

Abstract
High mobility group (HMG) box proteins are abundant and ubiquitous DNA binding proteins with a remarkable array of functions throughout the cell. The structure of the HMG-box DNA binding domain and general mechanisms of DNA binding and bending have been known for more than a decade. However, new mechanisms that regulate HMG-box protein intracellular translocation, and by which HMG-box proteins recognize DNA with and without sequence specificity, have only recently been uncovered. This review focuses primarily on the Sry-like HMG box family, HMGB1, and mitochondrial transcription factor A. For these proteins, structural and biochemical studies have shown that HMG-box protein modularity, interactions with other DNA binding proteins and cellular receptors, and post-translational modifications are key regulators of their diverse functions.
Keywords: High mobility group (HMG) box protein, DNA recognition, posttranslational modifications
High mobility group box proteins
Since the discovery of the high mobility group (HMG) proteins nearly forty years ago [1], their roles in the nucleus and mitochondria as architectural DNA binding proteins, in the cytoplasm as signaling regulators, and in the extracellular milieu as inflammatory cytokines have earned them a reputation as the ultimate utility player of the cell. The term ‘high mobility group’ originates from their discovery as proteins in the acid extracts of mammalian cellular chromatin that had high electrophoretic mobility [1]. The HMG proteins comprise three families [2, 3]: HMG-A [4], HMG-N [5], and HMG-box (HMGB) ([6–10]) proteins. The HMGB proteins are by far the largest group, playing essential roles in recognition and maintenance of DNA in DNA-dependent cellular processes [10, 11]. By contrast, it was especially surprising when, a dozen years ago, HMGB1 was found to act as an extracellular cytokine [12] and function in autophagic processes (reviewed in [13–15]). Here we review recent work that provides new insights into the mechanisms of molecular recognition and regulation that determine how the HMGB proteins can interact with different binding partners in such diverse cellular processes.
HMGB protein recognition of DNA
A conserved sequence of approximately 75 amino acids defines the HMG-box found in many transcription factors and chromosomal proteins [16]. HMGB proteins have single (Figure 1a) or multiple HMG boxes (Figure 1b). They are classified as either DNA sequence-specific or non-sequence-specific based on their ability to produce DNaseI footprints on specific DNA sequences [17]. Generally, transcription factors are sequence-specific and contain a single HMG box (Figure 1a, Table 1) [18]. However, non-sequence-specific single HMG boxes exist in Drosophila melanogaster (HMGD) [19], Saccharomyces cerevisiae (NHP6A) [20] (Figure 1a), and humans (PMS1) [21]. Tandem HMG box proteins (Figure 1b, Table 1) include HMGB1 through HMGB4, and mitochondrial transcription factor A (mtTFA/TFAM), among others (reviewed in [10, 22]). These generally recognize DNA non-sequence-specifically, except for TFAM, which interestingly has both sequence specific and non-sequence specific DNA binding properties [23, 24].
Figure 1. Alignments of single- and dual-domain high mobility group (HMG)-box proteins.

(a) Sequence alignment of the HMG domains of selected single HMG box proteins: structure specific recognition protein 1 (SSRP1), sex-determining region Y (SRY) [80], and the SRY-like box (Sox) family of HMGB proteins [81]. Primary site (1°, red font) intercalating residues and secondary site (‘2°’, green font) intercalating/hydrogen bonding residues are indicated. Conserved residues are shown in blue font. Green cylinders below the sequence alignment illustrate the regions of the proteins that form the alpha helices of the HMG box. (b) Sequence alignment of the HMG domains of selected dual HMG box proteins including mitochondrial transcription factor A (TFAM); the Saccharomyces cerevisiae homologue of TFAM, ABF2 (*numbering here begins after the 42 or 35 amino acid residue mitochondrial localization sequence for TFAM or ABF2, respectively); the Drosophila melanogaster dorsal repressor DSP1, and HMGB1-4. Primary and secondary intercalating residues and conserved residues are indicated as in (a). The cysteine residues that have been shown to form an intramolecular disulfide bond in HMGB1 are shown in magenta. The green unbroken cylinders below the sequence alignment indicate the amino acids that form the helical structures comprising the HMG boxes of TFAM, when bound to DNA. The broken green cylinder represents the linker region of TFAM that forms an alpha helix upon binding to DNA.
Table 1.
Summary of features of select human HMG box proteins.
| Protein | # of HMG boxes | DNA specificity | Function | Known Interacting Proteins | Known HMG-box Postranslational Modificationsa | Referencec |
|---|---|---|---|---|---|---|
| HMGB1 | 2 | NSS | Chromatin binding architectural, DNA repair, DAMP | TLR2, TLR4, RAGE, Beclin1, Progesterone receptor, RAG1, p53, p73 b | K3a^, K12a, S35p, S39p, K43a, K44a, K55a, K82a^, S100p, Y109p, K114u, K157a, K157u, Y162p, S181p^ | [11, 53, 63, 71] |
| HMGB2 | 2 | NSS | Chromatin binding, DNA repair | SET, Oct1, RAG1, p53 b | Y16p, S35p, K55a, K114u, K139a, K139u, K141u, K157a, K157u, Y162p | [6, 10] |
| UBF | 6 | NSS | Ribosomal RNA transcription | TATA binding protein, RRN3, Pol I, Casein kinase 2 α 1 b | T117p, T201p, K352a, S412p, S449p | [83] |
| TFAM | 2 | SS/NSS | Mitochondrial DNA transcription and protection | TFB2M, TFB1M, POLRMT b | S55p, K62u, K76u, K118u, S193p, K186u, K190u, S193p, S195p | [8] |
| Lef1 | 1 | SS | Wnt Signalling, Pre B Cell and T Cell development | Runt related transcription factor 2, CREB binding protein, SMAD2, SMAD3, SMAD7, Caudal type homeobox, ALX, PIASY b | K324u, K347u, Y363p | [84] |
| Sry | 1 | SS | Male sex determination | p300, Imp-β, CaM* | Y127p | [85] |
| Sox2 | 1 | SS | Stem Cell differentiation | Oct4 b | K73a | [66, 86] |
| Sox4 | 1 | SS | Neurological development | β-catenin, sydecan-binding protein 1 | Y123p, Y126p | [87] |
| Sox5 | 1 | SS | Cell fate determination | SMAD1, SMAD5, SMAD7 | K595a | [88] |
| Sox7 | 1 | SS | Embryo development, Wnt signalling repression | SMAD5, SMAD7 b | S76p, Y112p | [88] |
| Sox9 | 1 | SS | Skeletal development, male sex determination | p300, Imp-β, CaM, CRM1 b | K141a | [85] |
| Sox17 | 1 | SS | Embryo development, Wnt signalling repression | β-catenin | None | [87] |
| PMS1 | 1 | NSS | DNA mismatch repair | MLH1 | Y620p | [21] |
| TCF3 | 1 | SS | Neuronal cell differentiation | CREB binding protein, p300 b | K394u, Y396p | [89] |
Postranslational modifications abbreviations are as follows: p-phosphorylation, a-acetylation, s-sumolation, m-methylation, u-ubiquitination. Human postranslational modifications on HMG-box regions of the proteins were identified using PhosphoSitePlus [90].
These postranslational modifications do not occur on the the HMG box.
Several additional proteins not listed have been identifed to interact with the HMG protein in this row.
Refererences indicate protein-protein interactions.
The ability of the HMG-box proteins to bend DNA is requisite for their functions as transcription factors, DNA chaperones, and DNA repair agents. Much of the current understanding of the bending mechanism comes from studies of single HMG boxes (reviewed in [10, 18, 25]). To summarize, HMGB proteins preferentially bind to the minor groove of DNA using the HMG-box domain, characterized by three alpha helices forming an ‘L’ shaped structure [26] (Figure 1, Figure 2a, b). The HMG-box severely bends and underwinds DNA, using electrostatic and hydrophobic interactions to widen the minor groove and induce a bend towards the major groove. Importantly, HMG-box residues that intercalate DNA aid in stabilizing the distorted DNA structure (reviewed in [19]). The HMG boxes of both sequence-specific and non-sequence-specific proteins typically contain a non-polar DNA intercalating residue in the 1° site at the N terminus of alpha helix 1 (in red) (Figure 1, Figure 2). Non-sequence-specific HMG boxes have an additional non-polar intercalating residue in the 2° site at the N terminus of alpha helix 2 (in red). A residue at the same position in the HMG box of the sequence-specific HMGB proteins forms base-specific hydrogen bonds (in cyan) (Figure 1, Figure 2a, b). The presence of N-terminal and/or C-terminal tails composed of disordered stretches of basic and/or acidic residues can enhance the DNA binding and bending ability of HMGB proteins [22, 27, 28].
Figure 2. Structures of high mobility group (HMG)-box-DNA complexes.

(a) Non-sequence-specific HMGD bound to an unmodified DNA decamer (PDB ID 1QRV) [33]. (b) Sequence-specific Sox4 bound to a 16 base pair DNA oligomer from the Lama 1 gene (PDB ID 3U2B) [41]. The structure is orientated as if it was superimposed onto the HMG box of HMGD in panel (a). (c) Dual HMG box Sex determining region Y-HMGB1 Box B chimera (Sry.B) bound to 16 base pair DNA oligomer containing the cognate Sry binding site (PDB ID 2GZK) [29]. SRY is in green and the HMGB1 box B is in pink. (d) Native dual HMG box mitochondrial transcription factor A (TFAM) bound to a 28 base pair segment of the human mitochondrial light strand promoter (PDB ID 3TMM) [31]. The numbering for TFAM is based on the residual protein after loss of the 42 amino acid residue long mitochondrial localization sequence, indicated by a *. The structure is oriented as if box A was superimposed onto the SRY HMG box from the SRY.B chimera in panel (c). The non-polar intercalating residues are shown in red, and polar residues at the 1° or 2° intercalation sites are shown in cyan.
Tandem HMG box proteins
The majority of the non-sequence-specific HMG-box proteins have tandem HMG boxes whose structures have been notoriously difficult to study. However, the structure of the Sex determining region Y-HMGB1 box B chimera (Sry.B) bound to DNA [29], and two structures of mitochondrial transcription factor A (TFAM) DNA complexes [30, 31] have provided insights into DNA recognition by tandem HMG-box proteins. The Sry.B chimera structure was the first glimpse at tandem HMG-box DNA recognition. In it, the HMG boxes adopt a head-to-head configuration on the DNA and the ten amino acid linker region tracks loosely along the DNA minor groove (Figure 2c). The Sry HMG-box interactions with DNA resemble those in the Sry-DNA complex [32]. By contrast, the interactions of the HMGB1 HMG box B with DNA resemble HMGD and NHP6A bound to unmodified DNA, because intercalating residues 1° Phe 97 and 2° Ile 116 both contribute to DNA bending [20, 33]. This differs from the complex of HMGB1 box A with cisplatin-treated DNA, which lacks the 1° intercalating residue [34]. Each HMG box individually bends the DNA by approximately 90°, but the overall bending is only 101.5°, and thus is not additive; this is due to the spacing between the 2° intercalation sites, which are out of phase with the helical repeat of the DNA helix (Figure 2c). The lack of direct interactions between the two HMG boxes [29] might be due to the construction of the chimera, which includes a hybrid linker between the HMG boxes. This informative view of a tandem HMG-box-DNA complex does not exclude the possibility that the linker will function differently and the HMG boxes will bind interdependently in a native context.
Unlike the independence of Sry.B HMG-box interactions with DNA, the HMG boxes in TFAM bind to DNA in a highly cooperative fashion. TFAM induces a large DNA bend of approximately 180° on short segments (22 bp [30] and 28 bp [31]) of mitochondrial light strand promoter (LSP) DNA (Figure 2d). Although previous solution spectroscopic studies revealed the large DNA bend induced by TFAM [28], the finding that both HMG box A and box B bind to the DNA was somewhat surprising (Figure 2d), because box B does not bind with any observable affinity, independent of the rest of the protein [35, 36]. By contrast, other HMG boxes can bind to DNA independently of the other domains of the proteins in which they reside [37]. Two distinctive properties of TFAM, the phased intercalation mechanism and the structure of the long linker, might explain these unique features of DNA recognition.
Analysis of the HMG box intercalation motifs provides insight into the contributions of each HMG box to the structure of the TFAM-DNA complex. As indicated earlier, non-sequence-specific HMG boxes typically have non-polar 1° and 2° intercalating residues, whereas the sequence-specific HMG boxes have a non-polar 1° intercalating residue and a polar residue at the 2° site (Figure 1, 2a, b). By this classification, box A of TFAM would be sequence-specific, because 1° Leu 16* (numbering of starred [*] residues considers residue 1 to be the first residue of TFAM that is after the 42 amino acid mitochondrial localization sequence) intercalates the DNA and 2° Thr 35* forms hydrogen bonds with the DNA (Figure 1, Figure 2d) [36]. However, in box B of TFAM the 1° intercalation site (Asn 121*) forms hydrogen bonds with the DNA, and the 2° site (Leu 140*) intercalates the DNA. Therefore, TFAM overall does not conform to either HMG-box classification and is unique among HMGB proteins in its intercalation motif. The term ‘inverted motif’ was used to describe the DNA sequence of the motifs found at the major intercalation sites but can also be applied to the inverted tail-to-tail configuration of the HMG boxes that is requisite for creating the 180° bend in the DNA. This configuration generates a symmetry within the complex such that each HMG box bends the DNA by approximately 90° at positions along the DNA that are in phase with the DNA helical screw, and thus are additive [30]. The overall 180° bend is also supported by the formation of an additional alpha helix in the long (25 residue) linker region (Figure 1b). This basic helix binds to the minor groove of the DNA and neutralizes the negatively charged and compressed DNA phosphate backbone [30, 31]. The linker facilitates the interaction of box B with the DNA and might promote the internal cooperativity between box A and box B in the final structure. Thus, the structures of the Sry.B-DNA and the TFAM-DNA complexes revealed two key mechanisms for DNA recognition by tandem HMG boxes: the former showed how HMG boxes can bind DNA independently, and the latter revealed how a unique linker and an inverted motif can facilitate the cooperative assembly of tandem HMG boxes on DNA.
New mechanisms for specificity of DNA recognition in HMGB proteins
The thermodynamic basis of HMGB protein recognition of DNA has been well described (reviewed in [18, 25, 38]), and the determinants of sequence specificity for most transcription factors are generally guided by a simple model of base sequence independence [39]. However, recent studies of TFAM and Sry-related HMG-box (Sox) proteins have revealed more complex mechanisms for modulating sequence specificity. Although Sox proteins preferentially bind to a core sequence element, TTGT, high throughput protein binding microarray experiments found that secondary binding motifs, which differ among Sox family members, are governed by nucleotide ‘position interdependence’ [40]. For example, in the sequence X6YTTGT11, if nucleotide ‘Y’ is a T, Sox4 will have high affinity when nucleotide ‘X’ is a C, whereas if position ‘Y’ is an A, then A is preferred in the ‘X’ position [41]. An illustration of secondary motif recognition by position interdependence can be found by comparing the crystal structures of Sox4 to Sox17 bound to the same DNA sequence (Figure 2b, Figure 3a) [41, 42]. In Figure 3b, both proteins have similar interactions with the TTGT core sequence (shown in black), but subtle structural differences exist at the ‘XY’ secondary recognition site of the protein-DNA interface, as seen in the altered protein-DNA contacts shown for Sox17 in green [42, 43]. Specifically, Arg 74 and Asn 86 of the two proteins make different contacts with the ‘X’ and ‘Y’ nucleotides, respectively (Figure 3b). Differences in DNA recognition extend outside of the XYTTGT motif, where His 85 interacts with G12 and in Sox17 His 85 contacts A13 (Figure 3a). The molecular origins of this rearrangement in contacts are unclear, but differential sequence recognition through position interdependence might be key to distinguishing between secondary DNA binding motifs in the Sox protein family [41].
Figure 3. Specificity of DNA recognition.

(a) Sox17-DNA complex. The primary intercalating residue is shown in red, and the residue at the secondary intercalating site is shown in cyan. The bases ‘XY’, for which nucleotide interdependence has been observed, are shown in black, and the primary recognition site sequence is colored orange. (b) Two dimensional representation of the amino acids that contact the DNA in the crystal structures of Sox4 (PDB ID 3U2B) and Sox17 (PDB ID 3F27). The primary intercalating residue is Met 67 (red), and Sox17 amino acids that differ in DNA contacts from the Sox4 structure are indicated in green. Amino acid numbering is based on the Sox4 structure. Hydrogen bonds to the bases are indicated with blue lines and van der Waal’s contacts are shown as broken beige lines. The figure was generated using the program NUCPLOT [82]. (c) Sox2 (green) and the POU domains of Oct1 [51] (PDB ID 1GTO). The protein-protein interaction interface is circled in red. The DNA is colored as in (a).
TFAM is unique among HMGB proteins in that it both binds promoter DNA sequence-specifically and genomic DNA non-specifically [28, 31]. TFAM coats the entire mitochondrial genome and protects the DNA from oxidative stress by compacting the DNA into nucleoids [44]. In addition, TFAM specifically recognizes three promoter sequences in the mitochondrial genome: the LSP, the heavy strand promoter 1 (HSP1), and the heavy strand promoter 2 (HSP2), which bear little sequence similarity. Despite the observations that the promoter regions have different abilities to drive transcription [45], TFAM does not have significantly greater affinity for LSP DNA than for HSP1 or non-specific DNA [28], as the KD values are all in the 5–10 nM range. Spectroscopic studies have shown that TFAM bends 25 bp LSP and HSP1 promoter DNA with an estimated overall change in end-to-end distance of ~23 Å, and this distance for non-promoter DNA is ~15 Å, which gives estimated bend angles of 86°–180° and 63°–120°, respectively, depending on whether the DNA bend is modeled as a kink or a smooth bend [28]. Furthermore, deletion of the C-terminal tail of TFAM results in loss of promoter specificity [46] and a decrease in the DNA bend angle similar to that observed for TFAM bound to non-sequence-specific DNA [28]. Thus, binding affinity alone is not the driving force for determining the ability of TFAM to form a ‘specific complex’ capable of being footprinted on DNA.
The crystal structures of TFAM sequence-specifically bound to promoter DNA illustrate how a ‘specific complex’ can form when the correct base sequence is present [30, 31]. Given that the individual HMG boxes do not by themselves have sequence specificity, the intricate network of interactions between the two HMG boxes, the linker, and the DNA must be responsible for generating site-specificity. Figure 2d shows where Thr 35* at the 2° position in box A forms hydrogen bonds with a DNA base, and Asn 121* at the 1° position in box B forms hydrogen bonds with two adjacent thymines. The 1° intercalating residue in box A, the 2° intercalating residue in box B, and the alpha helical linker would be expected to stabilize the distorted DNA structure. Thus, the sequence-specific hydrogen bond contacts potentially ‘lock in’ a cooperatively formed highly bent conformation of the DNA, which is a hallmark of a ‘specific’ TFAM-DNA complex [30, 31]. How the C-terminal tail of TFAM, which promotes transcription [32] and bending of specific promoter DNA [28], also stabilizes the formation of this specific complex is not completely clear from the structures. However, if these special protein-DNA contacts do not form, such as for the incorrect DNA sequence, then TFAM can recognize a wide range of DNA sequences with comparable affinity and the conformation of the DNA adopts a less bent form [28]. Indeed, recent single molecule analysis of TFAM binding to much longer DNA sequences revealed that sliding, DNA melting and DNA compaction accompany the non-sequence specific binding mode of TFAM, and this also did not require the C-terminal tail [47]. Structural studies of TFAM with non-promoter DNA and further mutagenesis studies will provide needed insights on this mechanism.
Sequence positional interdependence could explain subtle differences in the DNA recognition, bending, and binding affinities of Sox and TFAM HMGB proteins for different DNA sequences. Interestingly, the Asn residues that occupy the 2° intercalation site in several Sox proteins are key sequence interdependence regulators (Figure 2b, Figure 3), which makes it tempting to speculate that Thr 35* of TFAM could play a similar role in regulating the sequence specificity through a sequence-positional-interdependence mechanism. Finally, the DNA recognition mechanisms discussed here also are likely to be influenced by other proteins.
HMG-box interactions with other proteins and chromatin
HMG box proteins are involved in a myriad of protein-protein interactions that regulate transcription, chromatin dynamics, immune response, development, and other cellular functions. Both the sequence-specific (Sox) and non-sequence-specific (HMGB1) HMG box proteins can facilitate the interactions of other transcription factors with DNA. For instance, several Sox proteins partner with Oct family members to function as key regulators of cell fate determination (Table 1) [48, 49]. The classical mechanism for co-regulation proposes that the HMG-box protein bends promoter DNA to bring other transcription factors in closer proximity [50]. However, structural analysis of the Oct1/Sox2/DNA complex revealed different molecular determinants of HMG box DNA recognition in conjunction with an Oct protein (Figure 3c) [51]. This crystal structure was obtained using the same Sox primary DNA recognition motif, CTTTTGT, as was used for Sox4 and Sox17 (Figure 3b, c) [41, 42]. The POU domain of the Oct protein class binds to a ATGCAAAT motif, and the relative spacing from the TTGT motif and the secondary DNA motifs determines the specificity of the Sox/Oct pair for DNA. Sox and Oct bind to DNA at adjacent sites and have a small protein-protein interaction interface, which explains how spacing could influence their cooperation. However, the Sox interactions with DNA are similar in the presence and absence of Oct proteins (Figure 3a, c). Further insights into the mechanism of this cooperation came from NMR studies of the Sox2 and Oct1 interactions with DNA, which showed that Sox2 binding to the adjacent DNA site stabilized the interaction of the POUS domain of Oct1 through combinatorial control involving direct protein-protein interactions on the DNA [51, 52]. Thus, a direct protein-protein interaction model operates in the Sox-Oct system for HMG-box mediated transcriptional enhancement in cells.
By contrast, HMGB1 co-regulates a wide variety of sequence-specific transcription factors by means that include the formation of ternary complexes with transcription factors and DNA, and ‘hit and run’ mechanisms whereby the HMG box pre-bends DNA to favor the binding and/or interaction of transcription factors with DNA or with each other (reviewed in [10]). Specific protein-protein interactions between the HMG boxes of HMGB1 and the C-terminal extension domain of progesterone receptor (PR) have been found to enhance PR promoter DNA binding [53]. Interestingly, this activity did not require the DNA bending capacity of HMGB1. However, the enhanced transcriptional activity of PR in cells did require the ability of HMGB1 to bend DNA. These results suggested a two phase synergistic recruitment model for HMGB1 activity, whereby the interaction of the two proteins in the vicinity of the specific promoter can increase the local concentration of HMGB1 for its subsequent activities in nucleosome remodeling [53].
HMG boxes and HMGB protein involvement in chromatin remodeling is widespread but is not well understood at the molecular level. Models for the action of HMGB proteins within the chromatin context focus on their ability to assist the activity of chromatin modifiers presumably by loosening the histone-DNA contacts within the nucleosome [10, 54] (Figure 4). HMGB1 can assist a variety of ATP-dependent chromatin remodeling machineries and histone chaperones in the process of ‘nucleosome sliding’, by a mechanism that involves HMGB1 binding and bending nucleosomal DNA [55]. By contrast, the nucleosome remodeling complexes, switching defective or sucrose nonfermenting (SWI/SNF in S. cerevisiae) or Brg1-associated factor (BAF in metazoans) [56], and histone chaperone FACT (facilitates chromatin transcription) complex, incorporate HMGB proteins as subunits within much larger remodeling machinery [54]. The complexity of subunits and tissue-specific distributions of the BAF family of remodelers is staggering, and the role of the HMG-box containing subunit is not clear [56]. However, the HMG box subunit (BAF57), which was not essential for the human BAF complex to function in remodeling and activation of many genes, was intriguingly necessary for the recruitment of calcium-calmodulin and the stimulation of specific TLR-4 gene expression in macrophages [57].
Figure 4. High mobility group (HMG) boxes in nucleosome remodeling.
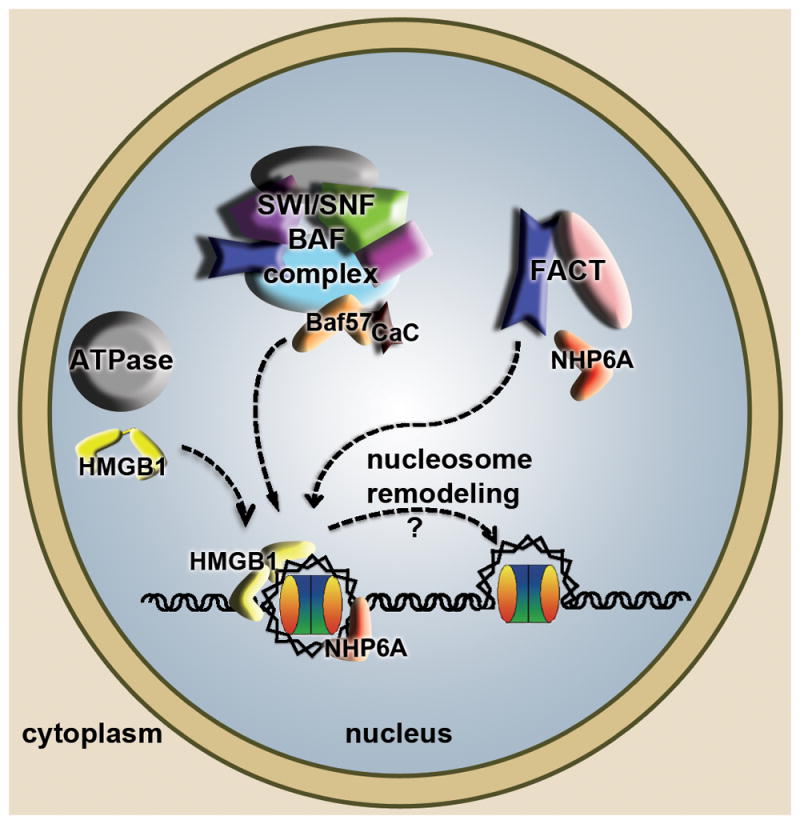
The diagram summarizes and combines findings from S. cerevisiae and other eukaryotes. HMGB proteins in metazoans (yellow) and NHP6A in S. cerevisiae (red), in their individual cellular contexts can bind to nucleosomal DNA and assist ATP-dependent chromatin remodelers (grey) in loosening DNA and providing access to other DNA dependent machineries. Related SWI/SNF (in S. cerevisiae) and BAF (in metazoans) complexes harbor an HMG box containing subunit BAF57 (orange) that promotes nucleosome remodeling, in some cases requiring binding of calcium calmodulin (CaC in black). The histone chaperone FACT has three subunits in S. cerevisiae (Spt16, Pob3, NHP6A), of which NHP6A promotes the association of the FACT complex with nucleosomes to facilitate remodeling.
FACT facilitates Pol II transcription elongation through nucleosomes and is important for eviction and redeposition of histones onto DNA [54]. In metazoans, FACT is composed of the Spt16 and SSRP subunits, of which SSRP has an HMG-box at its C terminus (Figure 1). The equivalent subunit to SSRP in S. cerevisiae, called Pob3, lacks the HMG-box motif, but the DNA binding and bending function is provided by the HMGB protein NHP6A (Figure 4). NHP6A promotes the association of Spt16/Pob3 with nucleosomes, which then become ‘remodeled’ into a looser form that has increased accessibility of the nucleosomal DNA to nucleases [58]. Biophysical analyses of metazoan FACT revealed that nucleosome remodeling occurs through multiple synergistic binding events [59]. Moreover, additional regulation of HMG-box activity has been observed within the larger metazoan protein, SSRP1 (in Drosophila melanogaster), in which the HMG box intramolecularly associates with an acidic intrinsically disordered region (AID), but can still interact with nucleosomal DNA [60]. Interestingly, phosphorylation of the AID region better competes with the HMG box and reduces the association of the HMG box with DNA, illustrating how this post-translational modification could influence the activity of FACT during embryogenesis [60].
The multiplicity of interactions of HMGB proteins with transcription factors, nucleosomes, and chromatin remodeling machinery is quite remarkable. For these functions, the DNA binding activity of the HMG box is almost certainly functionally relevant to HMGB protein activity within the nucleus.
Modulation of HMG-box cellular location through post-translational modification
Post-translational modification of DNA binding proteins has long been recognized as an important modulator of DNA binding activity, and the HMG box family of proteins is no exception [10]. Numerous posttranslational modifications have been identified in HMGB proteins (Table 1), including acetylation, phosphorylation, methylation, and oxidation [10]. However, the functional consequences of these modifications are only recently becoming understood. Here, we focus on the best understood posttranslational modifications of HMGB1.
HMGB1 is modified on several residues, with varying consequences on DNA binding and bending ability. Acetylation of Lys 3 in bovine HMGB1 increased the affinity for cis-platinated or UV damaged DNA, but decreased the DNA bending ability [61], and acetylation of Lys 82 in the linker region also influenced DNA bending [62]. Interestingly, phosphorylation of HMGB1 at Ser 181 by cyclin-dependent kinase 5 requires prior acetylation at Lys 3 [63]. This phosphorylation decreases DNA end-joining activity, which is normally promoted by Lys 3 acetylation alone, but had no effect on the HMGB1 DNA binding affinity [63]. Other modifications to HMGB1 have been identified (Table 1), and certainly other cases where modifications regulate HMGB1 function in a combinatorial manner remain to be discovered.
Perhaps most surprising is how HMGB1 post-translational modifications influence HMGB1 cellular location. Whereas acetylation of HMGB1 at Lys 3 increases DNA binding affinity, general lysine hyperacetylation and specific methylation of Lys 43 decrease its DNA binding activity and correspond with a change in its cellular location from the nucleus to the cytoplasm [64, 65]. Acetylation of Lys 75 (K73 in humans) in Sox 2 was also shown to promote transport from the nucleus to the cytoplasm (Figure 5), suggesting that acetylation of HMG boxes might serve as a transport signal for this entire class of proteins [66].
Figure 5. High mobility group box 1 (HMGB1) interactions in the cell.

The diagram summarizes and combines findings from studies of post-translational modifications and cysteine oxidation of HMGB1 that correlate with different functions and localization of HMGB1. The DNA-bound nuclear form of HMGB1 (yellow) is generally reduced, but phosphorylation (green ‘P’), methylation (cyan ‘Me’) and acetylation (blue ‘Ac’) can regulate DNA (in blue) binding activity. The cytoplasmic and extracellular forms of HMGB1 are generally more highly acetylated and oxidized than the nuclear form. Cytoplasmic HMGB1 can compete with Bcl-2 (grey) for binding to Beclin1 (blue), which promotes autophagy. Release of HMGB1 to the extracellular space, where it can act on receptors of the same as well as neighboring cells, results in different responses, depending on the receptor; receptor for advanced glyclation end products (RAGE, purple) promotes autophagy, whereas toll-like receptor 4 (TLR4, green) promotes cytokine release, and oxidized forms of HMBG1 can promote apoptosis. Reduced Cys 22, Cys 44 and Cys 106 are indicated by a black ‘C’. The oxidized Cys 22 and Cys 44 form a disulfide bond indicated by red ‘C-C’ and the oxidized C 106 sulfinic acid is represented by a black ‘C’ that has a red star outline. The specific placement of the marks is not meant to indicate where they are located in HMGB1.
In addition to enzyme-catalyzed modifications, HMGB1 function and cellular location are also modulated by the redox state of three conserved cysteine residues (Figure 1, Figure 5). HMGB1 contains two cysteines in box A, Cys 23 and Cys 45, and a third cysteine, Cys 106, in box B. Mutagenesis studies demonstrated that Cys 106 is a regulator of cellular localization [67]. A C106S substitution resulted in HMGB1 localization to the cytoplasm, whereas wild-type HMGB1 and mutants with the C23S and C45S substitutions localized to the nucleus, suggesting that oxidation of C106 regulates the nomadic behavior of HMGB1. Cys 23 and Cys 45 readily oxidize to form a disulfide bond that alters the structure of the HMG box [68] and results in a 10-fold decrease in the affinity (0.70 nM vs. 7.58 nM) for cis-platinated DNA [69]. This weaker binding affinity for damaged DNA has a different structural basis than oxidation of the HMGD primary intercalating methionine, which not only decreased binding affinity but altered the protein-induced DNA bend [70]. Interestingly, the Cys 23 and Cys 45 residues are conserved in HMGB1, HMGB2, and the Drosophila protein DSP1 (Figure 1b), and so it will not be surprising if these and potentially other HMG box proteins are regulated in a similar fashion.
Post-translational modifications and oxidation regulate HMGB translocation out of the cell, where it serves as a cytokine regulator of the immune response, tissue regeneration, chemotaxis, and inflammation [71]. Hyperacetylation of HMGB1 lysine residues leads to extracellular translocation of HMGB1 [72]. The hyperacetylation of HMGB1 is thought to be due to decreased histone deacetylase activity [65]. Active transport of HMGB1 also occurs in cells undergoing oxidative stress [73] or apoptosis [15], whereas necrosis allows HMGB1 to diffuse to the extra cellular space in a passive manner [74, 75].
Once in the extracellular space, the function of HMGB1 as a signaling molecule appears to be largely regulated by the oxidative state of the protein. The presence of extracellular reduced Cys 106 HMGB1 promotes interaction with the receptor for advanced glycation end products (RAGE) to induce autophagy, whereas oxidized HMGB1 induces apoptosis though a caspase-3/9 mitochondrial mediated mechanism [76]. Interestingly, cytoplasmic oxidized HMGB1 with a Cys 23/Cys 45 disulfide bond promotes autophagy by competing with the anti-apoptotic protein Bcl-2 for binding to the autophagy regulator Beclin-1 (Figure 5). Moreover, once outside of the cell, HMGB1 can interact with RAGE and Toll-like receptor 2 (TLR2) and TLR4, with the latter requiring reduction of Cys 106 [77, 78], thus eliciting a myriad of signals for immune response and inflammation. As these mechanisms of cytoplasmic and extracellular HMGB1 regulation by post-translational modification and oxidation are slowly being uncovered, it is clear that these are not unique to HMGB1. TFAM has recently been identified as a cytokine protein that interacts with RAGE and TLR9 [79]. Furthermore, several other members of the HMG-box family have the potential to function as immune modulators, and it will be interesting to see whether the redox state of these proteins also promotes changes in cellular distribution and or cytokine activity.
It is clear that post-translational modifications and redox state can drastically influence the function of HMGB proteins. Although a general mechanism of how posttranslational modifications alter HMG box protein function is emerging, the molecular mechanisms are not known.
Concluding remarks
The HMG box is a simple yet remarkably versatile DNA- and protein-binding module. Much is now known about the biophysical and structural aspects of single- and di-HMG box protein recognition of DNA and the mechanisms of DNA bending. Bending DNA not only involves DNA intercalation, but disorder-to-order transitions in linkers and tails that neutralize the negative charge of the distorted DNA. Specificity of DNA recognition is achieved not only through sequence-specific hydrogen bonding but also through sequence interdependence of DNA bases, as seen for the Sox family of HMG-box proteins. Future work investigating sequence-specific HMG box interactions with other secondary recognition sites should reveal how this occurs for other HMG-box family members. Another future challenge is to attain this level of insight into the molecular mechanisms of DNA recognition of the native tandem HMG box proteins, such as the HMGB1-4 and UBF. Although numerous interactions between HMG-box proteins and other proteins have been identified, there is still very little known about the nature of the interactions because they tend to be weak or have only been investigated in cells. Furthermore, how the HMG box functions in chromatin remodeling is still a mystery despite the initial discovery of HMGB1 as a chromosomal protein several decades ago. Similarly, an understanding of the effects of HMG-box protein posttranslational modifications on their interactions with DNA and other proteins is only beginning to emerge. With further proteomic analysis, the number of posttranslational modifications identified in HMG box proteins will continue to grow, as will the need for a better understanding of the molecular basis by which they alter HMG box functions within different cellular compartments and regulate translocation among them. The amazing range of activities of the HMG box proteins has truly earned them the title of the utility players of the cell.
Acknowledgments
Due to limitations on the number of references, we were unable to directly cite all of the relevant literature, and many relevant references can be found within the cited references. We are grateful to Sarah Roemer and Wallace Liu for their suggestions for the manuscript. We acknowledge support from the MDA and AHA to MEAC.
Literature Cited
- 1.Goodwin GH, et al. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem. 1973;38:14–19. doi: 10.1111/j.1432-1033.1973.tb03026.x. [DOI] [PubMed] [Google Scholar]
- 2.Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem Sci. 2001;26:152–153. doi: 10.1016/s0968-0004(00)01777-1. [DOI] [PubMed] [Google Scholar]
- 3.Reeves R. Nuclear functions of the HMG proteins. Biochim Biophys Acta. 2010;1799:3–14. doi: 10.1016/j.bbagrm.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sgarra R, et al. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim Biophys Acta. 2010;1799:37–47. doi: 10.1016/j.bbagrm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Gerlitz G. HMGNs, DNA repair and cancer. Biochim Biophys Acta. 2010;1799:80–85. doi: 10.1016/j.bbagrm.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas JO, Travers AA. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem Sci. 2001;26:167–174. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 7.Asin-Cayuela J, Gustafsson CM. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem Sci. 2007;32:111–117. doi: 10.1016/j.tibs.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Bonawitz ND, et al. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 9.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Thomas JO. HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans. 2001;29:395–401. doi: 10.1042/bst0290395. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 13.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang R, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soullier S, et al. Diversification pattern of the HMG and SOX family members during evolution. J Mol Evol. 1999;48:517–527. doi: 10.1007/pl00006495. [DOI] [PubMed] [Google Scholar]
- 17.Landsman D, Bustin M. A signature for the HMG-1 box DNA-binding proteins. Bioessays. 1993;15:539–546. doi: 10.1002/bies.950150807. [DOI] [PubMed] [Google Scholar]
- 18.Murphy FVt, Churchill ME. Nonsequence-specific DNA recognition: a structural perspective. Structure. 2000;8:R83–89. doi: 10.1016/s0969-2126(00)00126-x. [DOI] [PubMed] [Google Scholar]
- 19.Churchill ME, et al. Structural analysis of HMGD-DNA complexes reveals influence of intercalation on sequence selectivity and DNA bending. J Mol Biol. 2010;403:88–102. doi: 10.1016/j.jmb.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masse JE, et al. The S. cerevisiae architectural HMGB protein NHP6A complexed with DNA: DNA and protein conformational changes upon binding. J Mol Biol. 2002;323:263–284. doi: 10.1016/s0022-2836(02)00938-5. [DOI] [PubMed] [Google Scholar]
- 21.Leung WK, et al. Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J Biol Chem. 2000;275:15728–15732. doi: 10.1074/jbc.M908768199. [DOI] [PubMed] [Google Scholar]
- 22.Park S, Lippard SJ. Binding Interaction of HMGB4 with Cisplatin-Modified DNA. Biochemistry. 2012 doi: 10.1021/bi300649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol. 1988;8:3496–3509. doi: 10.1128/mcb.8.8.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alam TI, et al. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003;31:1640–1645. doi: 10.1093/nar/gkg251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Privalov PL, et al. The cost of DNA bending. Trends Biochem Sci. 2009;34:464–470. doi: 10.1016/j.tibs.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Weir HM, et al. Structure of the HMG box motif in the B-domain of HMG1. EMBO J. 1993;12:1311–1319. doi: 10.1002/j.1460-2075.1993.tb05776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Churchill ME, et al. Interactions of high mobility group box proteins with DNA and chromatin. Methods Enzymol. 1999;304:99–133. doi: 10.1016/s0076-6879(99)04009-4. [DOI] [PubMed] [Google Scholar]
- 28.Malarkey CS, et al. Transcriptional activation by mitochondrial transcription factor A involves preferential distortion of promoter DNA. Nucleic Acids Res. 2012;40:614–624. doi: 10.1093/nar/gkr787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stott K, et al. Structure of a complex of tandem HMG boxes and DNA. J Mol Biol. 2006;360:90–104. doi: 10.1016/j.jmb.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 30.Rubio-Cosials A, et al. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nat Struct Mol Biol. 2011;18:1281–1289. doi: 10.1038/nsmb.2160. [DOI] [PubMed] [Google Scholar]
- 31.Ngo HB, et al. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat Struct Mol Biol. 2011;18:1290–1296. doi: 10.1038/nsmb.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werner MH, et al. Molecular basis of human 46X,Y sex reversal revealed from the three-dimensional solution structure of the human SRY-DNA complex. Cell. 1995;81:705–714. doi: 10.1016/0092-8674(95)90532-4. [DOI] [PubMed] [Google Scholar]
- 33.Murphy FVt, et al. The structure of a chromosomal high mobility group protein-DNA complex reveals sequence-neutral mechanisms important for non-sequence-specific DNA recognition. Embo J. 1999;18:6610–6618. doi: 10.1093/emboj/18.23.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohndorf UM, et al. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 35.Wong TS, et al. Biophysical characterizations of human mitochondrial transcription factor A and its binding to tumor suppressor p53. Nucleic Acids Res. 2009;37:6765–6783. doi: 10.1093/nar/gkp750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gangelhoff TA, et al. Structural analysis and DNA binding of the HMG domains of the human mitochondrial transcription factor A. Nucleic Acids Res. 2009;37:3153–3164. doi: 10.1093/nar/gkp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller S, et al. Thermodynamics of HMGB1 interaction with duplex DNA. Biochemistry. 2001;40:10254–10261. doi: 10.1021/bi0100900. [DOI] [PubMed] [Google Scholar]
- 38.Rohs R, et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Stormo GD. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat Biotechnol. 2011;29:480–483. doi: 10.1038/nbt.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badis G, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jauch R, et al. Crystal structure of the Sox4 HMG/DNA complex suggests a mechanism for the positional interdependence in DNA recognition. Biochem J. 2012;443:39–47. doi: 10.1042/BJ20111768. [DOI] [PubMed] [Google Scholar]
- 42.Palasingam P, et al. The structure of Sox17 bound to DNA reveals a conserved bending topology but selective protein interaction platforms. J Mol Biol. 2009;388:619–630. doi: 10.1016/j.jmb.2009.03.055. [DOI] [PubMed] [Google Scholar]
- 43.Jauch R, et al. Conversion of Sox17 into a pluripotency reprogramming factor by reengineering its association with Oct4 on DNA. Stem Cells. 2011;29:940–951. doi: 10.1002/stem.639. [DOI] [PubMed] [Google Scholar]
- 44.Kaufman BA, et al. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Litonin D, et al. Human mitochondrial transcription revisited: only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J Biol Chem. 2010;285:18129–18133. doi: 10.1074/jbc.C110.128918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dairaghi DJ, et al. Addition of a 29 residue carboxyl-terminal tail converts a simple HMG box-containing protein into a transcriptional activator. J Mol Biol. 1995;249:11–28. doi: 10.1006/jmbi.1995.9889. [DOI] [PubMed] [Google Scholar]
- 47.Farge G, et al. Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat Commun. 2012;3:1013. doi: 10.1038/ncomms2001. [DOI] [PubMed] [Google Scholar]
- 48.Kondoh H, Kamachi Y. SOX-partner code for cell specification: Regulatory target selection and underlying molecular mechanisms. Int J Biochem Cell Biol. 2010;42:391–399. doi: 10.1016/j.biocel.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 49.Ambrosetti DC, et al. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol Cell Biol. 1997;17:6321–6329. doi: 10.1128/mcb.17.11.6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grosschedl R, et al. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 51.Remenyi A, et al. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev. 2003;17:2048–2059. doi: 10.1101/gad.269303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams DC, Jr, et al. Molecular basis for synergistic transcriptional activation by Oct1 and Sox2 revealed from the solution structure of the 42-kDa Oct1.Sox2.Hoxb1-DNA ternary transcription factor complex. J Biol Chem. 2004;279:1449–1457. doi: 10.1074/jbc.M309790200. [DOI] [PubMed] [Google Scholar]
- 53.Roemer SC, et al. Mechanism of high-mobility group protein B enhancement of progesterone receptor sequence-specific DNA binding. Nucleic Acids Res. 2008;36:3655–3666. doi: 10.1093/nar/gkn249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stillman DJ. Nhp6: a small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim Biophys Acta. 2010;1799:175–180. doi: 10.1016/j.bbagrm.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langst G, Becker PB. Nucleosome remodeling: one mechanism, many phenomena? Biochim Biophys Acta. 2004;1677:58–63. doi: 10.1016/j.bbaexp.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 56.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai D, et al. Induction of TLR4-target genes entails calcium/calmodulin-dependent regulation of chromatin remodeling. Proc Natl Acad Sci U S A. 2009;106:1169–1174. doi: 10.1073/pnas.0811274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xin H, et al. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol Cell. 2009;35:365–376. doi: 10.1016/j.molcel.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winkler DD, Luger K. The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J Biol Chem. 2011;286:18369–18374. doi: 10.1074/jbc.R110.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsunaka Y, et al. Phosphorylated intrinsically disordered region of FACT masks its nucleosomal DNA binding elements. J Biol Chem. 2009;284:24610–24621. doi: 10.1074/jbc.M109.001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ugrinova I, et al. In vivo acetylation of HMG1 protein enhances its binding affinity to distorted DNA structures. Biochemistry. 2001;40:14655–14660. doi: 10.1021/bi0113364. [DOI] [PubMed] [Google Scholar]
- 62.Elenkov I, et al. The DNA binding and bending activities of truncated tail-less HMGB1 protein are differentially affected by Lys-2 and Lys-81 residues and their acetylation. Int J Biol Sci. 2011;7:691–699. doi: 10.7150/ijbs.7.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ugrinova I, et al. Cyclin-dependent kinase 5 phosphorylates mammalian HMGB1 protein only if acetylated. J Biochem. 2011;149:563–568. doi: 10.1093/jb/mvr005. [DOI] [PubMed] [Google Scholar]
- 64.Ito I, et al. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 2007;282:16336–16344. doi: 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- 65.Bonaldi T, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baltus GA, et al. Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells. 2009;27:2175–2184. doi: 10.1002/stem.168. [DOI] [PubMed] [Google Scholar]
- 67.Hoppe G, et al. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 68.Sahu D, et al. Redox properties of the A-domain of the HMGB1 protein. FEBS Lett. 2008;582:3973–3978. doi: 10.1016/j.febslet.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park S, Lippard SJ. Redox state-dependent interaction of HMGB1 and cisplatin-modified DNA. Biochemistry. 2011;50:2567–2574. doi: 10.1021/bi2000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dow LK, et al. Oxidation of a critical methionine modulates DNA binding of the Drosophila melanogaster high mobility group protein, HMG-D. FEBS Lett. 1997;414:514–520. doi: 10.1016/s0014-5793(97)01059-4. [DOI] [PubMed] [Google Scholar]
- 71.Tang D, et al. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Evankovich J, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang D, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rovere-Querini P, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scaffidi P, et al. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 76.Tang D, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park JS, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 78.Yang H, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Julian MW, et al. Mitochondrial transcription factor a serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol. 2012;189:433–443. doi: 10.4049/jimmunol.1101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koopman P, et al. Expression of a candidate sex-determining gene during mouse testis differentiation. Nature. 1990;348:450–452. doi: 10.1038/348450a0. [DOI] [PubMed] [Google Scholar]
- 81.Lefebvre V, et al. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int J Biochem Cell Biol. 2007;39:2195–2214. doi: 10.1016/j.biocel.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luscombe NM, et al. NUCPLOT: a program to generate schematic diagrams of protein-nucleic acid interactions. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russell J, Zomerdijk JC. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci. 2005;30:87–96. doi: 10.1016/j.tibs.2004.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arce L, et al. Diversity of LEF/TCF action in development and disease. Oncogene. 2006;25:7492–7504. doi: 10.1038/sj.onc.1210056. [DOI] [PubMed] [Google Scholar]
- 85.Sim H, et al. Boys, girls and shuttling of SRY and SOX9. Trends Endocrinol Metab. 2008;19:213–222. doi: 10.1016/j.tem.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 86.Yuan H, et al. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995;9:2635–2645. doi: 10.1101/gad.9.21.2635. [DOI] [PubMed] [Google Scholar]
- 87.Sinner D, et al. Sox17 and Sox4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol Cell Biol. 2007;27:7802–7815. doi: 10.1128/MCB.02179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colland F, et al. Functional proteomics mapping of a human signaling pathway. Genome Res. 2004;14:1324–1332. doi: 10.1101/gr.2334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hyndman BD, et al. E2A proteins enhance the histone acetyltransferase activity of the transcriptional co-activators CBP and p300. Biochim Biophys Acta. 2012;1819:446–453. doi: 10.1016/j.bbagrm.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 90.Hornbeck PV, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]