

ABSTRACT
The opportunistic pathogen Pseudomonas aeruginosa produces two major cell surface lipopolysaccharides, characterized by distinct O antigens, called common polysaccharide antigen (CPA) and O-specific antigen (OSA). CPA contains a polymer of d-rhamnose (d-Rha) in α1-2 and α1-3 linkages. Three putative glycosyltransferase genes, wbpX, wbpY, and wbpZ, are part of the CPA biosynthesis cluster. To characterize the enzymatic function of the wbpZ gene product, we chemically synthesized the donor substrate GDP-d-Rha and enzymatically synthesized GDP-d-[3H]Rha. Using nuclear magnetic resonance (NMR) spectroscopy, we showed that WbpZ transferred one d-Rha residue from GDP-d-Rha in α1-3 linkage to both GlcNAc- and GalNAc-diphosphate-lipid acceptor substrates. WbpZ is also capable of transferring d-mannose (d-Man) to these acceptors. Therefore, WbpZ has a relaxed specificity with respect to both acceptor and donor substrates. The diphosphate group of the acceptor, however, is required for activity. WbpZ does not require divalent metal ion for activity and exhibits an unusually high pH optimum of 9. WbpZ from PAO1 is therefore a GDP-d-Rha:GlcNAc/GalNAc-diphosphate-lipid α1,3-d-rhamnosyltransferase that has significant activity of GDP-d-Man:GlcNAc/GalNAc-diphosphate-lipid α1,3-d-mannosyltransferase. We used site-directed mutagenesis to replace the Asp residues of the two DXD motifs with Ala. Neither of the mutant constructs of wbpZ (D172A or D254A) could be used to rescue CPA biosynthesis in the ΔwbpZ knockout mutant in a complementation assay. This suggested that D172 and D254 are essential for WbpZ function. This work is the first detailed characterization study of a d-Rha-transferase and a critical step in the development of CPA synthesis inhibitors.
IMPORTANCE This is the first characterization of a d-rhamnosyltransferase and shows that it is essential in Pseudomonas aeruginosa for the synthesis of the common polysaccharide antigen.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium that is ubiquitous in the environment and is an opportunistic pathogen that can cause life-threatening infections in humans whose defenses are compromised, e.g., those with immune deficiency, burn wounds, cancer, or cystic fibrosis (CF). Specific epidemic strains have been shown to cause local outbreaks and hospital-associated infections (1). In the airways of CF patients, P. aeruginosa thrives, adopting a biofilm lifestyle, and often becomes resistant to antibiotic treatment. Therefore, it is important to understand the mechanisms by which P. aeruginosa synthesizes its virulence factor, in order to develop new antibacterial strategies.
Lipopolysaccharides (LPS) on the outer membrane of P. aeruginosa are required for its survival against host defense mechanisms; hence, LPS is one of the major virulence factors (2–4). These bacteria are unusual in that they simultaneously synthesize two distinct forms of LPS differing in the O-antigen structures (5). Each of these O antigens is synthesized by a different pathway.
The heteropolymeric O antigen (O-specific antigen [OSA], formerly called B band) is the basis for serotyping of P. aeruginosa. The most commonly studied P. aeruginosa strain PAO1 belongs to the serotype O5 and has OSA repeating units containing two residues of mannosuronic acid derivatives with N-acetyl-d-fucosamine (d-FucNAc) at the reducing end (5, 6).
The other form of LPS contains the common polysaccharide antigen (CPA) (formerly called A band) as its O antigen, and this antigen is conserved in most serotypes of P. aeruginosa. The structure of CPA consists of a homopolymer of d-rhamnose (6-deoxy-d-mannose) (d-Rha) residues with the repeating unit [d-Rhaα1-2-d-Rhaα1-3-d-Rhaα1-3] (7, 8). d-Rha is a rare sugar in bacterial polysaccharides and is not found in human glycoconjugates. Interestingly, a different enantiomer of Rha, l-Rha, is a constituent of the LPS core oligosaccharide (2) of the same LPS and is also a component of the surfactant rhamnolipid of P. aeruginosa. Both OSA and CPA polysaccharides are presumably linked to the 3 position of l-Rha in the core oligosaccharide, and this l-Rha residue is α1-3 linked to glucose (9). Thus, P. aeruginosa can synthesize oligosaccharides containing both l-Rha and d-Rha.
Genetic studies revealed that the biosynthetic locus for CPA contains contiguous 8-gene and 5-gene operons (10). The presence of genes encoding the ABC transporter system Wzm/Wzt in the 8-gene operon led to the proposal that the biosynthetic pathway of CPA is ABC transporter dependent (11). In the model of this pathway, glycosyltransferases (GTs) are thought to transfer sugars from nucleotide sugars in the cytoplasm utilizing the membrane-bound undecaprenol-phosphate (P-Und) acceptor intermediate. The complete CPA is synthesized in the cytoplasm by the addition of individual sugar residues. It is also possible that one of these transferases is a polymerase that polymerizes the polysaccharide chain in a processive fashion, without releasing the intermediate products. Subsequent processing may require a chain termination event, such as methylation, and the transport of the long polysaccharide chain by the ABC transporter with the transmembrane domain Wzm and the nucleotide and carbohydrate binding domain Wzt to the periplasmic space (4, 11), where it is ligated to the outer core oligosaccharide bound to lipid A. The completed LPS is then transported to the outer membrane by the Lpt transporter complex (12).
The enzyme that initiates CPA polysaccharide synthesis in the PAO1 strain is thought to be sugar-phosphate transferase WbpL, a homolog of WecA that transfers GlcNAc-1-phosphate or GalNAc-1-phosphate to P-Und (2, 13, 14). The wbpL gene is present in the OSA biosynthesis gene cluster, and thus WbpL may also initiate OSA synthesis and transfer d-FucNAc-1-phosphate from UDP-d-FucNAc to P-Und. Three glycosyltransferase-encoding genes, wbpX, wbpY, and wbpZ, are localized in the 8-gene CPA biosynthesis operon. Rocchetta et al. (15) proposed that WbpZ adds a d-Rha residue to GlcNAc-PP-Und, followed by two d-Rhaα1-3 residues (to Rha) added by WbpY and then Rhaα1-2 (to Rha) by WbpX. The rmd and gmd genes in the CPA O-antigen gene cluster are involved in the synthesis of the GDP-d-Rha donor substrate (16). The biosynthetic pathway for GDP-d-Rha involves the conversion of GDP-d-Man to GDP-6-deoxy-d-lyxo-4-hexulose by GDP-d-mannose-4,6-dehydratase (GMD). The stereo-specific GDP-4-dehydro-6-deoxy-d-mannose reductase (RMD) then synthesizes GDP-d-Rha. In P. aeruginosa, the GMD enzyme also has RMD activity. The donor substrate for L-Rha-transferases, dTDP-l-Rha, is synthesized by a different pathway, from dTDP and glucose-1-phosphate (13, 17). Our group recently reported a novel synthesis of GDP-d-Rha and its use in assays for WbpZ d-Rha-transferase activity (18). In this paper, we report the synthesis of high yields of GDP-d-Rha as well as the enzymatic synthesis of radioactive GDP-d-Rha as donor substrates to extensively characterize WbpZ.
The three putative d-Rha-transferases WbpX, -Y, and -Z encoded by the CPA biosynthesis gene cluster in P. aeruginosa have been classified (CAZy data bank) within the very large GT4 family of retaining GTs with a predicted GT-B fold. WbpX shares 18% and WbpY shares 19% identity with WbpZ. WbpZ has four EX7E motifs, while WbpX has three and WbpY has two EX7E motifs, that appear to be conserved in α-Man-transferases (19, 20). WbpZ has two centrally located DXD motifs in the amino acid sequence (21) that may potentially contain a catalytic base. WbpY has several DXD motifs, but their presence is not immediately apparent in the WbpX sequence. Other bacteria also have putative Rha-transferases; for example, WbpZ from Serratia marcescens has 38% sequence identity to WbpZ from PAO1 (determined by UniProt) (see Table S1 and Fig. S1 in the supplemental material). The highest sequence identity (53.4%) was found in WejK encoded by the Escherichia coli O99 antigen synthesis gene cluster (22). The O99 antigen is similar to that of CPA of P. aeruginosa and has a backbone of d-Rhaα1-2-d-Rhaαα1-3-d-Rhaα1-3 with d-Glcα1-2 residues linked to Rha1-2 and Rha1-3. To the best of our knowledge, WejK has not been biochemically characterized, but based on the high sequence identity and similarity of the d-rhamnan structure shared by PAO1 and E. coli O99, one could speculate that WejK is an ortholog of WbpZ.
A number of putative Man-transferases in various bacteria also have significant homology to WbpZ. These include LpcC, GumH, and WejO. These transferases have one or more DXD or modified-DXD motifs, as well as one or more EX7E motifs. Those GT4 enzymes that have been characterized include Man-transferase PimB from Corynebacterium glutamicum with 17% identity, Man-transferase WbdA from E. coli O9 which has two GT domains and low overall sequence identity but significant homology in the N-terminal domain of the protein, as well as Man-transferases from E. coli O9a WbdB (18% identity) and WbdC from E. coli O8 (48% identity) involved in the synthesis of the Man-containing adaptor structure of Man polymers (19, 20).
Based on the high degree of identity, the function of WbpZ was anticipated to be the first enzyme to add a d-Rha residue to the lipid carrier (2). This would form an adaptor sugar structure upon which the CPA O-antigen repeating units are built. Based on their sequences and sizes, WbpZ, -Y, and -X are expected to have only one catalytic GT domain, being highly homologous to d-Rha-transferases and resembling Man-transferases. All three enzymes are essential, since mutants lacking the genes encoding any of the WbpZ, -Y, and -X proteins do not make the CPA polysaccharide (2, 14).
In this work, we showed that WbpZ is the second enzyme in the repeating-unit synthesis of CPA and transferred one d-Rha residue from GDP-d-Rha to the synthetic acceptor GlcNAcα-PO3-PO3-(CH2)11-O-phenyl (GlcNAc-PP-PhU). WbpZ is a nonprocessive α1,3-d-Rha-transferase, essential for the synthesis of the CPA antigen, with a relaxed acceptor and donor specificity. By mutational analysis we showed that three acidic amino acids of WbpZ are important for the activity and that two of these are essential for the synthesis of O antigen in PAO1. WbpZ therefore catalyzes a critical step in CPA synthesis and can be blocked by small-molecule inhibitors that could be developed into new antibacterial drugs.
MATERIALS AND METHODS
Materials.
Materials were purchased from Sigma-Aldrich, unless otherwise stated. d-Rhamnose was purchased from Carbosynth (Compton, United Kingdom). GDP-[3H]Man and other radioactive nucleotide sugars were from American Radiolabeled Chemicals. GlcNAc and GalNAc derivatives were synthesized as reported earlier (23–25). Anthracenyl derivatives (24) and potential inhibitors were synthesized as described previously (25–27). Glycopeptides were kindly donated by Hans Paulsen, University of Hamburg, Germany. The synthesis of benzylα-d-rhamnopyranoside (compound 1) (d-Rhaα-Bn) is described in detail in the supplemental material (28). GDP-d-Rha was synthesized as described previously (18) by a reaction in aqueous solution. To increase the yield of GDP-d-Rha, GMP-morpholidate and d-Rha-1-phosphate were coupled. Full details of the intermediate steps (29–31) are given in the supplemental material.
Enzymatic synthesis of GDP-d-[3H]Rha donor substrate.
Radioactive GDP-d-[3H]Rha was synthesized from GDP-[3H]Man using GMD and RMD, expressed in BL21 bacteria. The gmd gene from P. aeruginosa was in the pQE30 plasmid (Kanr) in E. coli M15 with pRER4 (Ampr). The rmd gene was from Aneurinibacillus thermoaerophilus in the pQE80 plasmid (Kanr). After culturing at 6 h with either 100 μg/ml ampicillin (GMD) or 50 μg/ml kanamycin (RMD), IPTG (isopropyl-β-d-thiogalactopyranoside) was added to make a 1 mM solution and proteins expressed overnight at room temperature. SDS-PAGE and Western blots (14, 23) showed that the 4,6-dehydratase GMD and reductase RMD were well expressed. Isolated bacteria were sonicated in 90% phosphate-buffered saline (PBS)–10% glycerol. First, 2.8 μmol GDP-[3H]Man was incubated with 1 ml GMD homogenate (15 mg protein), 10 μmol MnCl2, and 250 μmol Tris-HCl (pH 7.5) in a total of 2 ml for 2 h at 37°C. This was followed by the addition of 1 ml RMD homogenate (15 mg protein) and 500 μmol NADPH in a total volume of 3.25 ml. After incubation for 1 h at 37°C, 6 ml of acetonitrile was added, and mixtures were centrifuged at 8,000 × g for 10 min. The supernatant was subjected to high-pressure liquid chromatography (HPLC) using an analytical amine column and 50 mM KH2PO4 buffer (pH 5.2), showing a >95% conversion of GDP-[3H]Man to GDP-[3H]Rha, monitored by radioactivity and absorbance at 254 nm (A254). GDP-[3H]Rha eluted at 56 min and a small peak of GDP-[3H]Man at 63 min. Hence, the condition of 5 mM triethylammonium acetate buffer at pH 6 was appropriate to separate GDP-d-Man from GDP-d-Rha.
Cloning and expression of wild-type and mutant WpbZ.
The wbpZ gene in the pET30a+ plasmid (pET-wbpZ) was expressed as a C-terminally His-tagged WbpZ protein in Escherichia coli BL21 as described previously (18). Site-directed mutagenesis (SDM) was performed following the QuikChange mutagenesis protocol provided by the manufacturer (Agilent) with the primers listed in Table S2 in the supplemental material and pET-wbpZ as the template. Purification of wild-type or mutant forms of the C-terminally His-tagged WbpZ was performed using Ni2+-nitrilotriacetate (Ni-NTA) affinity chromatography, and SDS-PAGE and Western blots were carried out as described previously (23). To test the in vivo activity of the site-directed mutants of wbpZ in P. aeruginosa, the broad-host-range vector pHERD20T (32) was used. First, primers wbpZ-F-SacI (TGCGAGCTCAATGCGGGTACTGCACTTC) and wbpZ-R-XbaI (TGCTCTAGACTTACCGAGCGGCCTTCAC) were used to amplify the wild-type wpbZ gene from PAO1, which was inserted into the SacI and XbaI sites of pHERD20T to obtain pHERD-wbpZ. pHERD-wbpZ was then used as a PCR template to obtain the SDM mutants of wbpZ carried in the pHERD20T vector using the same set of primers listed in Table S2 in the supplemental material. To examine whether these SDM mutants of wbpZ were still functional in P. aeruginosa, the obtained vectors were then transformed into a wbpZ knockout mutant of P. aeruginosa PAO1 (wbpZ::Gm) (14) by electroporation. LPS samples from these strains were then extracted using the method of Hitchcock and Brown (33). LPS was characterized using SDS-PAGE, silver staining, and Western immunoblotting as described by Rocchetta et al. (14).
d-Rhamnosyltransferase assays.
Nonradioactive assays were carried out for analyses of reaction products by electrospray ionization mass spectrometry (ESI-MS) using an Orbitrap Velos Pro spectrometer (Thermo Scientific). The assay mixtures for WbpZ contained, in a total volume of 40 μl, 5 mM MnCl2, 0.1 mM Tris buffer (pH 7.5), bacterial homogenate (0.5 mg protein) 0.25 mM purified or crude synthetic GDP-d-Rha, and 0.2 mM GlcNAcα-PO3-PO3-phenylundecyl (GlcNAc-PP-PhU) as an acceptor substrate. In some assay mixtures, 2.5 mM dithiothreitol (DTT) was present. Mixtures were incubated at 37°C for 1 h. Seven hundred microliters of H2O was added and the mixture applied to a C18 Sep-Pak column, which was eluted with 4 ml H2O and 3 ml methanol (MeOH). Fractions eluted with MeOH were subjected to analysis by ESI-MS in the negative-ion mode. Assays using fluorescent anthracen-containing acceptor substrates were carried out similarly. The product eluted from Sep-Pak was purified by HPLC and fluorescence intensity measured at 415 nm (with an excitation wavelength at 247 nm).
Radioactive d-rhamnosyltransferase assays using GDP-[3H]Rha.
Standard assay mixtures contained, in a total volume of 40 μl, 10 μl WbpZ homogenate in 20% glycerol–80% PBS (0.5 to 1 mg protein) or purified enzyme, 0.2 mM acceptor substrate, 0.25 mM GDP-d-[3H]Rha, 0.125 M Tris-HCl (pH 9), and 0.25% Triton X-100. Mixtures were incubated for 1 h at 37°C, and product was isolated using C18 Sep-Pak or both Sep-Pak and HPLC. The amount of product was determined by scintillation counting. The kinetic parameters were determined using GraphPad Prism software.
Synthesis of large-scale WbpZ products.
Large-scale WbpZ products were produced from both GalNAc-PP-PhU and GlcNAc-PP-PhU as substrates. The 500× small-scale assay mixtures contained, in a total volume of 20 ml, 5 ml of WbpZ homogenate in 20% glycerol–80% PBS (500 mg protein), 2.5 μmol Tris-HCl (pH 9), 4 μmol acceptor substrate GalNAc-PP-PhU or GlcNAc-PP-PhU, 1 ml 5% Triton X-100, and 5 μmol GDP-d-Rha. Mixtures were incubated for 1 h at 37°C, and 20 ml cold H2O was added. Aliquots were applied to 40 C18 Sep-Pak columns which were washed with 4 ml H2O followed by 3 ml MeOH, which eluted the reaction product Rha-GalNAc/GlcNAc-PP-PhU. Fractions were concentrated by rotor evaporation, filtered, and further purified by reverse-phase HPLC, using a C18 column and acetonitrile-H2O (27:73) mixtures. Product eluted at 25 min and substrate at 32 min. Fractions were dried by rotor evaporation and lyophilization. After exchange with D2O, NMR spectra were collected using a 600-MHz Bruker NMR spectrometer.
RESULTS
Purification of WbpZ.
SDS-PAGE of the purified protein showed high expression and an enrichment of a protein band at an apparent molecular mass of 45 kDa, which corresponded to the calculated value of 43.5 kDa (see Fig. S2 in the supplemental material). Western blots probed with an anti-His tag antibody showed a single band at an apparent molecular mass of 45 kDa. Less than 20% of the enzyme was solubilized, and the purified enzyme from this cell fraction had very little activity. Subsequent assays were therefore carried out using the homogenate.
A hydrophobicity plot of the WbpZ protein shows that it is void of transmembrane domains. However, small hydrophobic sequences may help to bind lipid-like molecules. While in the presence of 2.5 mM DTT in the assay mixture, 0.125% octyl-glucoside had little effect, the inclusion of 0.125% NP-40 in the assay mixture stimulated the activity 2-fold. Triton X-100 in the assay mixture increased the activity maximally 2.5-fold at 0.25% (see Fig. S3 in the supplemental material). This suggested that the detergent helped to solubilize and/or activate the enzyme.
The time course experiment showed that the Rha-transferase reaction was linear up to 1 h of incubation of the enzyme-substrate reaction mixture. Initial rates were present at up to at least 15 μl enzyme homogenate (0.5 mg protein) per assay mixture. The optimal buffer pH was tested with MES (morpholineethanesulfonic acid) and Tris buffers in the assay mixture. Maximal activity was observed at the relatively high buffer pH 9 (see Fig. S4 in the supplemental material).
A series of divalent metal ions or EDTA at 5 mM was tested in the assay and compared to controls without addition (see Fig. S5 in the supplemental material). Slightly higher activities were seen with Mg2+ and Pb2+ salts, but Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Ca2+, or EDTA did not stimulate WbpZ activity. These observations suggest that WbpZ is not a metal ion-dependent GT.
Donor specificity.
The donor specificity of WbpZ using GlcNAc-PP-PhU as an acceptor was studied with nucleotide sugars GDP-d-[3H]Rha, GDP-[3H]Man, GDP-[3H]Fuc, UDP-[3H]Gal, UDP-[3H]GalNAc, UDP-[3H]GlcNAc, and CMP-[3H]sialic acid. While all other nucleotide sugars were inactive, GDP-Man had 16% of the activity of GDP-d-Rha as a donor substrate. In duplicate assays, less than 5% activity was observed with UDP-Glc. WbpZ therefore has dual activities and is mainly a d-Rha-transferase but also has Man-transferase activity at lower levels.
The apparent Km for GDP-d-Rha was 0.45 mM, with an apparent Vmax of 14.0 nmol/h/mg (see Fig. S6A in the supplemental material).
Acceptor substrate specificity.
The acceptor specificity of d-Rha-transferase was determined with a number of GalNAc- and GlcNAc derivatives. Compounds that had a diphosphate as the aglycone group were very active as acceptors for d-Rha. However, compounds lacking the diphosphate group or containing a diphosphate mimic were inactive. Thus, GlcNAcα-Bn and GalNAcα-Bn were not substrates, and the diphosphate group in the acceptor was critical for activity (Table 1). This requirement appears to be common to all GTs that catalyze the second step in O-antigen synthesis.
TABLE 1.
Acceptor specificity of d-Rha-transferase WbpZa
| Compound (concn, mM)b | Activity (%) |
|---|---|
| GlcNAcα-PO3-PO3-(CH2)11-O-Ph (0.1) | 100 |
| GlcNAcα-PO3-PO3-AnthrU (0.1) | 57 |
| GlcNAcα-PO3-PO3-(CH2)9-CH3 (0.1) | 80 |
| GlcNAcα-O-CO-CH2-CO-O-(CH2)11-O-Ph (0.5) | <1 |
| GalNAcα-PO3-PO3-(CH2)11-O-Ph (0.1) | 94 |
| GalNAcα-PO3-PO3-AnthrU (0.1) | 47 |
| GalNAcα-p-nitrophenyl (1) | <1 |
| GalNAcα-Bn (1) | <1 |
| 4-Deoxy-GalNAcα-Bn (1) | <1 |
| 3,4-Dideoxy-GalNAcα-Bn (1) | <1 |
| 6-Deoxy-GalNAcα-Bn (1) | <1 |
| GlcNAcα-Bn (1) | <1 |
| GlcNAcβ-Bn (1) | <1 |
| N-Butyryl-glucosamineβ-S-2-naphthyl (1) | <1 |
| GlcNAc (1) | <1 |
| GalNAc (1) | <1 |
| d-Rha (1) | <1 |
| d-Rhaα-Bn (1) | <1 |
| A-(GalNAcα)T (1) | <1 |
| Acetyl-(GalNAcα)T-NH2 (1) | <1 |
| AHGVT-(GalNAcα)SAPDTRPAPGSTAPPA (1) | <1 |
Neutral compounds were assayed by the AG1x8 method, and sugar-diphosphate-lipids were assayed by the C18 Sep-Pak method (23). Radioactive product was quantified by scintillation counting. Anthracene-containing compounds were also separated after the incubation by HPLC and then estimated by their fluorescence intensity (24).
AnthrU, anthracenyl-undecyl.
Since both synthetic GlcNAc-PP-PhU (100% activity) and GalNAc-PP-PhU (94% activity) were excellent acceptors for WbpZ, the enzyme does not have specificity for the configuration of the 4-hydroxyl of the sugar moiety. This is in contrast to the case for other bacterial Gal- and Glc-transferases that catalyze the second step in O-antigen synthesis and are specific for either GlcNAc or GalNAc. The apparent Km for GalNAc-PP-PhU was 0.1 mM, with an apparent Vmax of 12 nmol/h/mg (see Fig. S6B in the supplemental material). These kinetic values are rough approximations, since only homogenates were used in our experiments. The more precise kinetic data have to be confirmed once active and purified WbpZ enzyme has been obtained.
Compounds containing the fluorescent undecyl-anthracenyl group (GlcNAc-PP-AnthrU and GalNAc-PP-AnthrU) were also good acceptors, confirming the relaxed sugar specificity of WbpZ d-Rha-transferase for the acceptor. These compounds can be used in assays based on fluorescence measurements of enzyme products (24). The fluorescent anthracenyl compounds were applied to C18-HPLC (24% acetonitrile–76% H2O) to separate the substrate (25-min elution) from the product (35-min elution), which was quantified by fluorescence intensity at 415 nm. For the anthracenyl substrate, the assay buffer at pH 9 yielded the highest activity.
The addition of a single d-Rha residue (146 Da) to either GlcNAc-PP-PhU or GalNAc-PP-PhU due to the activity of WbpZ was confirmed by mass spectrometry (ESI-MS, negative-ion mode), showing an m/z of 772 for the d-Rha-GlcNAc-PP-PhU product [M − H]− and m/z 794 for the monosodium salt of GlcNAc-PP-PhU [M − H + Na]−. The product using GalNAc-PP-PhU showed similar spectra. No consistent m/z peaks at 918 or higher could be discerned, indicating that a single Rha residue had been transferred to GlcNAc-PP-PhU or GalNAc-PP-PhU by the nonprocessive d-Rha-transferase WpbZ.
The Man-transferase activity of WbpZ was tested with GlcNAc-PP-PhU and GalNAc-PP-PhU and replacing GDP-d-Rha with GDP-d-Man in the assay mixture. The two acceptors were equally active. The transfer of a single Man residue was confirmed by ESI-MS (negative-ion mode), showing m/z 788 [M − H]− and m/z 810 [M − H + Na]− for both Man-GlcNAc-PP-PhU and Man-GalNAc-PP-PhU and the lack of m/z 950 and higher (data not shown).
Verification of the Rhaα1-3 linkage by 600-MHz NMR.
To establish the new linkage in the enzyme products Rha-GlcNAc-PP-PhU and Rha-GalNAc-PP-PhU (Fig. 1), NMR parameters of d-Rha-transferase products dissolved in D2O were determined in one-dimensional (1D) and 2D NMR experiments. The chemical shifts were established by 2D correlation spectroscopy (correlation spectroscopy [COSY] and total correlation spectroscopy [TOCSY]) experiments (Fig. 2; see Fig. S7 in the supplemental material). In the Rha-GlcNAc-PP-PhU product, there was a nuclear Overhauser effect (NOE) between Rha H-1 and Rha H-2, as well as between Rha H-1 and GlcNAc H-3 (data not shown). The Rha H-1 signal was found at 5.05 ppm, indicative of Rha in an α linkage (Table 2). This establishes the Rhaα1-3GlcNAc-R linkage. In the spectrum of Rha-GalNAc-PP-PhU, the chemical shift values were similar but not identical to those of GlcNAc-PP-PhU. A strong NOE was seen in the rotating-frame nuclear Overhauser effect spectroscopy (ROESY) spectrum from Rha H-1 to Rha H-2 and to both GalNAc H-3 and H-4 (Fig. 2C). Since the GalNAc H-4 is in the equatorial position, it is close in space to the Rha H-1 that is linked 1-3 (Fig. 1). These NOE signals would not be seen if Rha were 1-4 or 1-6 linked. The carbon spectrum showed that GalNAc H-3 exhibited a significant downfield shift for the H-3 signal (by 5.6 ppm) in the d-Rha-transferase product, compared to the signal in the substrate GalNAc-PP-PhU (23). This was greater than the shifts for the other GalNAc protons. The NMR spectra therefore confirmed the d-Rhaα1-3GalNAc linkage.
FIG 1.

Structure of WbpZ reaction products and expected NOEs.
FIG 2.

(A) The 600-MHz COSY NMR spectrum of d-Rha-transferase product d-Rhaα1-3GlcNAcα-PO3-PO3-(CH2)11-O-phenyl. (B) The 600-MHz COSY NMR spectrum of d-Rha-transferase product d-Rhaα1-3GalNAcα-PO3-PO3-(CH2)11-O-phenyl. (C) The 600 MHz ROESY NMR spectrum of d-Rha-transferase product d-Rhaα1-3GalNAcα-PO3-PO3-(CH2)11-O-phenyl (0.3-s mixing time).
TABLE 2.
NMR (600 MHz) parameters of d-Rha-transferase products d-Rhaα1-3GlcNAcα-PO3-PO3-(CH2)11-O-phenyl and d-Rhaα1-3GalNAcα-PO3-PO3-(CH2)11-O-phenyl
| Position | Rha-GalNAc-PP-PhU |
Rha-GlcNAc-PP-PhU, 1H ppm | |
|---|---|---|---|
| 1H ppm | 13C ppm | ||
| Rha-1 | 4.94 (s) | 96.3 | 5.05 |
| Rha-2 | 3.94 (o) | 70.0 | 3.95 |
| Rha-3 | 3.65 (o) | 70.5 | 3.62 |
| Rha-4 | 3.43 (o.t, J = 9.2Hz) | 71.7 | 3.33 |
| Rha-5 | 3.59 (o) | 69.1 | 3.51 |
| Rha-6-Me | 1.30 (o) | 16.8 | 1.20 (o) |
| GlcNAc-1 | 5.39 (m) | ||
| GlcNAc-2 | 3.99 (o) | ||
| GlcNAc-3 | 3.79 | ||
| GlcNAc-4 | 3.55 (o) | ||
| GalNAc-1 | 5.53 (dd, J = 6.7,3.8) | 94.7 | |
| GalNAc-2 | 4.39 (br, dt, J = ∼10.4,2.4) | 47.9 | |
| GalNAc-3 | 3.97 (dd, J = 11.1,2.8) | 72.5 | |
| GalNAc-4 | 4.23 (br, d, J = ∼2) | 64.4 | |
| GalNAc-5 | 4.15 (dd, J = 7.0,5.4) | 71.9 | |
| GalNAc-6 | 3.78, 3.72 (o) | 61.0 | |
| GalNAc-N-acetyl | 2.07 (s) | ||
Inhibition of WbpZ.
In order to find suitable inhibitors with potential as adjuvant antibiotics, we tested WbpZ activity with N-butyryl-galactosamineα-benzyl, a weak inhibitor of mammalian core 1 β3Gal-transferase, and with N-butyryl-glucosamine βbenzyl, a potent inhibitor of mammalian β4-Gal-transferase (25), which did not inhibit WbpZ. Bis-imidazolium salts with specific lengths of aliphatic chains are selective GT inhibitors (27). Several of these compounds reduced WbpZ activity (Table 3). Bis-imidazolium salts having aliphatic spacer groups with 4 or 6 carbons had little effect on WbpZ activity. However, salts having spacer groups of 18 aliphatic carbons inhibited activity by 45%. The most potent inhibitor was the bis-imidazolium salt having a 20-carbon chain spacer length, with a 50% inhibitory concentration (IC50) of 0.3 mM. The mesylate salt with a 22-carbon chain spacer length had an IC50 of 0.7 mM.
TABLE 3.
Inhibition of d-Rha-transferase activity of WbpZa
| Compound | Inhibition of WbpZ (%)b |
|---|---|
| N-Butyryl-glucosamineβ-thio-2-naphthyl | <1 |
| N-Butyryl-galactosamineα-benzyl | <1 |
| 1,4-Bis-(3-methyl-1H-imidazolium-1-yl)butane dichloride | 6.7 |
| 1,6-Bis-(3-methyl-1H-imidazolium-1-yl)hexane dichloride | 22.0 |
| 1,18-Bis-(3-methyl-1H-imidazolium-1-yl)octadecane dichloride | 55.9 |
| 1,20-Bis-(3-methyl-1H-imidazolium-1-yl)eicosane dichloride | 73.3 |
| 1,22-Bis-(3-methyl-1H-imidazolium-1-yl)docosane dimesylate | 47.7 |
d-Rha-transferase assays were performed in duplicate determinations as described in Materials and Methods. The acceptor concentration in the assay mixtures was 0.1 mM GalNAc-PP-PhU, and the inhibitor compound concentration was 0.5 mM.
One hundred percent activity corresponds to 0.32 nmol/h/mg for WbpZ.
Role of specific amino acids in WbpZ.
WbpZ has a single Cys residue (Cys26). Site directed mutation of Cys26 to Ala had little effect on the activity of the enzyme, which indicated that Cys26 is not an essential residue. DTT was not able to stimulate the d-Rha-transferase activity using GlcNAc-PP-PhU as acceptor substrate, confirming that the single Cys residue does not play a major role in the protein structure or activity, or by forming inactivating disulfide bonds. Two DXD motifs, D172-X-D174 and D253-D254-X-D256, are found in the WbpZ sequence. Mutations of these Asp residues to Ala showed that three residues, D172, D254, and D256, were important for d-Rha-transferase activity using the GlcNAc-PP-PhU acceptor, since mutants showed greatly reduced activity. Similar results were obtained using GalNAc-PP-PhU as the acceptor substrate. All mutant enzymes were capable of being expressed, and hence there was not a problem with low protein expression. Mutation of the second Asp (D174) of the N-terminal DXD sequence or mutation of the first Asp (D253) of the DDXD sequence did not decrease the activity, showing that those residues are not as essential. While the D172A and D254A mutants exhibited only residual activity, the D256A mutant retained 15% of the enzymatic activity (Fig. 3).
FIG 3.

d-Rha-transferase activities of wild-type and mutant WbpZ enzymes. Mutants were expressed in parallel with wild-type WbpZ and assayed in bacterial homogenates as described in Materials and Methods, using GDP-d-Rha as a donor and GlcNAc-PP-PhU as the acceptor substrate. Assays using GalNAc-PP-PhU gave similar results.
Mutations affect O-antigen synthesis.
To examine whether these site-directed mutations of wbpZ affected the functions of this enzyme in vivo, the P. aeruginosa PAO1 wbpZ::Gm strain, which lacks CPA biosynthesis due to an insertion mutation in the wbpZ gene, was complemented with either the wild-type or the site-directed mutant wbpZ gene and analyzed for the ability to restore the biosynthesis of CPA LPS. It was found that recombinant forms of wbpZ with mutations C26A, D174A, D253A, and D256A were still functional in vivo and capable of rescuing CPA biosynthesis to the wild-type level. Thus, in spite of the much lower d-Rha-transferase activity of the D256A mutant observed in vitro, this mutant construct was obviously still sufficiently active to act in the CPA synthesis pathway. However, the D172A and D254A mutants of wbpZ were not able to rescue the CPA biosynthesis (Fig. 4). This indicates that the D172A and D254A residues are crucial for the in vivo activity of wbpZ during O-antigen synthesis.
FIG 4.
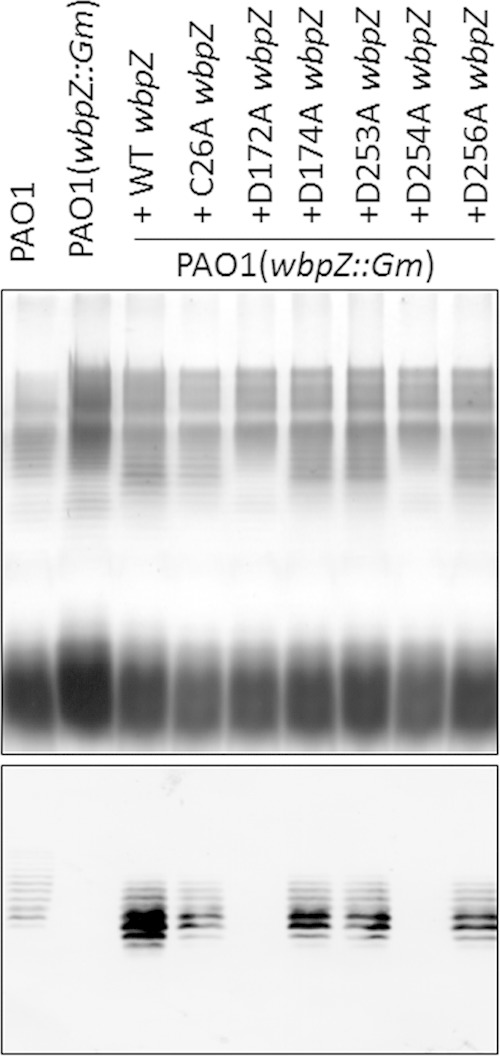
Complementation of the wbpZ knockout mutant with site-directed mutants of wbpZ. Top, silver-stained SDS-PAGE gel of extracted LPS; bottom, Western blot using monoclonal antibody (MAb) N1F10, which is specific for CPA LPS. The wbpZ knockout mutant PAO1 wbpZ::Gm lost the ability to synthesize CPA LPS as indicated by the lack of a Western blotting signal. When complemented with wild-type (WT) wbpZ, CPA biosynthesis is restored. The site-directed mutants C26A wbpZ, D174A wbpZ, D253A wbpZ, and D256A wbpZ were also able to restore CPA biosynthesis as indicated by the Western blot. However, the site-directed mutants D172A wbpZ and D254A wbpZ were not able to restore CPA biosynthesis, indicating that these two site-directed mutants were not functional in vivo.
DISCUSSION
The CPA polysaccharide antigen of P. aeruginosa is a unique homopolymer of d-Rha. It requires the synthesis of GDP-d-Rha from GDP-d-Man. Certain E. coli antigens contain l-Rha, which requires dTDP-l-Rha as a donor substrate (6). An exception is the E. coli O99 antigen (22), which has a backbone of d-Rha residues in a sequence resembling the d-Man backbone of the E. coli O9a antigen with 4 Man residues in the repeating unit. The d-Manα1-3GlcNAc-PP-Und adaptor structure of the O9a antigen is synthesized by α1,3-Man-transferase WbdC, which is followed by Man-transferase WbdB, which adds two more Manα1-3 residues to form the basic adaptor structure for the attachment of the Man polymer by WbdA. A similar adaptor structure, d-Rhaα1-3GlcNAc-, appears to be required for CPA synthesis. We have shown in this work that WbpZ, encoded by the CPA antigen gene cluster of PAO1, is the GDP-d-Rha:GlcNAc/GalNAc-diphosphate-lipid α1,3-d-Rha transferase that forms this adaptor. WbpZ is highly homologous to WejK in E. coli O99, which may also be an α1,3-d-Rha-transferase that synthesizes the O99 adaptor structure d-Rhaα1-3GlcNAc/GalNAc. WbpZ also has significant homology to Man-transferase WbdC from E. coli O8/O9.
It is not surprising that WbpZ, the second enzyme in the repeating-unit pathway, would require the diphosphate in the acceptor substrate (34, 35). However, the dual acceptor specificity of WbpZ is unusual and suggests that the adaptor could contain a GlcNAc or a GalNAc residue.
Both the d-Rha-transferase and the minor Man-transferase activities of WbpZ exhibited a relaxed acceptor specificity and required the diphosphate in the acceptor substrate, suggesting that these two activities are inherent in the same catalytic site, which remains to be proven by structural analysis of WbpZ.
Based on the similarities of the other GTs in the gene cluster, the biosynthesis of CPA is expected to continue with WbpY, which adds two d-Rhaα1-3 residues in succession to Rhaα1-3GlcNAc-, having an acceptor specificity for d-Rha. WbpX would then cooperate with WbpY and add d-Rha in α1-2 linkage to Rha to complete the synthesis of the repeating unit of CPA. This sequence of events would assemble the continuous repeating structures that form the final CPA polymer. After transfer of the polymer to the periplasm by the ABC transporter Wzt/Wzm, it is expected that the d-rhamnan is transferred to l-Rha in the outer core structure without the GlcNAc or GalNAc adaptor sugar by ligase WaaL (2, 4). After the transfer, GlcNAc/GalNAc-PP-Und may be flipped across the inner membrane to the cytoplasmic compartment to form another polysaccharide molecule. Alternatively, GlcNAc/GalNAc-phosphate may be cleaved by reversible GlcNAc/FucNAc-phosphotransferase WbpL, which is also involved in OSA biosynthesis. However, studies in Klebsiella showed that the GlcNAc residue is transferred together with the O antigen, thus leaving PP-Und behind for recycling (36).
We showed here by biochemical characterization that WbpZ is a retaining nonprocessive GDP-d-Rha:GalNAc/GlcNAc-diphosphate-lipid α1,3-d-Rha-transferase that catalyzes the second step in CPA O-antigen assembly in PAO1. Since mutations D172A and D254A, which drastically reduced WbpZ activity, resulted in the inability to complement in vivo O-antigen synthesis, these residues are critically involved in the enzyme activity and probably form essential bases from two different DXD motifs involved in nucleophilic attacks. This work also shows that there was a critical level of activity that could be measured in vitro and had to be maintained in vivo for biological activity.
Divalent metal ions were not required for WbpZ activity; thus, positively charged residues may be involved in binding the negatively charged donor and acceptor substrates, and the potential role of conserved Lys and Arg residues needs to be examined (see Fig. S1 in the supplemental material). In addition, the potential roles of the Glu residues (19, 20) of the three or four conserved EX7E motifs in WbpZ also need to be explored.
We have developed several chemical and enzymatic syntheses to effectively produce large amounts of the donor substrate for WbpZ, GDP-d-Rha, which is not yet commercially available. Thus, it is now feasible to characterize other d-Rha-transferases. According to the dual donor specificity of WbpZ and resemblance to other Man-transferases, WbpZ may also add a d-Man residue to GlcNAc/GalNAc-PP-Und. WbpY and WbpX also bear resemblance to Man-transferases, and it is therefore possible that a Man polymer could be assembled in PAO1. Accordingly, small amounts of Man have previously been detected in CPA preparations (2). Biochemical analyses of WbpY and -X would confirm this possibility. A cautionary note is that knowledge gained from structural and biochemical studies of GTs is necessary to determine the functions of uncharacterized GTs, which cannot be determined from sequence comparisons alone since a change of only a few amino acids can alter enzyme specificities and catalytic properties (37). It is hoped that future studies on the d-Rha-transferases will unravel the mechanisms of O-antigen biosynthesis by an important pathogen.
This study confirmed that the second step in the biosynthetic pathway of the O-antigen repeating unit is critical to form the O-antigen polymer (38). This second step is highly specific for the bacterial strain or serotype and a target for the prevention of O-antigen formation. We presented evidence that the second step catalyzed by WbpZ can be inhibited by a number of bis-imidazolium salts having aliphatic spacer chains of between 18 and 22 carbons. The inhibition is not as strong as that of other GTs (27, 35), which may be due to the fact that homogenates containing WbpZ were used. Inhibition of this critical step for LPS synthesis in P. aeruginosa could be a basis for drug development to prevent or treat P. aeruginosa infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Discovery grant from the Natural Science and Engineering Research Council (to I.B.) and operating grants from the Canadian Institutes of Health Research (to J.S.L. and to I.B. and W.A.S.).
We thank Francoise Sauriol (Queen's University) for running the NMR spectra.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02590-14.
REFERENCES
- 1.Pirnay JP, Bilocq F, Pot B, Cornelis P, Zizi M, Van Eldere J, Deschaght P, Vaneechoutte M, Jennes S, Pitt T, De Vos D. 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4:e7740. doi: 10.1371/journal.pone.0007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King JD, Kocincova D, Westman EL, Lam JS. 2009. Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun 15:261–312. doi: 10.1177/1753425909106436. [DOI] [PubMed] [Google Scholar]
- 3.Lam JS, Taylor VL, Islam ST, Hao Y, Kocíncová D. 2011. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front Microbiol 2:118. doi: 10.3389/fmicb.2011.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitfield C, Trent MS. 2014. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 5.Knirel YA, Bystrova OV, Kocharova NA, Zaehringer U, Pier GB. 2006. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res 12:324–336. doi: 10.1179/096805106X118906. [DOI] [PubMed] [Google Scholar]
- 6.Samuel G, Reeves P. 2003. Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr Res 338:2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Arsenault TL, MacLean DB, Zou W, Szarek WA. 1994. Smith-degradative studies on the polysaccharide portion of A-band lipopolysaccharide from a mutant (AK1401) of Pseudomonas aeruginosa strain PAO1. Can J Chem 72:1376–1382. doi: 10.1139/v94-172. [DOI] [Google Scholar]
- 8.Yokota S, Kaya S, Sawada S, Kawamura T, Araki Y, Ito E. 1987. Characterization of a polysaccharide component of lipopolysaccharide from Pseudomonas aeruginosa IID 1008 (ATCC 27584) as d-rhamnan. Eur J Biochem 167:203–209. doi: 10.1111/j.1432-1033.1987.tb13324.x. [DOI] [PubMed] [Google Scholar]
- 9.Poon KKH, Westman EL, Vinogradov E, Jin S, Lam JS. 2008. Functional characterization of MigA and WapR: putative rhamnosyltransferases involved in outer core oligosaccharide biosynthesis of Pseudomonas aeruginosa. J Bacteriol 190:1857–1865. doi: 10.1128/JB.01546-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hao Y, King JD, Huszczynski S, Kocincova D, Lam JS. 2013. Five new genes are important for common polysaccharide antigen biosynthesis in Pseudomonas aeruginosa. mBio 4(1):e00631–00612. doi: 10.1128/mBio.00631-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rocchetta HL, Lam JS. 1997. Identification and functional characterization of an ABC transport system involved in polysaccharide export of A-band lipopolysaccharide in Pseudomonas aeruginosa. J Bacteriol 179:4713–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freinkman E, Chng SS, Kahne D. 2011. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc Natl Acad Sci U S A, 108:2486–2491. doi: 10.1073/pnas.1015617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahim R, Burrows LL, Monteiro MA, Perry MB, Lam JS. 2000. Involvement of the rml locus in core oligosaccharide and O polysaccharide assembly in Pseudomonas aeruginosa. Microbiology 146:2803–2814. [DOI] [PubMed] [Google Scholar]
- 14.Rocchetta HL, Burrows LL, Pacan JC, Lam JS. 1998. Three rhamnosyltransferases responsible for assembly of the A-band d-rhamnan polysaccharide in Pseudomonas aeruginosa: a fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Mol Microbiol 28:1103–1119. doi: 10.1046/j.1365-2958.1998.00871.x. [DOI] [PubMed] [Google Scholar]
- 15.Rocchetta HL, Burrows LL, Lam JS. 1999. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol Mol Biol Rev 63:523–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King JD, Poon KKH, Webb NA, Anderson EM, McNally DJ, Brisson J-R, Messner P, Garavito RM, Lam JS. 2009. The structural basis for catalytic function of GMD and RMD, two closely related enzymes from the GDP-d-rhamnose biosynthesis pathway. FEBS J 276:2686–2700. doi: 10.1111/j.1742-4658.2009.06993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blankenfeldt W, Asuncion M, Lam JS, Naismith JH. 2000. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J 19:6652–6663. doi: 10.1093/emboj/19.24.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Tanaka H, Hindsgaul O, Lam JS, Brockhausen I. 2013. A convenient synthesis of GDP-d-rhamnose: the donor substrate for d-rhamnosyltransferase WbpZ from Pseudomonas aeruginosa PAO1. Bioorg Med Chem Lett 23:3491–3495. doi: 10.1016/j.bmcl.2013.04.051. [DOI] [PubMed] [Google Scholar]
- 19.Greenfield LK, Richards MR, Vinogradov E, Wakarchuk WW, Lowary TL, Whitfield C. 2012. Domain organization of the polymerizing mannosyltransferases involved in synthesis of the Escherichia coli O8 and O9a lipopolysaccharide O-antigens. J Biol Chem 287:38135–38149. doi: 10.1074/jbc.M112.412577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenfield LK, Richards MR, Li J, Wakarchuk WW, Lowary TL, Whitfield C. 2012. Biosynthesis of the polymannose lipopolysaccharide O-antigens from Escherichia coli serotypes O8 and O9a requires a unique combination of single- and multiple-active site mannosyltransferases. J Biol Chem 287:35078–35091. doi: 10.1074/jbc.M112.401000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. 2006. Structures and mechanisms of glycosyltransferases. Glycobiology 16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 22.Perepelov AV, Li D, Liu B, Senchenkova SN, Guo D, Shevelev SD, Shashkov AS, Guo X, Feng L, Knirel YA, Wang L. 2009. Structural and genetic characterization of Escherichia coli O99 antigen. FEMS Immunol Med Microbiol 57:80–87. doi: 10.1111/j.1574-695X.2009.00584.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Czuchry D, Liu B, Vinnikova AN, Gao Y, Vlahakis JZ, Szarek WA, Wang L, Feng L, Brockhausen I. 2014. Characterization of two UDP-Gal:GalNAc-diphosphate-lipid β1,3-galactosyltransferases WbwC from Escherichia coli serotypes O104 and O5. J Bacteriol 196:3122–3133. doi: 10.1128/JB.01698-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinnikova AN, Torgov VI, Utkina NS, Veselovsky VV, Druzhinina TN, Wang S, Brockhausen I, Danilov LL. 2015. The synthesis of P1-[11-(anthracen-9-ylmethoxy)undecyl]-P2-(2-acetamido-2-deoxy-α-d-glucopyranosyl)) diphosphate and the study of its acceptor properties in the enzymatic reaction catalyzed by a d-rhamnosyltransferase from Pseudomonas aeruginosa. Russian J Bioorg Chem 41:105–107. doi: 10.1134/S106816201501015X. [DOI] [PubMed] [Google Scholar]
- 25.Gao Y, Aryal RP, Ju T, Cummings RD, Gahlay G, Jarvis DL, Matta KL, Vlahakis JZ, Szarek WA, Brockhausen I. 2013. Acceptor specificities and selective inhibition of recombinant human Gal- and GlcNAc-transferases that synthesize core structures 1,2, 3 and 4 of O-glycans. Biochim Biophys Acta 1830:4274–4281. doi: 10.1016/j.bbagen.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riley J, Xu C, Brockhausen I. 2010. Synthesis of acceptor substrate analogs for the study of glycosyltransferases involved in the second step of the biosynthesis of O antigen repeating units. Carbohydr Res 345:586–597. doi: 10.1016/j.carres.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Gao Y, Vlahakis JZ, Szarek WA, Brockhausen I. 2013. Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg Med Chem 21:1305–1311. doi: 10.1016/j.bmc.2012.12.034. [DOI] [PubMed] [Google Scholar]
- 28.Guchhait G, Misra AK. 2011. Efficient glycosylation of unprotected sugars using sulfamic acid: a mild eco-friendly catalyst. Catalysis Commun 14:52–57. doi: 10.1016/j.catcom.2011.07.016. [DOI] [Google Scholar]
- 29.Horton D. 1966. 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranosyl chloride. Org Synth 46:1–4. doi: 10.15227/orgsyn.046.0001. [DOI] [Google Scholar]
- 30.Hardré R, Khaled A, Willemetz A, Dupré T, Moore S, Gravier-Pelletier C, Le Merrer Y. 2007. Mono, di and tri-mannopyranosyl phosphates as mannose-1-phosphate prodrugs for potential CDG-Ia therapy. Bioorg Med Chem Lett 17:152–155. doi: 10.1016/j.bmcl.2006.09.074. [DOI] [PubMed] [Google Scholar]
- 31.Wittmann V, Wong C-H. 1997. 1H-tetrazole as catalyst in phosphomorpholidate coupling reactions: efficient synthesis of GDP-fucose, GDP-mannose, and UDP-galactose. J Org Chem 62:2144–2147. doi: 10.1021/jo9620066. [DOI] [PubMed] [Google Scholar]
- 32.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu C, Liu B, Hu B, Han Y, Feng L, Allingham JS, Szarek WA, Wang L, Brockhausen I. 2011. Biochemical characterization of UDP-Gal:GlcNAc-pyrophosphate-lipid β-1,4-Galactosyltransferase WfeD, a new enzyme from Shigella boydii type 14 that catalyzes the second step in O-antigen repeating-unit synthesis. J Bacteriol 193:449–459. doi: 10.1128/JB.00737-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Liu B, Strum S, Schutzbach JS, Druzhinina TN, Utkina NS, Torgov VI, Danilov LL, Veselovsky VV, Vlahakis JZ, Szarek WA, Wang L, Brockhausen I. 2012. Biochemical characterization of WbdN, a β1,3-glucosyltransferase involved in O-antigen synthesis in enterohemorrhagic Escherichia coli O157. Glycobiology 22:1092–1102. doi: 10.1093/glycob/cws081. [DOI] [PubMed] [Google Scholar]
- 36.Vinogradov E, Frirdich E, MacLean LL, Perry MB, Petersen BO, Duus JØ Whitfield C. 2002. Structures of lipopolysaccharides from Klebsiella pneumoniae. Eluicidation of the structure of the linkage region between core and polysaccharide O chain and identification of the residues at the non-reducing termini of the O chains. J Biol Chem 277:25070–25081. doi: 10.1074/jbc.M202683200. [DOI] [PubMed] [Google Scholar]
- 37.Brockhausen I. 2014. Crossroads between bacterial and mammalian glycosyltransferases. Front Immunol doi: 10.3389/fimmu.2014.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riley JG, Menggad M, Montoya-Peleaz PJ, Szarek WA, Marolda CL, Valvano MA, Schutzbach JS, Brockhausen I. 2005. The wbbD gene of E. coli strain VW187 (O7:K1) encodes a UDP-Gal:GlcNAc α-pyrophosphate-R β1,3-galactosyltransferase involved in the biosynthesis of O7-specific lipopolysaccharide. Glycobiology 15:605–613. doi: 10.1093/glycob/cwi038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.