

Abstract
Myostatin regulates skeletal muscle size via the activin receptor IIB (ActRIIB). However, its effect on muscle energy metabolism and energy-dependent muscle function remains largely unexplored. This question needs to be solved urgently since various therapies for neuromuscular diseases based on blockade of ActRIIB signaling are being developed. Here, we show in mice, that 4-month pharmacological abrogation of ActRIIB signaling by treatment with soluble ActRIIB-Fc triggers extreme muscle fatigability. This is associated with elevated serum lactate levels and a severe metabolic myopathy in the mdx mouse, an animal model of Duchenne muscular dystrophy. Blockade of ActRIIB signaling downregulates porin, a crucial ADP/ATP shuttle between cytosol and mitochondrial matrix leading to a consecutive deficiency of oxidative phosphorylation as measured by in vivo Phophorus Magnetic Resonance Spectroscopy (31P-MRS). Further, ActRIIB blockade reduces muscle capillarization, which further compounds the metabolic stress. We show that ActRIIB regulates key determinants of muscle metabolism, such as Pparβ, Pgc1α, and Pdk4 thereby optimizing different components of muscle energy metabolism. In conclusion, ActRIIB signaling endows skeletal muscle with high oxidative capacity and low fatigability. The severe metabolic side effects following ActRIIB blockade caution against deploying this strategy, at least in isolation, for treatment of neuromuscular disorders.
Introduction
Skeletal muscle has inbuilt control mechanisms to prevent overgrowth. This function is executed, at least in part, by secreted molecules including members of the transforming growth factor-β (TGF-β) family, especially myostatin.1 Myostatin signals via its transmembrane activin receptor IIB (ActRIIB) and suppression of this pathway stimulates muscle growth.2,3 In the past few years, strategies have been developed to treat muscular dystrophies, muscle wasting, and cachexia by blocking the myostatin/ActRIIB pathway with first of many clinical trials already being concluded (ClinicalTrials.gov NCT01099761, NCT01519349, NCT01423110, NCT01669174, NCT01601600, and NCT01433263). However, it remains a matter of controversy whether the hypertrophic muscles that form as a result of blocking myostatin/ActRIIB signaling confer any functional benefit, because a number of groups have reported loss of specific force of larger muscles in myostatin knockout mice and a faster fatigability (Mstn−/−).4,5,6 In addition, myostatin knockout leads to a change of muscle contractile and metabolic characteristics towards a “glycolytic” phenotype,5,7,8 commonly attributed to a change in muscle specification during development. In contrast to the constitutive myostatin deficiency of Mstn−/− mice, postnatal treatment with soluble activin IIB receptor (sActRIIB-Fc) in adult mice blocks myostatin/ActRIIB signaling and increases muscle force without altering the fiber-type composition.9,10 Similar results have been obtained in the mdx mouse model of Duchenne muscular dystrophy (DMD).11,12 However, a recent transcriptome profiling demonstrated a downregulation of genes involved in oxidative phosphorylation and mitochondrial function following treatment with sActRIIB-Fc.13 Another study revealed a faster decline of muscle force following repetitive stimulation.14 Whether those changes reflect solely a change towards a faster muscle phenotype or a relevant mitochondrial dysfunction is presently unknown. In the view of ongoing clinical trials, we need to address the question of how myostatin blockade affects the metabolism of dystrophic muscle in mdx mice, which already has a preexisting deficit of mitochondrial function.15,16,17 Here we explored the hypothesis that ActRIIB signaling is a key regulator of oxidative metabolism in the adult muscle. We thus set out to systematically investigate in adult wild-type and mdx mice, how postnatal blockade of ActRIIB signaling using sActRIIB-Fc might affect muscle energy metabolism and energy-dependent muscle function. Our data conclusively show the importance of ActRIIB signaling as a pivotal link that acts to balance muscle size and strength against endurance capacity via optimization of energy metabolism.
Results
ActRIIB blockade in adult wild-type mice increases fatigability
Muscles of Mstn−/− mice, a model of constitutive inhibition of signaling via ActRIIB, exhibit a congenital fiber-type profile that is characterized by an increase in the number of fast “glycolytic” myosin heavy chain (MHCIIB) fibers and concomitant loss of “oxidative” (MHCI, MHCIIA) fibers, which entails changes in muscle function, exercise capacity, and muscle metabolism.4,7,8,18,19 To circumvent the effects of congenital fiber-type switching, we here inhibited ActRIIB signaling in adult wild-type mice with a soluble form of the activin receptor fused with the Fc-fragment of mouse IgG (sActRIIB-Fc). Four months of treatment promoted robust skeletal muscle growth together with a significant increase in total body weight, confirming previously published data (Supplementary Figure S1).20 Importantly, the increase in muscle mass was not accompanied by fiber-type conversion (Supplementary Figure S2). The absolute maximal force of EDL and soleus muscle increased in parallel with muscle size (Figure 1a,c). Specific maximal force was conserved, implicating a proportional increase of force and muscle mass (Figure 1b). However, sActRIIB-Fc treatment also increased muscle fatigue (Figure 1c,d) and mice exhausted precociously during incremental speed running tests (Figure 1e,g). Serum lactate, being already significantly increased at resting state, rose to pathological levels following incremental speed running (Figure 1f,g). The concept of “Critical Speed” accurately reflects the capacity for aerobic exercise and is based on the proportional relationship between “covered distance” and “time to exhaustion” at different velocities.21 During the 4-month treatment period, we found a steady decline in Critical Speed in the treatment and control group, however, the decline over time was by far larger in sActRIIB-Fc-treated animals as compared to phosphate-buffered saline (PBS)-treated mice (Figure 1h,i).
Figure 1.

Treatment of adult wild-type mice with soluble activin IIB receptor (sActRIIB-Fc). All tests were done after a 4-month treatment of the wild-type mice with either sActRIIB-Fc or PBS (controls). (a) Absolute maximal force (n = 10 for each condition), and (b) specific maximal force of EDL and soleus muscles (n = 10 for each condition). (c) Force recordings during the fatigue protocol over 180 seconds, and (d) fatigue resistance of EDL (n = 9 for PBS-treated mice and n = 8 for sActRIIB-Fc-treated mice) and soleus muscles (n = 10 for PBS-treated mice and n = 9 for sActRIIB-Fc-treated mice). (e) Running distances during incremental speed running until exhaustion (n = 5 for each condition). (f) Serum lactate levels at rest and 5 minutes after incremental speed running until exhaustion (n = 5 for each condition). (g) A plot depicting the relationship between travel distance until exhaustion during incremental speed running and serum lactate, which was measured 5 minutes after exhaustion, for individual mice (n = 5 for each condition). (h) Critical speed before and after 1, 2, and 4 months of treatment with sActRIIB-Fc in comparison to PBS-treated control mice (n = 5 for each condition). (i) A plot depicts the proportional relationship between distance run (y-axis) and time to exhaustion (x-axis) at different velocities. The slope of the regression line indicates the Critical Speed. Values are shown as means ± SEM. P values were calculated using the nonparametric U-test.
Severe exercise intolerance in dystrophic mdx mice following treatment with sActRIIB-Fc
In Duchenne muscular dystrophy and its mdx mouse model, oxidative metabolism is compromised due to membrane damage and the resulting intracellular calcium overload.15,16,17 Having shown that ActRIIB blockade decreased aerobic exercise capacity in wild-type mice, we now investigated what effect administration of sActRIIB-Fc would have on the metabolic phenotype of mdx mice. This information would have important clinical implications for the strategies to use ActRIIB blockade for the treatment of muscular dystrophies. Despite a massive increase in skeletal muscle mass after sActRIIB-Fc treatment (Supplementary Figure S1), absolute maximal force decreased in soleus muscle and most notably specific force in both EDL and soleus muscles (Figure 2a,b), serving as functional evidence for increased myopathic changes of sActRIIB-treated dystrophic mdx muscle. Interestingly, sActRIIB-Fc treatment did not cause any greater force decline during repetitive stimulation (Figure 2c,d). Electromyography excluded problems in neuromuscular transmission but revealed abnormal spontaneous potentials and the presence of complex repetitive discharges in both PBS and sActRIIB-Fc treatment groups of the mdx mice (Supplementary Figure S3). Such polyphasic potentials are characteristic of dystrophic mdx muscle.22 As voluntary motor activity seemed reduced when observing sActRIIB-Fc-treated mdx mice, we proceeded to analyze their exercise behavior. Remarkably, at the end of the treatment period, sActRIIB-Fc-treated mdx mice suffered from severe exercise intolerance associated with a pathological serum lactate increase (Figure 2e–g; Supplementary Video S1). It is important to note, that exercise capacity of mdx mice declined throughout the 4-month treatment period, however, to a far larger extent in sActRIIB-Fc-treated mdx mice than in the PBS-treated control group (Figure 2h,i).
Figure 2.

Treatment of adult mdx mice with soluble activin IIB receptor (sActRIIB-Fc). All tests were performed after a 4-month treatment of the mdx mice with either sActRIIB-Fc or PBS (controls). (a) Absolute maximal force (n = 9 for each condition for EDL muscles and n = 10 for each condition for soleus muscles), and (b) specific maximal force of EDL (n = 9 for each condition) and soleus muscles (n = 10 for each condition). (c) Force recordings during the fatigue protocol over 180 seconds of EDL and soleus muscles, and (d) fatigue resistance for EDL (n = 8 for each condition) and soleus (n = 10 for PBS-treated mice and n = 8 for sActRIIB-Fc-treated mice). (e) Running distance during incremental speed running until exhaustion (n = 5 for each condition). (f) Serum lactate levels at rest and 5 minutes after incremental speed running until exhaustion (n = 5 for each condition). (g) A plot depicts the relationship between travel distance until exhaustion during incremental speed running and serum lactate, which was measured 5 minutes after exhaustion, for individual mice (n = 5 for each condition). (h) Critical speed before and after 1, 2, and 4 months of treatment with sActRIIB-Fc in comparison to PBS (n = 5 for each condition). (i) A plot depicts the proportional relationship between distance run (y-axis) and time to exhaustion (x-axis) at different velocities. The slope of the regression line indicates the critical speed. Values are shown as means ± SEM. P values were calculated using the nonparametric U-test.
sActRIIB-Fc treatment affects muscle capillarization
The ability of mitochondria to produce ATP critically depends on the oxygen supply via tissue blood perfusion, thus a combination of hypoperfusion plus exercise-induced hypoxemia might explain the severe exercise intolerance. Treatment with sActRIIB-Fc caused a drop in capillary density especially in the oxidative soleus muscle from mdx mice with a subsequent increase in the capillary domain (Figure 3a–d). The increase of the capillary domain was also found in glycolytic EDL muscles of mdx mice, albeit to a lesser degree (Supplementary Figure S4a–d). Treatment of mice as well as of C2C12 myotubes with sActRIIB-Fc downregulated expression of Vegf-A suggesting an indirect negative effect of myostatin blockade on capillary formation (Figure 3e,f). Interestingly, Vegf-A expression in mdx mice was much lower than in wild-type mice, and treatment with sActRIIB-Fc did not decrease Vegf-A mRNA-abundance any further (Figure 3e), suggesting the presence of additional mechanisms for regulating capillary density. In this regard, the findings of Hayot et al.23 are of special interest, who reported an induction of myostatin expression in muscles of rats exposed to chronic hypoxia and in patients with chronic obstructive pulmonary disease. The authors interpreted these findings as a potential cause for the muscle wasting that is often seen in chronic obstructive pulmonary disease patients. These findings, however, could also be interpreted as a compensatory upregulation of myostatin to improve the metabolic functioning and to increase capillary density in a state of chronic hypoxia. It is of special interest that endothelial cells strongly express the mRNAs of transmembrane receptors (ActRIIA/B and ALK4/5) for myostatin or its homologs, whereas myostatin mRNA was only expressed at low levels (Figure 3g). Treatment of endothelial cells (Human umbilical vein endothelial cell line) with increasing dosages of recombinant myostatin in vitro increased the cell doubling time in culture, verifying a direct effect of myostatin or its homologs on endothelial cell proliferation (Supplementary Figure S4e), however, the exact ligands of muscle endothelial cell regulation in vivo remain to be determined.
Figure 3.
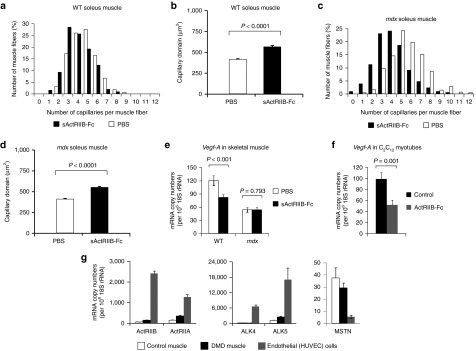
The effect of myostatin/ActRIIB signaling on vascularization. All investigations were done after a 4-month treatment of wild-type and mdx mice with either soluble activin IIB receptor (sActRIIB-Fc) or PBS (controls). (a–d) Capillarization of wild-type soleus (n = 430 fibers from PBS-treated muscles (n = 3) and n = 300 fibers from sActRIIB-Fc-treated muscles (n = 3)) and mdx soleus muscle (n = 605 fibers from PBS-treated muscles (n = 3) and n = 836 fibers from sActRIIB-Fc-treated muscles (n = 4)). Histograms in (a) and (c) depict the distribution of capillaries per muscle fiber (in (%)), whereas diagrams in (b) and (d) depict the capillary domain, the fiber area per capillary (in (µm2)). Values are depicted as means ± SEM. (e) Vegf-A relative mRNA copy numbers as expressed per 106 × 18S rRNA copies in wild-type TA muscle (n = 5 for each condition). (f) Vegf-A relative mRNA-copy numbers in C2C12 myotubes following 24 hours treatment with sActRIIB-Fc in comparison to control cultures (n = 3 for each condition). (g) Relative mRNA-copy numbers of MSTN and myostatin receptors ActRIIA/B and ALK4/5 in cultures of human umbilical vein endothelial cells (HUVEC) in comparison to muscle samples from a healthy control and a patient with Duchenne muscular dystrophy. Values are shown as means ± SEM. P values were calculated using the nonparametric U-test.
ActRIIB signaling regulates Pgc1α and Ppar transcription factors
The exercise intolerance and lactic acidosis following ActRIIB blockade suggests underlying changes in muscle metabolism, a hypothesis supported by previous transcriptome profiling.13 In agreement, we show that the copy numbers of Pgc1α and Pparβ, which are key transcription factors promoting oxidative metabolism in skeletal muscle, are downregulated after treatment with sActRIIB-Fc (Figure 4b,c) and following treatment of C2C12 myotubes with sActRIIB-Fc (Figure 4g). On the protein level, downregulation of Pgc1α was more pronounced in the mdx muscle if referred to desmin abundance (Figure 4a). Such loss of oxidative properties was accompanied by a compensatory activity increase of enolase, a key glycolytic enzyme (Figure 4d). Furthermore, mRNA levels of Pdk4, an inhibitor of pyruvate dehydrogenase (Pdh) and a regulatory switch of substrate utilization from glucose towards fatty acids,24 was strongly decreased following sActRIIB-Fc treatment in mdx mice (Figure 4e). We thus expected an inhibitory effect of sActRIIB-Fc treatment on β-oxidation and found a downregulation of Cpt1b mRNA levels (Figure 4f). Likewise, sActRIIB-Fc treatment of C2C12 myotubes reduced expression of genes controlling oxidative metabolism and β-oxidation within 24 hours of treatment (Figure 4g), implying a direct effect of myostatin signaling in the regulation of these genes.
Figure 4.
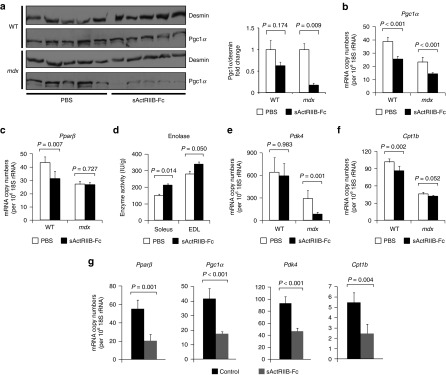
Effect of myostatin/ActRIIB signaling on muscle metabolic phenotype. All investigations were done after a 4-month treatment of wild-type (n = 5 for each condition) and mdx mice (n = 5 for each condition) with either sActRIIB-Fc or PBS (controls). (a) Western blots (left side) depict bands for Pgc1α referenced to Desmin. The bar chart (right side) depicts the quotients of Pgc1α/Desmin band densities. (b) Pgc1α and (c) Pparβ relative copy numbers in the TA muscle from wild-type and mdx mice as expressed per 106 × 18S rRNA copies. (d) Enolase enzymatic activity in wild-type EDL (n = 5 for each condition) and wild-type soleus muscles (n = 5 for each condition). (e,f) Relative mRNA copy numbers of genes involved in the regulation of the oxidative metabolism in TA muscle from wild-type and mdx mice and (g) from C2C12 myotubes following 24 hours treatment with sActRIIB-Fc in comparison to control cultures (n = 3 for each condition). Values are shown as means ± SEM. P values were calculated using the nonparametric U-test. sActRIIB-Fc, soluble activin IIB receptor.
We further focused our attention on the neuronal nitric oxide synthase (Nos1, nNos), because it is well known that the sarcolemmal presence of the Nos1 enzyme is strongly reduced in the absence of its binding partner dystrophin in patients with DMD and in mdx mice.25,26 The resulting dysregulation of NO synthesis entails a failure of contraction-induced vasodilatation as well as changes in the cellular calcium homeostasis associated with exacerbated postexercise fatigability, exercise-induced muscle edema, and cell necrosis.27,28 We thus wondered, whether sActRIIB-Fc treatment would influence sarcolemmal Nos1 expression, hence further compromising the pathophysiological effect of sActRIIB-Fc on vasculature and oxidative metabolism. As expected, we found a strong decrease of Nos1 mRNA copy numbers in mdx muscles of both treatment groups in comparison to wild-type muscle (Supplementary Figure S11a), which was paralleled by a strong decrease of sarcolemmal expression of Nos1 protein in mdx muscle (Supplementary Figure S11b). Furthermore, treatment with sActRIIB-Fc diminished Nos1 transcription in wild-type and mdx muscle (Supplementary Figure S11a). However, subsarcolemmal Nos1 protein content remained unchanged (Supplementary Figure S11b), and western blot did not reveal any changes in Nos1 protein levels in wild-type mice (Supplementary Figure S11c), whereas Nos1 protein levels in mdx mice were below detection levels (data not shown). This suggests that sActRIIB-Fc treatment unlikely aggravates NO dysregulation of dystrophin deficient muscle, although further experiments are required to ascertain or to exclude a role of the ActRIIB-receptor on NO signaling.
Reduced oxidative metabolism in mdx muscle following treatment with sActRIIB-Fc
It should be noted that the mRNA and protein levels of key regulatory genes (Pparβ, Pgc1α) important for oxidative metabolism (Figure 4a–c) were significantly lower in mdx than in wild-type mice, supporting previous findings that oxidative muscle metabolism is depressed in dystrophic muscle to some extent.15,29
We therefore studied in real-time the response of the oxidative metabolism to a standardized bout of exercise in anesthetized mdx mice either treated with PBS (controls) or sActRIIB-Fc. Muscle function and energy metabolism were assessed strictly noninvasively in calf muscle with an innovative experimental setup using phosphorus (31P) nuclear magnetic resonance spectroscopy (MRS).30 An exercise bout of 6 minutes consisting of repeated maximal isometric contractions was induced in vivo by transcutaneous electrostimulation. After induced repeated contractions fatigue levels (Figure 5a), intracellular acidosis (Figure 5b) as well as phosophocreatine (PCr) consumption (Figure 5c) were similar in both groups. However, the time constant of postexercise phosphocreatine resynthesis (τPCr) was significantly prolonged in sActRIIB-Fc-treated mdx mice (Figure 5d,e). Given that PCr synthesis during the postexercise recovery period relies exclusively on oxidative ATP synthesis, τPCr has largely been acknowledged as an important in vivo index of oxidative mitochondrial capacity. Hence, the prolonged τPCr demonstrates that sActRIIB-Fc treatment reduces oxidative metabolism in vivo.
Figure 5.

In vivo investigation of muscle function and oxidative metabolism in mdx calf muscles by noninvasive 31P-MRS. Investigations were done in mdx mice that were treated either with sActRIIB-Fc (n = 5) or with PBS (controls; n = 4). (a) The extent of force reduction was measured at the end of the 6 minutes period of electrostimulation and is represented as percent of the starting value. (b) The drop of intracellular pH and (c) phosphocreatine (PCr) concentration was determined at the end of the 6-minute in vivo electrostimulation period. (d) For each animal, the poststimulation time course of PCr was fitted to a mono-exponential function with a least mean-squared algorithm in order to calculate the PCr recovery time constant (τPCr): τPCr = −t/ln(PCrt/ΔPCr). (e) τPCr was significantly larger in sActRIIB-Fc-treated mdx mice (200 ± 39 versus 78 ± 20 seconds in the control group). Values are shown as means ± SEM. P values were calculated using the nonparametric U-test.
The MRS results pointed to an underlying functional deficit of the skeletal muscle respiratory chain complexes or β-oxidation in response to sActRIIB-Fc treatment. However, on contrary to our hypothesis, we found (i) largely unaffected ex vivo activities of isolated key mitochondrial enzymes (Krebs cycle: citrate synthase; respiratory chain: cytochrome C oxidase; and β-oxidation: hydroxyacyl-CoA-dehydrogenase) and (ii) similar succinate dehydrogenase and cytochrome C oxidase fiber profiles (Supplementary Figures S5–S7). In fact, cytochrome C oxidase and succinate dehydrogenase enzyme activities even appeared somewhat increased in EDL muscles (Supplementary Figures S5c,d, S6a, S7a), likely reflecting a compensatory increase in response to decreased aerobic energy production. Furthermore, mitochondrial DNA copy numbers remained largely unchanged following treatment with sActRIIB-Fc (Supplementary Figure S8a). The normal mitochondrial DNA copy numbers together with unaltered citrate synthase enzyme activities (Supplementary Figure S5a,b) let us conclude that ActRIIB blockade did not affect mitochondrial mass.
Treatment with sActRIIB-Fc downregulates porin expression in wild-type and mdx muscle
Given the abnormal postexercise τPCr along with normal respiratory chain activities, we wondered whether the ATP transport from the mitochondrial matrix into the cytosol of skeletal muscle cells could be affected, which might explain the diminished rate of aerobic energy production. Keeping with such hypothesis, decreased protein levels of Vdac3 had already been reported for mdx muscle, hinting towards a derangement of the ADP/ATP-shuttling system through the outer mitochondrial membrane via the voltage-dependent anion channels (Vdac, syn. porin).31,32,33 In line with these findings, a proteomic survey of differentially expressed proteins from wild-type and mdx mouse hearts had discovered a substantial loss of Vdac1 protein.34 Indeed, here we show that wild-type and to an even larger extent mdx muscles exhibited a considerable reduction of porin mRNA transcripts (Supplementary Figure S8a) and porin protein levels (Supplementary Figure S9) after sActRIIB-Fc treatment. This pushes the muscle even further into global mitochondrial dysfunction than dystrophin deficiency alone. Such secondary mitochondriopathy might explain the high lactic acidosis and rapid fatigability of sActRIIB-Fc-treated mdx mice.
Myopathic changes in mdx mice following treatment with sActRIIB-Fc
We next investigated the consequences of sActRIIB-Fc treatment on the extent of muscle dystrophy in mdx mice. Muscles from both sActRIIB-Fc and PBS-treatment groups revealed typical dystrophic changes comprising muscle fiber necrosis, regenerating fibers, fibers with central nuclei, inflammatory infiltrates, and increased fibrosis, which are difficult to quantify (Supplementary Figure S10a). Muscle degeneration is accompanied by a leak of cytoplasmic enzymes such as creatine kinase. We measured serum creatine kinase levels, which were largely elevated in mdx mice from both treatment groups; however, we did not detect any significant differences since interindividual variation was large (Supplementary Figure S10b). In mdx mice, muscle degeneration is followed by excessive regeneration with abundant splitting of regenerated fibers, which appear as small fiber profiles on transverse sections. Such excessive regeneration leads to an increase of muscle mass (see comparison between wild-type and mdx mice: Supplementary Figure S1). Following treatment with sActRIIB-Fc, muscles enlarged on average, if compared to PBS treatment, by ≈1.6-fold in mdx and by ≈1.3-fold in wild-type mice (Supplementary Figure S1). However, analysis of morphometric features of EDL muscles from sActRIIB-Fc-treated mdx mice, revealed a further increase in the number of small fiber profiles if compared to PBS-treated mdx mice (Supplementary Figure S10c). This finding suggestes that the excessive increase of muscle weight was triggered by abnormal regeneration and not by fiber hypertrophy. The soleus muscle of mdx mice, while not increasing its mass after sActRIIB-Fc treatment, exhibited an increased fiber size variation (Supplementary Figure S10d). In conclusion, the dystrophic phenotype of dystrophin deficient muscle persisted or even increased following treatment with sActRIIB-Fc.
Discussion
Myostatin/ActRIIB signaling exerts three major functions on skeletal muscle. (i) It acts to limit its size, (ii) promotes oxidative properties, and (iii) balances glucose versus fat utilization. The changes in muscle physiology in hypermuscular mice following treatment of adult mice with sActRIIB-Fc highlights the fact that myostatin/ActRIIB blockade confers little functional advantage over wild-type muscle due to its rapid fatigability. Improved muscle strength, however short lived, comes at the cost of increased fatigability and exercise intolerance, which is often seen in patients with mitochondrial disorders such as mitochondrial encephalopathy, lactic acidosis, stroke-like episodes (MELAS) or myoclonic epilepsy with ragged-red fibers (MERFF) syndrome.35 Interestingly, muscle cramps are frequently observed in whippet dogs with Mstn mutations.36 Moreover, “double muscle cattle”, several breeds of which have been identified to carry Mstn mutations,37,38 are prone to exercise induced lactic acidosis and severe rhabdomyolysis.39,40 In myostatin deficient animals, such exercise failure could be attributed to congenital fiber-type disproportion with a shift toward the expression of the fast IIB MHC isoform,4,18 which is well known to be associated with loss of oxidative properties of skeletal muscle and increased fatigability.5,8 In contrast to animals born with mutations in the Mstn gene, we show that blockade of ActRIIB signaling in adult wild-type mice beyond the period of muscle development does not have any impact on fiber-type composition, thus confirming previous reports.10
After sActRIIB-Fc treatment, the mice exhibit clinical signs of early muscle fatigue, exercise intolerance, and lactic acidosis—characteristic signs for a depression of β-oxidation, a shift toward anaerobic glycolysis, and ATP deficiency. Interestingly, whereas ex vivo mitochondrial respiratory chain enzyme activities and mitochondrial DNA copy numbers were within the normal range, in vivo 31P-MRS clearly demonstrated a downregulation of oxidative energy metabolism in sActRIIB-Fc-treated mdx mice. We further demonstrate a significant loss of the porin complex in sActRIIB-Fc-treated mice, pointing towards an underlying defect of ATP handling and ATP transport as one causative mechanism for the metabolic phenotype. A second aggravating factor, which further compromised exercise tolerance, is the decrease of capillary density and the increase of the capillary domain. Previous reports attributed such reduced capillary density to the increase of muscle fiber size.41 However, here we demonstrate a net numerical loss of capillaries per fiber following treatment with sActRIIB-Fc. This finding was most pronounced in mdx mice with a profound rarefication of the capillary bed in dystrophic muscle. In dystrophinopathies diminished sarcolemmal Nos1 results in dysregulation of the capillary adaptive response to exercise leading to functional muscle ischemia.42 Our protein analysis argues against a further aggravation of the capillary adaptive response by additional loss of Nos1 in sActRIIB-Fc-treated animals, despite the fact that Nos1 mRNA levels were clearly reduced. sActRIIB-Fc treatment of dystrophic mdx mice dramatically worsened the myopathic phenotype as shown by the large deficit in specific force. As lack of dystrophin per se alters mitochondrial function in DMD patients and mdx mice,15,16,29 the blockade of myostatin signaling initiates a vicious cycle resulting in severe secondary metabolic myopathy. We also found an increased fiber size variation after sActRIIB-Fc treatment of mdx mice pointing towards increased myopathic changes even at the tissue level.
In the past, several investigators have used different strategies of interfering with the ActRIIB receptor mediated signaling pathway in order to treat mdx mice and Golden Retriever muscular dystrophy (GRMD) dogs. This was done either through injection of sActRIIB-Fc,12,43 AAV-mediated gene transfer11 or antibodies directed against the ActRIIB receptor.44 Overall, the conclusions were optimistic about the usefulness of such strategy to treat dystrophinopathies. However, it should be noted that our results and conclusions differ from previously published work in various aspects. We think the reason for that mainly lies in the choice of endpoints to define success or failure of such a treatment. For DMD patients, clinically relevant and quantifiable improvements would comprise better performance in the 6-minute walk45 and improvement of respiratory function. This implies the ability of the patient's body to maintain a certain workload for a prolonged time period and not just to be able to produce single bouts of maximum short duration muscle activity as tested by tetanic muscle contractions11,12 or by the whole body tension method.43 In most studies, the increase of muscle size was taken as an endpoint,11,44 automatically assuming that big muscles are healthier muscles. This basic assumption is put into question by our results. None of the studies investigated endurance capacity, which evaluates the effect of a treatment on the physiology of the entire body over a longer time period and would thus be a relevant parameter that could translate into improvement of life quality in patients. Several studies, one using the identical sActRIIB-Fc compound,12 reported a small, but significant decline of creatine kinase values after ActRIIB blockade.11,12 We were unable to reproduce this finding, the reason for these differences remaining unresolved.
We show that myostatin controls the metabolic profile of skeletal muscle, and its blockade depresses the main molecular determinants of oxidative metabolism and β-oxidation. Interestingly, genetic inactivation of Pparβ, similar to myostatin/ActRIIB blockade, reduced oxidative properties of skeletal muscle.46 This adds further evidence that myostatin controls the muscle oxidative phenotype via peroxisome proliferator-activated receptors (PPAR) and Pgc1α, the downstream target of Pparβ, while the down-regulation of Pdk4 indicates a shift away from β-oxidation towards glucose metabolism. These quantitative polymerase chain reaction data are strongly corroborated by a recent transcriptome study following treatment with sActRIIB-Fc of wild-type mice.13 However, we lack direct evidence to ascertain a shift towards higher glucose metabolism, although the downregulation of Pdk4 and upregulation of Enolase can be counted as indirect indicators for such a change. The unfavorable combination of decreased vascularization and metabolic changes after ActRIIB blockade is likely to cause a rapid imbalance between increased cytosolic ATP hydrolysis and insufficient mitochondrial ATP synthesis during exhaustive exercise. The subsequent shift toward anaerobic glycolytic ATP synthesis explains the rapid fatigability and the pathologically increased lactate production.47 However, the metabolic adaption in response to myostatin may differ in diverse physiological and pathophysiological contexts, e.g., myostatin inhibition was reported to improve motor performance in aged mice.48 Further work is required to elucidate the metabolic function of myostatin in different disease situations and during ageing.
In conclusion, our results suggest that myostatin/ActRIIB signaling optimizes oxidative metabolism of skeletal muscle leading to lower muscle fatigability and amelioration of endurance capacity. Such fundamental functions of myostatin should be taken into account in the development of therapies based on myostatin/ActRIIB blockade. However, it should be kept in mind that our experimental design does not allow to determine which effects can be ascribed to myostatin blockade alone and which to the inactivation of other TGF-β family members such as bone morphogenetic proteins, growth and differentiation factors, and activins that may also be sequestered by the soluble ActRIIB. Further investigations are required to answer the question, such as whether emerging therapies based on PPAR agonists might be able to prevent such adverse effects of ActRIIB blockade on the oxidative metabolism and on exercise tolerance. Furthermore, dose regime studies could answer the question, whether short-term treatment or pulse treatment may circumvent secondary effects of myostatin/ActRIIB blockade on muscle metabolism.
Materials and Methods
Animals. Male mdx mice (on a C57BL/10ScSn background) were bred in the animal facility of the Medical Faculty of Paris VI and kept according to institutional guidelines. Wild-type male C57BL/6J control mice were purchased from Charles River (France). Two-month-old wild-type and mdx mice were injected twice weekly subcutaneously with 10 mg/kg with the rodent form of the soluble activin receptor IIB (sActRIIB-Fc; Acceleron Pharma, Cambridge, MA) for a total of 4 months before killing. The methods of sActRIIB-Fc synthesis have previously been described.49 All animal studies have been approved and were carried out under the laboratory and animal facility licenses A75-13-11 and A91-228-107.
Evaluation of the critical speed. Mice were subjected to three or four separate bouts of runs until exhaustion at various treadmill speeds (between 20 and 80 cm/second according to individual motor capacity, one run per day) according to previously published protocols.21 Critical speed, an index of the aerobic exercise capacity, was calculated from the slope (a) of the regression line, plotting the distance (y) against the time to exhaustion (x) from the different runs.
Blood lactate assessment during exhaustive exercise. Lactate concentrations were determined in blood samples collected from the tip of the tail using a Lactate pro LT device (Arkray, Kyoto, Japan) at rest before exercise (0 minute) and 5 minutes after treadmill running-induced exhaustion. Exhaustion was defined as the time point at which the mice were unable to run anymore and stayed on the grid despite repeated electric stimulation. The running test started at the lowest speed of 5 cm/second to allow a warm-up and then increased by 1 cm/second every 30 seconds until exhaustion. This protocol is illustrated by the Supplementary Video S1, which demonstrates the running test of sActRIIB-Fc-treated wild-type (left) and mdx mice (right) side by side. The starting speed of 5 cm/second was increased by 1 cm/second every minute until exhaustion of the mdx mouse at 19 cm/second after 14 minutes, while the wild-type mouse was still able to run at a speed of 40 cm/second.
Electromyographic examination. Electromyographic examination of the triceps brachialis, tibialis, gastrocnemius, and quadriceps femoris muscles was performed in mice anesthetized with isoflurane. Standard noninvasive needle electromyography was conducted on a Viking Quest EMG apparatus (Viasys, Nicolet Biomedical, Madison, WI) using concentric bipolar needle electrodes. Insertional activity and pathological spontaneous activity were recorded.
Measurement of contractile properties. Absolute maximal isometric tetanic force (P0) was measured during tetanic contractions (frequency of 50–100 Hz, train of stimulation of 1,500 ms for soleus and 750 ms for EDL). Specific maximal isometric force (sP0) was given as the quotient between force and muscle weight. For analysis of fatigue resistance, muscles were stimulated at 75 Hz for 500 ms, every 2 seconds over 3 minutes. See Supplementary Methods.
Western blot. Protein was extracted from frozen tibialis anterior muscle of wild-type and mdx mice and processed as described.50 Briefly, after homogenization of the muscle in radioimmunoprecipitation assay buffer with a proteinase inhibitor cocktail (Complete; Roche-Diagnostics, Mannheim, Germany) proteins were separated through denaturating sodium dodecyl sulphate polyacrylamide gel electrophoresis with the Laemmli system and blotted onto nitrocellulose membranes by the semidry method (Biometra, Göttingen, Germany). The blots were probed with anti-porin as primary antibody (VDAC 31HL, AB-2; Calbiochem, Darmstadt, Germany), anti-PGC1α (Santa-Cruz, Heidelberg, Germany), anti-Nos1 (Abcam, Paris, France), and corresponding peroxidase-labeled secondary antibodies. The desmin and GAPDH bands were used as loading control for muscle. Bands were visualized by chemiluminescence. The protein bands were quantified by measuring their integrated density within a rectangle that covered the entire individual band and subtracting the integrated density of an empty rectangle of exactly the same size in the vicinity using the ImageJ software (NIH, Bethesda, MD).
Enzyme measurements. Enolase, citrate synthase, cytochrome C oxidase, and hydroxyacyl-CoA-dehydrogenase activities were determined in extracts from frozen cryostat sections using a coupled enzyme assay as detailed in Supplementary Methods.
Histology. H&E, succinate dehydrogenase and cytochrome C oxidase staining were performed using routine histological protocols. The following primary antibodies were used for immunohistochemistry: anti-CD31 (Pharmingen, Ranges, France), anti-MHCIIA (SC-71, DSMZ, Braunschweig, Germany), anti-MHCI (BAD5, DSMZ), anti-Nos1 (Abcam), and antilaminin (Dako, Les Ulis, France) followed by secondary antibodies with various fluorophores (Alexa Fluor; Invitrogen, Saint Aubin, France) (see Supplementary Methods).
Morphometric analysis of capillary number and capillary domains. Cryosections of 12 µm of the EDL and soleus muscles of PBS- and sActRIIB-Fc-treated animals were stained with antilaminin to delineate the muscle fibers. Muscle capillaries were stained with anti-CD31. Fluorescent photographs were taken with a ×20 objective on a Microscope (Zeiss, AxioImager Z1, Marly-le-Roi, France) and saved as TIFF files. These images were projected on a flatscreen coupled with a graphic tablet, which enabled the manual retracing of the muscle fiber outlines and the counting of capillaries that were found around it. For the EDL, the fibers of the entire muscle cross section were analyzed, and for the soleus muscle, the fibers from 10 representative nonoverlapping visual fields. For each muscle fiber, we determined the cross-sectional plane (µm2) and counted the number of bordering capillaries. The capillary domain (µm2) for each fiber was calculated by dividing its cross-sectional plane by the number of bordering capillaries.
Cell culture. C2C12 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco 41966-029, Darmstadt, Germany) supplemented with 1% Pen/Strep and 20% fetal bovine serum (Gibco 10500-064) to semiconfluency at 37 °C and 5% CO2 for 2 days. Thereafter, the medium was replaced by DMEM plus 10% horse serum (Gibco 26050-088) to induce fusion into multinucleated myotubes. sActRIIB-Fc was added to a final concentration of 200 ng/ml to the culture medium of the myotubes. After 24 hours, the myotubes were harvested by trypsinization, washed and pelleted for RNA extraction. Human umbilical vein endothelial cells were grown to confluency, trypsinized and pelleted for RNA extraction. The doubling time of the cells was determined using the AlamarBlue reagents from Invitrogen. Briefly, cells were plated at 20% confluency and allowed to settle for 12 hours before introducing recombinant myostatin (R&D Systems, Wiesbaden, Germany). The cells were grown for 24 hours before addition of 0.1 V of AlamarBlue reagent and incubation at 37 °C for 20 minutes before photometric analysis. The cells were then washed and cultured in fresh medium containing myostatin. Cell proliferation was monitored every 24 hours for 4 days after initial introduction of myostatin. Cell number was determined by comparison of absorbance against a standard curve. All experiments were performed in triplicate.
Reverse transcription quantitative polymerase chain reaction. Real-time polymerase chain reaction was performed according the SYBR Green protocol (Applied Biosystems, Darmstadt, Germany) on the Eco real-time polymerase chain reaction System (Illumina, Eindhoven, The Netherlands) with a HotStart Taq polymerase (Applied Biosystems). For primer sequences, See Supplementary Methods. Fold changes were calculated according to the efficiency corrected −ΔΔCt method. The use of normal and of DMD muscle from patients was covered by the approval of the ethical review board of the Charité (#216/2001). All patients or their legal guardians provided written informed consent according to the Declaration of Helsinki.
In vivo MRS investigation of muscle function and oxidative metabolism. Mice were anesthetized with 4% isoflurane in 100% air at a flow of 3 l/minute and were placed into a home-built cradle specifically designed for the strictly noninvasive MRS investigation of muscle function and energetics.30 Throughout the experiment, anesthesia was maintained using a facemask continuously supplying 1.75% isoflurane in 33% O2 (0.2 l/minute) and 66% N2O (0.4 l/minute). Animal body temperature was controlled by a rectal probe and maintained at physiological values by a feedback loop that regulated an electrical heating blanket. MR spectra were recorded in the 4.7 T horizontal magnet of a 47/30 Biospec Avance MR system (Bruker, Karlsruhe, Germany) equipped with a Bruker 120-mm BGA12SL (200 mT/m) gradient insert. Calf muscle were electrostimulated transcutaneously to produce maximal repeated isometric contractions at a frequency of 1.7 Hz. Mechanical performance was measured using a foot pedal coupled to a force transducer. Concentrations of phosphorylated compounds and intracellular pH of the calf muscle were continuously measured with an elliptic (8 × 12 mm2) 31P-MRS surface coil during 6 minutes of rest, 6 minutes of electrostimulation and 16 minutes of recovery. MRS data were processed using a custom-written analysis program developed on the IDL software (Research System, Boulder, CO). In order to determine the time constant of postexercise phosphocreatine resynthesis (τPCr, an in vivo index of oxidative mitochondrial capacity), the time course of phosphocreatine concentrations during the poststimulation period was fitted to a monoexponential function with a least mean-squared algorithm (Figure 5d): τPCr = −t/ln(PCrt/ΔPCr), where ΔPCr is the extent of PCr depletion measured at the start of recovery period.
Statistical analysis. Data were analyzed and significance levels calculated using the nonparametric Wilcoxon–Mann–Whitney U-test, as stated in the legends and detailed in Supplementary Methods. Values are presented as means ± SEM. Significance levels were set at P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Effect of sActRIIB-Fc on body weight and muscle weight in wild-type and mdx mice. Figure S2. Effect of sActRIIB-Fc on fiber-type distribution of EDL and soleus muscles from wild-type and mdx mice. Figure S3. Effect of sActRIIB-Fc on EMG recordings of mdx mice. Figure S4. Effect of myostatin on capillaries and endothelial cell proliferation. Figure S5. Effect of sActRIIB-Fc on enzymes activities of mitochondrial respiratory chain in EDL and soleus muscles from wild-type and mdx mice. Figure S6. Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from wild-type mice. Figure S7. Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from mdx mice. Figure S8. Effect of sActRIIB-Fc on mitochondrial DNA copy number and transcription of the gene encoding the mitochondrial protein porin in muscles from wild-type and mdx mice. Figure S9. Effect of sActRIIB-Fc on the expression of the mitochondrial protein porin in muscles from wild-type and mdx mice. Figure S10. Effect of myostatin/ActRIIB signaling on muscle phenotype. Figure S11. Effect of sActRIIB-Fc on the expression of Nos1 in muscles from wild-type and mdx mice. Video S1. Effect of sActRIIB-Fc on incremental speed running in mdx mice. Methods
Acknowledgments
We acknowledge Acceleron Pharma for the gift of sActRIIB-Fc. This work was supported by the Association Française contre les Myopathies towards H.A., A.F., A.V., L.A., L.G., and E.M., Association Monegasque contre les Myopathies and the Parents Project France toward H.A. and C.H., Aktion Benni & Co toward H.A., the Deutsche Forschungsgemeinschaft and the Université Franco-Allemand towards K.R., H.A., and M.S. (as part of the MyoGrad International Graduate School for Myology DRK 1631/1 and CDFA-06-11), and NeuroCure (Exc 257) to M.S. M.S. is a member of mitoNET (01GM1113D) supported by the BMBF (Germany). The authors do not declare any conflict of interest. K.R., E.M., C.H., O.A., K.P., S.L., K.J., B.G., F.P.-R., L.A., A.V., D.F., S.B., A.F., M.S., and H.A. performed the experiments; M.S. contributed patient material; O.R. contributed new reagents; K.R., E.M., D.F., S.B., B.G., D.B. A.F., R.V.C., M.S., and H.A. analyzed the data; M.S. did the statistical analysis; K.R., E.M., B.G., D.B., M.S., and H.A. wrote the manuscript, K.P. revised the article critically for important intellectual content. All authors read the final version of the manuscript and gave final approval of the manuscript to be published.
Supplementary Material
Effect of sActRIIB-Fc on body weight and muscle weight in wild-type and mdx mice.
Effect of sActRIIB-Fc on fiber-type distribution of EDL and soleus muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on EMG recordings of mdx mice.
Effect of myostatin on capillaries and endothelial cell proliferation.
Effect of sActRIIB-Fc on enzymes activities of mitochondrial respiratory chain in EDL and soleus muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from wild-type mice.
Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from mdx mice.
Effect of sActRIIB-Fc on mitochondrial DNA copy number and transcription of the gene encoding the mitochondrial protein porin in muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on the expression of the mitochondrial protein porin in muscles from wild-type and mdx mice.
Effect of myostatin/ActRIIB signaling on muscle phenotype.
Effect of sActRIIB-Fc on the expression of Nos1 in muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on incremental speed running in mdx mice.
References
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsakas A, Mouisel E, Amthor H, Patel K. Myostatin knockout mice increase oxidative muscle phenotype as an adaptive response to exercise. J Muscle Res Cell Motil. 2010;31:111–125. doi: 10.1007/s10974-010-9214-9. [DOI] [PubMed] [Google Scholar]
- Qaisar R, Renaud G, Morine K, Barton ER, Sweeney HL, Larsson L. Is functional hypertrophy and specific force coupled with the addition of myonuclei at the single muscle fiber level. FASEB J. 2012;26:1077–1085. doi: 10.1096/fj.11-192195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baligand C, Gilson H, Ménard JC, Schakman O, Wary C, Thissen JP, et al. Functional assessment of skeletal muscle in intact mice lacking myostatin by concurrent NMR imaging and spectroscopy. Gene Ther. 2010;17:328–337. doi: 10.1038/gt.2009.141. [DOI] [PubMed] [Google Scholar]
- Savage KJ, McPherron AC. Endurance exercise training in myostatin null mice. Muscle Nerve. 2010;42:355–362. doi: 10.1002/mus.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akpan I, Goncalves MD, Dhir R, Yin X, Pistilli EE, Bogdanovich S, et al. The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int J Obes (Lond) 2009;33:1265–1273. doi: 10.1038/ijo.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena SM, Tomkinson KN, Monnell TE, Spaits MS, Kumar R, Underwood KW, et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol (1985) 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, et al. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve. 2010;42:722–730. doi: 10.1002/mus.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistilli EE, Bogdanovich S, Goncalves MD, Ahima RS, Lachey J, Seehra J, et al. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Pathol. 2011;178:1287–1297. doi: 10.1016/j.ajpath.2010.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F, King OD, Warsing LC, Powell RE, Emerson CP, Kunkel LM, et al. Gene expression profiling of skeletal muscles treated with a soluble activin type IIB receptor. Physiol Genomics. 2011;43:398–407. doi: 10.1152/physiolgenomics.00223.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Peekhaus N, Weber H, Adamski S, Murray EM, Zhang HZ, et al. Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J Gerontol A Biol Sci Med Sci. 2013;68:1181–1192. doi: 10.1093/gerona/glt030. [DOI] [PubMed] [Google Scholar]
- Jongpiputvanich S, Sueblinvong T, Norapucsunton T. Mitochondrial respiratory chain dysfunction in various neuromuscular diseases. J Clin Neurosci. 2005;12:426–428. doi: 10.1016/j.jocn.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Winkler K, Wiedemann FR, von Bossanyi P, Dietzmann K, Kunz WS. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol Cell Biochem. 1998;183:87–96. doi: 10.1023/a:1006868130002. [DOI] [PubMed] [Google Scholar]
- Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath S, Song K, Whittemore LA. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve. 2005;31:34–40. doi: 10.1002/mus.20175. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Macharia R, Otto A, Elashry MI, Mouisel E, Romanello V, et al. Exercise training attenuates the hypermuscular phenotype and restores skeletal muscle function in the myostatin null mouse. Exp Physiol. 2012;97:125–140. doi: 10.1113/expphysiol.2011.063008. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci U S A. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billat VL, Mouisel E, Roblot N, Melki J. Inter- and intrastrain variation in mouse critical running speed. J Appl Physiol (1985) 2005;98:1258–1263. doi: 10.1152/japplphysiol.00991.2004. [DOI] [PubMed] [Google Scholar]
- Han JJ, Carter GT, Ra JJ, Abresch RT, Chamberlain JS, Robinson LR. Electromyographic studies in mdx and wild-type C57 mice. Muscle Nerve. 2006;33:208–214. doi: 10.1002/mus.20455. [DOI] [PubMed] [Google Scholar]
- Hayot M, Rodriguez J, Vernus B, Carnac G, Jean E, Allen D, et al. Myostatin up-regulation is associated with the skeletal muscle response to hypoxic stimuli. Mol Cell Endocrinol. 2011;332:38–47. doi: 10.1016/j.mce.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Zhao G, Jeoung NH, Burgess SC, Rosaaen-Stowe KA, Inagaki T, Latif S, et al. Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;294:H936–H943. doi: 10.1152/ajpheart.00870.2007. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Iannaccone ST, Lau KS, Masters BS, McCabe TJ, McMillan K, et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydemann A, McNally E. NO more muscle fatigue. J Clin Invest. 2009;119:448–450. doi: 10.1172/JCI38618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. doi: 10.1038/nature07414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Siegel MP, Knowels G, Marcinek DJ. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum Mol Genet. 2013;22:153–167. doi: 10.1093/hmg/dds415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannesini B, Vilmen C, Le Fur Y, Dalmasso C, Cozzone PJ, Bendahan D. A strictly noninvasive MR setup dedicated to longitudinal studies of mechanical performance, bioenergetics, anatomy, and muscle recruitment in contracting mouse skeletal muscle. Magn Reson Med. 2010;64:262–270. doi: 10.1002/mrm.22386. [DOI] [PubMed] [Google Scholar]
- Guzun R, Gonzalez-Granillo M, Karu-Varikmaa M, Grichine A, Usson Y, Kaambre T, et al. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim Biophys Acta. 2012;1818:1545–1554. doi: 10.1016/j.bbamem.2011.12.034. [DOI] [PubMed] [Google Scholar]
- Braun U, Paju K, Eimre M, Seppet E, Orlova E, Kadaja L, et al. Lack of dystrophin is associated with altered integration of the mitochondria and ATPases in slow-twitch muscle cells of MDX mice. Biochim Biophys Acta. 2001;1505:258–270. doi: 10.1016/s0005-2728(01)00172-4. [DOI] [PubMed] [Google Scholar]
- Massa R, Marliera LN, Martorana A, Cicconi S, Pierucci D, Giacomini P, et al. Intracellular localization and isoform expression of the voltage-dependent anion channel (VDAC) in normal and dystrophic skeletal muscle. J Muscle Res Cell Motil. 2000;21:433–442. doi: 10.1023/a:1005688901635. [DOI] [PubMed] [Google Scholar]
- Lewis C, Jockusch H, Ohlendieck K. Proteomic Profiling of the Dystrophin-Deficient MDX Heart Reveals Drastically Altered Levels of Key Metabolic and Contractile Proteins. J Biomed Biotechnol. 2010;2010:648501. doi: 10.1155/2010/648501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen AJ, Schuelke M, Smeitink JA, Trijbels FJ, Sengers RC, Lucke B, et al. Muscle 3243A–>G mutation load and capacity of the mitochondrial energy-generating system. Ann Neurol. 2008;63:473–481. doi: 10.1002/ana.21328. [DOI] [PubMed] [Google Scholar]
- Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:e79. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JH, Robinson DW, Ashmore CR. Blood lactic acid and behaviour in cattle with hereditary muscular hypertrophy. J Anim Sci. 1972;35:1011–1013. doi: 10.2527/jas1972.3551011x. [DOI] [PubMed] [Google Scholar]
- Holmes JH, Ashmore CR, Robinson DW. Effects of stress on cattle with hereditary muscular hypertrophy. J Anim Sci. 1973;36:684–694. doi: 10.2527/jas1973.364684x. [DOI] [PubMed] [Google Scholar]
- Personius KE, Jayaram A, Krull D, Brown R, Xu T, Han B, et al. Grip force, EDL contractile properties, and voluntary wheel running after postdevelopmental myostatin depletion in mice. J Appl Physiol (1985) 2010;109:886–894. doi: 10.1152/japplphysiol.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Carlson C, Bruemmer K, Sesti J, Stefanski C, Curtis H, Ucran J, et al. Soluble activin receptor type IIB increases forward pulling tension in the mdx mouse. Muscle Nerve. 2011;43:694–699. doi: 10.1002/mus.21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014;34:606–618. doi: 10.1128/MCB.01307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans N, Klingels K, van den Hauwe M, Boons S, Verstraete L, Peeters C, et al. Six-minute walk test: reference values and prediction equation in healthy boys aged 5 to 12 years. PLoS One. 2013;8:e84120. doi: 10.1371/journal.pone.0084120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407–414. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Robergs RA, Ghiasvand F, Parker D. Biochemistry of exercise-induced metabolic acidosis. Am J Physiol Regul Integr Comp Physiol. 2004;287:R502–R516. doi: 10.1152/ajpregu.00114.2004. [DOI] [PubMed] [Google Scholar]
- LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–948. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- Morrison BM, Lachey JL, Warsing LC, Ting BL, Pullen AE, Underwood KW, et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217:258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. 2010;6:e1000874. doi: 10.1371/journal.pgen.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of sActRIIB-Fc on body weight and muscle weight in wild-type and mdx mice.
Effect of sActRIIB-Fc on fiber-type distribution of EDL and soleus muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on EMG recordings of mdx mice.
Effect of myostatin on capillaries and endothelial cell proliferation.
Effect of sActRIIB-Fc on enzymes activities of mitochondrial respiratory chain in EDL and soleus muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from wild-type mice.
Effect of sActRIIB-Fc on SDH and COX enzyme activity on muscle sections of EDL and soleus muscles from mdx mice.
Effect of sActRIIB-Fc on mitochondrial DNA copy number and transcription of the gene encoding the mitochondrial protein porin in muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on the expression of the mitochondrial protein porin in muscles from wild-type and mdx mice.
Effect of myostatin/ActRIIB signaling on muscle phenotype.
Effect of sActRIIB-Fc on the expression of Nos1 in muscles from wild-type and mdx mice.
Effect of sActRIIB-Fc on incremental speed running in mdx mice.