

ABSTRACT
Legionella pneumophila, the etiological agent of legionellosis, replicates within phagocytic cells. Crucial to biogenesis of the replicative vacuole is the Dot/Icm type 4 secretion system, which translocates a large number of effectors into the host cell cytosol. Among them is LegK2, a protein kinase that plays a key role in Legionella infection. Here, we identified the actin nucleator ARP2/3 complex as a target of LegK2. LegK2 phosphorylates the ARPC1B and ARP3 subunits of the ARP2/3 complex. LegK2-dependent ARP2/3 phosphorylation triggers global actin cytoskeleton remodeling in cells, and it impairs actin tail formation by Listeria monocytogenes, a well-known ARP2/3-dependent process. During infection, LegK2 is addressed to the Legionella-containing vacuole surface and inhibits actin polymerization on the phagosome, as revealed by legK2 gene inactivation. Consequently, LegK2 prevents late endosome/lysosome association with the phagosome and finally contributes to remodeling of the bacterium-containing phagosome into a replicative niche. The inhibition of actin polymerization by LegK2 and its effect on endosome trafficking are ARP2/3 dependent since it can be phenocopied by a specific chemical inhibitor of the ARP2/3 complex. Thus, LegK2-ARP2/3 interplay highlights an original mechanism of bacterial virulence with an unexpected role in local actin remodeling that allows bacteria to control vesicle trafficking in order to escape host defenses.
IMPORTANCE
Deciphering the individual contribution of each Dot/Icm type 4 secretion system substrate to the intracellular life-style of L. pneumophila remains the principal challenge in understanding the molecular basis of Legionella virulence. Our finding that LegK2 is a Dot/Icm effector that inhibits actin polymerization on the Legionella-containing vacuole importantly contributes to the deciphering of the molecular mechanisms evolved by Legionella to counteract the endocytic pathway. Indeed, our results highlight the essential role of LegK2 in preventing late endosomes from fusing with the phagosome. More generally, this work is the first demonstration of local actin remodeling as a mechanism used by bacteria to control organelle trafficking. Further, by characterizing the role of the bacterial protein kinase LegK2, we reinforce the concept that posttranslational modifications are key strategies used by pathogens to evade host cell defenses.
INTRODUCTION
Legionella pneumophila is the most common etiological agent of severe legionellosis pneumonia in humans. Pathogenic Legionella strains emerge in the environment after intracellular multiplication in amoebae. Bacteria are disseminated by water aerosols and, when inhaled into the lungs, engulfed by alveolar macrophages. Within environmental phagocytic cells and human macrophages, a functional Dot/Icm type 4 secretion system (T4SS) and the approximately 300 proteins it secretes (1) are absolutely required for Legionella to reroute its phagosome and trigger the biogenesis of a Legionella-containing vacuole (LCV), a rough endoplasmic reticulum-like compartment permissive for its intracellular replication (2–4). Recently, important investigative efforts have begun to decipher the individual contributions of protein effectors to the Legionella intracellular life-style. Most of this work has relied on the construction of deletion mutants for the corresponding T4SS substrates. However, because of functional redundancy between effectors, single gene deletions very rarely result in a virulence defect; consequently, to date, only a few T4SS effectors have been functionally characterized (for reviews, see references 5, to ,9). Among these are proteins that interfere with small GTPases of the early secretory cellular pathway (10–17), the endocytic pathway (18), or the retrograde vesicle trafficking (19) or target the innate immune response and host cell apoptosis pathways (7, 20,21).
Many Dot/Icm-secreted effectors display distinctive eukaryotic domains that include protein kinase domains (22, 23). In silico analysis of effector sequences and in vitro phosphorylation assays with purified proteins identified five functional protein kinases, designated LegK1 to -5, that are encoded by the epidemic L. pneumophila Lens strain (24). All of the Legionella protein kinases except LegK5 are Dot/Icm effectors. Of these kinases, LegK1 has been shown to induce activation of the NF-κB transcription factor and, consequently, genes with antiapoptotic functions (25). In vitro and cell-free reconstitution assays have shown that LegK1 phosphorylates the NF-κB inhibitor IκB. However, it is noteworthy that despite the role of LegK1 in activating the NF-κB pathway, a legK1 deletion mutant does not present a virulence defect. In contrast to LegK1, we previously reported that inactivation of the legK2 gene resulted in a significant decrease in L. pneumophila virulence toward amoeba and macrophages, highlighting the key role of this effector in Legionella virulence. More precisely, the legK2 mutant poorly evades endocytic degradation and results in delayed intracellular replication. Because a kinase-dead legK2 mutant exhibits the same virulence defects as the deletion mutant, we concluded that the protein kinase activity of LegK2 is directly involved in evading host cell defenses and in the establishment of a replicative niche (24).
Here, we aimed to determine the precise function of LegK2-dependent protein phosphorylation during L. pneumophila infection. We show that LegK2 interacts with the ARPC1B and ARP3 subunits of the actin nucleator ARP2/3 complex. We demonstrate that LegK2 phosphorylates these subunits in vitro and in cells. Finally, we establish that the LegK2-ARP2/3 interplay inhibits actin polymerization on the LCV and interferes with late endosome/lysosome trafficking toward the LCV. Thus, we show for the first time that host cell actin remodeling is essential for Legionella to perturb host cell endocytic vesicle trafficking and allow bacteria to escape cell defenses.
RESULTS
LegK2 specifically interacts with host cell ARP2/3 complex subunits.
To identify host cell proteins that interact with LegK2, we performed yeast two-hybrid screening. The construct pGBKT7-legK2, which encodes LegK2 fused to the DNA binding domain of GAL4 (GAL4BD-LegK2), was transformed into Saccharomyces cerevisiae strain AH109. The toxicity/self-activation of this construct was evaluated by mating yeast strain AH109(pGBKT7-legK2) and reporting yeast strain Y187 for histidine auxotrophy, transformed by the empty vector pACT2. While no toxicity was detected, as indicated by the growth of the diploid (Fig. 1A, SD-W-L, lane 6), self-activation was observed. Therefore, we made use of a competitive inhibitor of the reporter HIS3 gene product 3-aminotriazole (3AT). When AH109(pGBKT7-legK2) was grown in the presence of 3AT, self-activation was decreased but not completely abolished (Fig. 1A, SD-W-L-H+3AT, lane 6). The Y187 yeast strain transformed by a human normalized cDNA spleen library cloned into pACT2 was mated with AH109(pGBKT7-legK2), and diploids were screened for histidine auxotrophy in the presence of 10 mM 3AT. DNA sequences fused to the transcription-activating domain of GAL4 in vector pACT2 in the selected clones were amplified by PCR and sequenced. Reproducibly, several candidates for interaction with LegK2 were proteins involved in actin cytoskeleton assembly. Because the legK2 mutant phenotype alters host cell vesicle trafficking during infection, particular attention was given to proteins of this family, which included ARP3 and ARPC1B subunits of the actin filament nucleator ARP2/3 complex.
FIG 1 .

LegK2 interacts with ARPC1B and ARP3 subunits of the ARP2/3 complex. (A) Yeast two-hybrid assays of L. pneumophila protein kinase LegK1/LegK2 and ARPC1B/ARP3 subunits of the human ARP2/3 complex. Diploids from the mating of AH109(pGBKT7-legK1) or AH109(pGBKT7-legK2) with Y187(pACT2), Y187(pACT2-ARPC1B), or Y187(pACT2-ACTR3) were grown on SD medium without tryptophan (−W) or leucine (−L). Histidine auxotrophy was tested by plating 5 or 50 µl of diploids on SD-W-L medium without histidine (−H) in the presence of 10 mM 3-AT. (B) Affinity copurification of Flag-tagged ARP2/3 subunits with GST-tagged LegK2 protein. HEK293T cells were cotransfected with pDEST27, pDEST27-legK2, or pDEST27-legK2K112M and pCI-Neo3Flag-ARPC1B or -ACTR3. GST, GST-tagged LegK2, or LegK2K112M was purified on glutathione-agarose 4B, and purified fractions were immunoblotted with both anti-GST and anti-Flag antibodies. (C) Cellular localization of ARPC1B/ARP3 and LegK2/LegK1 proteins in HEK293T cells cotransfected with pDEST27, pDEST27-legK2, or -legK1 and pCI-Neo3Flag-ARPC1B or -ACTR3. The GST, GST-LegK2, and GST-LegK1 proteins were detected by immunofluorescence with anti-GST antibodies (green), and 3Flag-ARPC1B and 3Flag-ARP3 were detected with anti-Flag antibodies (red). Scale bars, 10 µm. (D) Quantitation of colocalization by Pearson coefficient. The Pearson coefficient was determined with the JACoP plugin of the ImageJ software and is expressed as the mean value calculated for 30 cells.
To confirm that ARP3 and ARPC1B interact with LegK2, cDNAs from each of these two genes, named ACTR3 and ARPC1B, respectively, were cloned into vector pACT2 and transformed into reporting yeast strain Y187. Y187(pACT2-ACTR3) and Y187(pACT2-ARPC1B) were mated with AH109(pGBKT7-legK2), and diploids were assayed for histidine auxotrophy in the presence of 10 mM 3AT. Both diploids were able to grow without histidine (Fig. 1A, SD-W-L-H+3AT, lanes 2 and 4), which confirms the interactions between LegK2 and ARP3 and ARPC1B subunits. To determine if the interactions between LegK2 and ARP3 or ARPC1B subunits are specific, similar binary yeast two-hybrid assays were performed with LegK1 from L. pneumophila, which is known to subvert the host cell NF-κB pathway independently of ARP3/ARPC1B subunits. The AH109(pGBKT7-legK1) strain was constructed and then mated with Y187(pACT2-ACTR3) and Y187(pACT2-ARPC1B). Diploids could not grow without histidine (Fig. 1A, SD-W-L-H+3AT, lanes 1 and 3), which indicates that LegK1 does not interact with these ARP2/3 complex subunits. Together, these results strongly suggest that LegK2 interactions with ARP3 and ARPC1B subunits are specific.
Next, we investigated interactions between LegK2 and ARP3/ARPC1B subunits in mammalian cells by affinity copurification. Vectors pDEST27-legK2 and pCI-Neo3Flag-ACTR3 or -ARPC1B were constructed to express N-terminally glutathione S-transferase (GST)-tagged LegK2 and N-terminally Flag-tagged ARP3 or ARPC1B, respectively, in mammalian cells. HEK293T cells were cotransfected with these vectors, and GST-LegK2 was purified with a glutathione-agarose matrix. We found that GST-LegK2 (90 kDa) was expressed and detected 24 h posttransfection and that Flag-tagged ARPC1B (45 kDa), but not Flag-tagged ARP3, was copurified with the Legionella protein kinase (Fig. 1B). The same experiment was performed with a vector expressing the kinase-dead variant GST-LegK2K112M, where the invariant lysine essential for donor-ATP binding is substituted. As expected, ARPC1B was also copurified with this catalytic variant of LegK2. As ARPC1B was not purified with GST, these data confirm that LegK2 specifically interacts with the ARPC1B subunit of the ARP2/3 complex in mammalian cells. Regarding ARP3, we propose that the interaction between LegK2 and ARP3 is too weak and/or transient to be detected by affinity copurification under our conditions.
The subcellular localization of the bacterial protein kinase LegK2 and ARP2/3 subunits was analyzed after transfection into HEK293T cells. pDEST27-legK2- or -legK1- and pCI-Neo3Flag-ACTR3- or -ARPC1B-cotransfected fibroblasts were stained with anti-GST and anti-Flag antibodies to detect GST-LegK2/LegK1 and Flag-ARP3/ARPC1B, respectively (Fig. 1C). Both antibodies labeled the cytoplasm and periphery of cells with similar staining patterns when cells were cotransfected with pDEST27-legK2 and pCI-Neo3Flag-ACTR3 or -ARPC1B, while anti-GST labeling in cells cotransfected with the empty vector or the LegK1 expressing vector was diffused in the cell. These observations suggest that LegK2 specifically colocalizes with ARPC1B and ARP3. To quantify the degree of colocalization of GST-tagged LegK2 and Flag-tagged ARP3/ARPC1B, we calculated the Pearson overlap coefficient for each antibody. Pearson coefficients of 0.87 for both ARPC1B and ARP3 support colocalization with LegK2 and are consistent with a physiological relevance of the interactions between LegK2 and the ARP2/3 complex subunits in mammalian cells (Fig. 1D).
Bacterial protein kinase LegK2 phosphorylates ARP3 and ARPC1B subunits.
Because LegK2 specifically interacts with ARP3 and ARPC1B subunits and as ARP3 and ARPC1B had been previously described to be phosphorylated on threonine residues in mammalian cells (26–28), we investigated whether either protein is a substrate of the bacterial protein kinase LegK2. The ARP2/3 complex was purified from the amoeba Acanthamoeba castellanii (Fig. 2A, lanes 1 and 6) and dephosphorylated by the dual-specificity Antarctic phosphatase (Fig. 2A, lanes 2 and 7). The dephosphorylated ARP2/3 complex was then incubated in the presence of ATP with purified recombinant GST-LegK2 or its kinase-dead variant GST-LegK2K112M. Proteins were separated by SDS-PAGE and probed with antiphosphothreonine antibodies. Western blot assays revealed that both ARP3 and ARPC1B were phosphorylated in vitro by the catalytically active enzyme GST-LegK2 (Fig. 2A, lanes 3 and 8), while no or weak phosphorylation was observed with the kinase-dead mutant (Fig. 2A, lanes 4 and 9). Antiphosphothreonine immunolabeling of ARP3 and ARPC1B subunits decreased after incubation of the complex with Antarctic phosphatase, thus demonstrating that ARP3 and ARPC1B were in vitro modified by LegK2-catalyzed phosphorylation (Fig. 2A, lanes 5 and 10).
FIG 2 .

Protein kinase LegK2 phosphorylates ARPC1B and ARP3 subunits. (A) In vitro phosphorylation assays of ARPC1B and ARP3 subunits by LegK2 detected by Western blot assay with antiphosphothreonine antibodies. The ARP2/3 complex purified from A. castellanii (lanes 1 and 6) and dephosphorylated (lanes 2 to 5 and 7 to 10) was incubated with purified GST-LegK2 (lanes 3, 5, and 8, 10) or its catalytic variant GST-LegK2K112M (lanes 4 and 9) in the presence of 100 µM ATP. Where indicated (lanes 5 and 10), ARP2/3 was further incubated with dual-specificity Antarctic phosphatase. ARP2/3 complex subunits were separated by SDS-PAGE, stained with Coomassie blue, or detected with antiphosphothreonine antibodies. (B) In vivo phosphorylation assays of ARPC1B and ARP3 subunits by LegK2. HEK293T cells were transfected with pCI-Neo3Flag-ARPC1B or -ACTR3 or cotransfected with pCI-Neo3Flag-ARPC1B or -ACTR3 and pDEST27-legK2 or -legK2K112M. 3Flag-ARPC1B and 3Flag-ARP3 were immunoprecipitated with anti-Flag antibodies. Expression of GST-LegK2 and GST-LegK2K112M in the total extract was checked by Western blot assay with anti-GST antibodies (lanes 3 and 4 and lanes 6 and 7). Immunoprecipitation (IP) of 3Flag-ARPC1B and 3Flag-ARP3 was checked by Western blot assay with anti-Flag antibodies (lanes 2 to 7). The phosphorylation level of 3Flag-ARPC1B and 3Flag-ARP3 was detected with antiphosphothreonine antibodies. Protein bands have been cut to present samples in an order that matches our comments in the text.
In order to confirm the physiological relevance of ARP2/3 subunit phosphorylation, the ARP3 and ARPC1B phosphorylation states were analyzed in mammalian cells ectopically expressing the legK2 gene or the kinase-dead variant. HEK293T cells were cotransfected with pDEST27-legK2 or -legK2K112M and pCI-Neo3Flag-ACTR3 or -ARPC1B. The GST-tagged protein kinases were well expressed at 24 h posttransfection, as revealed by immunoblot assays of whole-cell extracts with anti-GST antibodies (Fig. 2B, lanes 3 and 4 and lanes 6 and 7). Flag-tagged ARP2/3 subunits were immunoprecipitated with anti-Flag antibodies and then detected with anti-Flag or antiphosphothreonine antibodies. In immunoblot assays, we found that the ARP3 and ARPC1B proteins, well expressed and immunopurified in all of the samples, appeared as two bands with close apparent molecular weights in cells expressing LegK2 kinase (Fig. 2B, lanes 3 and 6). We next examined whether these two bands represent two distinct phosphorylation states of ARP3 and ARPC1B. We found that in the presence of LegK2, antibodies to phosphothreonine labeled ARP3 and ARPC1B (Fig. 2B, lanes 3 and 6). When cells were cotransfected with the LegK2K112M-expressing vector, a single Flag-tagged band was detected with anti-Flag antibodies while phosphorylated forms of ARP3 and ARPC1B were not detected with antiphosphothreonine antibodies (Fig. 2B, lanes 4 and 7). Thus, the bacterial protein kinase LegK2 is able to phosphorylate ARP3 and ARPC1B subunits of the ARP2/3 complex in mammalian cells.
LegK2 is an inhibitor of the ARP2/3 complex in vivo.
The effect of LegK2-dependent ARP2/3 phosphorylation on the actin nucleation activity of the complex was first investigated by measuring actin polymerization in vitro with the purified ARP2/3 complex and in mammalian cells. Proteins named nucleation-promoting factors (NPFs) are recognized as the primary activators of the ARP2/3 complex for actin nucleation. However, a recently identified requirement for activation of nucleation is phosphorylation of ARP2/3 subunits, in particular, the ARPC1B and ARP2 subunits (26–28). We used pyrenyl actin assembly assays to measure actin polymerization in the presence of the purified dephosphorylated ARP2/3 complex and GST-LegK2. No significant increase in actin polymerization was detected (see Fig. S1 in the supplemental material). On the basis of this finding, we hypothesized that LegK2 does not phosphorylate the sites of ARP2/3 subunits shown to activate the complex. To detect a putative inhibitory effect of LegK2-dependent ARP2/3 phosphorylation on actin nucleation, actin polymerization was then assessed in the presence of the purified endogenous phosphorylated ARP2/3 complex and GST-LegK2. Our data showed that rates of actin nucleation and polymerization were not modulated by LegK2 (see Fig. S1). Importantly, mass spectrometry analysis of the in vitro phosphorylated ARP2/3 samples did not allow the detection of phosphorylated peptides; these data highlight that only a very small fraction of the ARP2/3 complex was phosphorylated under these conditions, which could explain the lack of in vitro effect of LegK2-dependent ARP2/3 phosphorylation on actin ARP2/3 nucleation activity.
To address whether actin nucleation could be controlled by LegK2-dependent ARP2/3 phosphorylation in an in vivo context, actin polymerization was analyzed in human epithelial HeLa cells transfected with the empty vector peGFP or the peGFP-legK2 or peGFP-legK2K112M vector, which encodes GFP and an N-terminally GFP-fused LegK2 protein or its kinase-dead variant, respectively. Transfected cells were analyzed by confocal microscopy for F-actin labeling. Three types of cell labeling were categorized according to actin filament distribution: with long, straight actin fibers that are contained within the body of the cell (type 1), with reduced actin fibers that localize only at the periphery of the cell (type 2), and without noticeable actin fibers (type 3) (Fig. 3A). A total of 27.8% of the GFP-LegK2-expressing cells exhibited a type 3 phenotype, which is more than the 11 and 12% of type 3 phenotype observed among cells transfected with the empty vector and the vector expressing kinase-dead variant GFP-LegK2K112M, respectively (Fig. 3B). Thus, expression of active LegK2 protein kinase in mammalian cells inhibits actin fiber polymerization. Interestingly, expression of the kinase-dead variant resulted in an intermediate phenotype of reduced actin fibers, since cells transfected with the kinase-dead variant are characterized by a shift from type 1 to type 2, unlike empty-vector-transfected cells (41.5% of the cells are type 2 for the catalytic variant versus 20% for the empty vector). As previously proposed for other protein kinases (29–32), it can be assumed that the inactive form of LegK2 is still able to interact with ARP2/3 and that overexpression of this inactive variant results in the titration of its substrate, thus interfering with the cell actin cytoskeleton dynamic. Together, these data strongly support the notions that expression of LegK2 in mammalian cells inhibits the formation of actin stress fibers and that the protein kinase activity of LegK2 is partially responsible for this inhibition.
FIG 3 .

LegK2 protein kinase activity inhibits actin polymerization in an ARP2/3-dependent manner. (A) Actin filament distribution in GFP-LegK2-expressing HeLa cells. Polymerized actin was labeled with Alexa Fluor 594 phalloidin (red). Type 1 cells exhibit long and straight fibers in the body of the cell (arrow); type 2 is characterized by reduced and peripheral actin fibers (arrowhead); in type 3, neither central nor peripheral actin fibers are detected. Scale bars, 10 µm. (B) Quantitation of actin filament distribution in transfected HeLa cells. HeLa cells were transfected with the empty vector peGFP or vector peGFP-legK2 or peGFP-legK2K112M. The actin network was evaluated in 40 cells transfected with each vector. These data are representative of three independent experiments, and the error bars represent the standard deviations. (C) Cell invasion by Listeria and Listeria comet tail formation in GFP-LegK2-expressing mammalian cells. HeLa cells transfected with the vector peGFP or peGFP-legK2 were infected for 5 h at an MOI of 5 with L. monocytogenes and then processed for differential immunolabeling, in which extracellular bacteria were differentiated from total bacteria. Extracellular bacteria were labeled by a rabbit-derived polyclonal serum (R11) and an Alexa Fluor 647-conjugated secondary antibody (white labeling); cells were then permeabilized, and total bacteria were labeled with the same R11 serum and an Alexa Fluor 350-coupled secondary antibody (blue labeling). Actin was labeled with Alexa Fluor 546 phalloidin (red labeling). In the right panel, arrows indicate two extracellular bacteria, the dashed arrow highlights an intracellular bacterium with a short actin tail, and arrowheads point toward intracellular bacteria without actin comet tails. Bar, 5 µm. (D) Quantitation of cells with extracellular bacteria. peGFP-, peGFP-legK2-, or peGFP-legK2K112M-transfected HeLa cells (>100 for each transfection) with only extracellular bacteria were counted and compared to the total number of transfected cells with intra- and extracellular bacteria. Results are expressed as the percentage of HeLa cells displaying only extracellular L. monocytogenes. These data are representative of three independent experiments, and the error bars represent the standard deviations. (E) Quantitation of infected cells without actin comet tails. Cells were processed as indicated for panel C, and the number of infected HeLa cells in which actin tails were not detectable was determined and expressed as a percentage of the total. These data are representative of three independent experiments, and the error bars represent the standard deviations.
To establish whether LegK2 inhibits actin polymerization in an ARP2/3-dependent manner, we monitored the effect of LegK2 on two well-known ARP2/3-dependent processes, namely, cell invasion and actin comet tail formation by Listeria. Mammalian cells transfected with the empty peGFP vector or the peGFP-legK2 vector were infected with Listeria monocytogenes for 5 h. Actin was labeled with fluorescent phalloidin, and cells were processed for a differential immunolabeling protocol in which extracellular bacteria were differentiated from total ones (Fig. 3C). Cell transfection with peGFP does not hamper bacterial invasion, as reflected by the low number of extracellular versus intracellular bacteria (Fig. 3C, white versus blue channels). When quantified, only 2% of the cells displayed extracellular bacteria (Fig. 3D). Actin comet tail formation also takes place efficiently in GFP-expressing cells (Fig. 3C, red channel), since less than 3% of the infected cells do not exhibit these actin structures (Fig. 3E). In contrast, expression of GFP-LegK2 increases the number of extracellular bacteria (Fig. 3C, arrows) and reduces the number of intracellular ones (Fig. 3C, arrowhead), resulting in about 18% of the cells displaying extracellular L. monocytogenes (Fig. 3D). Moreover, in >20% of the infected GFP-LegK2-expressing cells, no actin comet tails are detected (Fig. 3E). These effects depend on the protein kinase activity of LegK2, since expression of the kinase-dead variant resulted in only 3.8% of the cells displaying extracellular bacteria (Fig. 3D) and 2.5% of the infected cells exhibiting no actin comet tail (Fig. 3E), which is not significantly different from what is observed in cells transfected with the empty vector peGFP. Together, these results therefore indicate that ectopic expression of active LegK2 protein kinase in mammalian cells inhibits ARP2/3-dependent cell invasion and actin comet tail formation by L. monocytogenes, leading to the conclusion that in cells, the protein kinase activity of LegK2 is an inhibitor of the actin nucleation activity of the ARP2/3 complex.
LegK2 localizes to the LCV.
To get insight into the role of LegK2 during Legionella infection, we first analyzed the dynamics of LegK2 localization in infected cells, i.e., after translocation into the host cell cytosol. To examine LegK2 localization, the amoeba Dictyostelium discoideum was infected with an L. pneumophila legK2 mutant strain transformed with a vector encoding N-terminally hemagglutinin (HA)-tagged LegK2. This strain does not encode the endogenous LegK2 protein that might compete with tagged LegK2 for translocation, localization, or function. Bacteria were immunolabeled with an antibody directed against the major outer membrane protein (MOMP) of L. pneumophila, and HA-tagged LegK2 protein was revealed with an anti-HA antibody at 0, 5, 10, 15, 20, 30, and 60 min after bacterium-amoeba contact (Fig. 4A and B). As a control, D. discoideum amoebae were also infected with the L. pneumophila dotA mutant strain transformed with the same vector encoding HA-tagged LegK2. As expected, HA-tagged LegK2 protein was not detected when amoebae were infected with the dotA mutant strain, confirming that LegK2 translocation is Dot/Icm dependent (Fig. 4A). In contrast, HA-tagged LegK2 protein was found on the legK2 mutant strain LCVs immediately after bacterial entry and with an increased frequency up to 15 min postcontact before disappearance and was only slightly detectable 20 min postcontact (Fig. 4B). This time-dependent decrease may be due to rapid proteolytic degradation. These results suggest that upon L. pneumophila infection, LegK2 function is very transient and localized mostly on LCVs. Because the ARP2/3 complex was identified by proteomic analysis on the nascent Legionella-containing phagosome (33–35), the transient localization of LegK2 on the LCV supports a direct effect of LegK2 on ARP2/3 activity and consequently on actin polymerization on the LCV.
FIG 4 .

LegK2 inhibits actin polymerization on the LCV. (A) HA-LegK2 detection on LCVs. D. discoideum was infected for 15 min at an MOI of 100 with dotA or legK2 mutant L. pneumophila transformed with a vector encoding N-terminally HA-tagged LegK2. The presence of bacteria was detected by an immunofluorescence assay with anti-MOMP antibodies (anti-Lp, red labeling), and the HA-LegK2 fusion is labeled with anti-HA antibodies (anti-HA, green labeling). Scale bars, 5 µm. (B) HA-LegK2 detection during LCV biogenesis. D. discoideum was infected at an MOI of 100 with legK2 mutant L. pneumophila transformed with a vector encoding N-terminally HA-tagged LegK2. HA-LegK2-positive LCVs were counted at 0, 5, 10, 15, 20, 30, and 60 min after bacterium-amoeba contact (>100 LCVs per time point). These data are representative of three independent experiments, and the error bars represent the standard deviations. (C) Actin polymerization on LCVs during Legionella infection. D. discoideum was infected for 15 min at an MOI of 100 with mCherry-labeled legK2 mutant L. pneumophila. Polymerized actin on LCVs was detected by labeling with phalloidin-FITC. Arrows show examples of actin-positive LCVs. Scale bar, 10 µm. (D) Detection of polymerized actin in legK2 mutant-containing vacuoles. Actin-positive vacuoles (n = >100) were counted for amoeba infected with WT L. pneumophila strain Lens, the derivative dotA and legK2 mutants, and the transformed legK2(plegK2) and legK2(plegK2cat) mutants. These data are representative of three independent experiments, and the error bars represent the standard deviations. **, P < 0.01. (E) Exclusive detection of LegK2 and actin on LCVs. D. discoideum was infected for 15 min at an MOI of 100 with L. pneumophila transformed with a vector encoding HA-tagged LegK2. Bacteria were detected with anti-Lp1 serum and an anti-rabbit secondary antibody (blue labeling), HA-LegK2 on the LCVs was labeled with an anti-HA antibody and an anti-mouse secondary antibody (red labeling), and actin was stained with phalloidin-FITC (green labeling). Arrows indicate LegK2-positive LCVs (without actin labeling), and the dotted arrow indicates an LCV without LegK2 on its surface that was consequently stained for actin. Scale bar, 10 µm. This micrograph is representative of 50 LCVs observed in two independent experiments.
LegK2 inhibits actin polymerization on the LCV.
To address the question of actin cytoskeleton remodeling during Legionella infection, the amoeba D. discoideum was infected with the mCherry-producing L. pneumophila Lens strain and the dotA and legK2 deletion mutant strains and polymerized actin was visualized with phalloidin-fluorescein isothiocyanate (FITC) at 15 min postinfection (Fig. 4C). Infection with L. pneumophila does not trigger significant cytoskeleton reorganization in host cells but most likely induces local actin remodeling on the LCV. While less than 2% of the wild-type (WT) bacterium-containing vacuoles were labeled with phalloidin, more that 20% of the dotA mutant-containing vacuoles were actin positive (Fig. 4D). Strikingly, 14.8% of the legK2 mutant-containing vacuoles were labeled with phalloidin, a percentage that is significantly higher than that for WT LCVs (Fig. 4D). To confirm that this effect is due to the legK2 deletion, a legK2 gene-expressing vector was introduced into the bacterium and found to complement the deletion. In complemented cells, only 3.4% of the LCVs were phalloidin positive. In contrast, the kinase-dead LegK2-expressing vector (plegK2cat) did not complement the legK2 deletion since 13.8% of the LCVs were phalloidin positive (Fig. 4D). Thus, LCVs containing an avirulent mutant impaired for the T4SS, such as the dotA mutant or the kinase-dead and legK2 deletion mutants, are decorated with polymerized actin. It is noteworthy that neither WT nor dotA or legK2 mutant LCVs are phalloidin positive when actin labeling is performed immediately after uptake. This is consistent with previous observations that cortical actin associated with the bacterial entry sites during phagocytosis disassemble from the Legionella-containing phagosome less than 1 min after engulfment of the bacteria (36). To further check that inhibition of actin polymerization is correlated with the presence of LegK2 on the LCV, D. discoideum amoebae were infected with an L. pneumophila strain transformed with a vector encoding HA-tagged LegK2. They were costained 15 min postinfection for LegK2 and actin with anti-HA antibodies and phalloidin-FITC, respectively (Fig. 4E). We observed that LegK2-positive vacuoles (red labeling) were not labeled with phalloidin (green labeling), while LCVs that did not exhibit LegK2 (blue labeling) were clearly actin positive (Fig. 4E). When quantified on 50 LCVs from two independent experiments, 100% of the LegK2-positive vacuoles lacked actin labeling. Together, these data support the hypothesis that LegK2 is translocated by the Dot/Icm T4SS into the host cell cytosol and its protein kinase activity triggers local inhibition of actin polymerization on the LCV after bacterial uptake.
LegK2 interferes with late endosome/lysosome trafficking to the LCV.
Actin-based intracellular movements of endosomes and phagosomes are dependent on a driving force provided by the ARP2/3 complex (37), and actin assembly on phagosome or late endocytic organelles provides tracks for fusion partner organelles to facilitate vesicle fusion (38). Taking into account (i) actin-dependent endosome trafficking, (ii) the localization of LegK2 and its role in the inhibition of actin polymerization on the LCV, and (iii) that a legK2 deletion mutant poorly evades endocytic degradation since only 10% of mutated bacteria survive after cell uptake (24), we hypothesized that LegK2 could be involved in the control of phagosome maturation, in particular, by inhibiting the fusion of the LCV with late endosomes.
D. discoideum was infected with the mCherry-labeled L. pneumophila Lens strain or the dotA or legK2 deletion mutant, and the late endosomal or lysosomal vacuolar H+-ATPase (V-ATPase) was labeled at 1 h postinfection by immunofluorescence with anti-VatA antibodies (Fig. 5A). As a control, we observed that while the WT Lens strain interferes with phagosome maturation, as demonstrated by only 11.8% VatA-positive LCVs, a T4SS-impaired dotA mutant strain is less able to inhibit the endocytic pathway since around 45% of the dotA mutant-containing vacuoles are labeled with VatA (Fig. 5B). About 28% of the legK2 mutant-containing vacuoles were also VatA positive, which is significantly higher than the 11.8% obtained in the case of WT LCVs. Transforming the legK2 mutant with a WT LegK2-encoding vector partially complements this endocytic pathway evasion defect by decreasing the percentage of VatA-positive vacuoles to 18.4% of the LCVs. In contrast, the kinase-dead LegK2-expressing vector (plegK2cat) did not complement the legK2 deletion since 33.9% of the LCVs were VatA positive (Fig. 5B). Thus, the protein kinase activity of LegK2 contributes to the blocking of late endosomal trafficking toward the LCV and consequently allows bacteria to escape the late endocytic pathway.
FIG 5 .
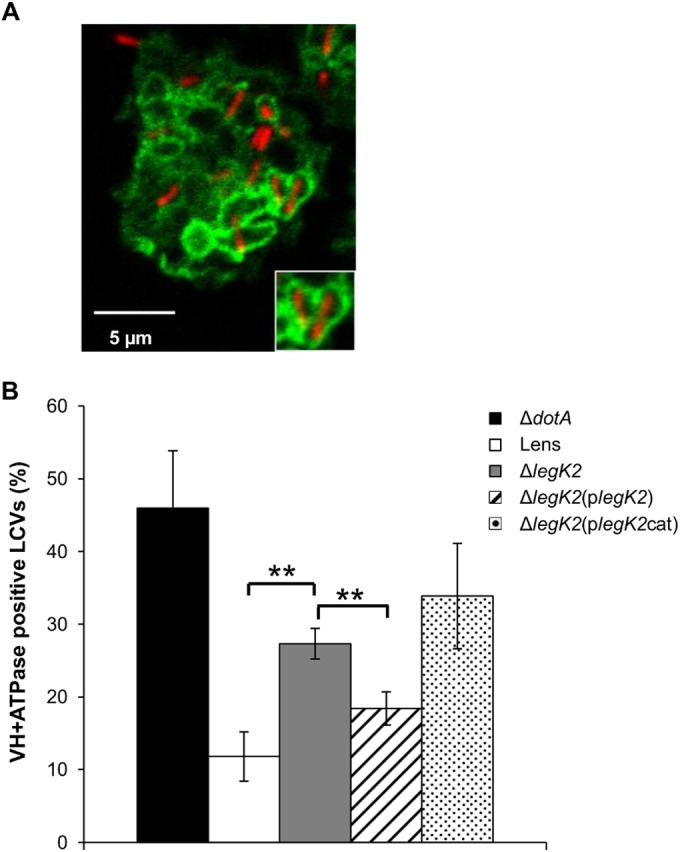
LegK2 inhibits late endosome trafficking to LCVs. (A) Acquisition of vacuolar proton ATPase on LCVs. D. discoideum was infected for 1 h at an MOI of 100 with mCherry-labeled legK2 mutant L. pneumophila. The presence of vacuolar H+-ATPase on LCVs was detected by an immunofluorescence assay with anti-VatA antibodies. The inset shows typical VatA-positive vacuoles. (B) Acquisition of vacuolar proton ATPase in legK2 mutant-containing vacuoles. VatA-positive vacuoles (n = >100) in amoebae infected with WT L. pneumophila Lens, the derivative dotA and legK2 mutants, and the transformed legK2(plegK2) and legK2(plegK2cat) mutants were counted. These data are representative of three independent experiments, and the error bars represent the standard deviations. **, P < 0.01.
LegK2-ARP2/3 interplay controls actin polymerization on the LCV to evade LCV-late endosome fusion.
The data presented above demonstrate that LegK2 interacts with and phosphorylates two subunits of the actin filament nucleator ARP2/3, and it localizes to the LCV to inhibit both actin polymerization and late endosome fusion with the LCV, thus contributing to escape of the bacteria from the late endocytic pathway. In order to establish whether these two major events controlled by LegK2 are related, i.e., whether LegK2 inhibits ARP2/3 nucleation activity to control actin polymerization on the LCV, D. discoideum was infected with the mCherry-labeled legK2 mutant strain in the presence of 100 µM CK-666, a specific inhibitor of the ARP2/3 complex shown to block Listeria actin tail formation (39). Pharmacological inhibition of ARP2/3 activity was preferred to ACTR3 and ARPC1B silencing in order to temporally control ARP2/3 inhibition and consequently avoid a pleiotropic effect of this inhibition. Infected cells were labeled with phalloidin, and vacuoles positive for polymerized actin were scored (Fig. 6A). The presence of CK-666 reverts the legK2 deletion mutant phenotype; i.e., it strongly decreases the presence of actin on the LCVs so that the percentage of actin-positive LCVs was similar to that obtained with WT LCVs. Moreover, when infected cells were immunolabeled with anti-VatA antibodies (Fig. 6B), addition of CK-666 resulted in a significant decrease in VatA-positive vacuoles, suggesting that CK-666 partially reverts the legK2 deletion mutant phenotype for the endocytic pathway evasion defect. Thus, chemical inhibition of the actin nucleator activity of ARP2/3 phenocopies the effect of LegK2 protein kinase, which suggests that LegK2 acts on ARP2/3 by inhibiting its normal actin nucleation activity. Together, these data show that the LegK2-ARP2/3 interplay results in inhibition of both actin polymerization on the LCV and late endosome trafficking toward the LCV and that these events contribute to bacteria evasion of the endocytic pathway.
FIG 6 .
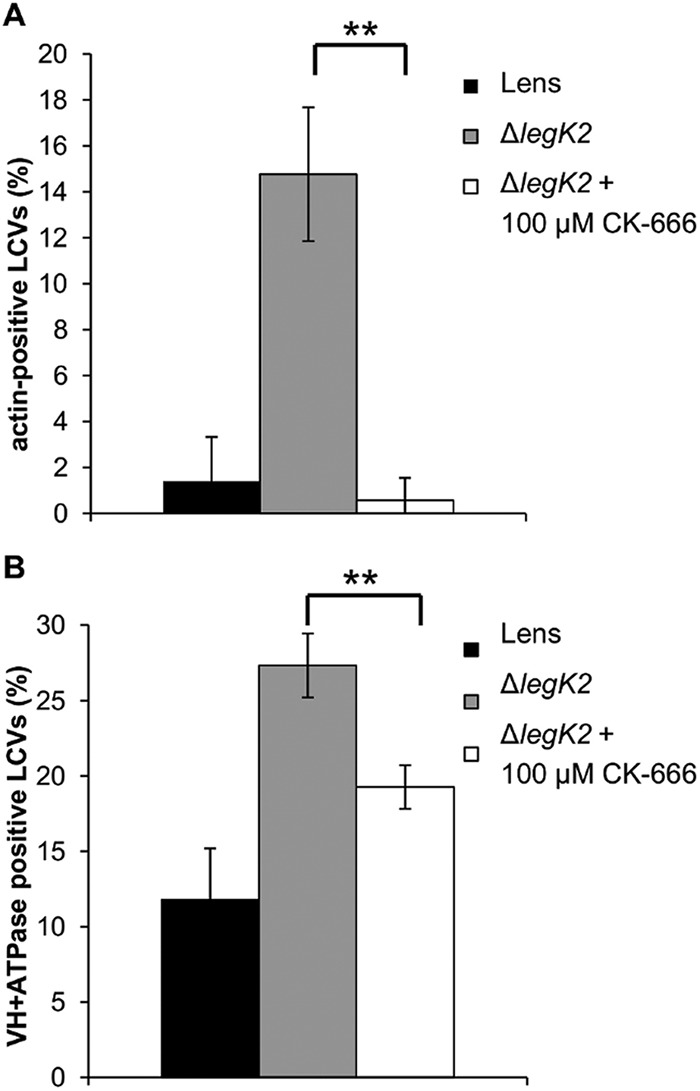
ARP2/3 chemical inhibition complements a legK2 gene deletion defect. (A) Actin polymerization on LCVs in the presence of CK-666. More than 100 LCVs were scored from each sample of phalloidin-FITC-labeled D. discoideum infected at an MOI of 100 with mCherry-labeled L. pneumophila Lens or the legK2 mutant in the absence or presence of 100 µM CK-666. These data are representative of three independent experiments, and the error bars represent the standard deviations. (B) Acquisition of vacuolar proton ATPase on LCVs in the presence of CK-666. More than 100 LCVs were scored for VatA immunolabeling in each sample of D. discoideum infected at an MOI of 100 with mCherry-labeled L. pneumophila Lens or the legK2 mutant in the absence or presence of 100 µM CK-666. These data are representative of three independent experiments, and the error bars represent the standard deviations. **, P < 0.01.
DISCUSSION
Host cell pathways targeted by the Legionella protein kinase LegK2 were screened by a two-hybrid assay in yeast cells, and several human proteins were identified as putative candidates for interaction with LegK2. As the legK2 mutant prevents evasion of endocytic degradation (24), we focused our studies on proteins involved in host cell organelle trafficking and cytoskeleton remodeling. Using biochemical and tissue culture assays, we demonstrated that LegK2 interacts with and phosphorylates the ARP3 and ARPC1B subunits of the actin filament nucleator ARP2/3 complex. These data highlight ARP2/3 phosphorylation as a mechanism for controlling ARP2/3 complex activity (26–28). Expression of active LegK2 inhibits host cell invasion and actin comet tail formation during Listeria infection, i.e., two well-documented ARP2/3-dependent actin polymerization processes, while the kinase-dead variant is unable to do so. Thus, we have robustly demonstrated that LegK2 is a potent inhibitor of the ARP2/3 complex and that the protein kinase activity of LegK2 is essential for this property.
During Legionella infection, rather than changing the overall level of actin polymerization, we observed that LegK2 interferes with local actin remodeling, most likely by being targeted to the LCV and inhibiting actin polymerization in this compartment. This local effect on actin polymerization is ARP2/3 dependent since it can be phenocopied by a specific chemical inhibitor of the ARP2/3 complex. Inhibition of actin polymerization on the LCV is consistent with comparative proteomics of phagosomes containing highly virulent L. pneumophila Corby versus the less virulent species L. hackeliae. These studies revealed that phagosomes containing nonreplicative L. hackeliae displayed more polymerized actin than those containing the virulent Legionella species (33). The authors of that study proposed that this change in virulence was due to actin degradation on the LCV containing the virulent L. pneumophila strain, but it also could result from inhibition of actin polymerization on the LCV. Moreover, it is noteworthy that the ARP2/3 complex, along with other actin cytoskeleton-associated proteins, is transiently enriched in the proteome of the nascent Legionella-containing phagosome without any significant changes between virulent and avirulent bacteria (33). These observations are consistent with our finding that LegK2 could control the activity of the ARP2/3 complex on the LCV rather than the recruitment of the actin nucleator complex.
Our finding that LegK2 is a key Dot/Icm T4SS effector of the control of actin polymerization remodeling on the LCV gives important insights to help understand the molecular mechanisms by which L. pneumophila evade endocytic degradation. We demonstrated that LegK2 contributes to L. pneumophila evasion of endosome trafficking toward the LCV and that this role is dependent on the interplay between LegK2 and ARP2/3 to inhibit actin polymerization on the LCV. Although Legionella evasion of the endocytic pathway is a main virulence-related event of the infection cycle of L. pneumophila, the molecular mechanisms involved in hijacking this pathway are limited to the demonstration that L. pneumophila secretes SidK, which inhibits V-ATPase activity, to decrease LCV acidification (18). Here, our results highlight the essential role of LegK2 activity in the inhibition of actin polymerization on the LCV, thus preventing fusion with late endosomes and finally rerouting the phagosome to a replicative niche.
Actin polymerization has recently been identified as a host cell target of two other Dot/Icm substrates, VipA (40) and Ceg14 (Lpg0437) (41). The Legionella VipA effector binds actin in vitro and directly polymerizes microfilaments without the requirement of additional proteins, a property distinct from those of other bacterial actin nucleators. Microscopy studies have revealed that VipA localizes to actin patches and early endosomes during macrophage infection (40). The Legionella effector Ceg14 cosediments with filamentous actin and inhibits in vitro actin polymerization. Ceg14 is toxic to yeast, and this toxicity can be alleviated by overexpression of the cytoskeleton-related protein profilin. Despite its molecular effect on actin polymerization, the role of Ceg14 in Legionella infection is still not documented (41). In future studies, it will be interesting to decipher whether LegK2, Ceg14, and VipA display synergic or antagonistic activities to temporally control actin polymerization on the LCV in order to control LCV biogenesis. Interestingly, whatever the precise mechanism controlled by each of these T4SS effectors, LegK2 function seems to be predominant since a legK2 single mutant is affected in virulence while a vipA or ceg14 mutant is altered neither for entry nor for replication in amoebae and macrophages, like most of the Dot/Icm effector mutants. Noteworthy, an unanswered question is whether the growth defect of the legk2 mutant is due only to its failure to inhibit actin nucleation around the phagosome. Nevertheless, VipA, Ceg14, and LegK2 effectors highlight the host cell actin cytoskeleton as a major target of Legionella, and we demonstrate here for the first time that actin remodeling is a critical determinant of an effective cycle of L. pneumophila infection.
Actin cytoskeleton remodeling is known to be targeted by numerous extra- and intracellular bacteria in order to (i) trigger bacterial uptake by nonphagocytic cells (42), (ii) move within the host cell cytosol and spread from cell to cell (43), or (iii) decorate the phagocytic vacuole with a polymerized actin meshwork (44). To date, actin remodeling targeted by bacteria to alter host cell organelle trafficking for successful infection, as we propose LegK2 to do, has not been described. Moreover, the ARP2/3 complex has been shown only to be hijacked by recruiting or mimicking NPFs but not shown to be modified by phosphorylation to control ARP2/3-dependent actin nucleation activity. Thus, both the role of actin remodeling and the molecular mechanism controlled by the Legionella effector LegK2 are unique and novel.
It is noteworthy that the atypical protein kinase SteC from Salmonella enterica serovar Typhimurium is required for F-actin meshwork formation around the Salmonella-containing vacuole (45). Although SteC has recently been shown to target another actin cytoskeleton effector protein, namely, the MAP kinase MEK (46), we note the similarity of both the enzymatic activities of the bacterial protein kinases LegK2 and SteC to the host cell actin cytoskeleton remodeling pathway targeted by these kinases. Similarly, the Yersinia pseudotuberculosis T3SS effector YpkA is an essential virulence determinant that disrupts the host actin cytoskeleton and inhibits phagocytosis. In the presence of actin, the kinase YpkA phosphorylates Gαq, which impairs guanine nucleotide binding by Gαq and results in the inhibition of multiple Gαq signaling pathways, among which are Rho GTPase activation and consequently actin stress fiber assembly (47). Although the molecular mechanisms controlled by these protein kinases are different in each case, the role of protein phosphorylation in the control of host cell actin remodeling emerges as a paradigm in bacterial pathogenesis.
MATERIALS AND METHODS
Cells, bacteria, and plasmids.
For the bacterial strains, cells, and plasmids used in this study, see Table S1 in the supplemental material.
L. pneumophila strains were grown at 30°C either on buffered charcoal yeast extract (BCYE) agar or in BYE liquid medium. Each medium was supplemented with 5 µg·ml−1 chloramphenicol when appropriate. L. monocytogenes strain EGDe.PrfA* (BUG 3057) was cultured in brain heart infusion broth at 37°C. Escherichia coli strains were grown at 37°C in LB medium supplemented with 100 µg·ml−1 ampicillin, 20 µg·ml−1 kanamycin, 100 µg·ml−1 spectinomycin, and 20 µg·ml−1 chloramphenicol. E. coli strains XL1-Blue and DH5α were used to maintain plasmids, and E. coli strain BL21(DE3)(pREP4-groESL) was used for recombinant protein overproduction.
S. cerevisiae strains AH109 and Y187 were grown at 30°C in rich YPD (yeast extract, peptone, dextrose) medium or in SD (synthetic dextrose) medium supplemented with the appropriate amino acids.
The WT amoeba D. discoideum strain AX2 (DBS0235534) was obtained from the Dicty Stock Center (http://dictybase.org). D. discoideum cells were grown axenically in HL5 medium at 22°C with 100 µg·ml−1 streptomycin and 66 µg·ml−1 penicillin G.
Human HEK293T fibroblasts and human epithelial HeLa cells (gift from INSERM U1111, Lyon, France) were maintained at 37°C in 5% CO2 in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% heat-inactivated fetal calf serum (FCS) and 50 µg·ml−1 gentamicin. HeLa CCL-2 cells (American Type Culture Collection) were cultured at 37°C in DMEM supplemented with 10% heat-inactivated FCS in a 10% CO2 atmosphere.
General DNA techniques and Gateway cloning.
For the oligonucleotides used in this study, see Table S2 in the supplemental material.
DNA constructions were performed with the Gateway recombinational cloning system as recommended by the manufacturer (Invitrogen) or by restriction-ligation molecular cloning. Plasmid DNA from E. coli was prepared by Plasmid Midi and Mini kits (Qiagen). PCR amplifications were carried out with Phusion polymerase as recommended by the manufacturer (Finnzymes). Site-directed mutagenesis experiments were performed with the QuikChange II site-directed mutagenesis kit (Stratagene). To obtain the kinase-dead mutant protein LegK2K112M defective in phosphate donor ATP binding and the kinase-dead mutant protein LegK2K112M/D209N defective in both phosphate donor ATP binding and phosphate transfer, nucleotide substitutions in the legK2 gene were performed with primer pairs 1/2 and 3/4 (see Table S2).
Vector construction.
The DNA fragments corresponding to the legK1 (lpl1545) and legK2 (lpl2066) coding sequences were PCR amplified by using genomic DNA of L. pneumophila Lens as the template and oligonucleotide pairs 5/6 and 7/8 (see Table S2), respectively. The coding sequences were inserted into the Gateway pDONR207 vector (Invitrogen) by in vitro recombination. The LegK1- and LegK2-encoding genes were then transferred from pDONR207 to pGBKT7 (Clontech), thus producing LegK proteins fused to the DNA binding domain of the GAL4 transcriptional activator in yeast cells. The LegK2-encoding gene was also transferred by Gateway cloning from pDONR207-legK2 to vectors pDEST27 and peGFP (Invitrogen) to produce GST-tagged LegK2 and N-terminally GFP-fused LegK2 proteins in mammalian cells, respectively. The legK2 gene from pXDC61-legK2 (gift from J. Allombert in our lab) was ligated into plasmid pMMB207c-HA*4 at the BamHI and KpnI restriction sites (gift from G. Frankel, London, United Kingdom [48]) to encode N-terminally HA-tagged LegK2 upon isopropyl-β-d-thiogalactopyranoside (IPTG) induction in L. pneumophila cells. The mutated legK2K112M/D209N gene was ligated into the SphI restriction site of pXDC50 to encode kinase-dead LegK2 under the control of its own promoter in L. pneumophila cells.
ARPC1B and ACTR3 cDNAs were obtained from the human ORFeome resource (hORFeome v3.1) in the pDONR223 Gateway vector. cDNAs were transferred by in vitro recombination from pDONR into yeast vector pACT2 (Clontech) to produce proteins fused with the transcription activator domain of GAL4. ARPC1B and ACTR3 cDNAs were in vitro recombined from pDONR223 vectors to the pCI-Neo3Flag vector (Invitrogen), thus producing 3Flag-tagged ARP2/3 complex subunits in mammalian cells.
Yeast two-hybrid screening and binary assays.
Vectors pGBKT7-legK1 and -legK2, which encode LegK proteins fused to the DNA binding domain of the GAL4 transcriptional activator, were transformed into bait yeast strain AH109. Bait cells were screened against a human normalized cDNA spleen library for systematic screening or against ACTR3 and ARPC1B for binary assays cloned into vector pACT2 transformed into prey yeast strain Y187, which produces human proteins fused to the transcriptional activator domain of GAL4. AH109(pGBKT7-legK) and Y187(pACT2-human cDNA), Y187(pACT2-ACTR3), or Y187(pACT2-ARPC1B) yeast cells were mated and subsequently plated onto a selective medium lacking histidine and supplemented with 10 mM 3AT to test the interaction-dependent transactivation of the HIS3 reporter gene.
Affinity copurification.
HEK293T cells were cotransfected with pDEST27, pDEST27-legK2, or pDEST27-legK2K112M and pCI-Neo3Flag-ARPC1B or -ACTR3 by using Lipofectamine 2000 (Invitrogen). Transfected cells were lysed at 24 h posttransfection in modified RIPA buffer (50 mM Tris HCl [pH 7.4], 100 mM NaCl, 10% glycerol, 1% NP-40), and GST-tagged LegK2 or LegK2K112M was purified on glutathione-agarose 4B (Macherey-Nagel). Purified fractions were immunoblotted with both anti-GST (13-6700; Invitrogen) and anti-Flag (clone M2; Sigma) antibodies to detect GST or GST-tagged LegK2 protein and Flag-tagged ARP2/3 subunits, respectively.
Protein localization in transfected mammalian cells.
HEK293T cells were transfected or cotransfected with empty pDEST27 or pDEST27-legK2/legK1 and/or pCI-Neo3Flag-ARPC1B/ACTR3 by using Lipofectamine 2000 (Invitrogen). At 24 h posttransfection, cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100. Flag-tagged ARPC1B and ARP3 proteins were immunolabeled with mouse anti-Flag antibodies conjugated with the fluorochrome Cy3 (A9594; Sigma). GST, GST-LegK2, or GST-LegK1 protein was labeled with a rabbit anti-GST antibody (A7340; Sigma) and detected with an anti-rabbit secondary antibody conjugated with the fluorochrome Alexa Fluor 488 (A11034; Molecular Probes). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss). Quantitation of images was performed with ImageJ software (National Institutes of Health). The Pearson coefficient was determined with a JACoP plugin (49) and expressed as the mean value calculated for 30 cells.
ARP2/3 complex in vitro phosphorylation assays.
GST-LegK2 and GST-LegK2K112M were overproduced in and purified from E. coli as previously described (24). The ARP2/3 complex was purified from A. castellanii as previously described (26, 50). Where indicated, ARP2/3 was dephosphorylated with the dual-specificity Antarctic phosphatase (New England Biolabs Inc.) as previously described (26). In vitro phosphorylation of 7.2 µg of the purified ARP2/3 complex was performed in the presence of 1 µg of GST-LegK2 or GST-LegK2K112M for 30 min at 37°C in 20 µl of a buffer containing 25 mM Tris HCl (pH 7.5), 5 mM MnCl2, 5 mM dithiothreitol, and 100 mM ATP. Reactions were stopped by the addition of an equal volume of 2× Laemmli loading buffer (51). Where indicated, the reaction was further incubated in the presence of 10 U of Antarctic phosphatase for 1 h at 30°C before the addition of Laemmli buffer. Proteins were then separated by SDS-PAGE and immunoblotted with an antiphosphothreonine monoclonal antibody (clone PTR-8; Sigma).
ARPC1B and ARP3 in cellulo phosphorylation assays.
HEK293T cells were transfected or cotransfected with vectors pCI-Neo3Flag-ARPC1B/ACTR3 and pDEST27-legK2 or -legK2K112M by using Lipofectamine 2000 (Invitrogen). Protein A/G resin was cross-linked with 10 µg of anti-Flag antibodies by using a Pierce Cross-link immunoprecipitation kit (Thermo Scientific). Transfected cells were lysed at 24 h posttransfection, and Flag-tagged ARP2/3 subunits were immunoprecipitated as recommended by the manufacturer. Immunoprecipitation supernatants were then incubated with glutathione-agarose 4B (Macherey-Nagel) and analyzed by Western blot assay with anti-GST antibodies to check the expression of the GST-tagged LegK2 and LegK2K112M protein kinases. Immunoprecipitated proteins were immunoblotted with both anti-Flag (clone M2; Sigma) and antiphosphothreonine (clone PTR-8; Sigma) antibodies to check the immunoprecipitation and phosphorylation level of the ARP2/3 subunits, respectively.
In vitro actin polymerization assays.
In vitro actin polymerization assays were performed with 5 µM monomeric actin containing 10% pyrene-labeled actin in KMEI (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole [pH 7.0]). Polymerization was determined in the absence or presence of the ARP2/3 complex at 50 nM, the VCA domain of N-WASP at 500 nM, and recombinant WT LegK2. Fluorescence was measured with a BioTek Synergy 2 plate reader with 365-nm excitation and 407-nm emission filters at 10-s intervals. The purified ARP2/3 complex was dephosphorylated by treatment with 1 U of Antarctic phosphatase (New England Biolabs, Inc., Ipswich, MA) in Tris-HipH buffer (50 mM Tris [pH 8.0], 1 mM MgCl2, 0.1 mM ZnCl2) for 1 h at 30°C. The activation and inhibition of the ARP2/3 complex by LegK2 were tested on the dephosphorylated and phosphorylated ARP2/3 complex, respectively.
Actin polymerization in transfected mammalian cells.
HeLa cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions with a vector encoding peGFP alone or fused with legK2 or legK2K112M. At 24 h posttransfection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 5 min at room temperature (RT), and stained with Alexa Fluor 594 phalloidin (Molecular Probes). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Listeria comet actin tail formation.
HeLa CCL-2 cells grown on coverslips were transfected for 18 h with the empty vector peGFP or with the peGFP-legK2 vector with the FuGENE transfection reagent (Roche) at a ratio of 2 µg of DNA to 6 µl of FuGENE per treatment. Bacteria were grown overnight at 37°C in a shaking device, and on the following day, 1 ml of bacterial culture was washed three times with 1 ml of phosphate-buffered saline (PBS) and the optical density at 600 nm was measured to estimate bacterial numbers. HeLa cells were infected at a multiplicity of infection (MOI) of 5 for 1 h in DMEM supplemented with 1% FCS, and then extracellular bacteria were killed by adding DMEM supplemented with 10% FCS and gentamicin at 10 µg·ml−1. After 5 h of infection, HeLa cells were fixed with 4% paraformaldehyde and extracellular L. monocytogenes bacteria were labeled by adding a rabbit-derived polyclonal serum (R11) and an Alexa Fluor 647-conjugated secondary antibody (Molecular Probes). Cells were then permeabilized for 4 min with 0.1% Triton X-100 in PBS, and total bacteria were labeled with the same R11 serum and an Alexa Fluor 350-conjugated secondary antibody (Molecular Probes). Actin was labeled with phalloidin Alexa Fluor 546-coupled reagent (Molecular Probes). Images were acquired with a 100× objective installed on an inverted wide-field Axiovert 200M microscope (Zeiss) equipped with an electron multiplication charge-coupled device Neo camera (Andor) and the MetaMorph imaging software (Molecular Devices).
D. discoideum infection by L. pneumophila for microscopic analysis.
D. discoideum cells were seeded onto sterile glass in MB (7.15 g ⋅ liter−1 yeast extract, 14.3 g ⋅ liter−1 peptone, 20 mM MES [pH 6.9]). Monolayers were infected at an MOI of 100 with bacteria grown for 4 days at 30°C. The plates were spun at 880 × g for 10 min and further treated for microscopic analysis.
LegK2 protein localization during infection.
D. discoideum cells were seeded onto sterile glass coverslips in MB. The L. pneumophila dotA or legK2 mutant strain encoding N-terminally HA-tagged LegK2 protein was grown for 21 h at 37°C. Two hours before use, HA-tagged LegK2 protein production was induced by the addition of 2 mM IPTG. Monolayers were infected at an MOI of 100. The plates were spun at 880 × g for 10 min, and cells were immediately fixed or incubated for 5, 10, 15, 20, 30, or 60 min at 25°C. Monolayers were fixed with 4% paraformaldehyde and permeabilized with 100% ice-cold methanol. The coverslips were then stained with anti-HA antibodies (3724S; Cell Signaling) and visualized with an Alexa Fluor 488-conjugated secondary antibody (A11034; Molecular Probes). Bacteria were immunolabeled with a rhodamine-conjugated anti-MOMP antibody (52). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Actin polymerization in infected cells.
D. discoideum on sterile glass coverslips was infected with Legionella as described above in the absence or presence of 100 µM CK-666, and cells were immediately fixed or incubated for 15 min at 25°C. Monolayers were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 5 min at RT. The coverslips were then stained with phalloidin-FITC (P5282; Sigma). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Detection of LegK2 and actin on LCVs.
.D. discoideum on sterile glass coverslips was infected with Legionella as described above, and cells were incubated for 15 min at 25°C. Monolayers were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 5 min at RT. The coverslips were then stained with mouse anti-HA antibodies (gift from S. Salcedo, Lyon, France) and visualized with a Dylight 594 (115-515-003; Jackson)-conjugated secondary antibody. Bacteria were immunolabeled with anti-Lp1 Paris/Lens strain serum and an Alexa Fluor 547-conjugated anti-rabbit secondary antibody (A21245; Molecular Probes), and actin was stained with phalloidin-FITC (P5282; Sigma). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Maturation of Legionella-containing phagosomes.
D. discoideum on sterile glass coverslips was infected with Legionella as described above. CK-666 (100 µM) was added 5 min after amoeba-Legionella contact, and plates were incubated for 1 h at 25°C. Monolayers were fixed with 4% paraformaldehyde, permeabilized with 100% ice-cold methanol for 2 min at RT. The coverslips were then stained with anti-VatA antibodies (221-35-2; gift of F. Letourneur, Lyon, France) and visualized with an Alexa Fluor 488-conjugated secondary antibody (A11029; Molecular Probes). Microscopy was carried out with a confocal laser scanning microscope (LSM510 Meta; Zeiss).
Statistical analysis.
The results were statistically analyzed with Student’s t test. The results obtained correspond to a comparison of the values obtained with the legK2 mutant and those obtained with the parental strain under the same conditions or of the values obtained in the absence or presence of CK-666, the ARP2/3 complex inhibitor.
SUPPLEMENTAL MATERIAL
LegK2 has no direct impact on ARP2/3 activity. Pyrene actin polymerization assays show that the ARP2/3 complex in the presence of 500 nM N-WASP-VCA (purple trace) nucleates actin faster than actin alone (black trace). The ARP2/3 complex dephosphorylated with dual-specificity Antarctic phosphatase (AP-ARP2/3) does not nucleate actin filaments in the presence of N-WASP-VCA (blue trace). The ARP2/3 complex phosphorylated by LegK2 (LegK2 + ARP2/3, orange trace) nucleated actin filaments at rates similar to those of the untreated ARP2/3 complex (purple trace). The nucleation activity of the phosphatase-treated ARP2/3 complex was not restored by LegK2 phosphorylation (LegK2 + AP-ARP2/3) (green trace). Download
Strains and plasmids used in this study.
Primers used in this study.
Supplemental references. Download
ACKNOWLEDGMENTS
We are grateful to F. Letourneur for helpful discussions about D. discoideum manipulations and for the gift of anti-VatA antibody and to G. Frankel for the gift of plasmid pMMB207c-HA*4. We thank the Dicty Stock Center for D. discoideum strains.
This work was performed within the framework of the LABEX ECOFECT (ANR-11-LABX-0042) of the Université de Lyon, within the program Investissements d’avenir (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR). This work was funded by the Centre National de la Recherche Scientifique (UMR 5308), the Institut National de la Recherche Médicale (U1111 and U604), the Université Lyon 1, the Fondation FINOVI, the Pasteur Institute, the European Research Council (advanced grant 233348), and the Institut National de la Recherche Agronomique (USC2020). The Ph.D. grant to C.M. was provided by the Programme Avenir Lyon Saint-Etienne (ANR-11-IDEX-0007) of the Université de Lyon, within the program Investissements d’avenir operated by the ANR. P.C. is a Howard Hughes Medical Institute senior international research scholar.
Footnotes
Citation Michard C, Sperandio D, Baïlo N, Pizarro-Cerdá J, LeClaire L, Chadeau-Argaud E, Pombo-Grégoire I, Hervet E, Vianney A, Gilbert C, Faure M, Cossart P, Doublet P. 2015. The Legionella kinase LegK2 targets the ARP2/3 complex to inhibit actin nucleation on phagosomes and allow bacterial evasion of the late endocytic pathway. mBio 6(3):e00354-15. doi:10.1128/mBio.00354-15.
REFERENCES
- 1.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews HL, Vogel JP, Isberg RR. 1998. Identification of linked Legionella pneumophila genes essential for intracellular growth and evasion of the endocytic pathway. Infect Immun 66:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol 7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 4.Segal G, Purcell M, Shuman HA. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc Natl Acad Sci U S A 95:1669–1674. doi: 10.1073/pnas.95.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isberg RR, O’Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cozy niche inside host cells. Nat Rev Microbiol 7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu L, Luo ZQ. 2013. Cell biology of infection by Legionella pneumophila. Microbes Infect 15:157–167. doi: 10.1016/j.micinf.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo ZQ. 2012. Legionella secreted effectors and innate immune responses. Cell Microbiol 14:19–27. doi: 10.1111/j.1462-5822.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol 26:261–283. doi: 10.1146/annurev-cellbio-100109-104034. [DOI] [PubMed] [Google Scholar]
- 9.Allombert J, Fuche F, Michard C, Doublet P. 2013. Molecular mimicry and original biochemical strategies for the biogenesis of a Legionella pneumophila replicative niche in phagocytic cells. Microbes Infect 15:981–988. doi: 10.1016/j.micinf.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Ingmundson A, Delprato A, Lambright DG, Roy CR. 2007. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450:365–369. doi: 10.1038/nature06336. [DOI] [PubMed] [Google Scholar]
- 11.Machner MP, Isberg RR. 2007. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318:974–977. doi: 10.1126/science.1149121. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S, Liu X, Arasaki K, McDonough J, Galán JE, Roy CR. 2011. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477:103–106. doi: 10.1038/nature10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. 2006. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol 8:971–977. doi: 10.1038/ncb1463. [DOI] [PubMed] [Google Scholar]
- 14.Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Arnold RJ, Luo ZQ. 2011. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc Natl Acad Sci U S A 108:21212–21217. doi: 10.1073/pnas.1114023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan Y, Luo ZQ. 2011. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475:506–509. doi: 10.1038/nature10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan Y, Luo ZQ. 2011. Take it and release it: the use of the Rab1 small GTPase at a bacterium’s will. Cell Logist 1:125–127. doi: 10.4161/cl.1.4.17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6:e1000822. doi: 10.1371/journal.ppat.1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, Johannes L, Hilbi H. 2013. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14:38–50. doi: 10.1016/j.chom.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Ge J, Shao F. 2011. Manipulation of host vesicular trafficking and innate immune defence by Legionella Dot/Icm effectors. Cell Microbiol 13:1870–1880. doi: 10.1111/j.1462-5822.2011.01710.x. [DOI] [PubMed] [Google Scholar]
- 21.Lippmann J, Müller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, Hellwig K, Kirschning CJ, Taylor GA, Barchet W, Bauer S, Suttorp N, Roy CR, Opitz B. 2011. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol 13:1668–1682. doi: 10.1111/j.1462-5822.2011.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lurie-Weinberger MN, Gomez-Valero L, Merault N, Glöckner G, Buchrieser C, Gophna U. 2010. The origins of eukaryotic-like proteins in Legionella pneumophila. Int J Med Microbiol 300:470–481. doi: 10.1016/j.ijmm.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Nora T, Lomma M, Gomez-Valero L, Buchrieser C. 2009. Molecular mimicry: an important virulence strategy employed by Legionella pneumophila to subvert host functions. Future Microbiol 4:691–701. doi: 10.2217/fmb.09.47. [DOI] [PubMed] [Google Scholar]
- 24.Hervet E, Charpentier X, Vianney A, Lazzaroni JC, Gilbert C, Atlan D, Doublet P. 2011. Protein kinase LegK2 is a type IV secretion system effector involved in endoplasmic reticulum recruitment and intracellular replication of Legionella pneumophila. Infect Immun 79:1936–1950. doi: 10.1128/IAI.00805-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, Liu L, Shao F. 2009. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci U S A 106:13725–13730. doi: 10.1073/pnas.0907200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeClaire LL, Baumgartner M, Iwasa JH, Mullins RD, Barber DL. 2008. Phosphorylation of the Arp2/3 complex is necessary to nucleate actin filaments. J Cell Biol 182:647–654. doi: 10.1083/jcb.200802145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanan A, LeClaire LL, Barber DL, Jacobson MP. 2011. Phosphorylation of the Arp2 subunit relieves auto-inhibitory interactions for Arp2/3 complex activation. PLOS Comput Biol 7:e1002226. doi: 10.1371/journal.pcbi.1002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R. 2004. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep 5:154–160. doi: 10.1038/sj.embor.7400079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebrin F, Chambaz EM, Bianchini L. 2001. A role for protein kinase CK2 in cell proliferation: evidence using a kinase-inactive mutant of CK2 catalytic subunit alpha. Oncogene 20:2010–2022. doi: 10.1038/sj.onc.1204307. [DOI] [PubMed] [Google Scholar]
- 30.Alarcon CM, Heitman J, Cardenas ME. 1999. Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Mol Biol Cell 10:2531–2546. doi: 10.1091/mbc.10.8.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. 2006. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol 5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohtoshi A, Miyake T, Arai K, Masai H. 1997. Analyses of Saccharomyces cerevisiae Cdc7 kinase point mutants: dominant-negative inhibition of DNA replication on overexpression of kinase-negative Cdc7 proteins. Mol Gen Genet 254:562–570. doi: 10.1007/s004380050452. [DOI] [PubMed] [Google Scholar]
- 33.Shevchuk O, Batzilla C, Hägele S, Kusch H, Engelmann S, Hecker M, Haas A, Heuner K, Glöckner G, Steinert M. 2009. Proteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int J Med Microbiol 299:489–508. doi: 10.1016/j.ijmm.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, Hilbi H. 2009. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10:76–87. doi: 10.1111/j.1600-0854.2008.00851.x. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann C, Finsel I, Otto A, Pfaffinger G, Rothmeier E, Hecker M, Becher D, Hilbi H. 2014. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell Microbiol 16:1034–1052. doi: 10.1111/cmi.12256. [DOI] [PubMed] [Google Scholar]
- 36.Lu H, Clarke M. 2005. Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell Microbiol 7:995–1007. doi: 10.1111/j.1462-5822.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- 37.Fehrenbacher KL, Boldogh IR, Pon LA. 2003. Taking the A-train: actin-based force generators and organelle targeting. Trends Cell Biol 13:472–477. doi: 10.1016/S0962-8924(03)00174-0. [DOI] [PubMed] [Google Scholar]
- 38.Kjeken R, Egeberg M, Habermann A, Kuehnel M, Peyron P, Floetenmeyer M, Walther P, Jahraus A, Defacque H, Kuznetsov SA, Griffiths G. 2004. Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol Biol Cell 15:345–358. doi: 10.1091/mbc.E03-05-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. 2009. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franco IS, Shohdy N, Shuman HA. 2012. The Legionella pneumophila effector VipA is an actin nucleator that alters host cell organelle trafficking. PLoS Pathog 8:e1002546. doi: 10.1371/journal.ppat.1002546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo Z, Stephenson R, Qiu J, Zheng S, Luo ZQ. 2014. A Legionella effector modulates host cytoskeletal structure by inhibiting actin polymerization. Microbes Infect 16:225–236. doi: 10.1016/j.micinf.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cossart P, Sansonetti PJ. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- 43.Gouin E, Welch MD, Cossart P. 2005. Actin-based motility of intracellular pathogens. Curr Opin Microbiol 8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 44.Unsworth KE, Way M, McNiven M, Machesky L, Holden DW. 2004. Analysis of the mechanisms of Salmonella-induced actin assembly during invasion of host cells and intracellular replication. Cell Microbiol 6:1041–1055. doi: 10.1111/j.1462-5822.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- 45.Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, Holden DW. 2008. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell Microbiol 10:20–30. doi: 10.1111/j.1462-5822.2007.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odendall C, Rolhion N, Förster A, Poh J, Lamont DJ, Liu M, Freemont PS, Catling AD, Holden DW. 2012. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12:657–668. doi: 10.1016/j.chom.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Navarro L, Koller A, Nordfelth R, Wolf-Watz H, Taylor S, Dixon JE. 2007. Identification of a molecular target for the Yersinia protein kinase A. Mol Cell 26:465–477. doi: 10.1016/j.molcel.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 48.Dolezal P, Aili M, Tong J, Jiang JH, Marobbio CM, Lee SF, Schuelein R, Belluzzo S, Binova E, Mousnier A, Frankel G, Giannuzzi G, Palmieri F, Gabriel K, Naderer T, Hartland EL, Lithgow T. 2012. Legionella pneumophila secretes a mitochondrial carrier protein during infection. PLoS Pathog 8:e1002459. doi: 10.1371/journal.ppat.1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolte S, Cordelières FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 50.Zalevsky J, Lempert L, Kranitz H, Mullins RD. 2001. Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr Biol 11:1903–1913. doi: 10.1016/S0960-9822(01)00603-0. [DOI] [PubMed] [Google Scholar]
- 51.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 52.Hilbi H, Segal G, Shuman HA. 2001. Icm/dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol 42:603–617. doi: 10.1046/j.1365-2958.2001.02645.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LegK2 has no direct impact on ARP2/3 activity. Pyrene actin polymerization assays show that the ARP2/3 complex in the presence of 500 nM N-WASP-VCA (purple trace) nucleates actin faster than actin alone (black trace). The ARP2/3 complex dephosphorylated with dual-specificity Antarctic phosphatase (AP-ARP2/3) does not nucleate actin filaments in the presence of N-WASP-VCA (blue trace). The ARP2/3 complex phosphorylated by LegK2 (LegK2 + ARP2/3, orange trace) nucleated actin filaments at rates similar to those of the untreated ARP2/3 complex (purple trace). The nucleation activity of the phosphatase-treated ARP2/3 complex was not restored by LegK2 phosphorylation (LegK2 + AP-ARP2/3) (green trace). Download
Strains and plasmids used in this study.
Primers used in this study.
Supplemental references. Download