitochondrial dysfunction and hepatic pathobiology.
MODIFICATION TO THE LIVER MITO-CHONDRIAL PROTEOME IN RESPONSE TO CHRONIC ALCOHOL CONSUMPTION
From the discussion above it is clear that increased NO and altered control of respiration by NO can lead to excessive ROS/RNS formation and the post-translational modification of mitochondrial proteins. Specifically, it has been shown that ROS, RNS, and reactive lipid species can modify critical amino acid residues thereby disrupting the catalytic function of proteins. As stated above, these toxic effects on proteins largely stem from the diffusion of NO into mitochondria and its reaction with O2●– to generate more reactive species, such as ONOO-, other secondary RNS, and reactive lipid products. Because alterations in the redox state of protein thiols are important in regulating mitochondrial function such as respiration and oxidant production[57,58], the identification of proteins with oxidized and/or modified thiol groups is critical for elucidating the mitochondrial defects that contribute to alcoholic liver disease.
A number of reversible and irreversible modifications to cysteine residues are known to occur upon interaction of free sulfhydryl groups (-SH) with ROS, RNS, and reactive lipids. Reversible modifications to thiols include the formation of nitrosothiols (P-SNO), sulfenic acids (P-SOH) and mixed disulfides (P-SSG). Cysteine residues can also be irreversibly oxidized to higher oxidation states such as sulfinic (P-SO2H) and sulfonic (P-SO3H) acids by ROS and RNS. Each of these modifications has the potential to elicit a unique biological response that may disrupt normal mitochondrial functions. Studies have linked the oxidation and modification of protein thiols with induction of the mitochondrial permeability transition, alterations in energy metabolism, and oxidant production[59]. Our laboratories have shown an alcohol-dependent loss of function of the mitochondrial low Km aldehyde dehydrogenase (ALDH) as a consequence of oxidative modification of thiols[60]. This is significant because inactivation of this important detoxification enzyme could potentially lead to increased levels of acetaldehyde and other reactive aldehyde species, which themselves have been shown to inactivate proteins through cysteine modifications[61,62]. Recently, Song and colleagues also demonstrated alcohol-dependent inactivation of ALDH and several β-oxidation enzymes via oxidation and nitrosation of thiols[63]. Taken together, these findings are consistent with the concept that modification of protein thiols may contribute to alcoholic steatosis and mitochondrial dysfunction through inactivation of proteins critical to the energy conservation pathways in liver.
While changes in the redox status of cysteine residues are known to affect mitochondrial activities, other types of post-translational modification should be considered as well. Tyrosine nitration (P-NO2) is generally thought to induce permanent loss of protein function[64] however recent studies suggest that tyrosine nitration may be oxygen-regulated, target-selective, and reversible such that it may function as a "nitrative signaling" process controlling mitochondrial energy metabolism[65,66]. Studies have also established that 4-hydroxynonenal (4-HNE), a reactive lipid, may play an important role in alcohol hepatotoxicity via modification of key signaling proteins[67,68] and inhibition of cytochrome c oxidase activity[69,70]. 4-HNE has also been shown to inhibit several of the matrix dehydrogenase enzymes[71,72] and proteins of the β-oxidation system[73]. Moreover, electrophilic lipids like 4-HNE and the cyclopentenone 15-deoxy-∆12,14-prostaglandin J2 have been shown to co-localize to mitochondria and induce mitochondrial ROS production[74]. These results provide strong evidence suggesting that reactive lipids may disrupt mitochondrial function through selective targeting of mitochondrial enzymes.
In addition to post-translational modifications, it is also proposed that changes in the levels (i.e. abundances) of proteins that comprise the mitochondrial proteome may also negatively affect mitochondrial bioenergetics leading to liver injury. Early studies by Coleman and Cunningham established a key link between the chronic alcohol-related defects in Complexes I, III, IV, and V and losses in the 13 mitochondrial encoded polypeptides and redox centers that comprise the oxidative phosphorylation system complexes[27,75,76]. Similarly, using a variety of proteomics approaches our laboratories have extended these findings to demonstrate a coordinated decrease in both mitochondrial and nuclear encoded subunits of the respiratory complexes, particularly those that comprise cytochrome c oxidase[12,13]. Moreover, proteomic analyses revealed that 40 additional mitochondrial proteins had altered levels in response to chronic alcohol consumption[13]. These studies are interesting in that, previously unidentified alterations in several key energy metabolism enzymes of β-oxidation, the TCA cycle, and amino acid metabolism, as well as several mitochondrial chaperones were found to be altered by chronic alcohol exposure. Importantly, these changes can also be linked to pathways with a clear impact on one of the primary pathologies of alcoholic liver disease, i.e. steatosis. These findings highlight the power of proteomics to detect previously unidentified alterations to "vulnerable" mitochondrial sub-proteomes following chronic alcohol consumption that contribute, in part, to the development of alcoholic liver disease (Figure 2). These alterations may include changes in protein levels as well as post-translational modifications to this susceptible sub-set of mitochondrial proteins.
Figure 2.
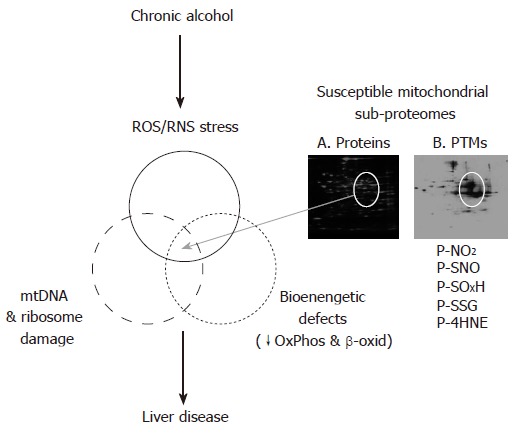
Overlapping alterations to the liver mitochondrial proteome contribute to the development of alcohol-induced liver disease. This figure illustrates the concept that there is a unique mitochondrial sub-proteome that when altered by chronic alcohol mediated mtDNA/ribosome damage, increased ROS/RNS production, and energy deficits contribute to the development of alcohol hepatotoxicity. These alterations involve changes in both protein levels (panel A) and post-translational modifications (PTMs, panel B) to this susceptible population of mitochondrial proteins.
In summary, mitochondria play a variety of roles in a number of essential cellular functions including energy production and homeostasis, redox cell signaling, and apoptosis. Disruption of mitochondrial function, manifested by the inability to maintain cellular ATP levels is critical to the development of chronic alcohol-induced liver injury. Moreover, as a primary source for the formation of ROS and RNS, the mitochondrion is recognized as a critical component involved in cellular stress responses. While the sequence of events leading to alcohol induced mitochondrial dysfunction and liver injury remains undefined, emerging evidence suggests that: (1) interaction of NO with the respiratory chain may predispose hepatocytes to hypoxic injury and (2) post-translational modifications of critical residues within mitochondrial proteins by reactive species may alter cellular functions including energy metabolism and redox signaling. Similarly, an alcohol-mediated increase in ROS/RNS can damage mitochondrial DNA and ribosomes resulting in decreased mitochondrial protein synthesis, which ultimately translates into a severe mitochondrial dysfunction. Investigations have provided novel mechanistic information regarding the impact of chronic alcohol consumption on the mitochondrial proteome; providing unique insight into the molecular mechanisms responsible for disease. This knowledge will clearly advance the design and testing of novel mitochondria-specific therapeutics in the treatment of alcoholic liver disease.
Footnotes
Supported by NIH/NIAAA grants AA13395 (VDU) and AA15172 (SMB)
S- Editor Ma N L- Editor Alpini GD E- Editor Liu Y
References
- 1.Alcohol-attributable deaths and years of potential life lost--United States, 2001. MMWR Morb Mortal Wkly Rep. 2004;53:866–870. [PubMed] [Google Scholar]
- 2.Day CP. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006;26:1021–1028. doi: 10.1111/j.1478-3231.2006.01323.x. [DOI] [PubMed] [Google Scholar]
- 3.Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Crocè L, Sasso F, Pozzato G, Cristianini G, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41:845–850. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Zayadi AR. Heavy smoking and liver. World J Gastroenterol. 2006;12:6098–6101. doi: 10.3748/wjg.v12.i38.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hézode C, Lonjon I, Roudot-Thoraval F, Mavier JP, Pawlotsky JM, Zafrani ES, Dhumeaux D. Impact of smoking on histological liver lesions in chronic hepatitis C. Gut. 2003;52:126–129. doi: 10.1136/gut.52.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pessione F, Ramond MJ, Njapoum C, Duchatelle V, Degott C, Erlinger S, Rueff B, Valla DC, Degos F. Cigarette smoking and hepatic lesions in patients with chronic hepatitis C. Hepatology. 2001;34:121–125. doi: 10.1053/jhep.2001.25385. [DOI] [PubMed] [Google Scholar]
- 7.Zein CO, Beatty K, Post AB, Logan L, Debanne S, McCullough AJ. Smoking and increased severity of hepatic fibrosis in primary biliary cirrhosis: A cross validated retrospective assessment. Hepatology. 2006;44:1564–1571. doi: 10.1002/hep.21423. [DOI] [PubMed] [Google Scholar]
- 8.Zhu K, Moriarty C, Caplan LS, Levine RS. Cigarette smoking and primary liver cancer: a population-based case-control study in US men. Cancer Causes Control. 2007;18:315–321. doi: 10.1007/s10552-006-0105-8. [DOI] [PubMed] [Google Scholar]
- 9.Marrero JA, Fontana RJ, Fu S, Conjeevaram HS, Su GL, Lok AS. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J Hepatol. 2005;42:218–224. doi: 10.1016/j.jhep.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Arai M. Effect of ethanol on the intestinal uptake of endotoxin. Nihon Shokakibyo Gakkai Zasshi. 1986;83:1060. [PubMed] [Google Scholar]
- 11.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 12.Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, et al. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol. 2006;291:G857–G867. doi: 10.1152/ajpgi.00044.2006. [DOI] [PubMed] [Google Scholar]
- 13.Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, et al. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem. 2004;279:22092–22101. doi: 10.1074/jbc.M402245200. [DOI] [PubMed] [Google Scholar]
- 14.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38:141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham CC, Coleman WB, Spach PI. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol. 1990;25:127–136. doi: 10.1093/oxfordjournals.alcalc.a044987. [DOI] [PubMed] [Google Scholar]
- 16.Bailey SM. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic Res. 2003;37:585–596. doi: 10.1080/1071576031000091711. [DOI] [PubMed] [Google Scholar]
- 17.Cahill A, Wang X, Hoek JB. Increased oxidative damage to mitochondrial DNA following chronic ethanol consumption. Biochem Biophys Res Commun. 1997;235:286–290. doi: 10.1006/bbrc.1997.6774. [DOI] [PubMed] [Google Scholar]
- 18.Cahill A, Cunningham CC. Effects of chronic ethanol feeding on the protein composition of mitochondrial ribosomes. Electrophoresis. 2000;21:3420–3426. doi: 10.1002/1522-2683(20001001)21:16<3420::AID-ELPS3420>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Patel VB, Cunningham CC. Altered hepatic mitochondrial ribosome structure following chronic ethanol consumption. Arch Biochem Biophys. 2002;398:41–50. doi: 10.1006/abbi.2001.2701. [DOI] [PubMed] [Google Scholar]
- 20.Bailey SM, Cunningham CC. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcohol Clin Exp Res. 1999;23:1210–1218. [PubMed] [Google Scholar]
- 21.Spach PI, Bottenus RE, Cunningham CC. Control of adenine nucleotide metabolism in hepatic mitochondria from rats with ethanol-induced fatty liver. Biochem J. 1982;202:445–452. doi: 10.1042/bj2020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivester P, Roberts LJ, Young T, Stafforini D, Vivian J, Lees C, Young J, Daunais J, Friedman D, Rippe RA, et al. Ethanol self-administration and alterations in the livers of the cynomolgus monkey, Macaca fascicularis. Alcohol Clin Exp Res. 2007;31:144–155. doi: 10.1111/j.1530-0277.2006.00276.x. [DOI] [PubMed] [Google Scholar]
- 23.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 24.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey SM, Cunningham CC. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology. 1998;28:1318–1326. doi: 10.1002/hep.510280521. [DOI] [PubMed] [Google Scholar]
- 26.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 27.Coleman WB, Cunningham CC. Effects of chronic ethanol consumption on the synthesis of polypeptides encoded by the hepatic mitochondrial genome. Biochim Biophys Acta. 1990;1019:142–150. doi: 10.1016/0005-2728(90)90136-r. [DOI] [PubMed] [Google Scholar]
- 28.Thayer WS, Ohnishi T, Rubin E. Characterization of iron-sulfur clusters in rat liver submitochondrial particles by electron paramagnetic resonance spectroscopy. Alterations produced by chronic ethanol consumption. Biochim Biophys Acta. 1980;591:22–36. doi: 10.1016/0005-2728(80)90217-0. [DOI] [PubMed] [Google Scholar]
- 29.Thayer WS, Rubin E. Molecular alterations in the respiratory chain of rat liver after chronic ethanol consumption. J Biol Chem. 1981;256:6090–6097. [PubMed] [Google Scholar]
- 30.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 31.Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol. 2003;64:1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- 32.Lambert AJ, Brand MD. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 34.Robin MA, Sauvage I, Grandperret T, Descatoire V, Pessayre D, Fromenty B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett. 2005;579:6895–6902. doi: 10.1016/j.febslet.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 35.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- 36.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kessova IG, Cederbaum AI. Mitochondrial alterations in livers of Sod1-/- mice fed alcohol. Free Radic Biol Med. 2007;42:1470–1480. doi: 10.1016/j.freeradbiomed.2007.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, Darley-Usmar VM. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40:565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 39.Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 2002;383:401–409. doi: 10.1515/BC.2002.044. [DOI] [PubMed] [Google Scholar]
- 40.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 41.Stewart VC, Heales SJ. Nitric oxide-induced mitochondrial dysfunction: implications for neurodegeneration. Free Radic Biol Med. 2003;34:287–303. doi: 10.1016/s0891-5849(02)01327-8. [DOI] [PubMed] [Google Scholar]
- 42.Brookes PS, Kraus DW, Shiva S, Doeller JE, Barone MC, Patel RP, Lancaster JR, Darley-Usmar V. Control of mitochondrial respiration by NO*, effects of low oxygen and respiratory state. J Biol Chem. 2003;278:31603–31609. doi: 10.1074/jbc.M211784200. [DOI] [PubMed] [Google Scholar]
- 43.Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 44.Nisoli E, Clementi E, Moncada S, Carruba MO. Mitochondrial biogenesis as a cellular signaling framework. Biochem Pharmacol. 2004;67:1–15. doi: 10.1016/j.bcp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Brown GC. Nitric oxide inhibition of cytochrome oxidase and mitochondrial respiration: implications for inflammatory, neurodegenerative and ischaemic pathologies. Mol Cell Biochem. 1997;174:189–192. [PubMed] [Google Scholar]
- 46.Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. Am J Physiol Cell Physiol. 2007;292:C1993–C2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 47.Poderoso JJ, Lisdero C, Schöpfer F, Riobó N, Carreras MC, Cadenas E, Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 48.Torres J, Darley-Usmar V, Wilson MT. Inhibition of cytochrome c oxidase in turnover by nitric oxide: mechanism and implications for control of respiration. Biochem J. 1995;312(Pt1):169–173. doi: 10.1042/bj3120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, Bailey SM, Darley-Usmar VM. Nitroxia: the pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med. 2005;38:297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 50.McKim SE, Gäbele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, Connor HD, Mason RP, Doll MA, Hein DW, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 51.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 52.Kozhukhar AV, Yasinska IM, Sumbayev VV. Nitric oxide inhibits HIF-1alpha protein accumulation under hypoxic conditions: implication of 2-oxoglutarate and iron. Biochimie. 2006;88:411–418. doi: 10.1016/j.biochi.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Arteel GE, Raleigh JA, Bradford BU, Thurman RG. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: role of Kupffer cells. Am J Physiol. 1996;271:G494–G500. doi: 10.1152/ajpgi.1996.271.3.G494. [DOI] [PubMed] [Google Scholar]
- 54.Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–926. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- 55.Eguchi H, Sato N, Matsumura T, Kawano S, Kamada T. In vivo estimation of oxygen saturation of hemoglobin in hepatic lobules in rats. Adv Exp Med Biol. 1988;222:591–596. doi: 10.1007/978-1-4615-9510-6_72. [DOI] [PubMed] [Google Scholar]
- 56.Yuki T, Thurman RG. The swift increase in alcohol metabolism. Time course for the increase in hepatic oxygen uptake and the involvement of glycolysis. Biochem J. 1980;186:119–126. doi: 10.1042/bj1860119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 58.Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 59.Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 60.Venkatraman A, Landar A, Davis AJ, Ulasova E, Page G, Murphy MP, Darley-Usmar V, Bailey SM. Oxidative modification of hepatic mitochondria protein thiols: effect of chronic alcohol consumption. Am J Physiol Gastrointest Liver Physiol. 2004;286:G521–G527. doi: 10.1152/ajpgi.00399.2003. [DOI] [PubMed] [Google Scholar]
- 61.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 62.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol. 2006;19:102–110. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- 63.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 64.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koeck T, Fu X, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. Rapid and selective oxygen-regulated protein tyrosine denitration and nitration in mitochondria. J Biol Chem. 2004;279:27257–27262. doi: 10.1074/jbc.M401586200. [DOI] [PubMed] [Google Scholar]
- 66.Koeck T, Stuehr DJ, Aulak KS. Mitochondria and regulated tyrosine nitration. Biochem Soc Trans. 2005;33:1399–1403. doi: 10.1042/BST0331399. [DOI] [PubMed] [Google Scholar]
- 67.Sampey BP, Carbone DL, Doorn JA, Drechsel DA, Petersen DR. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Mol Pharmacol. 2007;71:871–883. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 68.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J Biol Chem. 2007;282:1925–1937. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen JJ, Schenker S, Henderson GI. 4-hydroxynonenal levels are enhanced in fetal liver mitochondria by in utero ethanol exposure. Hepatology. 1997;25:142–147. doi: 10.1002/hep.510250126. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Robinson NC, Schenker S, Frosto TA, Henderson GI. Formation of 4-hydroxynonenal adducts with cytochrome c oxidase in rats following short-term ethanol intake. Hepatology. 1999;29:1792–1798. doi: 10.1002/hep.510290611. [DOI] [PubMed] [Google Scholar]
- 71.Humphries KM, Szweda LI. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 72.Humphries KM, Yoo Y, Szweda LI. Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry. 1998;37:552–557. doi: 10.1021/bi971958i. [DOI] [PubMed] [Google Scholar]
- 73.Hussain SN, Matar G, Barreiro E, Florian M, Divangahi M, Vassilakopoulos T. Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L996–1003. doi: 10.1152/ajplung.00337.2005. [DOI] [PubMed] [Google Scholar]
- 74.Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, Zanoni G, Vidari G, Morrow JD, Darley-Usmar VM. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2006;290:H1777–H1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- 75.Colell A, García-Ruiz C, Miranda M, Ardite E, Marí M, Morales A, Corrales F, Kaplowitz N, Fernández-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 76.Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta. 1991;1058:178–186. doi: 10.1016/s0005-2728(05)80235-x. [DOI] [PubMed] [Google Scholar]