

Abstract
Recent studies have highlighted the importance of an inhibitory phosphorylation site, Ser485/491, on the α-subunit of AMP-activated protein kinase (AMPK); however, little is known about the regulation of this site in liver and skeletal muscle. We examined whether the inhibitory effects of insulin on AMPK activity may be mediated through the phosphorylation of this inhibitory Ser485/491 site in hepatocytes, myotubes and incubated skeletal muscle. HepG2 and C2C12 cells were stimulated with or without insulin for 15-min. Similarly, rat extensor digitorum longus (EDL) muscles were treated +/− insulin for 10-min. Insulin significantly increased Ser485/491 p-AMPK under all conditions, resulting in a subsequent reduction in AMPK activity, ranging from 40% to 70%, despite no change in p-AMPK Thr172. Akt inhibition both attenuated the increase in Ser485/491 p-AMPK caused by insulin, and prevented the decrease in AMPK activity. Similarly, the growth factor IGF-1 stimulated Ser485/491 AMPK phosphorylation, and this too was blunted by inhibition of Akt. Inhibition of the mTOR pathway with rapamycin, however, had no effect on insulin-stimulated Ser485/491 p-AMPK. These data suggest that insulin and IGF-1 diminish AMPK activity in hepatocytes and muscle, most likely through Akt activation and the inhibitory phosphorylation of Ser485/491 on its α-subunit.
Keywords: Insulin, Growth factors, AMPK, PKB, Ser485/491
Introduction
Activation of the fuel-sensing enzyme AMP-activated protein kinase (AMPK)1 results in up-regulation of processes involved in energy generation, such as glucose transport and fatty acid oxidation. Importantly, in addition to this classical role, it is now clear that AMPK activation leads to a host of other benefits, including inhibition of lipid and protein synthesis, inflammation, ER and oxidative stress and increased mitochondrial biogenesis. Furthermore, it exerts these effects both by acute actions on certain molecules and by genetically regulating the expression of others [1,2].
Whereas there is ample evidence regarding physiological (starvation and exercise) and pharmacological (biguanides, thiazolidinediones, salicylate) activation of AMPK in skeletal muscle and liver [2,3], the mechanisms responsible for its physiological down-regulation in these tissues are less well understood. This is particularly relevant as AMPK activity is diminished in obese hyperinsulinemic rodents and humans (compared to their healthy counterparts) [4–6], and its inhibition is an early event in the development of insulin resistance in response to nutrient excess [7–9].
Insulin, in addition to other anabolic stimuli, has been reported to diminish AMPK activity, as assessed by the SAMS peptide assay, in liver and muscle [10,11]. Although Thr172 of the α-subunit is regarded as the main phosphorylation/activation site on AMPK, changes in AMPK activity are often observed in the absence of altered Thr172 phosphorylation. As such, AMPK activity may be impacted by one of several other phosphorylation sites with less defined functions [12]. For instance, the reduction in AMPK activity caused by anabolic signals can be due, at least in part, to the phosphorylation of the inhibitory Ser485/491 site on α1/α2, in a variety of tissues, including cardiomyocytes/heart [13,14], adipocytes [15], and vascular smooth muscle cells [16]. However, data on Ser485/491 p-AMPK in liver and skeletal muscle are limited. Ser485/491 phosphorylation by insulin and IGF-1 appears to be primarily through Akt/PKB, as inhibition of Akt attenuates this phosphorylation and maintains AMPK activity in various cell types [14–17]. In addition to the role of Akt in phosphorylating Ser485/491 AMPK in other tissues, it was recently shown that leptin signals through mTOR/p70S6K to directly phosphorylate α2 Ser491 AMPK and reduce AMPK activity independently of changes in Thr172 in the hypothalamus [18]. Despite these findings, the physiological regulation of AMPK Ser485/491 phosphorylation under anabolic conditions has not been addressed in two of the primary insulin responsive tissues, skeletal muscle and liver.
In this study we investigated whether the inhibitory effects of insulin on AMPK activity may be mediated through the phosphorylation of its Ser485/491 site using cultured hepatocytes and myotubes and incubated EDL muscle as models. We hypothesized that (1) insulin would stimulate phosphorylation of Ser485/491 AMPK and reduce AMPK activity in these cells, and (2) inhibition of Akt would attenuate insulin-stimulated Ser485/491 p-AMPK. In addition, we assessed whether mTOR inhibition would have the same effects.
Materials and methods
Materials
HepG2 and C2C12 cells were purchased from ATCC (Manassas, VA). DMEM, Penicillin–Streptomycin (PS), fetal bovine serum (FBS) and horse serum (HS) were from Invitrogen (Grand Island, NY). D-(+)-Glucose solution, 45%, insulin, insulin-like growth factor-1 (IGF-1), and Akt inhibitor VIII were purchased from Sigma–Aldrich (St. Louis, MO). Rapamycin was purchased from LC Laboratories (Woburn, MA), and wortmannin was from Adipogen (San Diego, CA). SAMS peptide was purchased from Abcam (Cambridge, MA) and P32 was from Perkin–Elmer (Boston, MA).
Cell culture studies
HepG2 cells were cultured in normal glucose (5.5 mM) DMEM supplemented with 10% FBS and 1% PS. Media was replaced every 24–48 h and cells were passaged upon reaching 80–90% confluence. Glucose and FBS-free DMEM, supplemented with 1% PS and glucose at a final concentration of 5.5 mM, was used for all experimental incubations. C2C12 myoblasts were cultured as described above, with the addition of 1% glutamine. At 80–90% confluence they were differentiated into myotubes in DMEM supplemented with 2% HS and 1% PS.
Muscle incubations
Protocols for animal use were reviewed and approved by the Institutional Animal Care and Use Committee of Boston University Medical Center and were in accordance with National Institutes of Health guidelines. Male Sprague–Dawley rats weighing 55–65 g were purchased from Charles River Breeding Laboratories (Wilmington, MA). Rats were maintained on a 12:12-h light–dark cycle in a temperature-controlled (19–21 °C) room and were fed standard rat chow and water ad libitum. Following an overnight fast, they were anesthetized with pentobarbital (6 mg/100 g body weight), and muscles were removed for incubation.
After their removal, rat extensor digitorum longus (EDL) muscles were first equilibrated for 20 min at 37 °C in oxygenated Krebs–Henseleit solution (95% O2/5% CO2) containing 5.5 mM glucose. They were then stimulated with or without insulin (10 mU/ml) for 10 min. Following incubations muscles were snap-frozen in liquid nitrogen, and stored at −80 °C until analysis.
Cell lysate preparation
Cells were washed once on ice with Dulbecco’s PBS, lysed in buffer containing 20 mM Tris–HCl – pH 8.0, 1% IGEPAL, 1 mM EGTA, 10 mM nicotinamide, 1 μM trichostatin A, 10 mM sodium butyrate, 1 mM PMSF, 1 × phosphatase inhibitor cocktail 3 (Sigma), and 1 × protease inhibitor cocktail containing 1 mM EDTA (Complete Mini, Roche, Basel, Switzerland), and removed from wells using a cell scraper with a polyethylene copolymer blade (Fisher Scientific). Cell debris was removed by centrifugation at 13,200 g for 10 min at 4 °C, and the supernatant was removed and stored at −80 °C until analysis. Protein concentration was assessed by the bicinchoninic acid method (BCA; Pierce Biotechnology, Inc., Rockford, IL).
SDS–PAGE western blot analysis
Protein expression and phosphorylation were determined in 15–30 μg of protein lysate using SDS–PAGE gel electrophoresis and immunoblotting. Primary antibodies for Acetyl-CoA carboxylase (ACC), AMPK, Thr172 phospho-AMPKα, Ser485 phospho-AMPKα1, Ser485/491 phospho-AMPKα1/α2, Ser2448 phospho-mTOR, mTOR, Thr389 phospho-p70S6K, Ser473 phospho-Akt, and Akt antibodies, as well as secondary horseradish peroxidase (HRP)-linked antibodies were purchased from Cell Signaling Technology (Danvers, MA). Ser79 phospho-ACC antibody was from Upstate/Millipore (Temecula, CA). Anti-β-actin was from Sigma–Aldrich (St. Louis, MO). Following transfer onto a polyvinylidene difluoride membrane, membranes were blocked in Tris–buffered saline (pH 7.5) containing 0.05% Tween-20 (v/v; TBST) and 5% non-fat dry milk (w/v) for 1 h at room temperature, followed by incubation in primary antibodies (1:1000) at 4 °C overnight. After washing, membranes were incubated in a secondary antibody conjugated to horseradish peroxidase at a 1:5000 dilution for 1 h at room temperature. Bands were visualized using enhanced chemiluminescence solution (ECL; Pierce Biotechnology, Inc., Rockford, IL), and densitometry was performed with Scion Image software.
AMPK activity assay
AMPK activity was assessed as previously described [8,19]. Briefly, AMPK α1 or α2 was immunoprecipitated from 500 μg of protein from cell lysates by incubation at 4 °C overnight on a roller mixer using AMPK α1 or α2-specific antibodies (1:80; Santa Cruz Biotechnology, Inc.) and protein A/G agarose beads (1:10; Santa Cruz Biotechnology, Inc). Following several washes, activity was measured in the presence of 200 μM AMP and 80 μM [γ-32P] ATP (2 μCi) using 200 μM SAMS peptide (Abcam) as a substrate. Label incorporation into the SAMS peptide was quantified using a Lab-Logic (Brandon, FL) scintillation counter.
Statistical analysis
Results are reported as means ± standard error of the mean (SEM). Statistical significance was determined by two-tailed unpaired Student’s t tests or ANOVA with Tukey’s post hoc test. A level of p < 0.05 was considered statistically significant.
Results and discussion
Insulin stimulates phosphorylation of Ser485/491 AMPK and reduces AMPK activity
We first set out to ascertain whether Ser485/491 AMPK was physiologically regulated in hepatocytes, myotubes, and incubated skeletal muscle. Of note, the predominant AMPK isoform in hepatocytes is the α1 subunit, whereas in skeletal muscle α2 predominates, and in C2C12 myotubes α1 and α2 are fairly equally expressed in vitro. For these reasons, we focused on the regulation of α1 Ser485 p-AMPK in HepG2 hepatocytes and α1/α2 Ser485/491 p-AMPK in myotubes and EDL muscle.
As demonstrated previously in cardiomyocytes [14,17] and adipocytes [15], we found that insulin treatment acutely increased p-AMPK Ser485 in HepG2 hepatocytes (Fig. 1A) and p-AMPK Ser485/491 in C2C12 myotubes (Fig. 1B) by >5-fold. Similarly, incubation of rat EDL muscle with insulin increased AMPK Ser485/491 phosphorylation by over 60% (Fig. 1C). The reason for the quantitative difference between in vitro results and those observed ex vivo in the EDL muscle is not entirely clear and warrants further investigation. Interestingly, insulin also significantly diminished AMPK activity, as measured by the SAMS peptide assay, by 36–64% in HepG2 hepatocytes, C2C12 myotubes, and incubated EDL muscles (Fig. 3), despite no change in p-AMPK Thr172 (Fig. 1). The phosphorylation of ACC Ser79 coincided with that of p-AMPK Thr172 in vitro, showing no change with insulin treatment, whereas in the incubated EDL muscle insulin diminished p-ACC Ser79 (Fig. 1).
Fig. 1.
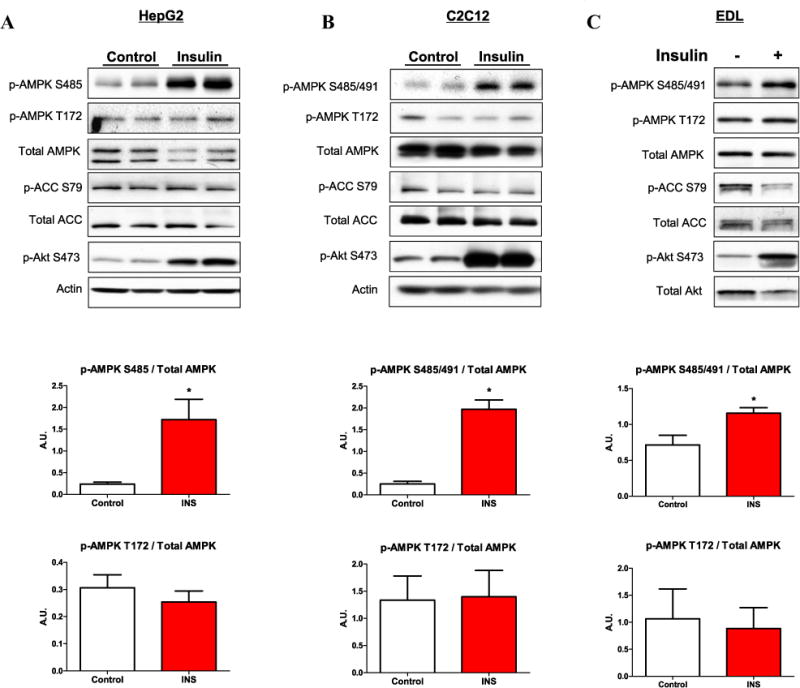
Insulin stimulates phosphorylation of AMPK Ser485 in HepG2 hepatocytes and Ser485/491 in C2C12 myotubes and incubated EDL muscle. HepG2 cells (A) and C2C12 myotubes (B) were cultured in normal glucose (5.5 mM), serum starved overnight, and stimulated with insulin (100 nM) for 15 min. Rat extensor digitorum longus (EDL) muscles were removed and equilibrated in Krebs–Henseleit buffer for 20 min, then stimulated with insulin (10 mU/ml) for 10 min (C). Following cell/tissue lysis, western blot analyses were performed, and representative blots are shown. Densitometry was used to quantify western blots. Phosphorylation of AMPK was normalized to total AMPK, and normalized data are displayed (shown in line with the corresponding cell type/tissue). Results are means ± SE (n = 3–6 per treatment). All experiments were performed in triplicate. *p < 0.05 vs. control.
Fig. 3.
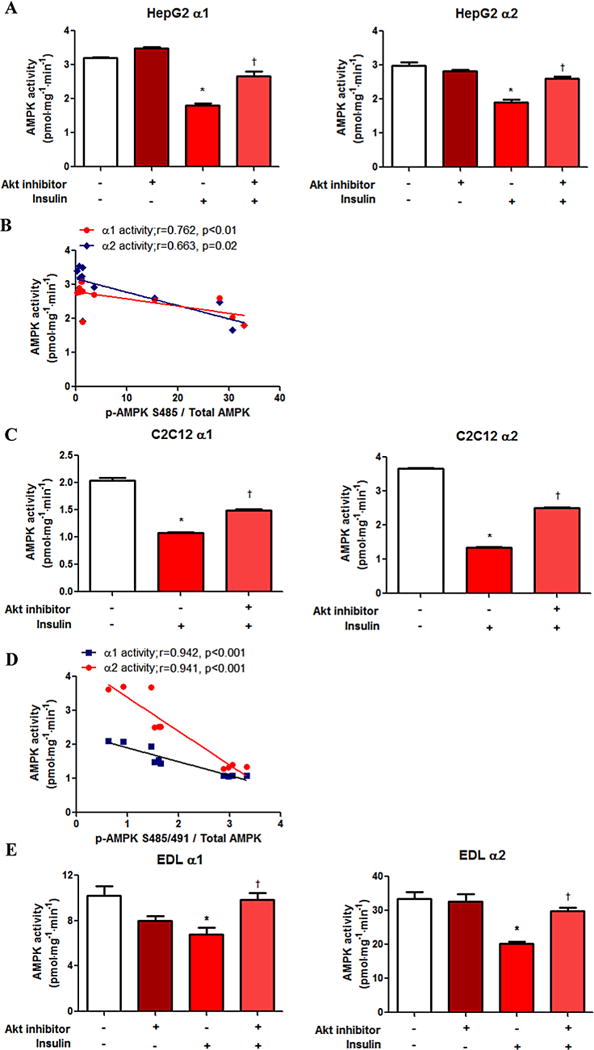
Insulin-induced inhibition of AMPK activity is partially prevented by Akt inhibition. AMPK α1 and α2 activities were assessed using the SAMS peptide assay, as described in the materials and methods section. HepG2 (A), C2C12 (C) and EDL (E) AMPK activity data are presented. Correlations between Ser485/491 AMPK phosphorylation data from western blot analysis (Fig. 2) and AMPK activity are shown (B and D).
Inhibition of Akt attenuates insulin-induced p-AMPK Ser485/491 and partially prevents the reduction in AMPK activity
The Akt pathway is involved in the phosphorylation of Ser485/491 in other tissues, including heart [13,14], brown adipose tissue [20], white adipocytes [15], and vascular smooth muscle [16] and tumor cells [21]. We tested the role of Akt on insulin’s effect on p-AMPK Ser485 in hepatocytes and Ser485/491 in myotubes. Pretreatment of the cells for 1 h with the Akt inhibitor, Akt inhibitor VIII, attenuated the insulin-stimulated increase in serine phosphorylation of AMPK in both cell types, despite no effect on p-AMPK Thr172 (Fig. 2A and B, respectively). This reduction in Ser485 in HepG2 cells or Ser485/491 p-AMPK in C2C12 myotubes and EDL muscle by Akt inhibition was accompanied by a significant attenuation in insulin’s effect on AMPK α1 and α2 activity. Taken together, insulin-induced phosphorylation of Ser485/491 of AMPK and inhibition of AMPK activity are both partially prevented by Akt inhibition (Fig. 3).
Fig. 2.
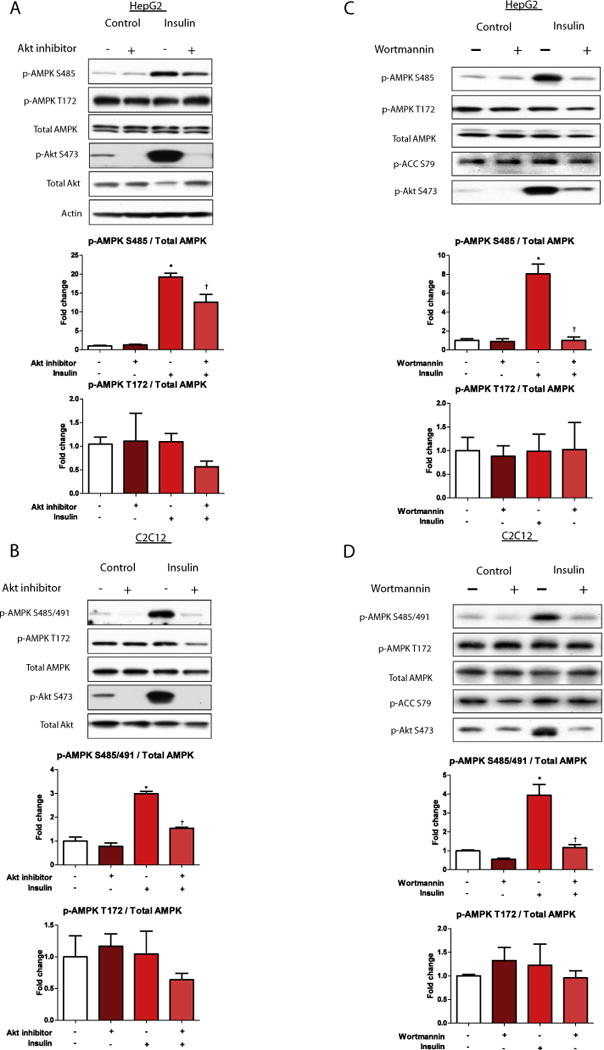
Inhibition of Akt attenuates insulin stimulated phosphorylation of AMPK Ser485/491. HepG2 (A) and C2C12 cells (B) were treated with or without Akt inhibitor VIII (250 μM) for 2 h, then stimulated with insulin (100 nM) for 15 min. Cells were lysed and subjected to western blot analysis. Representative western blots are shown. Quantification of western blots was performed using densitometry, and AMPK phosphorylation was normalized to total AMPK. Results are presented as fold-change in AMPK phosphorylation vs. control in HepG2 cells (A) and C2C12 cells (B). In Figures C (HepG2) and D (C2C12) cells were pre-treated with the PI3-kinase inhibitor wortmannin (10 nM) for 1 h, then stimulated with insulin (100 nM) for 10 min. Cells were processed and protein expression and phosphorylation were quantified as described in (A) and (B). Results are means ± SE (n = 3–6 per treatment). All experiments were performed in triplicate. *p < 0.05 indicates a significant effect of insulin vs. control, †p < 0.05 vs. insulin treatment alone.
These data suggest a dissociation between p-AMPK Thr172, the commonly used surrogate for activity, and AMPK activity assessed via the SAMS peptide assay, under conditions of insulin-induced AMPK inactivation. Similar results were reported by Dagon and colleagues in the hypothalamus, in which they showed that AMPK activity decreased when Ser491 p-AMPK was elevated by leptin, whereas p-AMPK Thr172 did not change [18]. Of note, in our study, the decline in AMPK activity caused by insulin was proportional to the increase in p-AMPK Ser485 (HepG2) or Ser485/491 (C2C12) in each case (Fig. 3B and D).
Wortmannin prevents insulin-induced p-AMPK Ser485/491
Inhibition of PI3K signaling by wortmannin, and subsequent reduction in Akt signaling recapitulated the results observed with the Akt inhibitor VIII in both HepG2 and C2C12 cells. Specifically, wortmannin pre-treatment prevented insulin-induced p-AMPK Ser485/491 without affecting Thr172 p-AMPK (Fig. 2C and D). Our results are limited to insulin’s effect on basal AMPK activity; however, others have shown that insulin antagonizes the activation of AMPK by various stimuli, including anoxia in heart [22], palmitate in adipocytes [23], and AICAR in hepatocytes and myotubes (Valentine, unpublished data).
Interestingly, although wortmannin and Akt inhibitor VIII had similar effects to diminish p-Akt Ser473, wortmannin also essentially eliminated the phosphorylation of AMPK at Ser485 in both cell types, whereas Akt inhibition only partially blunted this phosphorylation in HepG2 cells. This suggests a pathway independent of Akt may be involved in the phosphorylationof AMPK at Ser485, atleast in cultured hepatocytes, however the mechanism(s) involved remain to be determined.
IGF-1 mimics the effect of insulin on p-AMPK Ser485/491 and is attenuated by Akt inhibition
In addition to insulin, the anabolic factor insulin-like growth factor 1 (IGF-1) signals through the Akt pathway. Ning and colleagues recently demonstrated that IGF-1 dose-dependently increases Ser485 p-AMPK on the α1 AMPK subunit in vascular smooth muscle cells (VSMCs), an effect mediated by Akt [16]. Using a phosphodefective S485A mutant, the authors also demonstrated an inability of IGF-1 to diminish Thr172 p-AMPK, suggesting the involvement of Ser485 in AMPK downregulation by IGF-1, at least in VSMCs. In the present study we show for the first time that IGF-1 also increases the phosphorylation of Ser485/491 AMPK, in both hepatocytes (Fig. 4A and B) and myotubes (Fig. 4C and D). The effect of IGF-1 was dose-dependent, and the increase in Ser485/491 p-AMPK mirrored that for p-Akt Ser473 (data not shown). In addition, similar to its effect on insulin stimulation, inhibition of Akt attenuated the effect of IGF-1 on Ser485/491 p-AMPK (Fig. 4).
Fig. 4.
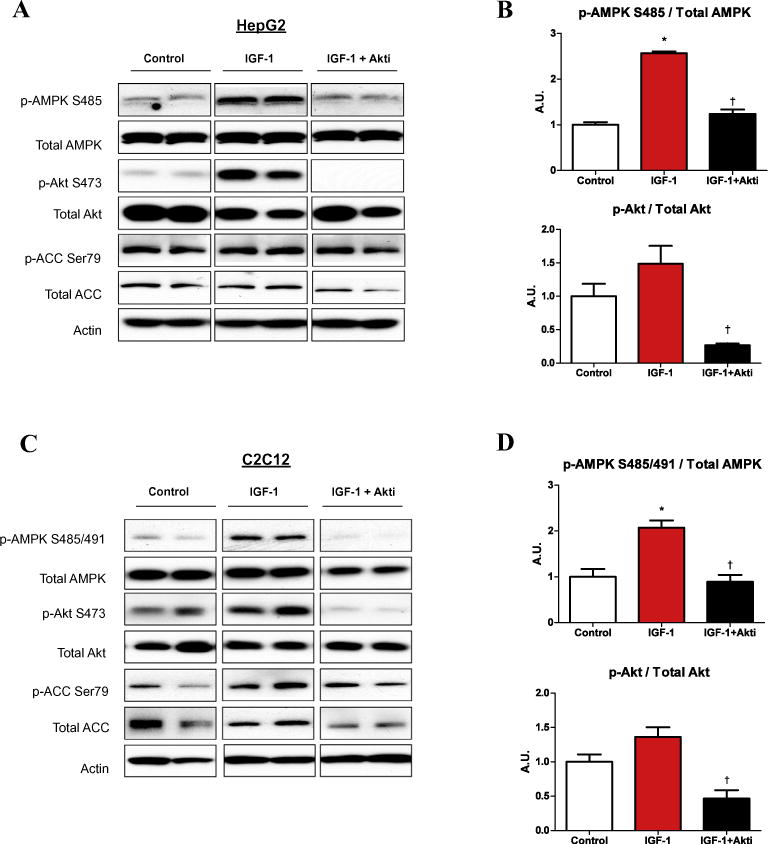
IGF-1 phosphorylation of AMPK Ser485/491 is prevented by Akt inhibition. HepG2 (A) and C2C12 cells (C) were serum starved overnight and treated with or without Akt inhibitor VIII (Akti; 250 μM) for 2 h, then stimulated with IGF-1 (20 ng/ml) for 15 min followed by whole cell lysis and analyzed by western blot. Representative western blots are shown (A and C). Densitometric analysis of western blots was used to quantify protein expression. Phosphorylation of proteins of interest were normalized to the corresponding total protein, and results are presented as fold-change compared to control (B and D). Results are means ± SE (n = 3–6 per treatment). All experiments were performed in triplicate. *p < 0.05 indicates a significant effect of IGF-1, †p < 0.05 for the effect of AktiVIII.
mTOR inhibition by rapamycin did not effect insulin-stimulated p-AMPK Ser485/491
As previously noted, it was shown that leptin can cause phosphorylation of p-AMPK Ser485/491 in the hypothalamus, through the mTOR/p70S6Kinase (p70S6K) pathway [18]. It is well established that insulin leads to similar increases in p70S6K (activity and p-p70S6K Thr389 in hepatocytes and myotubes [24,25] Fig. 5). Thus, we examined whether this pathway might also mediate the effect of insulin on Ser485/491 p-AMPK. Cells were pre-treated in the presence or absence of the mTOR inhibitor rapamycin for 1 h and then stimulated with insulin. As shown in Fig. 5, rapamycin inhibited the phosphorylation of mTOR and p70S6K by insulin; however, it had no effect on the increase in Ser485/491 p-AMPK caused by insulin (Fig. 5) or IGF-1 (data not shown). In keeping with these data, Beauloye and colleagues have demonstrated in heart the ability of insulin to inhibit AMPK activation by anoxia is prevented by pre-treatment with wortmannin, but not by rapamycin [22].
Fig. 5.
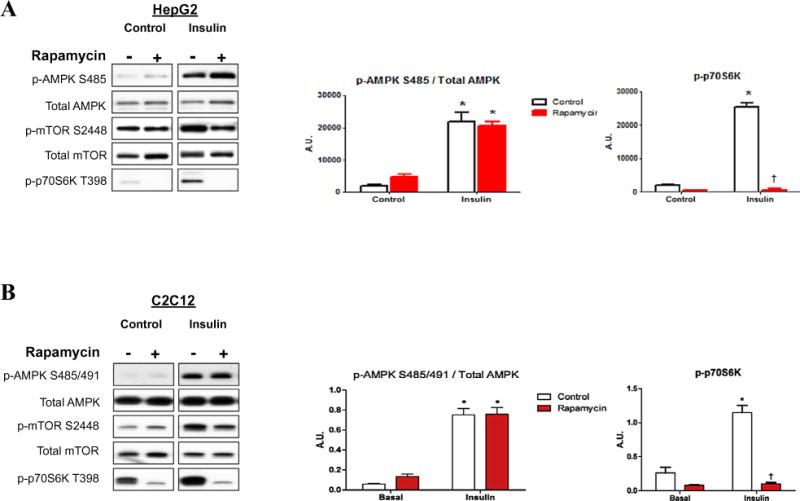
Insulin-stimulation of p-AMPK Ser485/491 is not affected by the mTOR inhibitor rapamycin. HepG2 (A) and C2C12 cells (B) were treated with or without rapamycin (50 nM) for 2 h, then stimulated with insulin for 15 min. Cell lysates were run on western blots and protein expression and phosphorylation were quantified using densitometry. Representative western blots are shown. Densitometry results are presented in graphs. Results are means ± SE (n = 3–6 per treatment). All experiments were performed in triplicate. *p < 0.05 indicates a significant effect of insulin, †p < 0.05 for the effect of rapamycin treatment.
Summary and conclusions
In this study, we show for the first time that insulin and IGF-1 can regulate the inhibitory phosphorylation of AMPK on its Ser485/491 of its α1/α2 site in both cultured hepatocytes and myotubes, and incubated EDL muscle. Our results also indicate that this effect of insulin is mediated through the Akt pathway, and not mTOR/p70S6K Similar findings were observed when these cells were stimulated with IGF-1.
It is well accepted that activation of AMPK enhances insulin sensitivity in a variety of tissues, including heart, liver, adipose tissue and skeletal muscle [26,27], whereas loss of AMPK contributes to insulin resistance [28,29]. Our data also highlight a potential mechanism by which hyperinsulinemia downregulates AMPK, through phosphorylation of Ser485/491 on its α-subunit, leading to diminished glucose uptake and exacerbation of insulin resistance. Furthermore, our data, along with those of others, suggest that there may be discordance between the commonly used surrogate measure of AMPK activity, its phosphorylation on Thr172, and AMPK activity measured by the SAMS peptide assay, at least under certain conditions in which Ser485/491 appears to play an inhibitory role [18].
These findings, along with previous results of others in heart [14] and adipocytes [15], suggest that insulin inhibits AMPK activity rapidly, with a peak in Ser485/491 AMPK phosphorylation occurring within minutes. Under conditions such as high glucose this event appears to take longer, and may occur through a different mechanism. Other kinases involved in the phosphorylation of AMPK Ser485/491 have been identified under various conditions, including the mTOR/p70S6K pathway [18] and protein kinase A (PKA) [30]. Likewise, our laboratory recently found that activation of protein kinase C (PKC) can cause phosphorylation of AMPK Ser485/491 and diminish its activity in myotubes[31]. Strategies to prevent this inhibitory phosphorylation may provide a novel approach to maintain AMPK function in the setting of hyperinsulinemia. Still to be determined is whether downregulation of AMPK by this mechanism occurs in states of chronic insulin resistance and what are the physiological and pathophysiological implications of this change.
Acknowledgments
This work was supported by grants from the National Institutes of Health, USA (DK19514 and DK67509 to N.R.) and T32 HL70024 to R.V.
Footnotes
Abbreviations used: AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin.
References
- 1.Ruderman N, Carling D, Prentki M, Cacicedo J. J Clin Invest. 2013;123 doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu XJ, Valentine RJ, Ruderman NB. Curr Obes Rep. 2014 doi: 10.1007/s13679-014-0095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. Diabetes Metab Syndr Obes. 2014 doi: 10.2147/DMSO.S43731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gauthier MS, O’Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, Gokce N, et al. Biochem Biophys Res Commun. 2011;404:382–387. doi: 10.1016/j.bbrc.2010.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, Farb M, et al. J Lipid Res. 2012;53:792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. J Endocrinol Diabetes Obes. 2013;1:1008. [PMC free article] [PubMed] [Google Scholar]
- 7.Kraegen EW, Saha AK, Preston E, Wilks D, Hoy AJ, Cooney GJ, Ruderman NB. Am J Physiol Endocrinol Metab. 2006;290:E471–E479. doi: 10.1152/ajpendo.00316.2005. [DOI] [PubMed] [Google Scholar]
- 8.Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, Ruderman NB. Diabetes. 2010;59:2426–2434. doi: 10.2337/db09-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Assifi MM, Suchankova G, Constant S, Prentki M, Saha AK, Ruderman NB. Am J Physiol Endocrinol Metab. 2005;289:E794–E800. doi: 10.1152/ajpendo.00144.2005. [DOI] [PubMed] [Google Scholar]
- 10.Witters LA, Kemp BE. J Biol Chem. 1992;267:2864–2867. [PubMed] [Google Scholar]
- 11.Gamble J, Lopaschuk GD. Metabolism. 1997;46:1270–1274. doi: 10.1016/s0026-0495(97)90229-8. [DOI] [PubMed] [Google Scholar]
- 12.Steinberg GR, Kemp BE. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 13.Soltys CL, Kovacic S, Dyck JR. Am J Physiol Heart Circ Physiol. 2006;290:H2472–H2479. doi: 10.1152/ajpheart.01206.2005. [DOI] [PubMed] [Google Scholar]
- 14.Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, et al. J Biol Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- 15.Berggreen C, Gormand A, Omar B, Degerman E, Goransson O. Am J Physiol Endocrinol Metab. 2009;296:E635–E646. doi: 10.1152/ajpendo.90596.2008. [DOI] [PubMed] [Google Scholar]
- 16.Ning J, Xi G, Clemmons DR. Endocrinology. 2011;152:3143–3154. doi: 10.1210/en.2011-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 18.Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. Cell Metab. 2012;16:104–112. doi: 10.1016/j.cmet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. J Biol Chem. 2002;277:32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 20.Pulinilkunnil T, He H, Kong D, Asakura K, Peroni OD, Lee A, Kahn BB. J Biol Chem. 2011;286:8798–8809. doi: 10.1074/jbc.M111.218719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Biochem J. 2014;459:275–287. doi: 10.1042/BJ20131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beauloye C, Marsin AS, Bertrand L, Krause U, Hardie DG, Vanoverschelde JL, Hue L. FEBS Lett. 2001;505:348–352. doi: 10.1016/s0014-5793(01)02788-0. [DOI] [PubMed] [Google Scholar]
- 23.Hebbachi A, Saggerson D. Biosci Rep. 2012;33:71–82. doi: 10.1042/BSR20120031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozma SC, Lane HA, Ferrari S, Luther H, Siegmann M, Thomas G. EMBO J. 1989;8:4125–4132. doi: 10.1002/j.1460-2075.1989.tb08597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somwar R, Sumitani S, Taha C, Sweeney G, Klip A. Am J Physiol. 1998;275:E618–E625. doi: 10.1152/ajpendo.1998.275.4.E618. [DOI] [PubMed] [Google Scholar]
- 26.Fujii N, Jessen N, Goodyear LJ. Am J Physiol Endocrinol Metab. 2006;291:E867–E877. doi: 10.1152/ajpendo.00207.2006. [DOI] [PubMed] [Google Scholar]
- 27.Hegarty BD, Turner N, Cooney GJ, Kraegen EW. Acta Physiol Oxf. 2009;196:129–145. doi: 10.1111/j.1748-1716.2009.01968.x. http://dx.doi.org/10.1111/j.1748-1716.2009.01968.x. Epub 02009 Feb 01919. [DOI] [PubMed] [Google Scholar]
- 28.Pehmoller C, Treebak JT, Birk JB, Chen S, MacKintosh C, Hardie DG, Richter EA, et al. Am J Physiol Endocrinol Metab. 2009;297:E665–E675. doi: 10.1152/ajpendo.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujii N, Ho RC, Manabe Y, Jessen N, Toyoda T, Holland WL, Summers SA, et al. Diabetes. 2008;57:2958–2966. doi: 10.2337/db07-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 31.Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. Pharmacological PKC activation inhibits AMPK by phosphorylation at Ser485/491 in muscle and liver cells. American Diabetes Association. 2014 Abstract. [Google Scholar]