

Abstract
IFI44 is an interferon-alfa inducible protein, and is associated with infection of several viruses. However, IFI44 elicits minimal antiviral effects on these viruses, and its exact role is still unknown. Here we show that IFI44 inhibits HIV-1 replication in vitro. Through depletion of endogenous IFI44 or overexpression of IFI44 we confirm that IFI44 suppresses HIV-1 LTR promoter activity and affects viral transcription. Furthermore, we find that IFI44 localizes to nuclei and binds to the HIV-1 LTR promoter in HIV-1 infected cells. Removing suppression of HIV-1 transcription benefits reactivation of HIV-1 proviruses for purging latent reservoirs. We demonstrate that depletion of endogenous IFI44 in J-LAT cells induces reactivation of latent HIV-1. Based on these results, we propose a model in which IFI44 is recruited to the HIV-1 LTR, which may suppress viral transcription and prevent reactivation of latent HIV-1. Our study suggests a previously unrecognized anti-HIV phenomenon for interferon-stimulated proteins.
Keywords: Interferon, ISG, IFI44, HIV-1, LTR, Transcription, Latency, Reactivation
Introduction
Type I interferon, IFN-α/β-binds to the interferon receptor and potently induces antiviral responses by initiating a signaling cascade that leads to the expression of thousands of interferon-stimulated genes (ISGs) and a generalized antiviral state. IFN-α can block HIV-1 replication at multiple steps including viral entry, reverse transcription, viral protein synthesis and processing, and viral assembly and release [1–7]. Several anti-HIV ISGs have recently been identified, including tetherin, TRIM5 alpha, APOBEC3 (apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3), and SAMHD1 (SAM domain and HD domain-containing protein 1) [8–10]. However, discovery of these ISGs is just tip of the iceberg, other ISGs with potential to antagonize HIV-1 intracellular replication are pending for identification and further investigation.
IFI44 expression is inducible by IFN-α/β but not by IFN-γ [11], and is associated with infections of several viruses. IFI44 was initially found as a hepatitis C virus (HCV) associated microtubular aggregate protein isolated from the hepatocytes of HCV-infected chimpanzees [12]. IFI44 is differentially expressed in rhinovirus (RV) infected primary bronchial epithelial cells derived from humans with asthma vs. normal subjects [13]. Early stage human papillomavirus (HPV) infections also induce IFI44 expression in a human keratinocyte line [14], seemingly through the expression of HPV E7 oncoprotein [15]. However, HIV-1 infection seems to down-regulate expression of a set of ISGs in primary macrophages, including IFI44 [16].
Although it is suggested that IFI44 may mediate the antiviral activity of type I interferon, there is no experimental data to support it, for example, in HCV infections [17, 18]. Recently, a study of IFN-β induced the inhibition of bunyamwera orthobunyavirus (BUNV) replication, demonstrating that overexpression of IFI44 leads to an approximately 10-fold reduction of BUNV replication in HEK293 cells [19]. However, the exact antiviral mechanism of IFI44 remains elusive. One possible link is that IFI44 may induce an anti-proliferative state in certain cell types in vitro [20], but still lacks direct evidence for how exactly it contributes to IFI44’s antiviral activity. It remains possible that IFI44 may exploit alternative strategies to restrict viral replication. Our earlier comprehensive siRNA screens identified IFI44 as a leading candidate of HIV-1 restriction factor [21]. We would like to further explore whether IFI44, as a general antiviral ISG, may also possess anti-HIV activity and its potential molecular mechanism.
Results
IFI44 depletion enhances HIV-1 infection
We first measured IFI44 expression in MAGI-HeLa, Jurkat, CD4+ and CD8+ T cells. Even without IFN treatment, IFI44 maintained a basal level of expression in these cells, although it seemed that IFI44 had higher expression in T cells compared to epithelial cells (Figure S1). Consistent with previous studies, IFI44 expression was highly inducible upon IFN-α stimulation, but to a lesser degree with IFN-γ (Figure S1). We were able to efficiently deplete basal endogenous expression of IFI44 using two sequence-unique siRNAs (Figure 1A). We infected IFI44-depleted MAGI-HeLa cells with HIV-1 IIIB and measured the intracellular HIV-1 capsid protein p24 expression by immunostaining using an anti-p24 antibody. We set up a fixed threshold of p24 signal from the FITC channel, and any cell with the p24 signal above the threshold was counted as an HIV-infected cell. HIV-1 infection rate was calculated by dividing the p24-expressing cells by the total cells (staining of nuclei with Hoechst). IFI44 depletion effectively increased the HIV-1 infection rate in MAGI-HeLa cells with or without IFN-α stimulation (Figure 1B). We also tested two VSV-G pseudo-typed viruses (HIV-NL4-3-GFP [Δ Env], MLV-GFP [Δ Env]) as well as lentiviral vectors harboring different promoters (LTR-GFP, CMV-ZsGreen [ZSG]), which were used in previous studies [21]. A threshold of GFP signal was decided to call out GFP-positive, virus-infected cells. GFP-positive cells were counted and normalized by total cell numbers to calculate viral infection rate for individual virus or viral vector. We noticed that IFI44 depletion did not affect MLV-GFP infection rate (Figure 1C). Furthermore, IFI44 depletion increased LTR-driven GFP expression but not CMV promoter (Figure 1D). These results indicate that IFI44 may target HIV-1 LTR promoter activity specifically. We further confirmed the anti-HIV effects of IFI44 in Jurkat cells. We generated pAPM lentiviral vectors expressing two sequence-unique IFI44 shRNAs, and transduced them individually to Jurkat cells. Both IFI44 shRNAs were able to deplete basal expression of IFI44 in Jurkat cells (Figure 1E), while enhancing infection rate of VSV-G pseudo-typed HIV-NL4-3-GFP [Δ Env] in these cells, measured by flow cytometry (Figure 1F).
Figure 1.

IFI44 depletion by RNAi increases HIV-1 infection. (A). MAGI-HeLa was transfected with sequence-unique IFI44 siRNA, siIFI44-1 or siIFI44-2, or non-targeting control siRNA (siNT). 72 hours post-transfection, total RNA was extracted for reverse transcription and quantitative real-time PCR to measure the IFI44 mRNA level. Data were normalized to the siNT-treated cells. (B). MAGI-HeLa was transfected with siIFI44-1, siIFI44-2, or non-targeting siNT. 48 hours post-transfection, cells were treated with IFN-α, or mock treated for 24 hours. Cells were then infected with HIV-IIIB wild-type viruses for 48 hours, and stained with anti-HIV-1 p24 CA antibody (anti-CA) and a FITC anti-mouse secondary antibody. Nuclei were stained using Hoechst. The percentage of infected cells was measured and normalized to the siNT-treated cells. (C, D). MAGI-HeLa was transfected with siIFI44-1, siIFI44-2, or siNT. 72 hours post-transfection, cells were infected with VSV-G pseudo-typed (C) HIV-NL4-3-GFP [Δ Env] or MLV-GFP virus [Δ Env], or (D) LTR-GFP or CMV-ZSG lentivirus for 48 hours. Cells were stained with Hoechst and the percentage of GFP-positive cells was measured and normalized to the siNT-treated cells. (E). Sequence-unique IFI44 shRNA, shIFI44-1, shIFI44-2, or control firefly luciferase shRNA (shFLuc) in pAPM vector was transduced in Jurkat T cells. Cells were subjected to RNA extraction, reverse transcription, and quantitative real-time PCR for measuring the IFI44 mRNA level. Data were normalized to the shFLuc-transduced cells. (F). Jurkat T cells stably expressing shIFI44-1, shIFI44-2, or shFLuc were infected with VSV-G pseudo-typed HIV-NL4-3-GFP [Δ Env] viruses for 48 hours. Cells were assessed using flow cytometry and the percentage of GFP-positive cells were calculated and normalized to the shFLuc-transduced cells. All results were presented as mean ± s.d. (n = 3); * P < 0.05 from t-test.
IFI44 expression reduces HIV-1 LTR activity
We also used a gain-of-function approach to test the anti-HIV activity of IFI44. We cloned HA-tagged IFI44 cDNA (HA-IFI44) into the pQCXIP retroviral vector. It was transduced to MAGI-HeLa, HEK293, and JLTRG cells. Cells were also transduced in parallel with empty pQCXIP vector as a negative control. Stable cells were selected by incubating with puromycin. HA-IFI44 expression was confirmed by either western blot to measure protein level (Figures 2A, 2C) or reverse transcription-coupled qPCR to measure mRNA level (Figure 2E). MAGI-HeLa cells expressing IFI44 significantly decreased HIV-IIIB infection rate with the presence or absence of IFN-α (Figure 2B). To rule out the possibility that this could be an artificial effect due to overexpression of IFI44, we measured the EBV lytic replication induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) and sodium butyrate (SB) in AGS/BX cells expressing IFI44 (Figure S2). The result demonstrated no obvious suppression of EBV replication, which indicates that IFI44-mediated suppression of HIV-1 replication is specific. We further performed an LTR-luciferase assay in HEK293 cells expressing IFI44. Consistent with our LTR-GFP expression results in IFI44-depleted cells (Figure 1D), we found that IFI44 expression significantly lowered the LTR-driven luciferase expression (Figure 2D). We also tested IFI44 anti-LTR effects in JLTRG cells, which are Jurkat cells with a stably integrated LTR-GFP construct but no TAT expression [22]. We transiently transfected a pcDNA vector expressing FLAG-tagged TAT (FLAG-TAT) into JLTRG cells expressing IFI44, and measured GFP-positive cells by flow cytometry. Similar to the LTR-luciferase results, IFI44 expression reduced the TAT-mediated LTR-GFP expression (Figure 2F).
Figure 2.

Exogenous expression of IFI44 suppresses HIV-1 viral gene expression. (A). pQCXIP-HA-IFI44 or pQCXIP empty vector was transduced into MAGI-HeLa cells. Cell lysate was prepared and analyzed for HA-IFI44 expression by western blot using an anti-HA antibody. (B). MAGI-HeLa cells stably expressing HA-IFI44 or empty vector were pretreated with IFN-α or mock-treated for 24 hours. Cells were infected with HIV-IIIB wild-type viruses for 48 hours and stained with an anti-HIV-1 p24 CA antibody (anti-CA) and Hoechst. The percentage of infected cells was measured and normalized to the vector-only cells. (C). pQCXIP-HA-IFI44 or pQCXIP empty vector was transduced into HEK293 cells. Cell lysate was prepared and analyzed for HA-IFI44 expression by western blot using an anti-HA antibody. (D). HEK293 cells stably expressing HA-IFI44 or empty vector were co-transfected with HIV-LTR firefly luciferase reporter and pRL-TK renilla luciferase control vectors, together with pcDNA-FLAG-TAT vector. Luciferase activity was measured 24 hours post-transfection and normalized to the empty vector cells. (E). pQCXIP-HA-IFI44 or pQCXIP empty vector was transduced into JLTRG cells. Total RNA was extracted for reverse transcription and quantitative real-time PCR to measure the IFI44 mRNA level. Data were normalized to the JLTRG-Vector cells. (F). JLTRG cells stably expressing HA-IFI44 or empty vector were transiently transfected with pcDNA-FLAG-TAT. 48 hours post-transfection, cells were assessed by flow cytometry, and the percentage of GFP-positive cells was normalized to the vector-only cells. All results were presented as mean ± s.d. (n = 3); * P < 0.05 from t-test.
IFI44 nuclear localization and association with LTR promoter
Our genetic studies indicate that IFI44 may specifically suppress HIV-1 LTR promoter activity (Figures 1D, 2D). However, earlier studies suggest that IFI44 may be a cytoplasmic protein [20]. Using a HEK293 cell line stably expressing IFI44, we confirmed that the majority of IFI44 was in the cytoplasm, but IFI44 localized to nuclei upon HIV-1 infection using VSV-G pseudo-typed HIV-1 NL4-3-GFP [Δ Env] virus (Figure 3A). This result is supported by the immunostaining of HEK293 cells showing increased nuclear localization of IFI44 upon HIV-1 infection (Figures S3A–B). We further confirmed that nuclear IFI44 bound specially to certain regions of HIV-1 LTR promoter by conducting a chromatin immuno-precipitation (ChIP) coupled to detection by semi-quantitative PCR (ChIP-PCR) experiment. IFI44 was strongly associated with the nuc-0, nuc-1, and nucleosome-free region (PPR) of the HIV-1 LTR, but not the nuc-2 region (Figures 3B, S3C). Considering that viral transcription is initiated within the PPR between nuc-0 and nuc-1 [23], we postulate that the presence of IFI44 at HIV-1 LTR may interfere with viral transcription initiation. However, GST-TAT pull-down assays demonstrated that IFI44 recruitment to HIV-1 LTR might not be mediated through interaction with HIV-1 TAT transactivator, indicating that IFI44 may be recruited to the HIV-1 LTR through an unidentified mechanism (Figures S3D–E).
Figure 3.

IFI44 enters the nuclei and associates with the HIV-1 LTR upon HIV-1 infection. (A). HEK293 cells stably expressing HA-IFI44 were infected with VSV-G pseudo-typed HIV-NL4-3-GFP [Δ Env] viruses (+HIV, MOI=5) or mock treated (−HIV) for 48 hours. Equal numbers of cells were subjected to cytoplasmic (cyt) and nuclear (nuc) fractionation. Extracted protein samples were separated by SDS-PAGE. HA-IFI44 cyt or nuc localization was analyzed by western blot using an anti-HA antibody. GAPDH protein level in cyt or nuc extraction was determined to indicate equal loading. E-cadherin (cytoplasm) and JMJD1A (nuclei) were stained to ensure the separation of cytoplasmic and nuclear proteins. (B). HEK293 cells stably expressing HA-IFI44 were infected with VSV-G pseudo-typed HIV-1 NL4-3-GFP [Δ Env] viruses. Cells were subjected to ChIP assays using an anti-HA antibody or a rabbit control IgG. Precipitated DNA samples were extracted and analyzed by PCR using primers amplifying the nuc-0, L1 nucleosome-free region (PPR), nuc-1, and nuc-2 regions of the HIV-1 LTR promoter.
IFI44 depletion reverses HIV-1 latency
Since IFI44 significantly suppresses HIV-1 transcription, we postulate that it may play a role in facilitating HIV-1 latency while preventing viral reactivation. We transduced two IFI44 shRNAs into several J-LAT cells, which are Jurkat cells harboring latently infected GFP-fused HIV proviruses [24]. To analyze the efficiency of HIV-1 reactivation, the population of GFP-expressing cells, indicating restoration of viral transcription, were sorted and analyzed by flow cytometry. We first confirmed the IFI44 shRNA knockdown efficiency by measuring basal IFI44 mRNA level in J-LAT A2 cells harboring a mini HIV genome (LTR-TAT-GFP) (Figure 4A). Interestingly, depletion of IFI44 reversed HIV-1 latency up to 2~3 folds in J-LAT A2 cells, which exerted the effect spontaneously without treatment of HIV-1 latency-reversing agents (LRAs) (Figures 4B–C). We also evaluated the effect of IFI44 depletion in another two J-LAT cell lines, 6.3 and 9.2, which harbor Env-deleted full-length HIV-1 NL4-3-GFP proviruses. We did not see any obvious effect on HIV-1 reactivation by depleting endogenous IFI44, but shRNA-mediated IFI44 knock down seemed to mildly benefit HIV-1 reactivation once cells were treated with Prostratin, a potent HIV-1 LRA, which activates the NF-κB pathway [25] (Figure S4).
Figure 4.
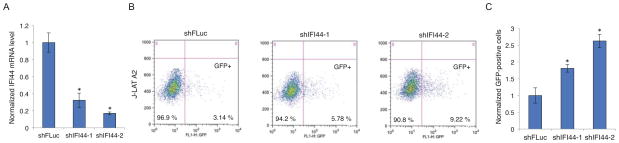
IFI44 depletion reverses HIV-1 latency in J-LAT A2 cells. (A). shIFI44-1, shIFI44-2, or shFLuc in pAPM vector was transduced into J-LAT A2 cells. Cells were subjected to RNA extraction, reverse transcription, and quantitative real-time PCR for measuring the IFI44 mRNA level. Data were normalized to the shFLuc-transduced cells. (B). Equal numbers of J-LAT A2 cells stably expressing shIFI44-1, shIFI44-2, or shFLuc were assessed by flow cytometry, and the percentage of GFP positive cells (bottom right quadrant) were measured. Data are one representative of three independent experiments. (C). Percentage of GFP positive cells from three independent flow cytometry assays for J-LAT A2 cells expressing shIFI44-1 or shIFI44-2 was averaged and normalized to that of J-LAT A2 cells expressing shFLuc. All results were presented as mean ± s.d. (n = 3); * P < 0.05 from t-test.
Discussion
Although it is clear that type I IFN induces potent cellular defense against viral infection mediated by up-regulation of ISGs, the exact function for most of them is unknown and demands further characterization. Only until recently ISGs have been scrutinized individually for determining their antiviral activity against a set of various virus families [26, 27]. In this work, we describe that one of the ISGs, IFI44, may reduce HIV-1 replication possibly through affecting viral transcription. We demonstrate that IFI44 may enter nuclei and bind with the HIV-1 LTR promoter to produce an inhibitory effect on HIV-1 transcription. Furthermore, our results using HIV-1 latently infected Jurkat cells suggest that IFI44 may play a role in facilitating HIV-1 latency while preventing reactivation due to its suppressive activity toward viral transcription.
In most of our studies, we measured the infection rate by deciding a threshold of the signal indicating HIV-1 infection, such as intracellular p24 or virus-fused GFP expression, and then counting the percentage of HIV-infected cell. We routinely use this as an indicator of viral intracellular replication, which provides a great dynamic range, to measure results of our siRNA screens and further genetic validations of host genes [21, 28, 29]. We show that either basal or IFN-α induced expression of IFI44 contributes to restriction of HIV-1 intracellular replication (Figure 1). This is not unusual, since studies of other ISGs demonstrate that even basal expression of ISGs still possess certain antiviral activity. For example, the high basal expression of an ISG, TRIM5α, in peripheral blood mononuclear cells (PBMCs) dramatically restricts mucosal transmission of simian immunodeficiency virus (SIV) more efficiently than when induced with type I IFN [30]. Certain host genes, including recently discovered tyrosine kinase nonreceptor 1 (TNK1), are required for maintenance of basal ISG expression [31]. Although IFI44 is implicated to play a role in regulating the replication of various viruses, it was never reported previously as a HIV-1 restriction factor. In addition, our findings seem to align with an earlier study, which shows that IFI44 expression is down-regulated in IFN-treated primary macrophages upon HIV-1 infection [16]. Above all, we conclude that IFI44 imposes an inhibitory effect on HIV-1 replication while evolutionarily HIV-1 may have developed a strategy to suppress the antiviral activity of IFI44 in order to benefit its replication, which is worthy of further investigation. However, we did not notice that HIV-1 infection led to IFI44 degradation (data not shown), so it is unlikely that HIV-1 proteins, such as Vif, Vpu, Vpr, and Vpx, may mask antiviral activity of IFI44, considering that these accessory proteins can associate with certain antiviral proteins and induce their proteasome-dependent degradation [32–35].
Our IFI44 localization experiments confirm that the majority of exogenously expressed IFI44 retains within the cytoplasm, which is consistent with an earlier study that describes IFI44 as a cytoplasmic protein that may associate with microtubules [20]. However, the surprising discovery from our study is that IFI44 partially diffuses into nuclei of HEK293 cells upon HIV-1 infection (Figures 3A, S3A–B). Hallen et al. [20] claimed that endogenous IFI44 was detected in the nuclei of nonstimulated human melanoma cell lines ME15 and D10 cells by immunostaining using a custom-made anti-IFI44 antibody. They failed to confirm the presence of IFI44 in nuclear fraction using the same antibody; therefore, IFI44 nuclear localization is inconclusive based on their studies. Using a HA epitope fused IFI44 cDNA, we clearly show that IFI44 partially localizes within nuclei, which is further enhanced upon HIV-1 infection. More importantly, we demonstrate that IFI44 in nuclear fraction is strongly associated with the HIV-1 LTR promoter region by ChIP-PCR assays (Figure 3B). This is critical evidence that may explain IFI44’s inhibitory effect on HIV-1 LTR activity and viral transcription (Figures 1D, 2D). We rule out the possibility that increased IFI44 localization in nuclei or IFI44 recruitment to LTR upon HIV-1 infection may be mediated by interaction with HIV-1 TAT protein, which encodes a nuclear localization sequence (NLS) [36] and associates with HIV-1 LTR through TAR RNA [37, 38], since our GST-TAT pull-down and FLAG-TAT co-immunoprecipitation assays failed to detect that TAT interacted with IFI44 (Figures S3D–E). We will continue to investigate exactly how HIV-1 infection facilitates IFI44 nuclear localization and by which means IFI44 is recruited to the HIV-1 LTR promoter to modulate its activity.
We also confirm that the presence of IFI44 may facilitate HIV-1 latency and prevent its reactivation, very likely through suppression of viral transcription. We tested cells harboring the mini-HIV genome (J-LAT A2) (Figure 4) and two J-LAT clones harboring Env-deleted full-length HIV genome (6.3, 9.2) (Figure S4). Our earlier studies suggest that the mini HIV-1 in J-LAT A2 is more susceptible to reactivation than the full-length HIV-1 in J-LAT clones, probably because sole induction of transcriptional initiation is not sufficient to potently reverse latency for full-length HIV-1. Latent stage of full-length HIV-1 may be subjected to other regulatory machineries. Since our data show that IFI44 is recruited to LTR (Figure 3B) and suppresses LTR promoter activity (Figures 1D, 2D), we expect that IFI44 may inhibit transcriptional initiation. Depletion of IFI44 is able to remove certain HIV transcriptional inhibition, which is more sufficient to reverse the mini HIV genome (J-LAT A2) than the full-length HIV (J-LAT 6.3 and 9.2). Type I IFN may not only restrict ongoing viral replication but also prevent the reactivation of HIV provirus. Thus, IFN-mediated inhibitory effects contribute substantially to establishment and/or maintenance of HIV-1 latency and persistent infection. An earlier study demonstrates that IFN-α suppresses HIV-1 gene expression in certain chronically infected cell lines [39]. Later, it is reported that by transferring human CD4+ T cells expressing IFN-β into a mouse model of persistent HIV infection results in reduced infection and a restoration of the CD4+ T cell population [40]. Recently, a study in a small group of AIDS patients suggests that IFN-α therapy may facilitate the reduction of HIV-1 proviral DNA level and clearance of HIV-1 latency reservoir in some individuals with well-controlled HIV-1 infection once antiretroviral therapy is interrupted [41, 42]. Furthermore, the antiretroviral capacity of IFN-α in vivo correlates with the increased expression of certain host restriction factors [43]. These results demonstrate that type I IFN treatment controls persistent HIV-1 infection in AIDS patients although the exact mechanism is still unclear. We postulate that IFI44 may be one of the mediators of the IFN-α antiviral effect, which partially suppresses latent HIV-1 from reactivation, and its role in the IFN-α regulation of HIV-1 persistence in vivo shall be further investigated in future.
In summary, we identify an IFN-α inducible protein, IFI44, as a HIV-1 restriction factor. HIV-1 infection may lead to IFI44 localization to nuclei and recruitment to the HIV-1 LTR promoter region, which could be a prerequisite for the suppression of HIV-1 transcription to promote its latency. HIV-1 latent reservoirs remain the major obstacle for HIV-1 elimination. Understanding host restrictive machineries facilitating HIV-1 latency, especially those induced by type I IFN, including IFI44, will be crucial for identifying new gene targets for purging latent HIV-1 reservoirs as a cure strategy.
Materials and Methods
Cell lines
The following cells were generously provided by the NIH AIDS reagent repository: MAGI-HeLa, Jurkat Clone E6-1 (Catalog # 177, [44]), J-LAT A2 (Cat. # 9854), 6.3 (Cat. # 9846), 9.2 (Cat. # 9848). MAGI-HeLa and HEK293 cells were cultured in Dulbecco’s modified eagle medium (DMEM, Life technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies); all Jurkat-derived cells were cultured in RPMI 1640 medium (Life Technologies) supplemented with 10% FBS. For interferon treatment, recombinant human interferon alpha (IFN-α, 2.5 × 105 units/ml) or human interferon gamma (IFN-γ, 2.5 ug/ml) (PBL Assay Science) was added to the medium and incubated for 24 hours. AGS/BX cells were cultured in Ham’s F-12K nutrient mixture supplemented with 10% FBS.
Viruses
The HIV-1 IIIB wild-type strain [45] was obtained from the NIH AIDS Reagent Repository. VSV-G pseudotyped viruses were created by co-transfecting HEK293T cells with pCG-VSV-G and the following viral constructs: HIV-1 NL4-3-Luc (Δ Env), HIV-1 NL4-3-GFP (Δ Env), HIV-1 LTR-GFP (LTR-GFP), CMV-ZSG (pPHAGE-CMV-ZSG), and MLV-GFP [45, 46]. GFP positive cells were either imaged using an Image Xpress Micro microscope (Molecular Devices) or sorted by flow cytometry (BD FACSAria II). pAPM pseudo-typed lentiviruses were created by co-transfecting HEK293T cells with psPAX2 and pMD2.G (Addgene #12260, #12259) with the subcloned pAPM shRNA constructs.
Antibodies
An anti-HIV-1 p24 mouse monoclonal antibody, mab-183, was obtained from the NIH AIDS Reagent Repository. Mouse (purified Clone HA.11, Covance) and rabbit (Bethyl Laboratories Inc.) HA antibodies were used for immunoblotting and immunostaining of HA-IFI44. A Texas Red conjugated goat anti-rabbit (Rockland) and a goat anti-mouse Alex-Fluor 488 (Life Technologies) secondary antibody were used for immunostaining. GAPDH, E-cadherin, and JMJD1A antibodies (Santa Cruz Biotechnology) were used for immunoblotting. A goat anti-mouse or anti-rabbit IgG-HRP (Santa Cruz Biotechnology) was used as secondary for immunoblotting. The rabbit HA antibody and a Rabbit IgG isotype control (Southern Biotech) were used for chromatin immunoprecipitation and co-immunoprecipitation assays.
Plasmids
pQCXIP-HA-IFI44 was constructed by subcloning IFI44 with an N-terminal HA peptide into the pQCXIP retroviral vector (Clontech) using AgeI and PacI sites. Pseudotyped viruses were packaged using pCG-VSV-G and pCG-Gag-Pol vectors. pQCXIP-FLAG-TAT was constructed by subcloning TAT with C-terminal FLAG peptide into the pQCXIP vector using NotI and BamHI sites [29]. pCDNA-FLAG-TAT was obtained from the NIH AIDS Repository. pAPM shRNA vectors were constructed by subcloning shRNA species into pAPM vector using EcorI and XhoI sites.
RNAi reagents
siRNAs were used at 50 nM final concentration using Oligofectamine transfection lipid (Life Technologies). The control siRNA contains a pool of Silencer Negative siRNAs (No.1-4, AM4611, AM4613, AM4615 and AM4641, Life Technologies). IFI44 siRNAs: si1, 5′-UAU ACU UCU CAG AUA UCC C -3′; si2, 5′-UCU AUC ACG AAU GUU ACC G -3′ (Life Technologies). shRNAs were subcloned into the pAPM shRNA expression vector. pAPM is a microRNA (mir30)-based shRNA lentiviral vector where both the puromycin N-acetyltransferase cDNA and the shRNA are expressed via the spleen focus-forming virus (SFFV) promoter [47]. Production of pAPM shRNA lentivirus has been described previously [48]. The control shRNA targets firefly luciferase (FLuc, 5′-TAC AAA CGC TCT CAT CGA CAA G -3′). IFI44 shRNAs: sh1 (5′-TCT ATC ACG AAT GTT ACC GTT C -3′); sh2 (5′-TAT ACT TCT CAG ATA TCC CAG T -3′).
Immunostaining
MAGI-HeLa cells (approximately 3000 per well) were reverse transfected with a 50 nM final concentration of siRNA using Oligofectamine (Life Technologies) in 96-well plates. At 72 hours post-transfection, cells were infected with HIV-IIIB in 150 uL fresh DMEM medium. At 48 hours post-infection, cells were fixed using 4% paraformaldehyde (Sigma-Aldrich) in 1x Dulbecco’s phosphate-buffered saline (DPBS, Life Technologies), permeabilized using 0.2% Triton X-100 (Sigma-Aldrich) in 1x DPBS, and stained for intracellular HIV-1 p24 expression using the anti-p24 antibody, mAB-183, and goat anti-mouse Alexa-Flour 488. Cellular DNA was stained using Hoechst 33342 (Life Technologies). Immunostained cells were imaged using an Image Xpress Micro microscope (Molecular Devices) and the images analyzed to determine percent infection and cell number (cell scoring module, Metamorph, Molecular Devices). To evaluate IFI44 nuclear translocation, HEK293 cells stably expressing HA-IFI44 were incubated with VSV-G pseudo-typed HIV-NL4-3-GFP virus, or mock-treated for 48 hours. Fixation and permeabilization were performed as aforementioned. Cells were stained for IFI44 using an anti-HA.11 monoclonal rabbit antibody, 16B12, followed by a Texas Red conjugated goat anti-rabbit secondary antibody. Cellular DNA was stained with Hoechst 33342. Immunostained cells were imaged using an Olympus ILL-100 inverted fluorescence microscope. Analyses of the nuclear region of cells were completed using ImageJ image analysis software (NIH). Hoechst and anti-IFI44 (Texas Red) images were stacked. The oval tool was used to create a circular area slightly smaller than one nuclei. The oval was laid directly on a nucleus, and a pixel measurement was made for the corresponding anti-HA-IFI44 image. Fifty nuclei were measured per stack.
Nuclear and cytoplasmic fractionation
HA-IFI44 expressing HEK293 cells were infected with VSV-G pseudo-typed HIV-NL4-3-GFP virus (Δ Env) (MOI=5), or mock treated for 48 hours. Cells were collected, washed with 1x DPBS and lysed in CE buffer (10 mM HEPES [pH 7.9], 60 mM KCl, 1 mM EDTA, 1 mM DTT, 0.3% NP-40) supplemented with protease inhibitor (Pierce). Nuclei were pelleted at 4°C and rinsed once with CE buffer without NP-40. Nuclei were lysed in NE buffer (20 mM HEPES [pH 7.9], 420 mM NaCl, 1.5 mM MgCl2, 20% glycerol, adding 0.5 mM PMSF and protease inhibitor tablet freshly) and incubated at 4°C for 10 minutes. Glycerol was added to the CE fraction to 20%. Cytoplasmic and nuclear fractions were analyzed by SDS-PAGE and western blot using E-cadherin, JMJD1A, GAPDH and HA antibodies.
GST-TAT pull-down
pGEX2TK-TAT1(86R) (NIH AIDS Reagent Repository) or pGEX empty plasmid was transformed into E. Coli BL21 strain. Overnight culture was induced with 0.1 mM isopropyl-1-thio-galactosidase (IPTG) at 30°C. Cells were pelleted, re-suspended in 10 ml ice-cold E. Coli lysis buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 0.5 mM EDTA, 5 mM MgCl2, and freshly added 0.2% NP-40, 2 mM DTT, 0.2 mM PMSF, 200 ug/ml lysozyme, protease inhibitor tablet), and incubated on ice for 1 hour. Lysate was sonicated briefly and cellular debris was pelleted. Pre-washed glutathione beads (Promega) were incubated with the supernatant at 4°C for 2 hours with rotation. Beads were washed three times each with wash buffer I (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 1 mM EGTA, 10% glycerol, 0.1% Triton X-100, and freshly added 0.1% beta-mercaptoethanol and 1mM PMSF) and wash buffer II (50 mM HEPES [pH 7.5], 100mM NaCl, 1mM EGTA, 10% glycerol, and freshly added 0.1% BME, 1mM PMSF). GST or GST-TAT beads were eluted in LDS sample buffer (Thermo Scientific) and analyzed by SDS-PAGE and coomassie blue staining (Bio-rad). Confluent HEK293 cells stably expressing HA-IFI44 in a 10 cm plate were harvested and lysed in 1 ml RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% Na deoxycholate, 1mM EDTA, and freshly added protease inhibitor tablet), and incubated for 10 minutes on ice. Lysate was sonicated briefly and cellular debris was pelleted. To pre-clear the lysate, empty glutathione beads were added and incubated with rotation at 4°C. Beads were magnetically removed and the pre-cleared lysate was then incubated with GST or GST-TAT coated beads overnight at 4°C with rotation. The beads were washed four times with 1x RIPA buffer. Precipitated protein samples were eluted to 1x LDS sample buffer and analyzed by SDS-PAGE and western blot using an HA antibody.
Protein co-immunoprecipitation (co-IP)
HEK293 cells stably expressing HA-IFI44 were transfected with pcDNA-FLAG-TAT plasmid on a 10 cm plate. As a positive control, pcDNA-FLAG-TAT was co-transfected with a pcDNA-V5-HDAC1 plasmid. 48 hours post-transfection, cells were harvested and lysed in 1 ml RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% Na deoxycholate, 1mM EDTA, and freshly added protease inhibitor tablet). Cell lysate was sonicated briefly, cellular debris was pelleted and the supernatant was aspirated to a new tube. A small aliquot was taken to run as input. To pre-clear, 40 uL protein A/G beads were added to the lysate and incubated for 2 hours at 4°C. The beads were magnetically removed, the lysate was split equally and 2 ug of mouse HA or IgG control antibody was added for overnight rotation at 4°C. 20 ul of protein A/G beads were added to each sample and incubated with rotation for 2 hours. The beads were washed for 3 times with 1x RIPA buffer and precipitated protein samples were eluted to 1 × LDS sample buffer and analyzed by SDS-PAGE and western blot using a rabbit anti-FLAG antibody (Sigma).
Flow Cytometry
J-LAT A2, 6.3, or 9.2 cells stably expressing shRNA were induced with 1 ug/ml prostratin (Sigma-Aldrich) or DMSO as a negative control. 24 hours post-induction, GFP positive cells were sorted using a FACSCalibur flow cytometer (Becton Dickinson). Results were analyzed using FlowJo software (v10.0.7). Jurkat T cells stably expressing shRNA were infected with VSV-G pseudo-typed HIV-1 NL4-3-GFP virus for 48 hours and then subjected to flow cytometry assay. JLTRG cells (from the NIH AIDS reagent repository) stably expressing HA-IFI44 or empty vector were transduced with pcDNA-FLAG-TAT. 48 hours post-transduction, cells were sorted. For AGS/BX cells stably expressing HA-IFI44 or empty vector, EBV lytic replication was induced with combined treatment of 12-o-tetradecanoyl phorbol-13-acetate (TPA, 100nM, Promega) and sodium butyrate (SB, 3mM, Fisher Scientific) for 24 hours and subjected to flow cytometry.
Quantitative real-time PCR
SYBR Green primers were designed to measure the mRNA level of the following genes. IFI44 forward, 5′-CCA CCG AGA TGT CAG AAA GAG -3′; IFI44 reverse, 5′-TGG TAC ATG TGG CTT TGC TC -3′. GAPDH forward, 5′-GCC TCT TGT CTC TTA GAT TTG GTC -3′; GAPDH reverse, 5′-TAG CAC TCA CCA TGT AGT TGA GGT -3′. Total mRNA extracted from cells (1 ug) was reverse-transcribed using random hexamers (0.1 uM) and SuperScript III Reverse Transcriptase following the manufacturer’s instruction (Life Technologies). The reverse-transcribed cDNA template was mixed with the primers (0.1 uM each) and 2x Platinum SYBR Green qPCR SuperMix (Life Technologies), which was subjected to real-time PCR analysis on a 7500 Fast PCR-System (Applied Biosystems). The following thermal cycles were used: 95 °C, 1 min / 95 °C, 5 sec; 60 °C, 30 sec; 40 cycles.
Luciferase assay
HEK293 cells stably expressing HA-IFI44 or empty vector were transfected with a pcDNA-TAT vector, a HIV-1 LTR luciferase reporter construct (NIH AIDS Reagent Repository), along with pRL-TK (Promega) as an internal transfection control. Cells were processed using the Dual-Glo Luciferase Assay System (Promega). Plates were read using a Luminoskan Ascent Microplate Luminometer (Thermo Fisher Scientific). Relative light units (RLU) were measured by normalizing the signal from firefly luciferase with that from Renilla luciferase.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays followed a protocol previously described [49] with minor changes. HEK293 cells expressing HA-IFI44 were transduced with pseudo-typed HIV-1 NL4-3-GFP. 48 hours post-infection, paraformaldehyde (Fisher Scientific) was added to the culture media to 0.5% and cells were incubated at room temperature. To stop the cross-linking reaction, glycine (Fisher Scientific) was added (125 mM final concentration). Cells were washed twice in ice-cold PBS and re-suspended in CE buffer (10 mM HEPES-KOH [pH 7.9], 60 mM KCl, 1 mM EDTA, 1 mM DTT, 0.5% NP-40, protease inhibitor tabled added freshly) at a concentration of 107 cells/ml. Nuclei were pelleted and re-suspended in SDS lysis buffer (50 mM Tris-HCl [pH 8.1], 1% SDS, 10 mM EDTA, added protease inhibitor tablet freshly) at a concentration of 5 × 107 cells/ml and sonicated (model 120 sonic dismembrator, Fisher Scientific) for 2 minutes at 50% output with 1 second pulsing. Sonicated nuclear extract was diluted 1:10 in ChIP dilution buffer (16.7 mM Tris HCl [pH 8.1], 0.01% SDS, 1% Triton X-100, 1.2 mM EDTA, 150 mM NaCl, protease inhibitor tabled added freshly) and split for each immunoprecipitation and input control. Input control was temporarily stored at −80°C. 5ug anti-HA or RIgG isotype control was added and samples were incubated at 4°C rotating overnight. Protein A/G magnetic beads (Thermo Scientific Pierce) were pre-incubated for 1 hour with 0.5 mg/ml BSA (Fisher Scientific) and 0.125 mg/ml sonicated herring sperm DNA (Promega) in PBS. 50 ul of the pre-incubated beads were added to each sample and incubated for 2 hours at 4°C with rotation. Beads were magnetically recovered and washed once in low-salt immune complex wash buffer (20 mM Tris-HCl [pH 8.1], 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl), once in high-salt immune complex wash buffer (20 mM Tris-HCl [pH 8.1], 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl), once in LiCl buffer (10 mM Tris-HCl [pH8.1], 0.25 M LiCl, 1% NP-40, 1% Na deoxycholate, 1 mM EDTA), and twice in TE buffer (10 mM Tris-HCl [pH 8.1], 0.1 mM EDTA). Input, RIgG and HA-IFI44 samples were incubated at 65°C with 20 ul of 5M NaCl for 6 hours and at 50°C for 2 hours with 1 ul of 20mg/ml proteinase K (Life Technology), 10 ul of 0.5M EDTA and 40 ul of 0.5M Tris-HCl. DNA samples were extracted using phenol: chloroform (Fisher Scientific) and recovered with standard ethanol precipitation. Pelleted DNA samples were re-suspended in nuclease-free water and subjected to semi-quantitative PCR assays using primers recognizing various regions of HIV-1 LTR promoter region [50]: Nuc-0 forward, 5′-GAA GGG CTA ATT TGG TCC CA -3′; Nuc-0 reverse, 5′-GAT GCA GCT CTC GGG CCA TG -3′. L1(PPR) forward, 5′-CGA GAG CTG CAT CCG GAG TA -3′; L1(PPR) reverse, 5′-AGC TTT ATT GAG GCT TAA GC -3′. Nuc-1 forward, 5′-AGT AGT GTG TGC CCG TCT GT -3′; Nuc-1 reverse, 5′-TTG GCG TAC TCA CCA GTC GC -3′. Nuc-2 forward, 5′-ATT TTG ACT AGC GGA GGC TA -3′; Nuc-2 reverse, 5′-ACA GCC TTC TGA TGT CTC TA -3′. DNA template and primers were mixed with 2x PCR master mix (Thermo Scientific). The following thermal cycles were used: 98°C, 30 sec / 98°C, 5 sec; 50°C, 10 sec; 72°C, 15 sec / 72°C 1 min; 30 cycles. PCR amplicons were separated on a 2% agarose gel in 1x Lithium Borate buffer (0.8 g/L lithium hydroxide monohydrate, 3.6 g/L boric acid, pH 8.0) and analyzed using the Gel Doc XR BioRad imaging system (Universal Hood II).
Supplementary Material
Figure S1. IFI44 expression is induced by IFN-α. (A). MAGI-HeLa, Jurkat T, CD4+ T, and CD8+ T cells were subjected to RNA extraction, reverse transcription, and quantitative real-time PCR for measuring basal IFI44 mRNA level. Data were normalized to the MAGI-HeLa cells. MAGI-HeLa (B) or Jurkat T (C) cells were treated with IFN-α, IFN-γ, or mock treated for 48 hours. Total RNA was extracted for reverse transcription and quantitative real-time PCR for measuring the IFI44 mRNA level. Data were normalized to the mock-treated cells. All results were presented as mean ± s.d. (n = 3); * P < 0.05 from t-test.
Figure S2. Exogenous expression of IFI44 has no effect on EBV replication. (A). pQCXIP-HA-IFI44 or pQCXIP empty vector was transduced in GFP-fused EBV infected AGS gastric cells (AGS/BX). Total RNA was extracted for reverse transcription and quantitative real-time PCR to measure the IFI44 mRNA level. Data were normalized to the empty-vector cells (n = 3, mean ± s.d.; *, P < 0.05; t-test). (B). AGS/BX cells stably expressing HA-IFI44 or empty vector were treated with TPA/SB for 24 hours to induce EBV lytic replication. GFP positive cells were assessed using flow cytometry. Data are one representative of three independent experiments.
Figure S3. IFI44 enters nuclei upon HIV-1 infection but does not bind with TAT. (A). HEK293 cells stably expressing HA-IFI44 were infected with VSV-G pseudo-typed HIV-NL4-3-GFP [Δ Env] viruses (+HIV) or mock treated (-HIV) for 48 hours. Cellular localization of HA-IFI44 was determined through staining using a mouse anti-HA antibody and a Texas Red anti-mouse secondary antibody. Nuclei were stained using Hoechst. (B). Fluorescence intensity at the Texas Red channel in nuclei was quantified using ImageJ software. Data were averaged from analysis of fifty cells (n = 50, mean ± s.d.; *, P < 0.05; t-test). (C). An illustration of the nuc-0, L1 nucleosome-free region (PPR), nuc-1, and nuc-2 regions of HIV-1 LTR promoter, adapted from [50]. (D). HA-IFI44 cell lysate was incubated with GST-TAT or GST beads. Associated proteins were eluted in laemmli protein sample buffer. The cell lysate (input) and protein elutes were separated by SDS-PAGE for western blot using an anti-HA antibody. (E). HEK293 cells stably expressing HA-IFI44 were transfected with pcDNA-FLAG-TAT vector. Meanwhile, pcDNA-V5-HDAC1 and pcDNA-FLAG-TAT vectors were co-transfected into HEK293 cells. Cell lysates were incubated with either anti-HA (HA-IFI44) or anti-V5 (V5-HDAC1) antibody for immunoprecipitations. Associated proteins were eluted in laemmli protein sample buffer. Protein elutes were separated by SDS-PAGE for western blot using an anti-FLAG (FLAG-TAT) antibody.
Figure S4. IFI44 depletion synergizes with Prostratin to reverse HIV-1 latency in J-LAT 6.3 and 9.2 cells. shIFI44-2 or shFLuc in pAPM vector was transduced into J-LAT 6.3 (A) or 9.2 (B) cells. Equal numbers of J-LAT 6.3 or 9.2 cells stably expressing shIFI44-2 or shFLuc were treated with Prostratin or DMSO for 24 hours and the percentage of GFP positive cells were assessed by flow cytometry. Data are one representative of three independent experiments.
Research Highlights.
IFI44 inhibits HIV-1 intracellular replication in vitro.
IFI44 suppresses HIV-1 LTR promoter activity and affects viral transcription.
IFI44 localizes to nuclei and binds to the HIV-1 LTR promoter region in cells infected with HIV-1.
IFI44 promotes HIV-1 latency and suppresses its reactivation in HIV-1 latently infected cell lines.
Acknowledgments
This work is supported by a grant from NIH/NIAID (R21AI116180) to J.Z. and a grant from the Bill and Melinda Gates Foundation to S.J.E. J.Z. is grateful to the Harvard CFAR, and supported in part by the UR CFAR grant from NIH/NIAID (P30AI078498) and the UR Center for Integrative Bioinformatics and Experimental Mathematics (CIBEM) pilot grant. We also thank Stephen Dewhurst (URMC) and Abraham Brass (UMass) for helpful discussions. S.J.E. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baca-Regen L, et al. Alpha interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes. J Virol. 1994;68(11):7559–65. doi: 10.1128/jvi.68.11.7559-7565.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coccia EM, Krust B, Hovanessian AG. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J Biol Chem. 1994;269(37):23087–94. [PubMed] [Google Scholar]
- 3.Fernie BF, Poli G, Fauci AS. Alpha interferon suppresses virion but not soluble human immunodeficiency virus antigen production in chronically infected T-lymphocytic cells. J Virol. 1991;65(7):3968–71. doi: 10.1128/jvi.65.7.3968-3971.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gendelman HE, et al. Regulation of HIV replication in infected monocytes by IFN-alpha. Mechanisms for viral restriction. J Immunol. 1990;145(8):2669–76. [PubMed] [Google Scholar]
- 5.Gendelman HE, et al. Restriction of HIV replication in infected T cells and monocytes by interferon-alpha. AIDS Res Hum Retroviruses. 1990;6(8):1045–9. doi: 10.1089/aid.1990.6.1045. [DOI] [PubMed] [Google Scholar]
- 6.Hansen BD, et al. Loss of infectivity by progeny virus from alpha interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J Virol. 1992;66(12):7543–8. doi: 10.1128/jvi.66.12.7543-7548.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barr SD, Smiley JR, Bushman FD. The interferon response inhibits HIV particle production by induction of TRIM22. PloS Pathog. 2008;4(2):e1000007. doi: 10.1371/journal.ppat.1000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malim MH, Bieniasz PD. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb Perspect Med. 2012;2(5):a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strebel K, Luban J, Jeang KT. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009;7(48) doi: 10.1186/1741-7015-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris RS, Hultquist JF, Evans DT. The restriction factors of human immunodeficiency virus. J Biol Chem. 2012;287(49):40875–83. doi: 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitamura A, et al. Induction of the human gene for p44, a hepatitis-C-associated microtubular aggregate protein, by interferon-alpha/beta. Eur J Biochem. 1994;224(3):877–83. doi: 10.1111/j.1432-1033.1994.00877.x. [DOI] [PubMed] [Google Scholar]
- 12.Honda Y, et al. Isolation and purification of a non-A, non-B hepatitis-associated microtubular aggregates protein. J Gen Virol. 1990;71(9):1999–2004. doi: 10.1099/0022-1317-71-9-1999. [DOI] [PubMed] [Google Scholar]
- 13.Bochkov YA, et al. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3(1):69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaczkowski B, et al. Integrative analyses reveal novel strategies in HPV11,-16 and -45 early infection. Sci Rep. 2012;2(515) doi: 10.1038/srep00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boccardo E, et al. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor-alpha. Carcinogenesis. 2010;31 (3):521–31. doi: 10.1093/carcin/bgp333. [DOI] [PubMed] [Google Scholar]
- 16.Wie SH, et al. HIV downregulates interferon-stimulated genes in primary macrophages. J Interferon Cytokine Res. 2013;33(2):90–5. doi: 10.1089/jir.2012.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balan V, et al. Modulation of interferon-specific gene expression by albumin-interferon-alpha in interferon-alpha-experienced patients with chronic hepatitis C. Antivir Ther. 2006;11(7):901–8. [PubMed] [Google Scholar]
- 18.Liu C, et al. Anti-hepatitis C virus activity of albinterferon alfa-2b in cell culture. Hepatol Res. 2007;37(11):941–7. doi: 10.1111/j.1872-034X.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 19.Carlton-Smith C, Elliott RM. Viperin, MTAP44, and protein kinase R contribute to the interferon-induced inhibition of Bunyamwera Orthobunyavirus replication. J Virol. 2012;86(21):11548–57. doi: 10.1128/JVI.01773-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallen LC, et al. Antiproliferative activity of the human IFN-alpha-inducible protein IFI44. J Interferon Cytokine Res. 2007;27(8):675–80. doi: 10.1089/jir.2007.0021. [DOI] [PubMed] [Google Scholar]
- 21.Zhu J, et al. Comprehensive Identification of Host Modulators of HIV-1 Replication using Multiple Orthologous RNAi Reagents. Cell Reports. 2014;9(2):752–66. doi: 10.1016/j.celrep.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ochsenbauer-Jambor C, et al. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques. 2006;40(1):91–100. doi: 10.2144/000112072. [DOI] [PubMed] [Google Scholar]
- 23.Verdin E, Paras PJ, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12(8):3249–59. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22(8):1868–77. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams SA, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279(40):42008–17. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 26.Schoggins JW, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505(7485):691–5. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–5. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J, et al. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Reports. 2012;2(4):807–16. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aamer HA, et al. Resistance to simian immunodeficiency virus low dose rectal challenge is associated with higher constitutive TRIM5α expression in PBMC. Retrovirology. 2014;11:39. doi: 10.1186/1742-4690-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ooi EL, et al. Novel antiviral host factor, TNK1, regulates IFN signaling through serine phosphorylation of STAT1. Proc Natl Acad Sci U S A. 2014;111(5):1909–14. doi: 10.1073/pnas.1314268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 33.Douglas JL, et al. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J Virol. 2009;83(16):7931–47. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romani B, Cohen EA. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol. 2012;2(6):749–57. doi: 10.1016/j.coviro.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahn J, et al. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem. 2012;287(15):12550–8. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Efthymiadis A, Briggs LJ, Jans DA. The HIV-1 Tat nuclear localization sequence confers novel nuclear import properties. J Biol Chem. 1998;273(3):1623–8. doi: 10.1074/jbc.273.3.1623. [DOI] [PubMed] [Google Scholar]
- 37.Selby MJ, Peterlin BM. Trans-activation by HIV-1 Tat via a heterologous RNA binding protein. Cell. 1990;62(4):769–76. doi: 10.1016/0092-8674(90)90121-t. [DOI] [PubMed] [Google Scholar]
- 38.Dingwall C, et al. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (TAR) RNA in vitro. Proc Natl Acad Sci U S A. 1989;86(18):6925–9. doi: 10.1073/pnas.86.18.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poli G, et al. Interferon-alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science. 1989;244(4904):575–7. doi: 10.1126/science.2470148. [DOI] [PubMed] [Google Scholar]
- 40.Vieillard V, et al. Transfer of human CD4(+) T lymphocytes producing beta interferon in Hu-PBL-SCID mice controls human immunodeficiency virus infection. J Virol. 1999;73(12):10281–8. doi: 10.1128/jvi.73.12.10281-10288.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azzoni L, et al. Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J Infect Dis. 2013;207(2):213–22. doi: 10.1093/infdis/jis663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNamara LA, Collins KL. Interferon alfa therapy: toward an improved treatment for HIV infection. J Infect Dis. 2012;207(2):201–3. doi: 10.1093/infdis/jis667. [DOI] [PubMed] [Google Scholar]
- 43.Pillai SK, et al. Role of retroviral restriction factors in the interferon-α-mediated suppression of HIV-1 in vivo. Proc Natl Acad Sci U S A. 2012;109(8):3035–40. doi: 10.1073/pnas.1111573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiss A, Wiskocil RL, Stobo JD. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J Immunol. 1984;133(1):123–8. [PubMed] [Google Scholar]
- 45.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 46.Valle-Casuso JC, et al. TNPO3 is Required for HIV-1 Replication After Nuclear Import but Prior to Integration and Binds the HIV-1 Core. J Virol. 2012 doi: 10.1128/JVI.00451-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pertel T, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472(7343):361–5. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernasconi R, et al. Cyclosporine A-sensitive, cyclophilin B-dependent endoplasmic reticulum-associated degradation. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0013008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jadlowsky JK, et al. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol Cell Biol. 2014;34(11):1911–28. doi: 10.1128/MCB.01013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lusic M, et al. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22(24):6550–61. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IFI44 expression is induced by IFN-α. (A). MAGI-HeLa, Jurkat T, CD4+ T, and CD8+ T cells were subjected to RNA extraction, reverse transcription, and quantitative real-time PCR for measuring basal IFI44 mRNA level. Data were normalized to the MAGI-HeLa cells. MAGI-HeLa (B) or Jurkat T (C) cells were treated with IFN-α, IFN-γ, or mock treated for 48 hours. Total RNA was extracted for reverse transcription and quantitative real-time PCR for measuring the IFI44 mRNA level. Data were normalized to the mock-treated cells. All results were presented as mean ± s.d. (n = 3); * P < 0.05 from t-test.
Figure S2. Exogenous expression of IFI44 has no effect on EBV replication. (A). pQCXIP-HA-IFI44 or pQCXIP empty vector was transduced in GFP-fused EBV infected AGS gastric cells (AGS/BX). Total RNA was extracted for reverse transcription and quantitative real-time PCR to measure the IFI44 mRNA level. Data were normalized to the empty-vector cells (n = 3, mean ± s.d.; *, P < 0.05; t-test). (B). AGS/BX cells stably expressing HA-IFI44 or empty vector were treated with TPA/SB for 24 hours to induce EBV lytic replication. GFP positive cells were assessed using flow cytometry. Data are one representative of three independent experiments.
Figure S3. IFI44 enters nuclei upon HIV-1 infection but does not bind with TAT. (A). HEK293 cells stably expressing HA-IFI44 were infected with VSV-G pseudo-typed HIV-NL4-3-GFP [Δ Env] viruses (+HIV) or mock treated (-HIV) for 48 hours. Cellular localization of HA-IFI44 was determined through staining using a mouse anti-HA antibody and a Texas Red anti-mouse secondary antibody. Nuclei were stained using Hoechst. (B). Fluorescence intensity at the Texas Red channel in nuclei was quantified using ImageJ software. Data were averaged from analysis of fifty cells (n = 50, mean ± s.d.; *, P < 0.05; t-test). (C). An illustration of the nuc-0, L1 nucleosome-free region (PPR), nuc-1, and nuc-2 regions of HIV-1 LTR promoter, adapted from [50]. (D). HA-IFI44 cell lysate was incubated with GST-TAT or GST beads. Associated proteins were eluted in laemmli protein sample buffer. The cell lysate (input) and protein elutes were separated by SDS-PAGE for western blot using an anti-HA antibody. (E). HEK293 cells stably expressing HA-IFI44 were transfected with pcDNA-FLAG-TAT vector. Meanwhile, pcDNA-V5-HDAC1 and pcDNA-FLAG-TAT vectors were co-transfected into HEK293 cells. Cell lysates were incubated with either anti-HA (HA-IFI44) or anti-V5 (V5-HDAC1) antibody for immunoprecipitations. Associated proteins were eluted in laemmli protein sample buffer. Protein elutes were separated by SDS-PAGE for western blot using an anti-FLAG (FLAG-TAT) antibody.
Figure S4. IFI44 depletion synergizes with Prostratin to reverse HIV-1 latency in J-LAT 6.3 and 9.2 cells. shIFI44-2 or shFLuc in pAPM vector was transduced into J-LAT 6.3 (A) or 9.2 (B) cells. Equal numbers of J-LAT 6.3 or 9.2 cells stably expressing shIFI44-2 or shFLuc were treated with Prostratin or DMSO for 24 hours and the percentage of GFP positive cells were assessed by flow cytometry. Data are one representative of three independent experiments.