

Abstract
AIM: To elucidate the sequential gene expression profile in AGS cells co-cultured with wild-type Helicobacter pylori (H pylori) as a model of H pylori-infected gastric epithelium, and to further examine the contribution of cag-pathogenicity islands (cagPAI)-coding type IV secretion system and the two pathways, nuclear factor kappa B (NF-kB) and extracellular signal-regulated kinases (ERK) on wild-type H pylori-induced gene expression.
METHODS: Gene expression profiles induced by H pylori were evaluated in AGS gastric epithelial cells using cDNA microarray, which were present in the 4 600 independent clones picked up from the human gastric tissue. We also analyzed the contribution of NF-kB and ERK signaling on H pylori-induced gene expression by using inhibitors of specific signal pathways. The isogenic mutant with disrupted cagE (△cagE) was used to elucidate the role of cagPAI-encoding type IV secretion system in the gene expression profile.
RESULTS: According to the expression profile, the genes were classified into four clusters. Among them, the clusters characterized by continuous upregulation were most conspicuous, and it contained many signal transducer activity-associated genes. The role of cagPAI on cultured cells was also investigated using isogenic mutant cagE, which carries non-functional cagPAI. Then the upregulation of more than 80% of the induced genes (476/566) was found to depend on cagPAI. Signal transducer pathway through NF-kB or ERK are the major pathways which are known to be activated by cagPAI-positive H pylori. The role of these pathways in the whole signal activation by cagPAI-positive H pylori was analyzed. The specific inhibitors against NF-kB or ERK pathway blocked the activation of gene expression in 65% (367/566) or 76% (429/566) of the genes whose activation appealed to depend on cagPAI.
CONCLUSION: These results suggest that more than half of the genes induced by cagPAI-positive H pylori depend on NF-kB and ERK signaling activation, and these pathways may play a role in the gene expression induced by host-bacterial interaction which may associate with H pylori-related gastro-duodenal diseases.
Keywords: Helicobacter pylori; Cag-pathogenicity islands; cDNA microarray, Cluster analysis; Signal transduction
INTRODUCTION
Helicobacter pylori (H pylori) is the causative agent for various gastro-duodenal diseases[1] and has been classified as a definite carcinogen by the International Agency for Research on Cancer[2]. Previous studies have shown that the eradication of H pylori virtually eliminates the recurrence of peptic ulcer diseases, suppresses the recurrence of gastric cancer after mucosal resection, and cures mucosa-associated lymphoid tissue lymphoma[3-10]. The molecular mechanisms underlying these H pylori-related diseases have been extensively studied.
The cag-pathogenicity island (cagPAI), a cluster of about 28 genes, is one of the best known virulence factors; it encodes a type IV secretion system that transports CagA protein, peptidoglycan and possibly other molecules into host epithelial cells[11-16]. Transported CagA binds to molecules such as SH2 domain-containing protein-tyrosine phosphatase-2 (SHP-2), growth factor receptor bound 2 (Grb2), and cortactin[17-19]. Then it induces epithelial disruption as well as cytoskeletal changes[20], which might result in inducing ulcer diseases and gastric cancer[21,22].
The cagPAI is also associated with activation of the transcription factors nuclear factor kappa B (NF-kB) and activator protein-1 (AP-1)[23-26], two key regulators of the expression of various inflammatory genes. NF-kB acts as a transcriptional modulator in the host response to bacterial invasion[27-33]. Its activation may in turn increase inflammation by inducing cytokines similar to the stimulation of interleukin-1 (IL-1) and tumor necrotizing factor-alpha (TNF-α)[34], and induce cell proliferation via activating anti-apoptotic effect[35]. The attachment of H pylorito gastric epithelial cells also induces the rapid activation of p38, c-Jun NH2-terminal kinase, and mitogen-activated protein kinase (MAPK)[36]; the downstream effectors of these signals include oncogene c-fos and its promoter element serum response element (SRE)[37,38]. These are possibly involved in cellular proliferation and survival[39-41], which might be associated with gastric carcinogenesis.
Some of the host cell responses to infection with H pylori induce many gene expressions and appear to function in contradictory directions, such as inflammation and cell proliferation, or apoptosis and anti-apoptosis[35]; the relative importance of each response is thus not immediately obvious. Recently, cDNA microarray analysis has been used to comprehensively characterize the individual responses in host cells[42,43]. The authors and coworkers have previously profiled H pylori-induced gene expression in vitro[44-51] and in vivo[52-54]. However, the contribution of the cagPAI-encoding type IV secretion system and the importance of intracellular signaling activation have not been fully elucidated.
In this study, we analyzed sequential gene expression profile in AGS cells co-cultured with wild-type H pylori as a model of H pylori-infected gastric epithelium. Furthermore, we examined the contribution of cagPAI-coding type IV secretion system and the two pathways, NF-kB and extracellular signal-regulated kinases (ERK) on wild-type H pylori-induced gene expression.
MATERIALS AND METHODS
Bacterial strains and cell culture
The H pylori TN2 strain, which is positive for entire cagPAI, and vacA, was generously denoted by Dr. M. Nakao (Takeda Chemical Industries, Ltd, Osaka, Japan). The isogenic cagE mutant TN2-△cagE was prepared by insertion of a kanamycin-resistant gene into the cagE gene locus of TN2, as described previously[55]. These strains were cultured on Columbia agar with 50 mL/L horse blood and Dent's selective antibiotic supplement (Oxoid, Basingstoke, UK) at 37 °C for 3 d under microaerobic conditions (Campy-Pak Systems; BBL, Cockeysville, MD, USA). The bacterial stocks were stored at -80 °C in Brucella broth with 50 mL/L fetal bovine serum (FBS) containing 160 mL/L glycerol. For co-culture experiments, H pylori was cultured in Brucella broth containing 7.5% FBS for 24 h, pelleted, resuspended in cell culture medium without FBS, and used to inoculate the host cells at a multiplicity of infection of 50 to 100:1 according to the previous reports[56,57]. At each time point, the AGS cells were collected by scraping, isolated by centrifugation, and stored at -80 °C for subsequent isolation of total RNA. Human gastric cancer cells (AGS; ATCC CRL 1739), established from poorly differentiated gastric adenocarcinoma, were maintained in Ham's F12 supplemented with 10% FBS. The medium was replaced with serum-free Ham's F12 at 24 h before the inoculation of the AGS cells with H pylori.
Treatment of AGS cells with specific NF-kB and ERK inhibitors
AGS cells were treated with ammonium pyrolidinedithi-ocarbamate (APDC; 400 mmol/L), which inhibits NF-kB activation, or with the ERK inhibitor PD98059 (25 mmol/L; Calbiochem, San Diego, CA, USA)[58] for 60 min before infection with H pylori, as described above. Total RNA was extracted from the AGS cells at 1.5, 3, 6, and 12 h of the co-culture period, as described above.
RNA extraction
Total cellular RNA was extracted using an acid guanidium thiocyanate-phenol-chloroform method, according to the manufacturer's instructions (ISOGEN Reagents; Nippon Gene, Tokyo, Japan), and column chromatography (RNeasy; Qiagen, Tokyo, Japan). The poly(A) mRNA was isolated from the total RNA using the Oligotex-dT30 mRNA purification kit (Takara Shuzo Co., Tokyo, Japan). The integrity of the purified mRNA was confirmed by agarose gel electrophoresis with ethidium bromide staining.
cDNA microarray
A cDNA microarray containing 4 600 cDNAs was made as previously described[44,59-61]. Briefly, human cDNAs were purchased from Research Genetics (Invitrogen Japan K.K., Tokyo, Japan). PCR-amplified cDNA products were mixed with nitrocellulose in dimethyl sulfoxide just before printing and were then spotted onto carbodiimide-coated glass slides using a robotics system (SPBIO-2000, Hitachi Software Engineering, Yokohama, Japan). In this study, we spotted 4 600 sequence-validated cDNAs on the array, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and beta-actin to serve as an internal standard and the luciferase gene of Photinus pyralis as a negative control.
Sequential changes in gene expression in vitro
For the analysis of the time course of infection-induced gene expression in AGS cells, Cy5-labeled fluorescent cDNAs were prepared from 2 mg mRNA isolated from the infected AGS cells after 1.5, 3, 6, and 12 h of co-culture. The Cy5-labeled sample probes were then applied to the microarray slides for analysis. The Cy3-labeled control probes were similarly prepared from mRNA isolated from uninfected AGS cells. Two independent experiments were performed, and the array data were subject to a simple algorithm (see Data analysis) to set a lower bound threshold and to normalize for unequal incorporation label. Then average ratio was calculated and they were used for the following analysis.
Data analysis
The fluorescence signals from the microarrays were quantified with IMAGENE ver. 4.01 (Biodiscovery, Los Angeles, CA, USA) and analyzed with GENESIGHT LIGHT (Biodiscovery). Briefly, a cDNA spot was included in the analysis only if the raw fluorescence signal intensity was at least 1.5 times that of the local background. The signals were normalized between the arrays using a correction factor calculated from the sum intensity of all spots. To control the uneven incorporation of the fluorescent dyes, the Cy5 and Cy3 fluorescence intensities for each spot were adjusted so that the mean Cy3:Cy5 ratio was equal to 1.0. We adjusted the raw fluorescence ratios by log transformation, median centering, and normalization to a mean of zero and a variance of one. Changes in expression (Cy3:Cy5 fluorescence ratio) with a factor of 3 or greater in either direction were considered significant, and if the maximum minus minimum values of the log-transformed fluorescence ratios were greater than Supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sport, and Culture of Japan
1.0. (Full microarray data are deposited in the Gene Expression Omnibus at NCBI. The platform ID numbers are GPL 1303, and the sample ID numbers are GSM 25915-20). We performed cluster analysis using the average linkage method with uncentered correlation as the distance function by using the Cluster program (ver. 2.11) with TreeView (ver. 1.50) (http://rana.stanford.edu/software)[62]. The Onto-Express program (http://vortex.cs.wayne.edu/Projects.html) was used to perform functional characterization accompanied by the computation of significance values for each functional category, allowing significant biological processes to be distinguished from random events[63-65].
Reverse transcriptase-polymerase chain reaction (RT-PCR)
To validate the results obtained in the microarray experiments, first-strand cDNA was synthesized using 1 mg of total RNA, 1 mmol/L of oligo-dT primer, and reverse transcriptase (Superscript II; Invitrogen, Carlsbad, CA, USA). Each cDNA sample was amplified by PCR using specific primers, as the following, for 10 min at 95 °C for initial denaturing, followed by 35-40 cycles of 95 °C for 30 s, 52-54 °C for 30 s and 72 °C for 30 s, yielding products of approximately 300-500 bp. The PCR products were examined, with appropriate molecular size markers, by agarose gel electrophoresis and ethidium bromide staining. The primer pairs used for PCR analysis are listed in Table 1.
Table 1.
Primer sequences for RT-PCR
| Genes | Forward | Reverse |
| IL-8 | 5’-GCT TTC TGA TGG AAG AGA GC-3’ | 5’-GGC ACA GTG GAA CAA GGA CT-3’ |
| c-fos | 5’-GTC AAG AGC ATC AGC AGCA T-3’ | 5’-TCG GGG TAG GTG AAG ACG AA-3’ |
| HNRPDL | 5’-GTT TCA GAG GAC CTG GAA TA-3’ | 5’-TCA CTA CCC TAG ACA CCG CA3’ |
| Vinculin | 5’-CAA GTG TGA CCG AGT GGA CC-3’ | 5’-TTG GTA TCA ATG GCT TCG TC-3’ |
| DSS1 | 5’-CAA GTC TCT ATG GTA GCG TCA GC-3’ | 5’-ACC ATG TTT CTC TAG TTC AG-3’ |
| NDRG1 | 5’-GGC GCG ACC TGG AGA TGAG-3’ | 5’-CTA GCA GGA GAC CTC CAT GG-3’ |
| EMK-1 | 5’-GAG ATG GAG GTG TGC AAA CT-3’ | 5’-TGG TTT AGG CGA AAT ACT CT-3’ |
| GAPDH | 5’-ACC ACA GTC CAT GCC ATC AC-3’ | 5’-TCC ACC ACC CTG TTG CTG TA-3’ |
RESULTS
Sequential changes in gene expression in AGS cells co-cultured with cagPAI-positive H pylori
We used microarray technology to characterize the time course of changes in gene expression induced by wild-type H pylori in AGS cells infected in vitro and obtained 3 228 genes that were valid for the analysis. Changes in expression with a factor of 3 or greater in either direction were considered significant. Of the 3 228 genes, about 20% (641 genes) were altered significantly (Figure 1A). The percentage of the significantly altered genes at each time point is shown in Figure 1B. The number of upregulated genes reached their peak at 3 h after infection, while the downregulated genes peaked at 6 h after infection. It implicates that H pylori-associated phenomena like inflammatory changes following the induction of IL-8 or c fos might occur as the early event in vitro. Hierarchical clustering was used to classify the genes into upregulated and downregulated groups (Figure 1C). Each group was further divided into two minor clusters, resulting in a total of four clusters, designated as C-1 to C-4, based on the time course of expression induced by H pylori. Genes in both C-1 and C-2 were upregulated during the early phase (1.5-3 h); however, the genes in C-1 were downregulated in the late phase (6-12 h), while those in C-2 remained upregulated. The C-1 cluster contained 764 genes which include c fos, DSS1 (DELETED IN SPLIT-HAND/SPLIT-FOOT 1 REGION), NF-kB p65 subunit, Rac1 (Rho family small GTP-binding protein), and MAP2K1 (mitogen-activated protein kinase kinase 1). The C-2 cluster contained 943 genes which include Interleukin-8, NFKBIA (factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha), VCL (vincullin), DUSP1 (dual specificity phosphatase 1), and NDRG1 (N-myc downstream regulated gene 1). The sequential changes in the expression of 2 of C-1 genes and 4 of C-2 genes were confirmed by RT-PCR (Figure 1D). In contrast, the expression of the genes in clusters C-3 (950 genes) showed downregulation at early-phase of infection, and C-4 (651 genes) were downregulated at late phase of infection. At late phase of infection, other functional events were modulated by response to these early events. We next performed to characterize the clusters to identify their function.
Figure 1.
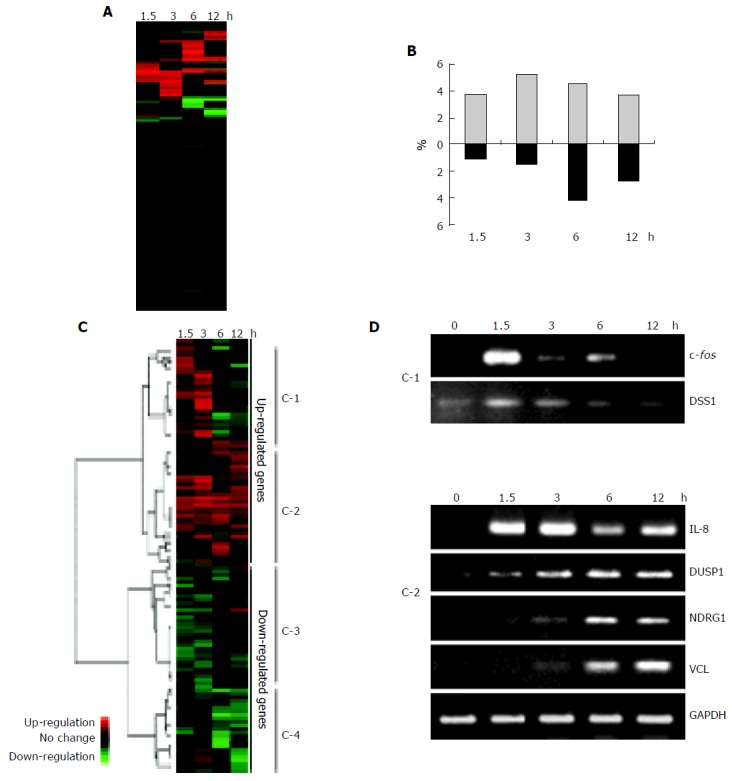
A: Schematic representation of the genes affected by wild-type H pylori-infection during 12 h in AGS cells. Red or green indicates up- or downregulation, respectively. About 20% of all the analyzed genes were significantly altered during infection experiments in vitro. Changes in expression with a factor of 3 or greater in either direction were considered significant; B: The percentage of the altered genes at each time point. The upregulated genes reached maximum number at 3 h after wild-type H pylori-infection, but the downregulated genes peaked at 6 h after infection; C: Cluster diagram of gene expression in AGS cells. The rows correspond to 3 228 genes for which the level of expression in AGS was changed by infection with wild-type H pylori, and the columns represent the various time points (1.5, 3, 6, and 12 h after wild-type H pylori-infection from left to right, respectively). Red or green indicates up- or downregulation, respectively, compared with the expression level in uninfected AGS cells. The dendrogram on the left and the horizontal distances between the nodes represent the statistical similarities between the neighboring genes and clusters. The genes were divided into two large clusters, comprising upregulated or downregulated genes. Each of the large clusters contained two smaller clusters, resulting in four clusters (C-1 to C-4; see Results for description); D: Validation of the microarray results by RT-PCR. Total RNAs from wild-type H pylori-infected AGS cells were isolated at the indicated times and subjected to reverse transcription and PCR with specific primers. The bands of the size corresponding to the expected length of the amplified fragment for each specific transcript were analyzed by agarose gel electrophoresis, along with GAPDH as a loading control.
Gene expression profile and annotated function
The functional profiles of the H pylori-induced changes in gene expression in vitro are shown in Table 2. The Onto-Express program was used for functional profiling as described in Materials and Methods, using hypergeometric distribution to determine the significance of differences (n≥5, P<0.05). This result indicated that the transiently upregulated C-1 cluster included genes that participate in transcription regulator activity, such as Smad4, c fos, and RelA, and others involved in chaperon activity, such as DNAJB1 (DnaJ (Hsp40) homolog, subfamily B, member 1) and CD74 antigen (invariant polypeptide of major histocompatibility complex, class II antigen-associated), more frequently than did the other clusters. The C-2 cluster, the members of which were upregulated during both the early and late phases, contained genes related to signal transduction activity, such as TNF receptor-associated factor 4, and those related to structural molecular activity, such as VCL and ARPS2. The genes in the C-3 and C-4 clusters were downregulated by H pylori. The C-3 cluster contained genes related to antioxidant activity, such as TXNRD1 (thioredoxin reductase 1) and GPX (glutathione peroxidase) 1/2, and the C-4 cluster contained genes encoding lyase activity, such as GLO1 and ENO1, and chaperone proteins. The genes undergoing most striking upregulation during H pylori infection in vitro are listed in Table 3.
Table 2.
The functional profiles of the H pylori-induced changes in gene expression in vitro. Cluster represents the assigned cluster numbers in Figure 1C. Functions were defined by using the Onto-Express program, using hypergeometric distribution to determine the significance of differences (n≥5, P<0.05, see Materials and methods)
| Cluster | Function | No. of the genes (Total) | P-value |
| C-1 | Chaperon activity | 12 (31) | 0.019 |
| Transcription regulator activity | 29 (92) | 0.033 | |
| C-2 | Apoptosis regulator activity | 7 (9) | <0.01 |
| Cell adhesion molecule activity | 18 (39) | <0.01 | |
| Signal transducer activity | 47 (132) | 0.034 | |
| Transcription regulator activity | 9 (20) | 0.037 | |
| Structural molecular activity | 40 (113) | 0.049 | |
| C-3 | Antioxidant activity | 6 (8) | <0.01 |
| C-4 | Lyase activity | 8 (10) | 0.011 |
| Chaperon activity | 10 (31) | 0.037 |
Table 3.
Genes undergoing most striking upregulation during H pylori-infection in vitro. Values show the mean change in gene expression from two separate microarray experiments in vitro(shown at “1.5, 3, 6, and 12 h”. Significantly upregulated values are shown in bold (above threefold)
| Acc. Number | Gene Symbol | Name | Function | 1.5h | 3h | 6h | 12h |
| U79243 | LZTR1 | leucine-zipper-like transcriptional regulator 1 | transcription | 5.08 | 3.08 | 0.95 | 2.30 |
| X61118 | LMO2 | LIM domain only 2 (rhombotin-like 1) | tumor associated | 2.53 | 16.24 | 0.28 | 1.04 |
| BC004247 | RAC1 | ras-related C3 botulinum toxin substrate 1 (rho family small GTP binding protein Rac1) | adhesion/cytoskeleton | 1.13 | 4.78 | 0.89 | 1.29 |
| AF005043 | PARG | poly (ADP-ribose) glycohydrolase | metabolism | 0.58 | 0.62 | 14.72 | 0.53 |
| U86782 | POH1 | 26S proteasome-associated pad1 homolog | RNA/Protein processing | 2.55 | 3.59 | 7.24 | 3.56 |
| M62839 | APOH | apolipoprotein H (beta-2-glycoprotein I) | immunity | 0.49 | 3.49 | 0.65 | 1.47 |
| BC004980 | SLC25A1 | solute carrier family 25 (mitochondrial carrier; citrate transporter) member 1 | metabolism | 4.07 | 0.81 | 0.95 | 1.54 |
| Z26649 | PLCB3 | phospholipase C beta 3 (phosphatidylinositol-specific) | tumor associated | 2.49 | 3.32 | 0.75 | 2.52 |
| M62399 | RELA | v-rel reticuloendotheliosis viral oncogene homolog A nuclear factor of kappa light polypeptide gene enhancer in B-cells 3 p65 (avian) | apoptpsis | 2.84 | 3.24 | 1.43 | 1.13 |
| BC000665 | TCP1 | t-complex 1 | RNA/Protein processing | 1.73 | 3.21 | 1.05 | 1.41 |
| V01512 | c-fos | V01512|HSCFOS Human cellular oncogene c-fos (complete sequence). | tumor associated | 37.81 | 2.48 | 0.57 | 0.91 |
| BC000117 | GMDS | GDP-mannose 46-dehydratase | metabolism | 2.63 | 4.37 | 2.28 | 3.75 |
| L29218 | CLK2 | CDC-like kinase 2 | growth/maintenance | 1.46 | 4.03 | 1.96 | 0.60 |
| BC000771 | NTRK1 | neurotrophic tyrosine kinase receptor type 1 | tumor associated | 3.33 | 3.47 | 1.42 | 3.59 |
| X68836 | MAT2A | methionine adenosyltransferase II alpha | metabolism | 3.14 | 2.95 | 0.85 | 1.28 |
| X94232 | MAPRE2 | microtubule-associated protein RP/EB family member 2 | growth/maintenance | 0.58 | 3.23 | 0.69 | 0.29 |
| X80200 | TRAF4 | TNF receptor-associated factor 4 | signal transduction | 3.20 | 3.02 | 3.67 | 1.08 |
| D76444 | ZFP103 | zinc finger protein 103 homolog (mouse) | growth/maintenance | 1.38 | 2.63 | 3.99 | 1.78 |
Contribution of cagPAI-coding type IV secretion system in host gene expression
The contribution of cagPAI-coding type IV secretion system in host gene expression was examined by comparing the pattern of expression induced by the wild-type H pylori vs the isogeniccagE mutant strain. We tried to compare the gene expression profile 3 h post-infection when the number of the upregulated genes were most frequent, and when interleukin-8 (IL-8) which is the gene induced by the functional type IV secretion system was upregulated. Five hundred and sixty-six genes on the microarray were upregulated at least threefold by the wild-type H pyloriinfection 3 h post-infection. Four hundred and seventy-six out of five hundred and sixty-six genes (86%) required cagPAI-coding type IV secretion system for their upregulation, whereas only 90 out of 566 genes (14%) were upregulated without functional type IV secretion system (Figure 2A). For example, IL-8, RelA, VCL, and Rac1 were upregulated 3- to 15-fold by infection with the wild-type H pylori, while the cagE mutant induced only one to threefold increases from the control level. On the other hand, some signal transduction-associated genes were significantly upregulated by the cagE mutant infection such as STC2, HKR3, and LZTR1. These genes might be induced by the cag-independent virulence factor.
Figure 2.
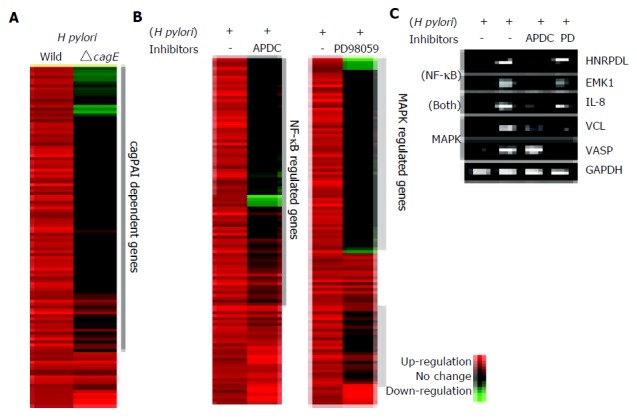
A: The contribution of the cagPAI-coded type IV secretion system to the gene expression profile in AGS cells. Hierarchical clustering of genes induced by H pylori with or without a functional type IV secretion system (wild type or cagE mutant, respectively). Left lane; gene expression profile of AGS cells co-cultured with wild-type H pylori relative to control. Right lane; gene expression profile of AGS cells co-cultured with the isogenic mutant cagE. Only the 566 significantly upregulated genes at 3 h are shown. About 84% of the genes were induced only by the wild-type H pylori infection. These genes were supposed to be induced by the presence of cagPAI-coded type IV secretion system. B: The contribution of NF-kB or ERK pathways to the gene expression profile. Inhibitors specific for NF-kB or ERK were added to AGS cells co-cultured with H pylori. The gene expression profiles in the presence or absence of the inhibitors were compared. C: The changes in the expression of some representative genes were confirmed by RT-PCR. All genes were induced by wild-type H pylori infection, and suppressed by the incubation with inhibitors of specific signal pathways.
Contribution of NF-kB and ERK pathways in H pylori-induced gene expression
It has been reported that wild-type H pylori activates NF-kB and ERK pathways; however, the contribution of these signaling pathways on gene expression profiles has not been investigated. Our analysis of the sequential changes in the gene expression profile revealed that signal transducer activity-associated genes show the continuous expression infected with wild-type H pylori. We thus analyzed the contribution of NF-kB and ERK signaling on H pylori-induced gene expression by using specific inhibitors as previously described[23,36]. As a control sample, we used AGS cells without adding inhibitor reagents because it could avoid the downregulation of the gene expression of nontreated AGS cells by the inhibitors. Among the 566 genes upregulated by wild-type H pylori, the expression of 367 genes (65%) was suppressed by pre-incubation with APDC, an inhibitor of NF-kB (Figure 2B). The suppressed genes included IL-8, NFKB1A, HNRPDL (heterogeneous nuclear ribonucleoprotein D-like protein) and EMK1 (ELKL motif kinase). On the other hand, pre-incubation with PD98059, an inhibitor of ERK, suppressed the expression of 429 genes (76%). The effects of the inhibitors on several genes were confirmed by RT-PCR (Figure 2C). Among the 566 upregulated genes, 315 genes (56%) were upregulated under NF-kB and ERK signaling activation, and 481 genes (85%) were upregulated under NF-kB and/or ERK signaling activation (Figure 3). HNRPDL was induced at 3.5-fold by wild-type H pylori infection, 1.2-fold with APDC, and 2.2-fold with PD98059, and the same suppression was confirmed by RT-PCR.
Figure 3.
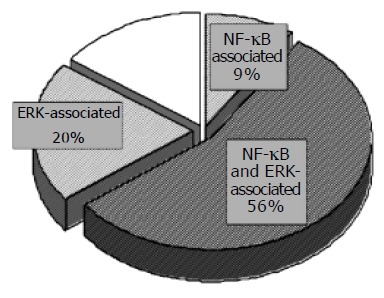
Schematic representation of the distribution of genes affected by wild-type H pylori infection. The complete upregulated genes at 3 h after infection is represented by the circle as a whole. The expression of 367 genes (65%) of the 566 genes upregulated by wild type H pylori was suppressed by pre-incubation with APDC, an inhibitor of NF-kB. On the other hand, pre-incubation with PD98059, an inhibitor of ERK, suppressed the expression of 429 genes (76%) of the 566 genes upregulated by wild type H pylori. Expression of 475 of the 566 genes (84%) was induced under NF-kB and/or ERK signaling activation, whereas changes in the remaining 16% are NF-kB or ERK signaling independent.
It was reported that the activation of NF-kB and ERK by H pylori is dependent on the presence of cagPAI. Thus, we compared the gene expression profiles under the presence or absence of cagPAI-coding type IV secretion system, and we analyzed the contribution of NF-kB or ERK on their gene expression (Figure 4). About 66% (315 genes) of the 476 upregulated genes were induced under NF-kB and/or ERK activation, while 34% (161 genes) of the 476 upregulated genes were dependent on cagPAI but not on either NF-kB or ERK.
Figure 4.
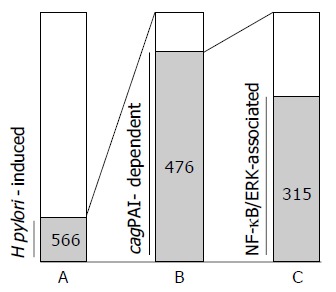
Characterization of the upregulated genes co-cultured with H pylori for 3 h (n = 566). A: The bar represents the number of 566 upregulated genes among 3 228 analyzed genes (566 genes). B: The genes induced by cagPAI-positive H pylori infection among the 566-H pylori-induced genes (476 genes). C: The contribution of NF-kB or ERK signaling pathway among thecagPAI-dependent gene expression. Among the 476-cagPAI-dependent genes, 66% (315 genes) were also involved in the NF-kB- and/or ERK-signaling activation, whereas 34% (161 genes) of 476 genes were dependent on cagPAI but not involved in either NF-kB or ERK signaling activation.
DISCUSSION
H pylori is now recognized as a definite exogenous carcinogen; hence intensive research effort is currently devoted to invest-igations of the molecular mechanism of H pylori-related gastric pathogenesis. In particular, recent reports have shown the importance of cagPAI as a potent bacterial virulence factor and the activation of NF-kB and MAPK signaling as the corresponding host responses[23,36]. However, the relative importance of these factors in gastric pathogenesis among all the events that are raised under H pylori infection has not been well established. In the present study, we showed that the genes upregulated by wild-type H pylori infection were mostly induced under the presence of cagPAI-coding type IV secretion system and that the expression of these genes mostly occurred via NF-kB and/or ERK activation in vitro.
Firstly, we analyzed the sequential gene expression changes in cultured cells. The experiments in vitro are supposed to simulate the environment in the acute phase of infection and cannot necessarily represent the conditions in vivo of chronic infection in which atrophic gastritis and/or gastric cancer actually develops. Therefore we analyzed the sequential changes of gene expression in vitro to seek the time-dependent characters of gene function. The genes in the C-1 cluster, which were upregulated during the early phase of infection, included interesting genes which might affect the pathogenesis of H pylori infection. c fos and DSS1 were included in this cluster. The expression of c fos is regulated by ERK (MAPK) signaling and H pyloriactivates the proto-oncogene c fos through SRE transactivation[38,66]. DSS1 has not previously been reported to be associated with H pylori infection. It has been shown to directly interact with BRCA2 and may play a role in the completion of the cell cycle[67]. The genes in C-2, which were persistently upregulated in vitro, contained the genes associated with intracellular signal transduction or cytoskeletal change, including Rho-family GTPases rac1 and cdc42. The activation of them are known to be dependent on cagPAI[68]; Rac1 in turn regulates the vacuolation caused by the vacA virulence factor of H pylori[69]. NDRG1 was upregulated during the late phase of infection as shown in Figure 1D; the protein encoded by this gene is a cytoplasmic protein involved in stress responses, hormone responses, cell growth, and differentiation[70].
We show here that the upregulation of a large part of the genes depends on the presence of functional type IV secretion system. CagPAI codes a type IV secretion system which transports CagA inside the host cells[11-15]; the transported CagA activates SRE[37]. Guilemin et al[16] reported that cagA gene, which encodes an effector molecule secreted by the type IV secretion system, induced the expression of many of the cytoskeletal genes. Recently, it has been reported that one of the effectors for NF-kB activation is peptidoglycan and CagA is probably not involved[37,71,72]. In addition, cagE, which codes a structural component of the type IV secretion system, is required for the expression of many immune responsive genes. These results suggest that several distinct molecules are involved in cagPAI-dependent upregulation. Moreover, the interaction of some molecules of the secretion system with the molecules of the host cell might directly activate some intracellular signaling cascades.
The microarray analysis is useful to identify the downstream genes in the specific intracellular signaling pathway because it could detect a large number of the gene expression at a time. Then we analyzed the alteration of the gene expression by the inhibitor reagents against the signaling molecules to identify the genes which are induced through NF-kB or ERK signaling pathway. In this study, we have shown that the majority of the H pylori-induced gene were upregulated under NF-kB and/or ERK activation[23,36] NF-kB activation in vivo is associated with gastric inflammation via the production of cytokines, IL-8[24] and is associated with cell survival induced by c-IAP-2 mediated anti-apoptosis[35]. Among the genes affected by NF-kB activation, HNRPDL and EMK1 were identified in the current microarray analysis and were confirmed by RT-PCR to be suppressed by NF-kB inhibition. HNRPDL is reported to be a member of the family of heterogeneous nuclear ribonucleoproteins (hnRNPs) that function in mRNA biogenesis and mRNA metabolism[73]. EMK (ELKL motif kinase) belongs to a small family of serine/threonine protein kinases involved in the control of cell polarity, microtubule stability, and cancer[74].
ERK signaling is activated by H pylori infection and leads to the expression of some oncogenes, such as c fos and cyclin-D1[75,76]. The upregulated genes under ERK-signaling activation included VCL and VASP, which are involved in integrin-mediated cell adhesion. Jawhari et al[77] reported that E-cadherin contributes to the development and progression of the neoplastic phenotype in gastric carcinoma. Kuroda et al[78] reported that the activation of Rho-Ras signaling induces the dissociation of cell-cell adhesion and it is associated with the cell migration and metastasis of carcinoma cells. Recently, Cottet et al[79] identified that H pylori-induced Rho-Ras signaling proteins.
In conclusion, we characterized the gene expression induced by H pylori using cDNA microarray, and we determined the contribution of cagPAI-coding type IV secretion system and NF-kB/ERK signaling pathways on the most part of the gene expression induced by H pylori. In the future, we should elucidate how the NF-kB and/or ERK contribute to the development of gastric inflammation in vivo and characterize the gastric mucosal gene expression in human beings which may help us to understand the gastric pathogenesis induced by H pylori infection.
ACKNOWLEDGMENTS
We thank Mitsuko Tsubouchi for her excellent technical assistance.
Footnotes
Science Editor Guo SY Language Editor Elsevier HK
Supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sport, and Culture of Japan
References
- 1.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 2.Schistosomes , liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [Google Scholar]
- 3.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 4.Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132–1136. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- 5.Graham DY, Lew GM, Klein PD, Evans DG, Evans DJ, Saeed ZA, Malaty HM. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann Intern Med. 1992;116:705–708. doi: 10.7326/0003-4819-116-9-705. [DOI] [PubMed] [Google Scholar]
- 6.Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de Boni M, Isaacson PG. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342:575–577. doi: 10.1016/0140-6736(93)91409-f. [DOI] [PubMed] [Google Scholar]
- 7.Forbes GM, Glaser ME, Cullen DJ, Warren JR, Christiansen KJ, Marshall BJ, Collins BJ. Duodenal ulcer treated with Helicobacter pylori eradication: seven-year follow-up. Lancet. 1994;343:258–260. doi: 10.1016/s0140-6736(94)91111-8. [DOI] [PubMed] [Google Scholar]
- 8.Sung JJ, Lin SR, Ching JY, Zhou LY, To KF, Wang RT, Leung WK, Ng EK, Lau JY, Lee YT, et al. Atrophy and intestinal metaplasia one year after cure of H. pylori infection: a prospective, randomized study. Gastroenterology. 2000;119:7–14. doi: 10.1053/gast.2000.8550. [DOI] [PubMed] [Google Scholar]
- 9.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 10.Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 11.Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, Naumann M, Meyer TF. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 12.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 13.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 17.Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of helicobacter pylori CagA protein activities. Mol Cell. 2002;10:745–755. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 18.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 19.Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol. 2003;161:249–255. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peek RM, Moss SF, Tham KT, Pérez-Pérez GI, Wang S, Miller GG, Atherton JC, Holt PR, Blaser MJ. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–868. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 22.Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterology. 2004;127:514–523. doi: 10.1053/j.gastro.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997;113:1099–1109. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- 24.Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160:2401–2407. [PubMed] [Google Scholar]
- 25.Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, Matsushima K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun. 1997;65:3218–3224. doi: 10.1128/iai.65.8.3218-3224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J Biol Chem. 2000;275:16064–16072. doi: 10.1074/jbc.M000959200. [DOI] [PubMed] [Google Scholar]
- 27.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 28.Baeuerle PA. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 30.Sha WC. Regulation of immune responses by NF-kappa B/Rel transcription factor. J Exp Med. 1998;187:143–146. doi: 10.1084/jem.187.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neurath MF, Becker C, Barbulescu K. Role of NF-kappaB in immune and inflammatory responses in the gut. Gut. 1998;43:856–860. doi: 10.1136/gut.43.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Münzenmaier A, Lange C, Glocker E, Covacci A, Moran A, Bereswill S, Baeuerle PA, Kist M, Pahl HL. A secreted/shed product of Helicobacter pylori activates transcription factor nuclear factor-kappa B. J Immunol. 1997;159:6140–6147. [PubMed] [Google Scholar]
- 33.Eckmann L, Smith JR, Housley MP, Dwinell MB, Kagnoff MF. Analysis by high density cDNA arrays of altered gene expression in human intestinal epithelial cells in response to infection with the invasive enteric bacteria Salmonella. J Biol Chem. 2000;275:14084–14094. doi: 10.1074/jbc.275.19.14084. [DOI] [PubMed] [Google Scholar]
- 34.Noach LA, Bosma NB, Jansen J, Hoek FJ, van Deventer SJ, Tytgat GN. Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand J Gastroenterol. 1994;29:425–429. doi: 10.3109/00365529409096833. [DOI] [PubMed] [Google Scholar]
- 35.Yanai A, Hirata Y, Mitsuno Y, Maeda S, Shibata W, Akanuma M, Yoshida H, Kawabe T, Omata M. Helicobacter pylori induces antiapoptosis through buclear factor-kappaB activation. J Infect Dis. 2003;188:1741–1751. doi: 10.1086/379629. [DOI] [PubMed] [Google Scholar]
- 36.Keates S, Keates AC, Warny M, Peek RM Jr, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag and cag- Helicobacter pylori. J Immunol. 1998;66:5552–5559. [PubMed] [Google Scholar]
- 37.Hirata Y, Maeda S, Mitsuno Y, Tateishi K, Yanai A, Akanuma M, Yoshida H, Kawabe T, Shiratori Y, Omata M. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology. 2002;123:1962–1971. doi: 10.1053/gast.2002.37044. [DOI] [PubMed] [Google Scholar]
- 38.Mitsuno Y, Maeda S, Yoshida H, Hirata Y, Ogura K, Akanuma M, Kawabe T, Shiratori Y, Omata M. Helicobacter pylori activates the proto-oncogene c-fos through SRE transactivation. Biochem Biophys Res Commun. 2002;291:868–874. doi: 10.1006/bbrc.2002.6530. [DOI] [PubMed] [Google Scholar]
- 39.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 40.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 41.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 42.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 43.Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda S, Otsuka M, Hirata Y, Mitsuno Y, Yoshida H, Shiratori Y, Masuho Y, Muramatsu M, Seki N, Omata M. cDNA microarray analysis of Helicobacter pylori-mediated alteration of gene expression in gastric cancer cells. Biochem Biophys Res Commun. 2001;284:443–449. doi: 10.1006/bbrc.2001.5006. [DOI] [PubMed] [Google Scholar]
- 45.Cox JM, Clayton CL, Tomita T, Wallace DM, Robinson PA, Crabtree JE. cDNA array analysis of cag pathogenicity island-associated Helicobacter pylori epithelial cell response genes. Infect Immun. 2001;69:6970–6980. doi: 10.1128/IAI.69.11.6970-6980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci USA. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bach S, Makristathis A, Rotter M, Hirschl AM. Gene expression profiling in AGS cells stimulated with Helicobacter pylori isogenic strains (cagA positive or cagA negative) Infect Immun. 2002;70:988–992. doi: 10.1128/IAI.70.2.988-992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim JW, Kim H, Kim KH. Cell adhesion-related gene expression by Helicobacter pylori in gastric epithelial AGS cells. Int J Biochem Cell Biol. 2003;35:1284–1296. doi: 10.1016/s1357-2725(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 49.Sepulveda AR, Tao H, Carloni E, Sepulveda J, Graham DY, Peterson LE. Screening of gene expression profiles in gastric epithelial cells induced by Helicobacter pylori using microarray analysis. Aliment Pharmacol Ther. 2002;16 Suppl 2:145–157. doi: 10.1046/j.1365-2036.16.s2.4.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang HT, Li ZH, Yuan JP, Zhao W, Shi XD, Tong SQ, Guo XK. Effect of Helicobacter pylori VacA on gene expression of gastric cancer cells. World J Gastroenterol. 2005;11:109–113. doi: 10.3748/wjg.v11.i1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan JP, Li T, Li ZH, Yang GZ, Hu BY, Shi XD, Shi TL, Tong SQ, Guo XK. mRNA expression profiling reveals a role of Helicobacter pylori vacuolating toxin in escaping host defense. World J Gastroenterol. 2004;10:1528–1532. doi: 10.3748/wjg.v10.i10.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen S, Felley CP, Bouzourene H, Reimers M, Michetti P, Pan-Hammarström Q. Inflammatory gene profiles in gastric mucosa during Helicobacter pylori infection in humans. J Immunol. 2004;172:2595–2606. doi: 10.4049/jimmunol.172.4.2595. [DOI] [PubMed] [Google Scholar]
- 53.Mills JC, Syder AJ, Hong CV, Guruge JL, Raaii F, Gordon JI. A molecular profile of the mouse gastric parietal cell with and without exposure to Helicobacter pylori. Proc Natl Acad Sci USA. 2001;98:13687–13692. doi: 10.1073/pnas.231332398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller A, O'Rourke J, Grimm J, Guillemin K, Dixon MF, Lee A, Falkow S. Distinct gene expression profiles characterize the histopathological stages of disease in Helicobacter-induced mucosa-associated lymphoid tissue lymphoma. Proc Natl Acad Sci USA. 2003;100:1292–1297. doi: 10.1073/pnas.242741699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogura K, Maeda S, Nakao M, Watanabe T, Tada M, Kyutoku T, Yoshida H, Shiratori Y, Omata M. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med. 2000;192:1601–1610. doi: 10.1084/jem.192.11.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, Akanuma M, Shiratori Y, Omata M. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119:97–108. doi: 10.1053/gast.2000.8540. [DOI] [PubMed] [Google Scholar]
- 57.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 58.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Otsuka M, Kato M, Yoshikawa T, Chen H, Brown EJ, Masuho Y, Omata M, Seki N. Differential expression of the L-plastin gene in human colorectal cancer progression and metastasis. Biochem Biophys Res Commun. 2001;289:876–881. doi: 10.1006/bbrc.2001.6047. [DOI] [PubMed] [Google Scholar]
- 60.Otsuka M, Aizaki H, Kato N, Suzuki T, Miyamura T, Omata M, Seki N. Differential cellular gene expression induced by hepatitis B and C viruses. Biochem Biophys Res Commun. 2003;300:443–447. doi: 10.1016/s0006-291x(02)02861-9. [DOI] [PubMed] [Google Scholar]
- 61.Mori M, Shimada H, Gunji Y, Matsubara H, Hayashi H, Nimura Y, Kato M, Takiguchi M, Ochiai T, Seki N. S100A11 gene identified by in-house cDNA microarray as an accurate predictor of lymph node metastases of gastric cancer. Oncol Rep. 2004;11:1287–1293. [PubMed] [Google Scholar]
- 62.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA. Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res. 2003;31:3775–3781. doi: 10.1093/nar/gkg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khatri P, Draghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using onto-express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 65.Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 66.Yang YL, Xu B, Song YG, Zhang WD. Overexpression of c-fos in Helicobacter pylori-induced gastric precancerosis of Mongolian gerbil. World J Gastroenterol. 2003;9:521–524. doi: 10.3748/wjg.v9.i3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 68.Churin Y, Kardalinou E, Meyer TF, Naumann M. Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in Helicobacter pylori infection. Mol Microbiol. 2001;40:815–823. doi: 10.1046/j.1365-2958.2001.02443.x. [DOI] [PubMed] [Google Scholar]
- 69.Hotchin NA, Cover TL, Akhtar N. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J Biol Chem. 2000;275:14009–14012. doi: 10.1074/jbc.c000153200. [DOI] [PubMed] [Google Scholar]
- 70.van Belzen N, Dinjens WN, Diesveld MP, Groen NA, van der Made AC, Nozawa Y, Vlietstra R, Trapman J, Bosman FT. A novel gene which is up-regulated during colon epithelial cell differentiation and down-regulated in colorectal neoplasms. Lab Invest. 1997;77:85–92. [PubMed] [Google Scholar]
- 71.Crabtree JE, Xiang Z, Lindley IJ, Tompkins DS, Rappuoli R, Covacci A. Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J Clin Pathol. 1995;48:967–969. doi: 10.1136/jcp.48.10.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma SA, Tummuru MK, Miller GG, Blaser MJ. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63:1681–1687. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsuchiya N, Kamei D, Takano A, Matsui T, Yamada M. Cloning and characterization of a cDNA encoding a novel heterogeneous nuclear ribonucleoprotein-like protein and its expression in myeloid leukemia cells. J Biochem. 1998;123:499–507. doi: 10.1093/oxfordjournals.jbchem.a021964. [DOI] [PubMed] [Google Scholar]
- 74.Espinosa L, Navarro E. Human serine/threonine protein kinase EMK1: genomic structure and cDNA cloning of isoforms produced by alternative splicing. Cytogenet Cell Genet. 1998;81:278–282. doi: 10.1159/000015046. [DOI] [PubMed] [Google Scholar]
- 75.Hirata Y, Maeda S, Mitsuno Y, Akanuma M, Yamaji Y, Ogura K, Yoshida H, Shiratori Y, Omata M. Helicobacter pylori activates the cyclin D1 gene through mitogen-activated protein kinase pathway in gastric cancer cells. Infect Immun. 2001;69:3965–3971. doi: 10.1128/IAI.69.6.3965-3971.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitsuno Y, Yoshida H, Maeda S, Ogura K, Hirata Y, Kawabe T, Shiratori Y, Omata M. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut. 2001;49:18–22. doi: 10.1136/gut.49.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jawhari AU, Noda M, Farthing MJ, Pignatelli M. Abnormal expression and function of the E-cadherin-catenin complex in gastric carcinoma cell lines. Br J Cancer. 1999;80:322–330. doi: 10.1038/sj.bjc.6690358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuroda S, Fukata M, Nakagawa M, Fujii K, Nakamura T, Ookubo T, Izawa I, Nagase T, Nomura N, Tani H, et al. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin- mediated cell-cell adhesion. Science. 1998;281:832–835. doi: 10.1126/science.281.5378.832. [DOI] [PubMed] [Google Scholar]
- 79.Cottet S, Corthésy-Theulaz I, Spertini F, Corthésy B. Microaerophilic conditions permit to mimic in vitro events occurring during in vivo Helicobacter pylori infection and to identify Rho/Ras-associated proteins in cellular signaling. J Biol Chem. 2002;277:33978–33986. doi: 10.1074/jbc.M201726200. [DOI] [PubMed] [Google Scholar]