

Abstract
Colonic bicarbonate (HCO3−) secretion is a well-established physiological process that is closely linked to overall fluid and electrolyte movement in the mammalian colon. These present studies show that extracellular calcium-sensing receptor (CaSR), a fundamental mechanism for sensing and regulating ionic and nutrient compositions of extracellular milieu in the small and large intestine, regulates HCO3− secretion. Basal and induced HCO3− secretory responses to CaSR agonists were determined by pH stat techniques used in conjunction with short-circuit current measurements in mucosa from rat distal colon mounted in Ussing chambers. R568, a specific CaSR activator, stimulated lumen Cl−- and short-chain fatty acid (SCFA)-dependent HCO3− secretion but inhibited cyclic nucleotide-activated HCO3− secretion. Consequently, at physiological conditions (either at basal or during lumen acid challenge) when electroneutral Cl−/HCO3− and SCFA/HCO3− exchangers dominate, CaSR stimulates HCO3− secretion; in contrast, in experimental conditions that stimulate fluid and HCO3− secretion, e.g., when forskolin activates electrogenic cystic fibrosis transmembrane conductance regulator-mediated HCO3− conductance, CaSR activation inhibits HCO3− secretion. Corresponding changes in JHCO3 (μeq·h−1·cm−2, absence vs. presence of R568) were 0.18 ± 0.03 vs. 0.31 ± 0.08 under basal nonstimulated conditions and 1.85 ± 0.23 vs. 0.45 ± 0.06 under forskolin-stimulated conditions. Similarly, activation of CaSR by R568 stimulated Cl−- and SCFA-dependent HCO3− secretion and inhibited cAMP-dependent HCO3− secretion in colon mucosa of wild-type mice; such effects were abolished in CaSR-null mice. These results suggest a new paradigm for regulation of intestinal ion transport in which HCO3− secretion may be fine-tuned by CaSR in accordance with nutrient availability and state of digestion and absorption. The ability of CaSR agonists to inhibit secretagogue-induced intestinal HCO3− secretion suggests that modulation of CaSR activity may provide a new therapeutic approach to correct HCO3− deficit and metabolic acidosis, a primary cause of morbidity and mortality in acute infectious diarrheal illnesses.
Keywords: calcium-sensing receptor, bicarbonate secretion, short-chain fatty acid, diarrhea
acute infectious diarrheal illnesses persist as an important worldwide public health problem. Approximately 600,000 children die each year, not necessarily because of an infection, but as a result of the associated dehydration and metabolic acidosis (4, 31). Reducing fluid and bicarbonate (HCO3−) losses from acute diarrhea offers a major opportunity for improving child health globally.
The extracellular calcium-sensing receptor (CaSR) (7) is a well-conserved G protein-coupled cell surface receptor that is expressed in diverse tissues in mammalian (8) and marine (32) species. CaSR is a key regulator of tissue responses, not only for calcium homeostasis (7), but also for fluid balance (37, 38) and osmotic regulation (32). The primary physiological ligand for the CaSR is extracellular ionized calcium (Ca2+o), providing a mechanism for Ca2+o to function as a first messenger. The CaSR also functions as a general sensor of the extracellular milieu because of allosteric modification of Ca2+o affinity by polyamines, L-amino acids, oligo-peptides, pH, and ionic strength (46).
The CaSR is widely expressed in the gastrointestinal tract, including the epithelial cells that line the entire small and large intestine (9, 14, 16, 23) and regulate ion and fluid transport, thus raising the possibility that these receptors may have important roles in regulation of intestinal fluid and electrolyte movement in both health and disease. In enteric epithelial cells, CaSR has been identified on both the apical and basolateral membranes of human (16, 44) and rat colonocytes (9, 16). Receptors in both membrane domains of these polarized epithelia are functionally active and can be activated by Ca2+o (14, 16), amino acids and peptides (29, 49), polyamines (14, 16), and the specific pharmacological CaSR agonist R568 (25). In colonic crypt cells, CaSR activation from either mucosal or serosal side inhibits net fluid secretion (14, 16, 25) and cyclic nucleotide accumulation (25) induced by synthetic/natural secretagogues. These secretagogues include forskolin (42) and guanylin (17), which generate cAMP and cGMP, respectively. CaSR activation also blocks the effects of bacterial enterotoxins (25) such as cholera toxin (21), a potent activator of membrane-bound adenylyl cyclase leading to elevated intracellular levels of cAMP, and heat stable Escherichia coli enterotoxin STa (19), which enhances cytosolic cGMP accumulation through the guanylyl cyclase C-type guanylin receptor. In the rat proximal and distal colon, activation of CaSR by R568 inhibits secretagogue-induced Cl− secretion (12), either directly via inhibition of epithelial cell basolateral Na+-K+-2Cl− cotransporter (25) or indirectly via the enteric nervous system (12). In perfused colonic crypts, it also promotes apical Na+/H+ exchanger activity (25); thus it may stimulate Na+ absorption in these cells. However, it remains to be elucidated whether CaSR regulates apical anion conductive pathway (e.g., cystic fibrosis transmembrane conductance regulator, CFTR).
Few studies have examined the role for the CaSR in regulating intestinal HCO3− secretion, which has been critically implicated in the pathophysiology of acute infectious diarrhea. For example, enhanced intestinal HCO3− secretion/loss associated with cholera and many other acute diarrheal illnesses can result in severe HCO3− deficit and metabolic acidosis (22, 34). The latter is another of the most common causes (in addition to dehydration and systemic volume depletion) of the morbidity and mortality associated with these clinical conditions. Whether CaSR activation inhibits secretagogue-induced HCO3− secretion as it does for the secretagogue-induced fluid and Cl− secretion is not known.
Regulated HCO3− secretion is also required for mucosal defense against luminal acid (via neutralization) in the upper gastrointestinal (GI) tract and bacteria (via stimulation of mucus secretion and maintenance of intestinal barrier function) in the lower GI tract, the defect of which has been shown to be a risk factor for peptic ulcer diseases (2, 20, 28) and intestinal inflammation (24, 51, 52). The role of CaSR in basal and acid-induced HCO3− secretion is unknown although a recent study with gut-specific CaSR knockout mice suggests that this receptor is crucial for mucosal defense and immunity (15).
HCO3− secretion in the intestine can be the result of one or more HCO3− transporters. In rat distal colon, at least three distinct mechanisms of HCO3− secretion have been described (47, 48): 1) lumen Cl−-dependent HCO3− secretion associated with a brush-border Cl−/HCO3− exchange, 2) lumen short-chain fatty acid (SCFA)-dependent HCO3− secretion mediated by a SCFA/HCO3− exchange, and 3) cyclic nucleotide-induced HCO3− secretion as a result of activation of an anion-conductive pathway (e.g., CFTR). To date, there is no information whether CaSR regulates one or more of these HCO3− transport mechanisms.
The present study was initiated to examine the CaSR effects on HCO3− secretion ex vivo in isolated intact colonic mucosa of rats and mice using pH stat and short-circuit current (Isc) measurements. Our experiments were designed in such a way that effects on basal, acid-induced, and secretagogue-induced HCO3− secretions were examined. Basal and acid-induced secretion was studied to provide information that may mimic physiological states, whereas secretagogue-induced secretion experiments were used to assess potential relevance of CaSR regulation in diarrhea. To provide insight into the mechanisms by which CaSR may regulate different HCO3− secretory processes, the effects of R568 on the Cl−/HCO3− and SCFA/HCO3− exchanges (which are electroneutral and thus can be measured by pH stat) and electrogenic HCO3− movement (thus can be measured by Isc and pH stat) were examined, both in rats and in CaSR wild-type and mutant mice. We have previously demonstrated that CaSR agonists can inhibit fluid/Cl− secretion (10, 12, 14, 16, 25) and mucosa inflammation (11, 15). These present studies show that CaSR agonists can also exert their effects on mucosal biology and physiology through differentially regulating HCO3− secretion.
MATERIALS AND METHODS
Animals and Tissue Preparations
Experiments were performed using nonfasting male Sprague-Dawley rats and C57BL/6 mice. Rats weighing 100–400 g were obtained from Charles River Laboratories. Mice lacking CaSR expression in intestinal epithelial cells (CaSR−/−) and their CaSR+/+ littermates were bred and maintained in house at the University of Florida Communicore Animal Facility. CaSR−/− mice were generated as previously described (36). Briefly, CaSR flox/flox mice were bred with transgenic mice expressing Cre recombinase under the control of the villin 1 promoter and genotyped before all experiments after an approximate 10–12 generations. Mice were used at 5–10 wk of age in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care. Animals were fed and maintained on regular chow (Harlan) with free access to water before death. Animals were killed with standard CO2 inhalation and killed by cervical dislocation. The colons were isolated, cut open along the mesenteric border into a flat sheet, and flushed with ice-cold Ringer solutions. Mucosa from the distal colon were carefully hand-stripped off of serosal, muscular, and submucosal layers as described (27), and a pair of adjacent mucosal segments was incised and mounted into Ussing chambers. In some experiments, stripped mucosa was incised longitudinally into two equal pieces to facilitate comparisons between control and treatment (see below). Either way, Isc and transepithelial resistance differences between these two adjacent tissues were <15%. The use of animals as well as the protocol for isolating colon tissues were approved by the Institutional Animal Care and Use Committee (IACUC no. 201307567) at the University of Florida.
Ussing Chamber Setup
Stripped mucosal sheets were mounted into Ussing chambers (window area = 0.3–0.5 cm2; Physiologic Instruments). The mucosal side of tissue was bathed with an unbuffered HCO3−-free Cl− Ringer solution (see Table 1 for detailed composition) circulated by a gas lift with 100% O2 while the serosal side was bathed with buffered Cl− Ringer solution (pH 7.4) that contained 25 mM HCO3− and gassed with 5% CO2-95% O2. Each side contained 3–5 ml of solution, and the temperature of the solution was adjusted to and maintained at 37°C by heated water-jacketed reservoirs. Experiments were performed under short-circuit conditions (Voltage-Current Clamp, VCC MC8; Physiologic Instruments) to maintain the transepithelial potentials at 0 mV, except for a brief interruption at 20-s intervals for recording of open-circuit potential (VT, mV).
Table 1.
The composition of Ringer solutions
| Serosa |
Lumen |
||||
|---|---|---|---|---|---|
| Cl−/HCO3− HCO3−-free |
Cl−-free | Cl− | SCFA | ||
| Cl− | 123 | 123 | 123 | ||
| HCO3− | 25 | ||||
| Isethionate | 25 | 147 | 25 | 122 | |
| Isobutyrate | 25 | ||||
| SO4− | 1 | 1 | |||
Compositions of constituents of all solutions are provided in mM; pH of all Cl−-containing solutions was adjusted to 7.4 with HCl, whereas that of Cl−-free solutions was adjusted with H2SO4. HCO3−-containing solutions were gassed with 95% O2-5% CO2; HCO3−-free solutions were gassed with 100% O2. All solutions contained (in mM) 140.8 Na+, 5 glucose, 5.2 K+, 0.5 Ca2+, and 0.5 Mg2+. Of note, the anion isethionate is not a short-chain fatty acid (SCFA) and was used to substitute Cl− and/or HCO3−.
Two types of approaches were used in the measurement of colonic HCO3− secretion, namely measurement of lumen pH or pH stat titration of net alkalinization by colonic mucosa. Except for limited experiments that require direct measurements of lumen pH (e.g., Experiment 1 and Experiment 3 below), luminal pH was maintained at 7.4 by the continuous infusion of 1 mM HCl or H2SO4 in the case of lumen Cl−-free conditions under the automatic control of a pH stat system (Bi-burette TIM 856 pH meter; Radiometer Analytical). The amount of the acid delivered per unit time per surface area was used to quantitate HCO3− secretion by the mucosa. Measurements were recorded continuously, and mean values for consecutive 5- or 10-min periods were averaged. The rate of HCO3− secretion (JHCO3) is expressed as μeq·h−1·cm−2. The Isc was measured in microamperes (μA) and converted into μeq·h−1·cm−2. Tissue resistance (R, Ω/cm2) was calculated from Ohm's law.
Experimental Design
Seven experiments for HCO3− secretion were conducted.
Lumen alkalinization response to [Ca2+]o.
Two equal pieces of mucosa from each colon that were obtained by longitudinal division along the antimessenteric border were mounted into two Ussing chambers. One piece was bathed with Cl− Ringer solution that contained 1.2 mM [Ca2+]o, while the other piece was bathed with Cl− Ringer solution that contained 0.5 mM [Ca2+]o. Initially, tissues were bathed, both luminally and serosally, in HCO3−-free solution. After 15–30 min, when stabilization was achieved and basal lumen pH recordings performed, HCO3−-free Ringer solution in the serosal side was replaced by 25 mM HCO3−-containing Ringer solution, and changes in lumen pH were monitored and recorded for 15–30 min. In some tissues, carbachol (CCH) was added to the serosal side before the experiment was concluded.
Basal HCO3− secretion.
Two adjacent sheets of mucosa from each colon were mounted into two Ussing chambers and bathed luminally with HCO3−-free Cl− Ringer solution and serosally with HCO3−-containing Cl− Ringer solution. After 15–30 min, when Isc and JHCO3 had stabilized, the CaSR agonist, R568, was added to the serosal or mucosal side of tissue, and changes in Isc and JHCO3 during the 15–30-min period ensuing after the addition of the agonist were determined.
Acid-induced HCO3− secretion.
Colon mucosa from each animal was divided longitudinally into two equal pieces and mounted into two Ussing chambers. Acid (hydrochloride) was added to the lumen to lower pH by ∼0.3–0.4 units, and changes in lumen pH were monitored. In response to luminal addition of acid, HCO3− is secreted, and pH in the lumen rises. When lumen pH rose above 7.4, another acid challenge was applied. After 15–30 min, when lumen pH responses had stabilized, one piece of mucosa was treated with R568 in the serosal bathing solution, while the other piece of mucosa was treated with vehicle control, and changes in lumen pH responses during the 30–45-min period ensuing after the addition of the agonist were determined. Initial studies demonstrated that, under these experimental conditions, the tissue tolerated acid challenges for at least 60 min without significantly compromising tissue responses and integrity. Initial studies also established that acid-induced pH recovery reflects HCO3− secretion from tissue, as additions of acid to no-tissue controls or tissue controls without the presence of serosal HCO3− did not generated significant pH recovery. A lowering of 0.3–0.4 pH units was used because this is the range of luminal pH by which this part of the intestine normally varies (33). Initial rates of acid-induced pH recovery were used for comparison and were expressed as pH unit recovered·min−1·cm−2.
Secretagogue-induced HCO3− secretion.
To examine CaSR effect on stimulated HCO3− secretion, a model of forskolin-induced HCO3− secretion was employed to mimic cholera toxin. The secretagogue forskolin was used to increase tissue cAMP content rapidly, as the effect of cholera toxin will not be seen for 1–2 h. Tissues were divided and treated as in the basal HCO3− secretion experiments except that, after 15–30 min, when Isc and JHCO3 had stabilized, forskolin was added to the serosal side of tissue for 15–30 min until Isc and JHCO3 had plateaued before R568 was then added to the serosal solution. Changes in Isc and JHCO3 during the 15–30-min period ensuing after the addition of the agonist were determined. Rodent distal colon displays Na+, K+, and Cl− currents, in addition to HCO3− conductance. To reduce current interference from non-HCO3− conductance, before the recording of HCO3− secretory responses, a cocktail containing 10 μM amiloride and 5 mM barium was added to the mucosal side to selectively inhibit Na+ and K+ conductance, and 100 μM bumetanide was added to the serosal side to inhibit Cl− secretory current (26, 41). Preliminary studies indicate that these transport inhibitors did not significantly affect JHCO3 response although Isc was inhibited by amiloride and stimulated by barium (see Table 2).
Table 2.
Effect of transport inhibitors on basal HCO3− secretion in rat distal colon
| ΔJHCO3, μEq·h−1·cm−2 | ΔIsc, μA/cm2 | |
|---|---|---|
| Amiloride, 10 μM, lumen | 0.01 ± 0.01 (3)ns | −6.3 ± 1.0 (3)† |
| Barium, 5 mM, lumen | 0.02 ± 0.06 (3)ns | 4.7 ± 0.7 (3)† |
| Bumetanide, 100 μM, serosa | 0.03 ± 0.08 (3)ns | −2.3 ± 4.7 (3)ns |
| DIDS, 100 μM, lumen | −0.08 ± 0.04 (4)* | 4.6 ± 3.9 (4)ns |
| NPPB, 100 μM, lumen | −0.17 ± 0.06 (3)* | −10.2 ± 0.8 (3)† |
Data shown are means ± SE (n).
P < 0.05;
P < 0.01; ns P > 0.05 vs. control (before treatment). DIDS, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid; NPPB, 5-nitro-2-(3-phenylpropylamino)benzoic acid.
Cl−/HCO3− exchange.
Mucosa from each colon was divided longitudinally into two equal pieces and mounted into two Ussing chambers. One piece of mucosa was bathed luminally with zero HCO3− Ringer solution that contained Cl−, while the other piece of mucosa was bathed luminally with zero HCO3− Ringer solution that did not contain Cl−. Both pieces of mucosa were bathed serosally with Ringer solution that contained Cl− and HCO3−. After 15–30 min, when JHCO3 had stabilized, R568 or vehicle control was added to the serosal or mucosal side of tissue, and changes in JHCO3 during the 15–30-min period ensuing after the addition of the agonist or vehicle were determined and averaged. When inhibitor was used, it was added at 30 min before the agonist. Cl−/HCO3− exchange activity was calculated as ΔJHCO3 (the difference between JHCO3 in the presence minus absence of luminal Cl−) (18, 48).
SCFA/HCO3− exchange.
Unless specifically described elsewhere, colon segments were prepared and treated as in the Cl−/HCO3− exchange studies except that they were bathed luminally with zero HCO3− Ringer solution that either contained or did not contain 25 mM isobutyrate. SCFA/HCO3− exchange activity was calculated as ΔJHCO3 (the difference between JHCO3 in the presence minus absence of luminal isobutyrate) (18, 48).
Cyclic nucleotide-dependent HCO3− secretion.
Colon segments were prepared and treated as in the secretagogue-induced HCO3− secretion experiments except that they were bathed luminally with HCO3−-free Ringer solution that contained no isobutyrate and no Cl−. Cyclic nucleotide-dependent HCO3− secretion was calculated as ΔJHCO3 (the difference between JHCO3 after minus JHCO3 before the addition of forskolin) (18, 48). When inhibitor was used, it was added at 30 min before R568 or forskolin.
Chemicals and Solutions
Forskolin, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), glibenclamide, 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), amiloride, barium, and bumetanide were obtained from Sigma and GlyH-101 from Santa Cruz Biotechnology, whereas R-568 was from Tocris Bioscience. All stock solutions were prepared in DMSO. The detailed composition of Ringer solutions used in these studies is listed in Table 1. A combination of 5% CO2-95% O2 was used to oxygenate Ringer solutions that contained HCO3−, whereas 100% O2 was used to oxygenate those solutions that did not contain HCO3−.
Statistical Analysis
Values are expressed as means ± SE. ΔJHCO3, ΔIsc, and ΔpH recovery rates refer to stimulated peak responses minus basal control levels. Data were analyzed by one-way ANOVA followed by Holm-Sidak's post hoc test or, when appropriate, by the paired or unpaired two-tailed Student's t-test using Microsoft Excel 2010 for Windows or GraphPad Prism version 6 for Windows (GraphPad Software). P < 0.05 was considered significant.
RESULTS
Basal HCO3− Secretion
Lowering extracellular Ca2+ concentration reduces and activation of CaSR by R568 enhances basal HCO3− secretion.
The initial experiments performed examined the role of extracellular Ca2+ in modulation of HCO3− secretion under basal (nonstimulated) conditions. Mucosa was first bathed with HCO3−-free, Cl− Ringer solution before 25 mM HCO3− was added to the serosa. As shown in Fig. 1, there was no change in lumen pH; thus lumen alkalinization did not occur in the absence of a serosa-to-lumen-directed HCO3− gradient. Consistent with HCO3− secretion, subsequent addition of HCO3− ion to serosal side induced significant lumen alkalinization [Fig. 1A; means ± SE (n) absence vs. presence of HCO3− ion: −0.011 ± 0.002 (3) vs. 0.035 ± 0.002 (3) pH U·min−1·cm−2, P < 0.01]. CCH, a known secretagogue for HCO3− secretion, was used as a positive control. CCH induced an initial transient increase followed by a sustained increase in lumen pH (Fig. 1A) characteristic of HCO3− secretion associated with cholinergic receptor activation (43).
Fig. 1.
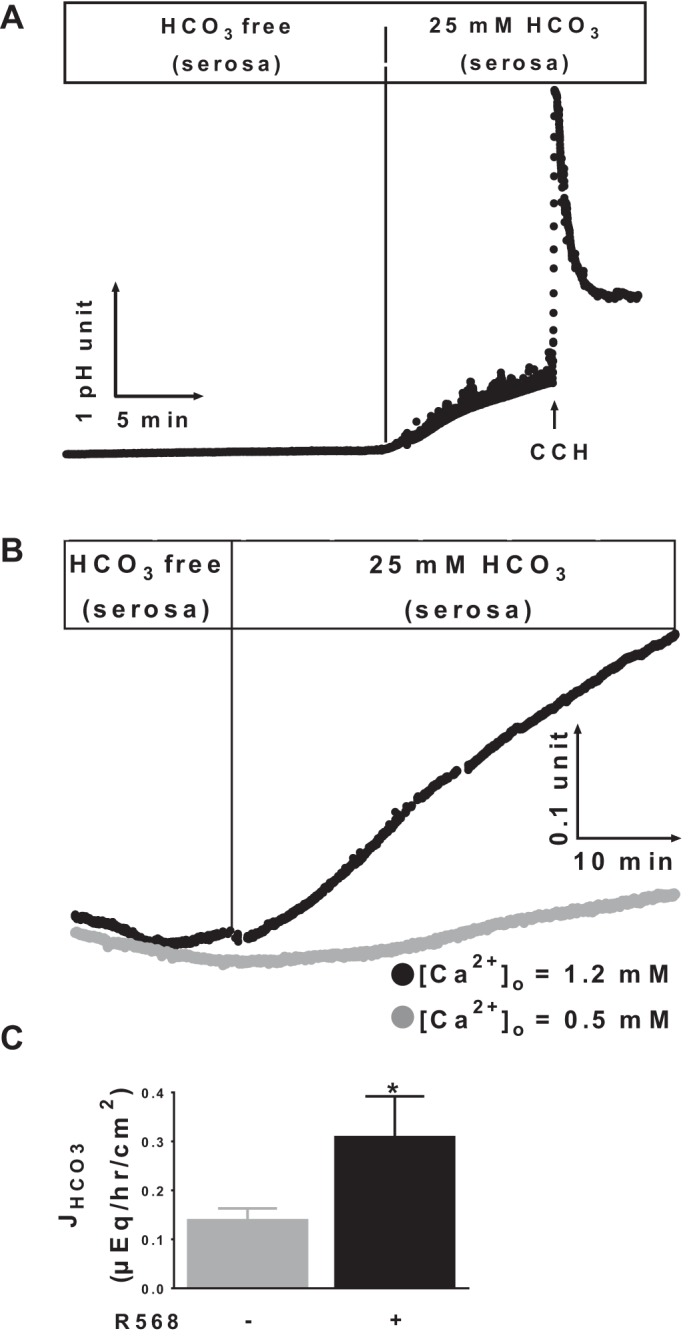
Effects of extracellular Ca2+ and calcium-sensing receptor (CaSR) agonist R568 on basal HCO3− secretion. A and B: representatives of 3 recordings of luminal pH responses of colonic mucosa to absence and presence of a serosa-to-lumen-directed HCO3− gradient and carbachol (CCH, 100 μM, serosa) (A) and normal vs. low [Ca2+]o (B). The presence or absence of HCO3− in the lumen or serosa is shown as indicated. C: serosal to mucosal JHCO3 peak responses to R568 (10 μM, serosa). The basal tissue resistance (Ω/cm2) and Isc (μeq·h−1·cm−2) were 69 ± 7 and 1.44 ± 0.32 at 0.5 mM [Ca2+]o vs. 72 ± 6 and 1.10 ± 0.10 at 1.2 mM [Ca2+]o (P > 0.05). Data are means ± SE of 5 experiments. *P < 0.05 vs. control (with no R568).
Experiments were then performed to examine whether CaSR regulates basal HCO3− secretion. For this, the concentration of Ca2+ in the buffer was lowered from 1.2 mM to 0.5 mM, and HCO3− secretory response was compared. Extracellular Ca2+ is a physiological ligand of CaSR. Previous studies have shown that this maneuver reduces CaSR activity and that, at 0.5 mM Ca2+ concentration, CaSR is only minimally stimulated (12, 14, 16, 25). Reduction of [Ca2+]o from 1.2 mM to 0.5 mM significantly reduced lumen alkalinization rates [Fig. 1B; [Ca2+]o = 1.2 vs. 0.5 mM: 0.031 ± 0.002 (3) vs. 0.013 ± 0.002 (3) pH U·min−1·cm−2, P < 0.01]. As extracellular Ca2+ is also a known determinant of paracellular permeability of intestinal epithelium, additional experiments were performed to exclude the possibility that the reduced rate of lumen alkalinization noted at reduced [Ca2+] was not simply caused by HCO3− “back leak” secondary to altered paracellular integrity; thus, transepithelial electrical resistances (TEER) were measured. No significant differences in TEER were noted between normal and reduced Ca2+-treated tissues (see details in Fig. 1 legend). Thus it is likely that extracellular Ca2+ (and CaSR) regulates transcellular and not paracellular HCO3− movement.
To further assess the role of CaSR as a regulator of HCO3− secretion, another set of experiments measured the rate of serosal to mucosal HCO3− flux (JHCO3) and HCO3− secretory Isc and tested the effect of R568, a specific pharmacological CaSR agonist. A relatively low basal JHCO3 was observed in the absence of R568. Addition of R568 to serosa (Fig. 1C) significantly stimulated JHCO3. Similar but slightly less pronounced effects were also noted when R568 was added to lumen solutions (not shown). Such stimulatory changes were not observed in Isc [absence vs. presence of R568: 1.36 ± 0.40 (5) vs. 1.48 ± 0.44 (5) μeq·h−1·cm−2, P > 0.05]. These data suggested that CaSR stimulates electroneutral HCO3− secretion.
Acid-Induced HCO3− Secretion
Activation of CaSR by R568 enhances acid-induced HCO3− secretion.
Acid-induced HCO3− secretion is a known mechanism that intestinal mucosa utilizes as a defense against acid-induced damage (2, 20, 28). Colonic mucosa is exposed to luminal acidic environment, generated as a result of bacteria fermentation of undigested carbohydrates (33, 45). To assess whether CaSR affects acid-induced HCO3− secretion, colonic tissues were challenged by additions of acid (HCl) to lower pH in the lumen, rates of pH recovery were monitored and recorded, and effect of R568 was examined. Figure 2A shows changes in the rates of pH recovery in the presence vs. absence of R568. The Δrate changes at peak responses above basal levels were calculated and are summarized in Fig. 2B. R568 induced a transient but significant stimulation of acid-induced HCO3− secretion.
Fig. 2.
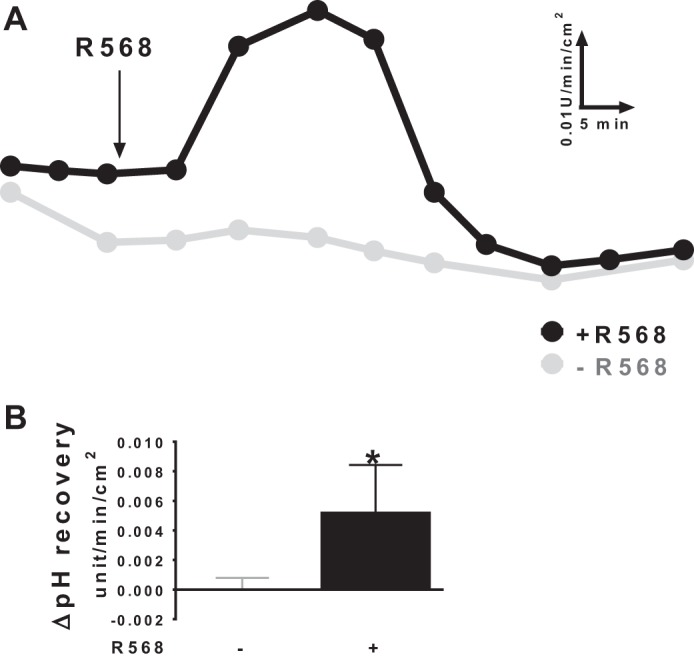
Effect of CaSR agonist R568 on acid-induced HCO3− secretion. A: representatives of pH recovery responses in the presence vs. absence of R568 (10 μM, serosa). The addition of R568 is shown as indicated. B: summary of ΔpH recovery rates (stimulated peak values above basal levels before additions of R568 or vehicle). Basal levels of acid-induced pH recovery were 0.015 ± 0.001 pH U·min−1·cm−2. The tissue resistance (Ω/cm2) and Isc (μeq·h−1·cm−2) at the end of 60-min experiments were 50 ± 12 and 1.74 ± 0.20 without challenge and 46 ± 10 and 1.97 ± 0.23 with acid challenges (P > 0.05). Data are means ± SE of 5 experiments. *P < 0.05 vs. control (with no R568).
Secretagogue-Induced HCO3− Secretion
Activation of CaSR by R568 inhibits secretagogue-induced HCO3− secretion.
HCO3− secretion is markedly increased in cholera and other secretagogue-induced diarrheal diseases (22, 34). To assess whether CaSR stimulates HCO3− secretion under these diseased conditions, the R568 effect was examined in a model of secretagogue-induced secretory diarrhea. In this study, forskolin was used to stimulate HCO3− secretion, and HCO3− secretory response was monitored by measuring HCO3− secretory rate (JHCO3) (Fig. 3A) and by recording Isc (Fig. 3C). Forskolin stimulated both JHCO3 and Isc; subsequent addition of R568 did not increase but rather decreased forskolin-induced HCO3− secretion.
Fig. 3.

Effect of CaSR agonist R568 on secretagogue-induced HCO3− secretion and HCO3− current. Shown are JHCO3 (A) and Isc (C) responses to forskolin (FSK, 500 nM, serosa) ± R568 (10 μM, serosa), assayed in lumen Cl−-containing Ringer solution. The changes induced by FSK in the absence vs. presence of R568 are summarized in B (ΔJHCO3FSK) and D (ΔISCFSK). The basal tissue resistance and Isc under these conditions were as follows: 68 ± 4 Ω/cm2 and 1.51 ± 0.22 μeq·h−1·cm−2. Data are means ± SE of 8 experiments. **P < 0.01 vs. control (with no R568); ##P < 0.01 vs. FSK.
Cl−-Dependent HCO3− Secretion
Activation of CaSR stimulates Cl−/HCO3− exchange activity.
Because the basal and acid-induced HCO3− secretion were assayed in Cl−-containing Ringer, it is likely that the CaSR effects reflect stimulation of a Cl−-dependent HCO3− secretory mechanism such as Cl−/HCO3− exchange (48). To address this possibility, Cl−/HCO3− exchange activity was measured and is shown in Fig. 4. Figure 4A demonstrates HCO3− secretory responses to the presence vs. absence of lumen Cl−. In agreement with previous studies (47, 48), a luminal Cl−-dependent HCO3− secretory mechanism (Cl−/HCO3− exchange) was observed. The latter was partially abolished by pretreatment with 100 μM DIDS added to lumen side (data not shown). Activation of CaSR by R568, added to serosal side, resulted in stimulation of the Cl−/HCO3− exchange activity (Fig. 4B). This Cl−-dependent HCO3− secretion was significantly higher in the presence than in the absence of R568 (Fig. 4B). A similar but less pronounced stimulatory effect of R568 was noted when this agonist was added to the mucosal solution (data not shown).
Fig. 4.
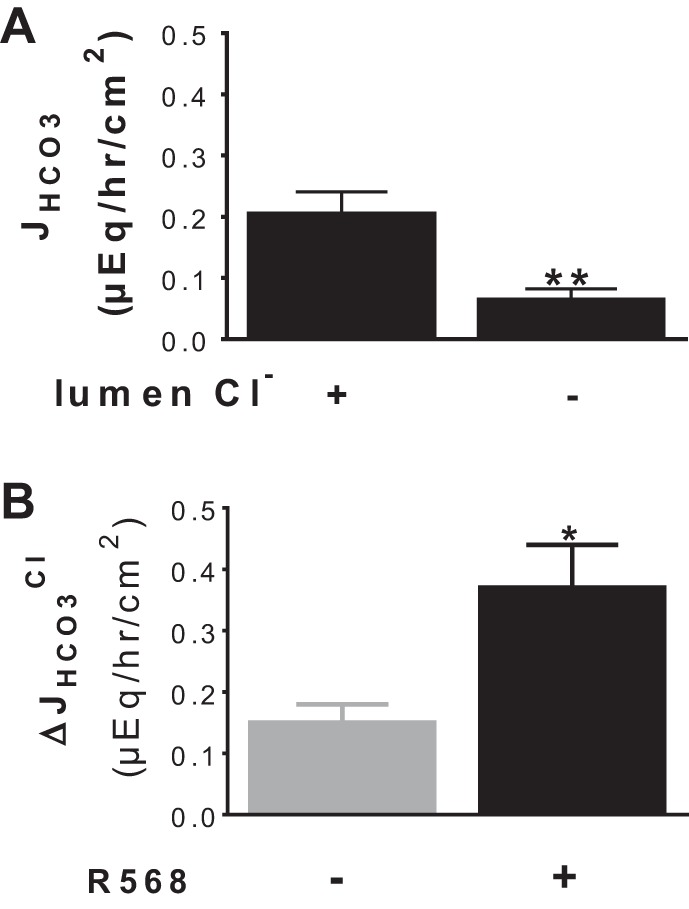
Activation of CaSR stimulates Cl−-dependent HCO3− secretion. A: JHCO3 responses to the presence vs. absence of lumen Cl−. B: ΔJHCO3Cl (i.e., Cl−-dependent HCO3− secretion) responses to the presence vs. absence of R568 (10 μM, serosa). The basal tissue resistance (Ω/cm2) and Isc (μeq·h−1·cm−2) were 68 ± 11 and 1.44 ± 0.32 in the presence vs. 95 ± 15 and 1.01 ± 0.08 in the absence of lumen Cl− (P > 0.05). Data are means ± SE of 3–5 experiments. *P < 0.05 and **P < 0.01 vs. control (1st bar).
SCFA-Dependent HCO3− Secretion
Activation of CaSR stimulates SCFA/HCO3− exchange activity.
SCFA are present in the colon and induce HCO3− secretion via the SCFA/HCO3− exchange (47, 48). To assess whether CaSR stimulates HCO3− secretion under these conditions, the next series of experiments examined isobutyrate-dependent HCO3− secretion and its response to R568. Figure 5, A and B, shows representative tracings, and Fig. 5C presents a summary of HCO3− secretory responses to the presence vs. absence of lumen SCFA (25 mM isobutyrate) with vs. without a serosa-to-mucosa-directed HCO3− gradient. SCFA-dependent HCO3− secretion was present and required serosal HCO3−, consistent with HCO3− secretion mediated by SCFA/HCO3− exchange. Similar to the R568 effect on the Cl−/HCO3− exchange, isobutyrate-dependent HCO3− secretion was significantly stimulated by activation of CaSR by R568 (Fig. 5D).
Fig. 5.

Activation of CaSR stimulates SCFA-dependent HCO3− secretion. Two equally longitudinally divided pieces of mucosa from same colon were mounted into 2 Ussing chambers. 1 piece was treated first in the presence then absence of lumen short-chain fatty acid (SCFA), whereas the other piece was treated first in the presence then absence of serosa HCO3−. Shown are representative recordings (A and B) and quantitative summary (C) of these JHCO3 responses. The ΔJHCO3SCFA (i.e., SCFA-dependent HCO3− secretion) was calculated, and its responses to the presence vs. absence of R568 (10 μM, serosa) are shown in D. The basal tissue resistance (Ω/cm2) and Isc (μeq·h−1·cm−2) were 126 ± 26 and 0.71 ± 0.17 in the presence vs. 95 ± 5 and 1.01 ± 0.08 in the absence of lumen 25 mM isobutyrate (P > 0.05). Data are means ± SE of 3–5 experiments. *P < 0.05 and **P < 0.01 vs. control (1st bar).
cAMP-Dependent HCO3− Secretion
Activation of CaSR by R568 inhibits cAMP-dependent HCO3− secretion.
In contrast to R568 stimulation of basal HCO3− secretion, HCO3− secretion was inhibited by R568 under forskolin-stimulated condition (see Fig. 3). It is uncertain how R568 produces this inhibition. Because R568 inhibited neither Cl−-dependent nor SCFA-dependent HCO3− secretion (see Figs. 4–5), it is unlikely that the effect of the CaSR agonist is via inhibition of either of these anion exchanges. Rather, a Cl−/SCFA-independent HCO3− transport mechanism might be responsible. One such mechanism is a cyclic nucleotide-dependent electrogenic channel (e.g., CFTR)-mediated HCO3− secretion (48). Thus, to address this latter possibility, stimulation of HCO3− secretion by forskolin and its inhibition following addition of R568 were reassessed in lumen Cl−/SCFA-free solutions. As shown in Fig. 6A, a low rate of HCO3− transport was noted under basal condition before the addition of forskolin. Addition of forskolin significantly stimulated HCO3− secretion. The subsequent addition of R568 almost completely reversed forskolin-induced HCO3− secretion.
Fig. 6.

Activation of CaSR inhibits cAMP-dependent HCO3− secretion and HCO3− current. Shown are JHCO3 (A) and Isc (C) responses to FSK (500 nM, serosa) ± R568 (10 μM, serosa), assayed in lumen Cl−-free Ringer solution. The changes induced by FSK in the absence vs. presence of R568 are summarized in B (ΔJHCO3FSK) and D (ΔISCFSK). The basal tissue resistance and Isc under these conditions were 103 ± 3 Ω/cm2 and 0.87 ± 0.05 μeq·h−1·cm−2. Data are means ± SE of 4–7 experiments. **P < 0.01 vs. control (1st bar); ##P < 0.01 vs. FSK alone (2nd bar).
Similar changes were observed in experiments that determined changes in Isc (Fig. 6C). Forskolin stimulated Isc; subsequent addition of R568 inhibited HCO3− secretion. Removal of the serosa to mucosa HCO3− gradient significantly diminished both basal and forskolin-stimulated Isc (data not shown); in the absence of serosal HCO3−, forskolin and R568 failed to stimulate or to inhibit Isc, respectively (data not shown). Pretreatment with luminal glibenclamide (100 μM) or GlyH-101 (10 μM), CFTR channel blockers, and NPPB (100 μM), an anion channel inhibitor, added either before the addition of forskolin or R568, abolished the forskolin stimulatory and R568 inhibitory effects on Isc (data not shown). As a consequence, these results suggest that R568 inhibits cAMP-dependent, glibenclamide/GlyH-101/NPPB-sensitive electrogenic HCO3− secretion.
Effect of CaSR Knockout
R568 fails to stimulate Cl−- and SCFA-dependent and inhibit cAMP-dependent HCO3− secretion in colon of CaSR-null mouse.
R568 is a specific pharmacological agonist that has been widely used to stimulate CaSR. To verify that the effects of R568 occurred via the CaSR, additional studies on the effect of R568 were performed in intestinal epithelium-specific CaSR knockout mice (see Fig. 7). In this study, intestinal epithelium-specific CaSR knockout mice were used together with their wild-type littermates. Activation of CaSR by R568 stimulated Cl−- and SCFA-dependent HCO3− secretion and inhibited cAMP-dependent HCO3− secretion in colon mucosa of wild-type mice (Fig. 7, A–C); such effects were abolished in CaSR-null mice (Fig. 7, D–F). These results indicate that the R568 effects occur via activating the CaSR in the intestinal epithelium.
Fig. 7.

R568 fails to stimulate Cl−- and SCFA- dependent and inhibit cAMP-dependent HCO3− secretion in colons of CaSR-null mice. Colonic mucosa from wild-type (CaSR+/+) (A–C) and knockout (CaSR−/−) mice (D–F) were isolated and treated, and activities of the 3 HCO3− transporters were assayed as in rats. Data are means ± SE of 4–6 experiments. *P < 0.05 vs. control (1st bar).
DISCUSSION
These present results provide the basis for a new model for regulation of HCO3− secretion in the mammalian colon in that activation of CaSR by R568 stimulated basal and acid-induced HCO3− secretion, but, in contrast, R568 inhibited cyclic nucleotide-mediated HCO3− secretion. Our studies also indicate that the enhancement of HCO3− secretion is mediated via stimulation of electroneutral Cl−/HCO3− and SCFA/HCO3− exchanges that are localized on the apical membrane of colonic surface epithelial cells; in contrast, the CaSR inhibitory action is a consequence of CaSR inhibition of a cAMP-dependent, lumen glibenclamide/GlyH-101/NPPB-sensitive electrogenic HCO3− secretory process primarily located in the crypt cells. A model for this differential regulation of colonic HCO3− secretion by CaSR is depicted in Fig. 8.
Fig. 8.

Cellular model of CaSR regulation of HCO3− secretion in colonocytes of rat distal colon. Top: R568 acting via CaSR causes enhancement of HCO3− secretion in surface epithelial cells by stimulation of luminal Cl−-dependent HCO3− secretion via apical Cl−/HCO3− exchange and stimulation of luminal SCFA-dependent HCO3− secretion mediated by apical SCFA/HCO3− exchange. Bottom: CaSR reduces HCO3− secretion in the crypt epithelial cells through inhibition of a cAMP-dependent HCO3− secretory process that may involve a 5-nitro-2-(3-phenylpropylamino)benzoic acid/glibenclamide-sensitive apical anion channel such as cystic fibrosis transmembrane conductance regulator (CFTR) and/or a basolateral HCO3− entry mechanism(s). +, stimulation; -, inhibition.
According to the present model of colonic ion function (50), absorptive processes are primarily localized to surface cells, whereas secretory processes are primarily present in crypt cells. Because Cl−-dependent HCO3− exchange and SCFA-dependent HCO3− exchange are present only in surface cells and are absent in crypts [see Refs. 47 and 48 and also in review by Binder, et al. (3)], Cl−-dependent HCO3− secretion and SCFA-dependent HCO3− secretion are most likely both surface cell functions. In contrast, CFTR-mediated HCO3− secretion is generally considered to represent a crypt cell function. Thus, in addition to mediating colonic HCO3− secretion, the primary function of these two anion exchanges in these absorptive surface cells is to absorb solutes/electrolytes. SCFA absorption is mediated by SCFA/HCO3− exchange, and NaCl absorption is the result of Cl−/HCO3− exchange coupled to Na+/H+ exchange, which is also localized in surface cells. The ability of CaSR agonists to stimulate both anion exchanges (see Figs. 4, 5, and 7) as well as Na+/H+ exchange (25) suggests that, in addition to stimulation of HCO3− secretion, CaSR may function as a mechanism to enhance electrolyte and fluid absorption. Interestingly, activation of CaSR also stimulates colonic acid-induced HCO3− secretion (see Fig. 2). One could speculate that this latter function may also neutralize H+ from bacterial fermentation and/or Na+/H+ exchange, further increasing solute absorption.
Consistent with a recent in vivo study in rat perfused duodenum (1), these present studies demonstrated that CaSR activation stimulated basal HCO3− secretion in ex vivo colonic mucosa. These findings may have important physiological significance, as HCO3− secretion is an integral part of mucosal defense mechanisms. HCO3− secretion is required for mucin secretion by goblet cells to establish a layer of mucus overlying the epithelium (35), an initial defense barrier that limits pathogen invasion. Defects in HCO3− secretion have been shown to impair the formation of the mucus layer and compromise the integrity of the intestinal barrier, leading to bacteria translocation and development of intestinal inflammation (24, 51, 52). Thus the ability for CaSR to stimulate HCO3− secretion under basal conditions suggests that, through modulating mucus secretion and barrier function, this well-conserved nutrient-sensing receptor may play a role in intestinal immune function. Indeed, mice deficient in CaSR with deficient regulated HCO3− secretion in the colon have altered barrier integrity, enhanced bacteria translocation, and increased inflammation (15, 30), whereas enteral nutrients, including the CaSR-activating nutrients/minerals, calcium, spermine, and tryptophan, have been shown to improve intestinal permeability and immunity (39, 40) and inflammation (5, 6, 11, 13).
Importantly, both HCO3− and Cl− secretion are markedly induced in cyclic nucleotide-mediated secretory diarrheas (e.g., cholera) (22, 34). Although these secretory responses may be helpful in enhancement of the defensive mucus layer so as to limit pathogen invasion and also to flush out toxins, overproduction and secretion of these anions by the intestine under these pathological conditions is harmful and may result in dehydration, alkali deficit, and metabolic acidosis (22, 34). Systemic volume depletion and metabolic acidosis are the two major causes of death associated with acute diarrheal illnesses, especially in infants and young children. The ability of CaSR agonists both to inhibit cyclic nucleotide-stimulated Cl− (10, 12, 14, 16, 25) and HCO3− secretion (see Figs. 6 and 7) and to promote Cl− and SCFA absorption (see Figs. 4, 5, and 7) as well as Na+ absorption (25) suggests that this class of drugs may provide a unique therapeutic approach to prevent and treat these potentially lethal diarrheal illnesses. Because CaSR agonists are naturally occurring nutrients, CaSR-based antidiarrheal therapies would be of particular utility among actively growing infants and children (13).
In the present study, the net increases in Isc induced by forskolin (ΔIscFSK) were greater than those in net JHCO3 (ΔJHCO3FSK) [compare the gray-colored columns in Figs. 3D and 6D (mean values: 3.6 and 1.5 μeq·h−1·cm−2) vs. Figs. 3B and 6B (mean values: 1.7 and 0.2 μeq·h−1·cm−2)]. These differences cannot be explained by a non-steady-state flux period, as all measurements were made after 15–30 min when both Isc and JHCO3 had stabilized and were in steady state. The most likely explanation is that a component of ΔIscFSK represents forskolin-induced Cl− secretion even though serosal bumetanide was present in both experiments. Serosal bumetanide was employed to prevent (or at least to reduce) such a contribution from forskolin-induced Cl− secretion. It is known that, in rat distal colon, bumetanide does not completely suppress Cl− secretion induced by forskolin (41). Only ∼70% of such Cl− secretion was inhibited by bumetanide; the remainder of the Cl− secretion was mediated by a Cl−/HCO3− exchange located in the basolateral membrane of colonocytes (41). Consistent with this, we found that the non-HCO3− portion of ΔIscFSK was greater when a serosa to mucosa transepithelial Cl− gradient was present than when a Cl− gradient was absent (compare 88% in Fig. 6 vs. 53% in Fig. 3).
Although most experiments were performed with a reduced concentration of Ca2+o to minimize background activation of the receptor before R568 addition, under normal Ca2+o condition, the effects of R568 were qualitatively similar, albeit with a slightly less pronounced effect (also see Fig. 6 of Ref. 12). As a result, the data from the present study suggest that the calcimimetic R568 may have physiological relevance and clinical utility. Indeed, this same class of drug has been employed successfully to inhibit parathyroid hormone secretion in hyperparathyroid patients, where Ca2+o in the serum can be either <1.0 mM (secondary hyperparathyroidism) or >1.5 mM (primary hyperparathyroidism) [see a review by Tfelt-Hansen and Brown (46)].
In summary, the present in vitro studies confirmed the presence of at least three distinct mechanisms for HCO3− secretion in rodent distal colon, i.e., lumen Cl−-dependent HCO3− secretion, SCFA-dependent HCO3− secretion, and cAMP-activated HCO3− secretion. Furthermore, CaSR agonists differently regulate HCO3− secretion, depending on the physiological state of the intestine and the specific transporter in question. During physiological conditions when electroneutral Cl−/HCO3− and SCFA/HCO3− exchanges dominate, CaSR enhances HCO3− secretion; however, in experimental conditions that result in stimulation of fluid and HCO3− secretion that also occurs in cholera in which electrogenic CFTR-mediated HCO3− conductance is dominant, CaSR also inhibits HCO3− secretion. We suggest that both of these two regulatory processes induced by CaSR are potentially beneficial. Whereas the stimulatory effect may help expand the mucus layer, the inhibition of channel-mediated HCO3− secretion may be of particular clinical significance, as it may reduce and minimize HCO3− losses in diarrhea.
GRANTS
This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health K08HD079674 (S. Cheng), Children's Digestive Health and Nutrition Foundation award 00102979 (S. Cheng), and Children's Miracle Network (S. Cheng). S. Cheng was a recipient of the North American Society of Pediatric Gastroenterology, Hepatology, and Nutrition Foundation's “Fellow to Faculty Transition Award in Inflammatory Bowel Disease” 2012–2013.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.T., M.P., and S.X.C. conception and design of research; L.T., M.P., and S.X.C. performed experiments; L.T., M.P., and S.X.C. analyzed data; L.T., H.J.B., and S.X.C. interpreted results of experiments; L.T. and S.X.C. prepared figures; L.T. and S.X.C. drafted manuscript; L.L., H.J.B., and S.X.C. edited and revised manuscript; W.C. and S.X.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We extend our gratitude to Sadasivan Vidyasagar, Liangjie Yin, and Xiangrong Sun for technical support on pH stat measurements, Megan McIntyre, Tao Huang, Peng Yi, and Suman Donepudi for technical assistance on short-circuit current recordings, and Svea Cheng for artistic assistance in figure design.
REFERENCES
- 1.Akiba Y, Watanabe C, Mizumori M, Kaunitz JD. Luminal L-glutamate enhances duodenal mucosal defense mechanisms via multiple glutamate receptors in rats. Am J Physiol Gastrointest Liver Physiol 297: G781–G791, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen A, Flemstrom G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Cell Physiol 288: C1–C19, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Binder HJ, Rajendran V, Sadasivan V, Geibel JP. Bicarbonate secretion: a neglected aspect of colonic ion transport. J Clin Gastroenterol 39: S53–S58, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Black R, Cousens S, Johnson H, Lawn J, Rudan I, Bassani D, Jha P, Campbell H, Walker C, Cibulskis R, Eisele T, Liu L, Mathers C. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375: 1969–1987, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Bovee-Oudenhoven IM, Lettink-Wissink ML, Van Doesburg W, Witteman BJ, Van Der Meer R. Diarrhea caused by enterotoxigenic Escherichia coli infection of humans is inhibited by dietary calcium. Gastroenterology 125: 469–476, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Bovee-Oudenhoven IM, Wissink ML, Wouters JT, Van Der Meer R. Dietary calcium phosphate stimulates intestinal lactobacilli and decreases the severity of a salmonella infection in rats. J Nutr 129: 607–612, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366: 575–580, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81: 239–297, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyay N, Cheng I, Rogers K, Riccardi D, Hall A, Diaz R, Hebert SC, Soybel DI, Brown EM. Identification and localization of extracellular Ca2+-sensing receptor in rat intestine. Am J Physiol 274: G122–G130, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Cheng CY, Petrova E, Stahl M, Cheng SX. Calcium-sensing receptor inhibits cholera toxin-induced anion secretion by intestine via enteric nervous system (Abstract). J Pediatr Gastroenterol Nutr 55: E20, 2012. [Google Scholar]
- 11.Cheng SX. Calcium-sensing receptor in the gut: evidence for its role in mediating known nutritional therapy for inflammatory bowel disease (Abstract). J Pediatr Gastroenterol Nutr 55: E70, 2012. [Google Scholar]
- 12.Cheng SX. Calcium-sensing receptor inhibits secretagogue-induced electrolyte secretion by intestine via the enteric nervous system. Am J Physiol Gastrointest Liver Physiol 303: G60–G70, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng SX, Bai HX, Gonzalez-Peralta R, Mistry PK, Gorelick FS. Calcium ameliorates diarrhea in immunocompromised children. J Pediatr Gastroenterol Nutr 56: 641–644, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng SX, Geibel J, Hebert S. Extracellular polyamines regulate fluid secretion in rat colonic crypts via the extracellular calcium-sensing receptor. Gastroenterology 126: 148–158, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Cheng SX, Lightfoot YL, Yang T, Zadeh M, Tang L, Sahay B, Wang GP, Owen JL, Mohamadzadeh M. Epithelial CaSR deficiency alters intestinal integrity and promotes proinflammatory immune responses. FEBS Lett 588: 4158–4166, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng SX, Okuda M, Hall A, Geibel JP, Hebert SC. Expression of calcium-sensing receptor in rat colonic epithelium: evidence for modulation of fluid secretion. Am J Physiol Gastrointest Liver Physiol 283: G240–G250, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Currie MG, Fok KF, Kato J, Moore RJ, Hamra FK, Duffin KL, Smith CE. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci USA 89: 947–951, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dohgen M, Hayashi H, Yajima T, Suzuki Y. Stimulation of bicarbonate secretion by luminal short-chain fatty acid in the rat and human colon in vitro. Jpn J Physiol 44: 519–531, 1994. [DOI] [PubMed] [Google Scholar]
- 19.Field M, Graf LH, Laird WJ, Smith PL. Heat-stable enterotoxin of Escherichia coli: in vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc Natl Acad Sci USA 75: 2800–2804, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flemstrom G, Isenberg JI. Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol Sci 16: 23–28, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Flores J, Sharp G. The activation of adenylate cyclase by cholera toxin: possible interaction with the nucleotide regulatory site. Ciba Found Symp 42: 89–108, 1976. [DOI] [PubMed] [Google Scholar]
- 22.Fordtran JS. Speculations on the pathogenesis of diarrhea. Fed Proc 26: 1405–1414, 1967. [PubMed] [Google Scholar]
- 23.Gama L, Baxendale-Cox LM, Breitwieser GE. Ca2+-sensing receptors in intestinal epithelium. Am J Physiol 273: C1168–C1175, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Garcia MA, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest 119: 2613–2622, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geibel J, Sritharan K, Geibel R, Geibel P, Persing JS, Seeger A, Roepke T, Deichstetter M, Prinz C, Cheng S, Martin D, Hebert S. Calcium-sensing receptor abrogates secretagogue-induced increases in intestinal net fluid secretion by enhancing cyclic nucleotide destruction. Proc Natl Acad Sci USA 103: 9390–9397, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haas M, McManus TJ. Bumetanide inhibits (Na/K/2Cl) co-transport at a chloride site. Am J Physiol 245: C235–C240, 1983. [DOI] [PubMed] [Google Scholar]
- 27.Holmberg C, Perheentupa J, Launiala K. Colonic electrolyte transport in health and in congenital chloride diarrhea. J Clin Invest 56: 302–310, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isenberg JI, Selling JA, Hogan DL, Koss MA. Impaired proximal duodenal mucosal bicarbonate secretion in patients with duodenal ulcer. N Engl J Med 316: 374–379, 1987. [DOI] [PubMed] [Google Scholar]
- 29.Liou A, Sei Y, Zhao X, Feng J, Lu X, Thomas C, Pechhold S, Raybould H, Wank S. The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to L-phenylalanine in acutely isolated intestinal I cells. Am J Physiol Gastrointest Liver Physiol 300: G538–G546, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macleod RJ. Extracellular calcium-sensing receptor/PTH knockout mice colons have increased Wnt/β-catenin signaling, reduced non-canonical Wnt signaling, and increased susceptibility to azoxymethane-induced aberrant crypt foci. Lab Invest 93: 520–527, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Moore S, Lima AA, Guerrant R. Infection: Preventing 5 million child deaths from diarrhea in the next 5 years. Nat Rev Gastroenterol Hepatol 8: 363–364, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Nearing J, Betka M, Quinn S, Hentschel H, Elger M, Baum M, Bai M, Chattopadyhay N, Brown EM, Hebert SC, Harris HW. Polyvalent cation receptor proteins (CaRs) are salinity sensors in fish. Proc Natl Acad Sci USA 99: 9231–9236, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nugent SG, Kumar D, Rampton DS, Evans DF. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 48: 571–577, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell DW, Solberg LI, Plotkin GR, Catlin DH, Maenza RM, Formal SB. Experimental diarrhea. 3. Bicarbonate transport in rat salmonella enterocolitis. Gastroenterology 60: 1076–1086, 1971. [PubMed] [Google Scholar]
- 35.Quinton PM. Role of epithelial HCO3− transport in mucin secretion: lessons from cystic fibrosis. Am J Physiol Cell Physiol 299: C1222–C1233, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey OCW, Bikle D, Rozengurt N, Young SH, Rozengurt E. Negative cross-talk between calcium-sensing receptor and β-catenin signaling systems in colonic epithelium. J Biol Chem 287: 1158–1167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 92: 131–135, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sands JM, Naruse M, Baum M, Jo I, Hebert SC, Brown EM, Harris HW. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest 99: 1399–1405, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schepens MA, Rijnierse A, Schonewille AJ, Vink C, Brummer RJ, Willemsen LE, van der Meer R, Bovee-Oudenhoven IM. Dietary calcium decreases but short-chain fructo-oligosaccharides increase colonic permeability in rats. Br J Nutr 104: 1780–1786, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Schepens MA, Vink C, Schonewille AJ, Dijkstra G, van der Meer R, Bovee-Oudenhoven IM. Supplemental calcium attenuates the colitis-related increase in diarrhea, intestinal permeability, and extracellular matrix breakdown in HLA-B27 transgenic rats. J Nutr 139: 1525–1533, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Schultheiss G, Horger S, Diener M. The bumetanide-resistant part of forskolin-induced anion secretion in rat colon. Acta Physiol Scand 164: 219–228, 1998. [PubMed] [Google Scholar]
- 42.Seamon KB, Daly JW. Forskolin: a unique diterpene activator of cyclic AMP-generating systems. J Cyclic Nucleotide Res 7: 201–224, 1981. [PubMed] [Google Scholar]
- 43.Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca(2+)-dependent HCO3− secretion. J Physiol 505: 411–423, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheinin Y, Kallay E, Wrba F, Kriwanek S, Peterlik M, Cross HS. Immunocytochemical localization of the extracellular calcium-sensing receptor in normal and malignant human large intestinal mucosa. J Histochem Cytochem 48: 595–602, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Talbot C, Lytle C. Segregation of Na/H exchanger-3 and Cl/HCO3− exchanger SLC26A3(DRA) in rodent cecum and colon. Am J Physiol Gastrointest Liver Physiol 299: G358–G367, 2010. [DOI] [PubMed] [Google Scholar]
- 46.Tfelt-Hansen J, Brown E. The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit Rev Clin Lab Sci 42: 35–70, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Vidyasagar S, Barmeyer C, Geibel J, Binder HJ, Rajendran VM. Role of short-chain fatty acids in colonic HCO3− secretion. Am J Physiol Gastrointest Liver Physiol 288: G1217–G1226, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Vidyasagar S, Rajendran VM, Binder HJ. Three distinct mechanisms of HCO3− secretion in rat distal colon. Am J Physiol Cell Physiol 287: C612–C621, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Chandra R, Samsa L, Gooch B, Fee B, Cook JM, Vigna S, Grant A, Liddle R. Amino acids stimulate cholecystokinin release through the Ca2+-sensing receptor. Am J Physiol Gastrointest Liver Physiol 300: G528–G537, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welsh MJ, Smith PL, Fromm M, Frizzell RA. Crypts are the site of intestinal fluid and electrolyte secretion. Science 218: 1219–1221, 1982. [DOI] [PubMed] [Google Scholar]
- 51.Xiao F, Juric M, Li J, Riederer B, Yeruva S, Singh AK, Zheng L, Glage S, Kollias G, Dudeja P, Tian DA, Xu G, Zhu J, Bachmann O, Seidler U. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(-) secretion in murine ileocolonic inflammation. Inflamm Bowel Dis 18: 101–111, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao F, Yu Q, Li J, Johansson ME, Singh AK, Xia W, Riederer B, Engelhardt R, Montrose M, Soleimani M, Tian DA, Xu G, Hansson GC, Seidler U. Slc26a3 deficiency is associated with loss of colonic HCO3 secretion, absence of a firm mucus layer and barrier impairment in mice. Acta Physiol (Oxf) 211: 161–175, 2013. [DOI] [PubMed] [Google Scholar]