

ABSTRACT
Pseudomonas aeruginosa uses N-acyl-homoserine lactone (AHL)-dependent quorum sensing (QS) systems to control the expression of secreted effectors. These effectors can be crucial to the ecological fitness of the bacterium, playing roles in nutrient acquisition, microbial competition, and virulence. In this study, we investigated the metabolic consequences of AHL-dependent QS by monitoring the metabolic profile(s) of a lasI rhlI double mutant (unable to make QS signaling molecules) and its wild-type progenitor as they progressed through the growth curve. Analysis of culture supernatants by 1H-nuclear magnetic resonance (1H-NMR) spectroscopy revealed that at the point where AHL concentrations peaked in the wild type, the metabolic footprints (i.e., extracellular metabolites) of the wild-type and lasI rhlI mutant diverged. Subsequent gas chromatography-mass spectrometry (GC-MS)-based analysis of the intracellular metabolome revealed QS-dependent perturbations in around one-third of all identified metabolites, including altered concentrations of tricarboxylic acid (TCA) cycle intermediates, amino acids, and fatty acids. Further targeted fatty acid methyl ester (FAME) GC-MS-based profiling of the cellular total fatty acid pools revealed that QS leads to changes associated with decreased membrane fluidity and higher chemical stability. However, not all of the changes we observed were necessarily a direct consequence of QS; liquid chromatography (LC)-MS analyses revealed that polyamine levels were elevated in the lasI rhlI mutant, perhaps a response to the absence of QS-dependent adaptations. Our data suggest that QS leads to a global readjustment in central metabolism and provide new insight into the metabolic changes associated with QS during stationary-phase adaptation.
IMPORTANCE Quorum sensing (QS) is a transcriptional regulatory mechanism that allows bacteria to coordinate their gene expression profile with the population cell density. The opportunistic human pathogen Pseudomonas aeruginosa uses QS to control the production of secreted virulence factors. In this study, we show that QS elicits a global “metabolic rewiring” in P. aeruginosa. This metabolic rerouting of fluxes is consistent with a variety of drivers, ranging from altered QS-dependent transcription of “metabolic genes” through to the effect(s) of global “metabolic readjustment” as a consequence of QS-dependent exoproduct synthesis, as well as a general stress response, among others. To our knowledge, this is the first study of its kind to assess the global impact of QS on the metabolome.
INTRODUCTION
The gammaproteobacterium Pseudomonas aeruginosa is widely distributed in the environment (1, 2). The organism is a human pathogen and is responsible for a range of acute infections, urinary tract infections, and respiratory infections (3). In addition, it is also associated with certain chronic infections, such as those that often develop in the airways of cystic fibrosis patients (4, 5). Such exquisite adaptability to different niches requires a complex regulatory hierarchy and an extremely versatile metabolism; indeed, P. aeruginosa is the ultimate generalist (6, 7). The bacterium can grow on a wide variety of substrates, although it has a particular predilection for amino acids (and alanine in particular [8]).
P. aeruginosa is the archetypal “secretor”; it exports an arsenal of proteins and small molecules, which collectively allow the bacteria to both shape and infer their environment (7, 9). The expression of many secreted virulence factors and secondary metabolites (as well as many other genes) is under the control of an N-acyl homoserine lactone (AHL)-dependent quorum sensing (QS) system (10, 11). The P. aeruginosa QS system is somewhat more complex than most, involving two interconnected AHL-dependent signaling pathways (12, 13). At the top of the hierarchy in P. aeruginosa is the Las signaling system (14, 15). In the Las system, N-3-oxo-dodecanoyl-l-homoserine lactone (OdDHL) is synthesized by LasI. Once OdDHL has accumulated beyond a critical threshold concentration in the culture, it binds to a transcriptional regulator, LasR, thereby eliciting a set of LasR-dependent gene expression changes. Subordinate to the Las signaling system is the Rhl signaling system (16). Here, N-butanoyl-l-homoserine lactone (BHL) is synthesized by the signal synthase RhlI. BHL binds to the response regulator RhlR, eliciting a further set of gene expression changes (some of which overlap with the Las pathway). In laboratory growth conditions, these two arms of the P. aeruginosa AHL QS system are intimately connected and regulate one another (14, 17). Strains defective in Las or Rhl signaling show reduced virulence, and considerable research has been concentrated on developing antimicrobials that interfere with these systems (18–23).
Population-level coordination of gene expression (mediated through QS) and metabolic plasticity both appear to be crucial to the lifestyle of P. aeruginosa, especially during the invasion of new niches. Not surprisingly then, several lines of evidence suggest that QS may impinge on the metabolic network of the cell. For example, transcriptomic and proteomic studies have pinpointed QS-dependent changes at key branchpoints in central metabolism (24, 25), and metabolic functions are represented among the “core” list of QS-regulated genes in P. aeruginosa (7, 18, 26). However, it is difficult to draw quantitative conclusions about the metabolic consequences of such gene expression changes without measuring the metabolites involved (7, 18, 27–29).
Recent work suggests that QS-mediated regulation of metabolic functions may have important (and sometimes unexpected) phenotypic consequences. For example, recent work by Goo et al. (30) has shown that in Burkholderia species, at least one metabolic change that is directly regulated by QS confers a clear benefit on the population. Oxalate synthesis (which is upregulated by QS in Burkholderia species) counteracts alkalization of the medium as the cell density rises. One interpretation of these findings is that the organism is “anticipating” problems associated with overcrowding, as the culture approaches carrying capacity. QS-dependent control of metabolism may also have more subtle effects. For example, QS controls the production of both “public” goods (i.e., secreted factors that can also be exploited by the non-QS bacteria that cohabit the niche) and “private” goods (i.e., “nonshared” intracellular metabolites). Dandekar et al. (31) showed that by bringing the synthesis of intracellular (private) goods under QS control, a metabolic “incentive” to cooperate is generated, which penalizes the appearance of “social cheats” (lasR mutants). Nevertheless, and in spite of the wealth of data accruing that suggests that QS and metabolism are tightly linked, to the best of our knowledge, no previous workers have attempted to quantify the global impact of QS on the metabolic profile of the population.
In the current work, we used nuclear magnetic resonance (NMR) spectroscopy, gas chromatography-mass spectrometry (GC-MS), and liquid chromatography-mass spectrometry (LC-MS) to quantitatively profile low-molecular-weight metabolites in a signal-null lasI rhlI mutant and in its wild-type progenitor. Given the relative simplicity of the extracellular metabolome, 1H-NMR spectroscopy was used to monitor the metabolic footprint of cultures throughout the growth curve. This revealed a clear divergence in the metabolic trajectory of the wild type compared to the lasI rhlI mutant which coincided with the peak in AHL signaling molecule concentration in the wild type. Since the sensitivity of 1H-NMR analysis is relatively limited, to gain a deeper insight into QS-dependent changes in the metabolome, we used a higher-resolution approach (GC-MS) to measure the metabolite profile in cellular fractions at key time points bracketing the metabolic divergence. This, in turn, prompted the use of more-targeted analyses (fatty acid methyl ester [FAME] GC-MS and LC-MS methods) to further investigate the impact of QS on fatty acid, polyamine, and amino acid metabolites. Taken together, the results of these analyses revealed that AHL-dependent QS (or its consequences) results in global perturbations in most major domains of metabolism.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains, plasmids, and bacteriophage used in this study are listed in Table 1. All experiments were conducted using either P. aeruginosa strain PAO1 or a lasI rhlI mutant derived from this strain. We constructed the lasI rhlI mutant “fresh” using previously described suicide plasmids (32). This resulted in a lasI rhlI mutant strain with insertional inactivations of both the lasI and rhlI signal synthase genes. QS mutants of P. aeruginosa are known to accumulate secondary mutations in other global regulators, e.g., vfr or algR. To ensure that the double mutant was otherwise isogenic with the wild-type progenitor and to reduce the possibility of unlinked accrued secondary mutations being responsible for the observed metabotype(s), we transduced the relevant markers into a wild-type strain using bacteriophage ϕPA3 as described in reference 33.
TABLE 1.
Bacterial strains, plasmids, and bacteriophage used in this study
| Bacterial strain, plasmid, or bacteriophage | Description or relevant genotype or phenotypea | Reference or source |
|---|---|---|
| P. aeruginosa strains | ||
| PAO1 | Wild-type PAO1 from the laboratory of John S. Mattick (University of Queensland, Australia) | 32 |
| PAO1 ΔlasI::Gmr | PAO1 with the Gmr cartridge inserted into the unique EcoRI site of lasI | This study |
| PAO1 ΔrhlI::Tetr | PAO1 with the Tcr cartridge inserted into the unique EcoRI site of rhlI | This study |
| PAO1 ΔlasI::Gmr ΔrhlI::Tetr | Double mutant generated by using ϕPA3 to transduce (first) the Tetr marker from strain PAO1 ΔlasI::Tetr and (second) the Gmr marker from strain PAO1 ΔrhlI::Gmr to PAO1 | This study |
| E. coli strains | ||
| S17-1 | thi pro hsdR recA chr::RP4-2 | 71 |
| JM109 | F′ traD36 proA+B+ lacIq Δ(lacZ)M15 Δ(lac-proAB) glnV44 e14− gyrA96 recA1 relA1 endA1 thi hsdR17 | 37 |
| Plasmids | ||
| pSB219.8A | pRIC380 carrying lasI::Gmr on the SpeI fragment from pSB222.7A | 32 |
| pSB224.12B | pRIC380 carrying rhlI::Tcr on the SpeI fragment from pSB224.8A | 32 |
| pSB536 | BHL biosensor | 37 |
| pSB1075 | OdDHL biosensor | 38 |
| pQF50 | Broad-host-range transcriptional fusion vector; Cbr lacZ incP | 72 |
| pβ01 | pQF50 derivative containing lasB-lacZ transcriptional fusion | 73 |
| pβ02 | pQF50 derivative containing rhlA-lacZ transcriptional fusion | 73 |
| Bacteriophage ϕPA3 | P. aeruginosa generalized transducing phage | 33 |
Abbreviations: Tcr, tetracycline resistance; Gmr, gentamicin resistance; Cbr, carbenicillin resistance.
Unless otherwise indicated, bacteria were grown in alanine-glycerol-salts medium supplemented with yeast extract (AGSY medium) (56 mM alanine, 17 mM K2HPO4, 86 mM NaCl, 100 μM CaCl2, 10 mM MgSO4, 5 μM FeCl2, 7.5 μM ZnCl2, 0.5% [vol/vol] glycerol, 3 g/liter yeast extract [pH 7.0]). Specific growth rates were calculated by linear regression in R 3.1 (34) by fitting log-transformed optical density at 600 nm (OD600) data during the exponential portion of growth from 0 h to 3 h. Analysis of covariance (ANCOVA) was used to test for significant differences in growth rates.
Planktonic cell cultures were prepared by washing cells from overnight cultures in fresh medium, resuspending them to an OD600 of 0.05 in 50 ml fresh medium, and then incubating them in 500-ml conical flasks at 37°C with vigorous aeration in an orbital shaker (300 rpm). All samples for metabolomic analysis were snap frozen in liquid nitrogen immediately after extraction and stored at −80°C. Each biological replicate was sampled hourly to generate the data presented in Fig. 1, 2, and 3. A different bacterial culture was used to generate the data points underlying the data shown in Fig. 4 and 5 in order to provide sufficient material for analysis.
FIG 1.
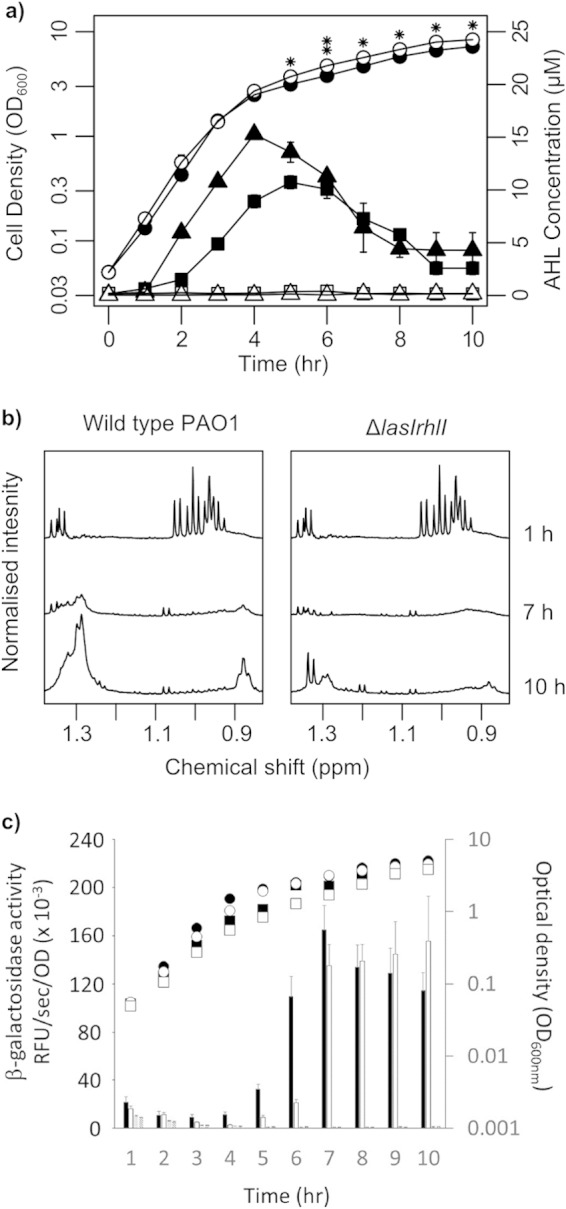
Cell growth and extracellular metabolic profiles of wild-type P. aeruginosa and the lasI rhlI mutant. (a) Growth curves (circles), OdDHL production (triangles), and BHL production (squares) by the wild type (filled symbols) and the lasI rhlI mutant (open symbols). Data are presented as means ± standard deviations (error bars) for six biological replicates. (b) Uptake of nutrients and secretion of secondary metabolites is observable in 1H-NMR spectra of bacterial supernatants. (c) QS-dependent promoter activity in the wild type and lasI rhlI mutant. Promoter activity was measured as β-galactosidase activity encoded by pβ01 (the PlasB reporter) and pβ02 (the PrhlA reporter). β-Galactosidase activity symbols: black bars, P. aeruginosa PAO1 carrying pβ01; white bars, PAO1(pβ02); stippled bars, lasI rhlI mutant carrying pβ01; hatched bars, lasI rhlI mutant(pβ02). Growth curve symbols: filled circles, PAO1(pβ01); filled squares, PAO1(pβ02); open circles, lasI rhlI mutant(pβ01); open squares, lasI rhlI mutant(pβ02).
FIG 2.
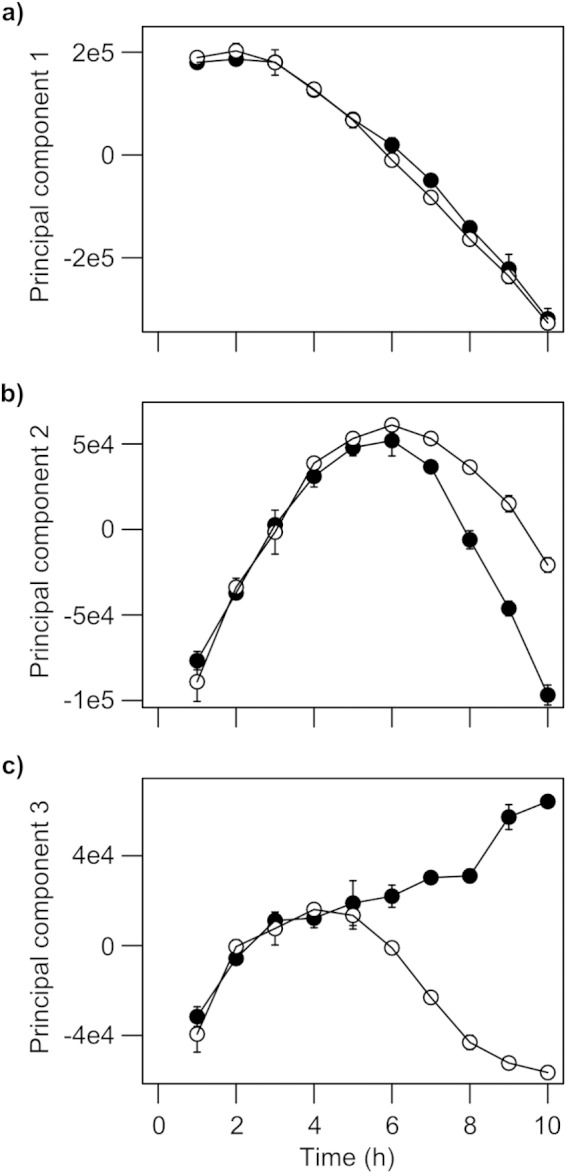
Latent patterns in 1H-NMR spectroscopy metabolic footprints of wild-type P. aeruginosa (●) and the lasI rhlI mutant (○) revealed by principal component analysis. Principal components 1, 2, and 3 explain 86%, 6.5%, and 2.9% of variation in the data (R2), respectively. Data are presented as means ± standard deviations (error bars) for six biological replicates.
FIG 3.

Concentrations of alanine (a) and acetate (b) measured by 1H-NMR spectroscopy in culture supernatants of wild-type P. aeruginosa (●) and the lasI rhlI mutant (○). Data are presented as means ± standard deviations for six biological replicates. Values that are significantly different by two-tailed Student's t test are indicated by asterisks as follows: *, P ≤ 0.05; **, P ≤ 0.01.
FIG 4.

Principal component analysis of the intracellular metabolic profiles of wild-type P. aeruginosa (filled symbols) and the lasI rhlI mutant (open symbols). Samples were taken at an OD600 of 1.5 (circles), 2.5 (squares), 4.5 (triangles pointing up), and 6.5 (triangles pointing down). Metabolic profiles were obtained by MSTFA derivatization followed by GC-MS analysis of the aqueous phase of methanol-chloroform extractions. Principal components 1 and 2 explain 33% and 17% of variation in the data (R2), respectively.
FIG 5.
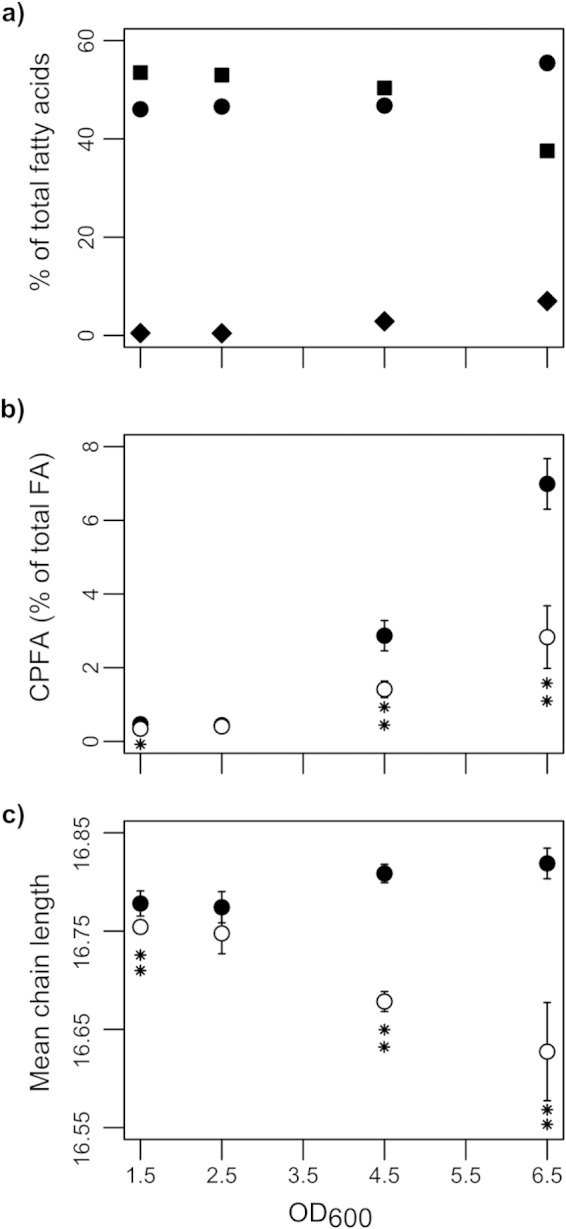
Fatty acid profiles of wild-type P. aeruginosa (filled symbols) and lasI rhlI mutant (open symbols) cells as determined by fatty acid methyl esterification (FAME) and subsequent GC-MS analysis. (a) Saturated fatty acids (circles), cis-unsaturated fatty acids (squares), and cyclopropane fatty acids (diamonds) in the wild type. (b) Cyclopropane fatty acids (CPFAs). (c) The average carbon chain length (ACCL) of fatty acids. Data are presented as means ± standard deviations for six biological replicates. Values that are significantly different by two-tailed Student's t test are indicated by asterisks as follows: *, P ≤ 0.05; **, P ≤ 0.01. The data in the figure are based on data presented in Table 2.
Complementation assays were performed using synthetic AHLs (kindly provided by James Hodgkinson and David Spring, Department of Chemistry, University of Cambridge, United Kingdom). OdDHL and BHL were dissolved in dimethyl sulfoxide (DMSO) and added to growth media at final concentrations of 15 μM and 10 μM, respectively. An equal volume of DMSO was added to the noncomplemented cultures.
Transcriptional fusion assays.
Transcriptional activity from the lasB and rhlA promoters was assayed using promoter-lacZ fusions. The plasmids pQF50, pβ01, and pβ02 (Table 1) (kindly supplied by Junichi Kato, Graduate School of Advanced Sciences of Matter, Hiroshima University, Japan) were introduced into P. aeruginosa by the protocol of Choi et al. (35). Cultures were grown in 50-ml AGSY medium (containing 250 μg/ml carbenicillin) at 37°C with good aeration (shaking at 250 rpm). β-Galactosidase activity was measured by the protocol of Ramsay et al. (36). The fluorogenic substrate 4-methylumbelliferyl-β-d-galactoside (catalog no. M1095; Melford) was used at a final concentration of 0.25 mg/ml. Fluorescence was measured using a SpectraMax Gemini XPS fluorescence plate reader (Molecular Devices, Sunnyvale, CA) as follows: 360-nm excitation; 450-nm emission; cutoff, 435 nm; reading every 30 s for 20 min at 37°C. β-Galactosidase activity was calculated as relative fluorescence units (RFU) per second and normalized to the OD600 of the corresponding sample.
Quantification of extracellular metabolites and AHLs.
Cells and cell debris were removed by centrifugation (6,000 × g, 20 min, 4°C). A 1.5-ml aliquot of the upper part of the supernatant was filtered (0.2-μm pore size) (Sartorius Minisart Sterile EO filters; Sartorius AG, Göttingen, Germany), and 1 ml of this aliquot was dried overnight in a Speed Vac (Savant) and then stored at −80°C.
To quantify BHL, Escherichia coli JM109(pSB536) was grown overnight at 37°C with shaking in LB broth containing 50 μg/ml carbenicillin. This overnight culture was then subcultured into LB with a 1:10 dilution (37). Once the subculture had grown to an OD600 of 0.5, 100 μl was transferred to a 96-well sterile white opaque microtiter plate (Greiner Bio-One, Germany) containing 100 μl sterile supernatant from the test culture. The microtiter plate was incubated statically at 30°C, and bioluminescence readings were taken after 3 h using a luminometer (Lucy1; Anthos Labtec Instruments, Austria). The same protocol was used to monitor OdDHL with the following modifications: E. coli JM109(pSB1075) was grown overnight at 30°C with 10 μg/ml tetracycline (38), the microtiter plate was incubated at 30°C, and bioluminescence readings were taken after 4 h of incubation. Synthetic OdDHL and BHL (kindly provided by James Hodgkinson and David Spring, Department of Chemistry, University of Cambridge, United Kingdom) were used to generate standard curves. Neither reporter strain generated detectable signal in response to their noncognate AHL.
1H-NMR spectroscopy of extracellular metabolites.
Dried samples were reconstituted in 600 μl D2O buffered with sodium phosphate (pH 7.0, 0.24 M) supplemented with 1 mM sodium 3-trimethylsilyl-2,2,3,3-tetradeuteriopropionate (TSP) (Cambridge Isotope Laboratories Inc., Andover, MA) and then transferred to 5-mm-internal-diameter NMR tubes for analysis. Samples were analyzed using a Bruker Avance II+ spectrometer operating at 500.13 MHz for the 1H frequency (Bruker, Coventry, United Kingdom) equipped with a 5-mm broad-band inverse probe (Bruker). Spectra were acquired using a conventional solvent suppression pulse sequence based on a one-dimensional (1D) nuclear Overhauser spectroscopy (NOESY) pulse sequence, which was used to saturate the residual water 1H signal (39). The solvent suppression pulse was applied during the relaxation delay (2 s) and mixing time (150 ms). The spin-lattice relaxation time (T1) was 3 μs. A total of 128 transients were collected into 32,768 data points over a spectral width of 16.0 ppm at 300K. Spectra were processed using Advanced Chemistry Development (ACD) SpecManager 1D NMR processor (version 8; ACD Inc., Toronto, Canada). Spectra were multiplied by an exponential window function and then Fourier transformed from the time to frequency domain (line broadening = 0.3 Hz). Spectra were phased, baseline corrected, and referenced to the TSP singlet at δ = 0 ppm, and integrated between 0.1 and 4.45 ppm and 4.60 and 9.60 ppm over a series of 0.01 ppm integral regions (thereby excluding the TSP and H2O resonances). Each spectral region was normalized to the integral of the TSP singlet at δ = 0 ppm. Metabolites were identified by comparison with in-house reference data.
GC-MS analysis.
Cellular metabolites were extracted using a modified version of the methanol-chloroform extraction described in reference 39. A volume of bacterial culture equivalent to 2.9 × 1010 viable cells was pelleted by centrifugation (5,000 × g, 5 min, 4°C), washed twice with 10 ml of 0.9% NaCl, then resuspended in 900 μl of a 2:1 (vol/vol) mixture of methanol and chloroform. The cells were further disrupted by tip sonication (24 2.5-s bursts on ice). Chloroform (300 μl) and water (300 μl) were added, and the samples were mixed by vortexing (30 s). Insoluble material was pelleted by centrifugation (6,000 × g, 20 min). The aqueous (top) and organic (bottom) fractions were separated, and aliquots (300-μl and 200-μl volume, respectively) were dried overnight in a Speed Vac concentrator (Savant). Aliquots of the aqueous phase of chloroform-methanol cell extracts were derivatized first with methoxyamine hydrochloride in pyridine (30 μl, 20 mg ml−1 solution, 17 h, 22°C) and then with N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) (30 μl, 1 h, 22°C). An aliquot (50-μl volume) of the derivatized sample was added to 50 μl hexane before analysis. GC-MS analysis was performed using a Thermo Trace GC Ultra gas chromatograph coupled to a Thermo Trace DSQ single quadrupole mass spectrometer (Thermo Scientific, Waltham, MA). One microliter of the derivatized sample was injected onto a 5%-phenyl-95%-dimethylpolysiloxane Zebron column (30 m [length] by 0.25 mm [internal diameter]; 0.25-μm film thickness [df]) (Phenomenex, Inc., Torrance, CA). A 35-min chromatography method was used with an initial temperature of 70°C, ramped to 130°C (at 10°C min−1), then to 230°C (at 5°C min−1), and finally to 310°C (at 20°C min−1).
For total fatty acid profiling, aliquots of the organic phase of chloroform-methanol cell extracts were dissolved in chloroform-methanol (1:1; 500-μl volume) and boron trifluoride in methanol (10%; 125-μl volume) and heated to 80°C for 90 min (39, 40). Samples were vortexed with deionized water (200 μl) and hexane (400 μl). The organic (top) layer was removed, dried, and reconstituted in 100 μl hexane prior to analysis. Two microliters of derivatized sample was injected onto a 100% polyethylene glycol Zebron column (30 m by 0.25-mm-internal diameter; 0.25 μm df) (Phenomenex, Inc., Torrance, CA). Analytes were separated using a 38-min chromatography method with an initial temperature of 60°C, ramped to 150°C (at 10°C min−1), and finally to 230°C (at 4°C min−1).
All GC-MS data were processed using Qual Browser and Quan Browser of Xcalibur version 2.0 (Thermo Scientific, USA). The peaks were individually integrated and normalized to the total peak area for each spectrum. Metabolites were identified by comparison of retention times and MS fragmentation patterns with in-house reference data and libraries available in the National Institute of Standards and Technology (NIST) Mass Spectral Search Program (version 2.0). Commercially available mixed FAME standards were used to aid assignment of FAMEs (food industry FAME mix; Sigma-Aldrich, United Kingdom).
LC-MS assay of amino acid and polyamine metabolites.
Amino acid and polyamine analytes were derivatized with chloroformate using the EZ:faast amino acid analysis (AAA) MS kit (Phenomenex, United Kingdom) and analyzed using methods reported in reference 41. Briefly, aliquots of the aqueous phase of chloroform-methanol cell extracts were reconstituted in 100 μl phosphate-buffered saline (PBS) containing homoarginine, methionine-D3, and homophenylalanine as internal standards. Following extraction and derivatization using the EZ:faast kit, analytes were reconstituted in 100 μl of a 1:2 (vol/vol) mixture containing 10 mM ammonium formate in water and 10 μM ammonium formate in methanol. Analytes were separated using a Waters Acquity UPLC system (Waters, United Kingdom) and mass spectrometric data collected using a Waters Quattro Premier XE Triple Quadrupole mass spectrometer equipped with an electrospray source and operated in positive ionization mode as described in reference 41. Multiple reaction monitoring (MRM) was used to quantify analytes, monitoring specific transitions for each analyte. At least 15 data points across each analyte peak were used for quantification. A sample containing known concentrations of chemical standards for each analyte was run at the beginning and end of the LC-MS analysis to enable calibration. Raw data were processed using Waters QuanLynx software version 4.1 (Waters, USA). Responses were calculated relative to the internal standard homophenylalanine, and concentrations were calculated from mean-averaged calibration curves of each analyte.
Data analysis.
The order of all samples was randomized prior to data analysis. Unpaired Student's t tests (with no equal-variance assumption) were used to calculate statistical significance. The Benjamini-Hochberg correction, α = 0.1, was applied to control the false discovery rate (42).
Principal component analysis (PCA) was performed using SIMCA-P+ 11.5 (Umetrics, Umeå, Sweden). NMR data were Pareto scaled: each variable was mean centered and then multiplied by 1/Sk, where Sk is the standard deviation of the variable. This scaling increases the contributions of variables with low means to the model, without introducing excessive noise. Metabolic perturbations were identified by analysis of the corresponding loadings plots. The robustness of all principal component models was assessed using R2 values (i.e., the fraction of variance explained by the components).
RESULTS
The metabolic footprints of P. aeruginosa PAO1 and a signal-deficient lasI rhlI mutant diverge at the point where AHL concentrations peak in the wild type.
Cultures of P. aeruginosa PAO1 and of an isogenic lasI rhlI mutant were grown aerobically in alanine-glycerol-salts medium supplemented with yeast extract (AGSY medium [43]), and the profiles of OdDHL and BHL were measured (Fig. 1a). The two strains grew at the same rate during exponential growth (growth rate [μ] = 1.12 h−1), but during early stationary phase (between 4 and 6 h), the lasI rhlI mutant achieved an optical density which remained ca. 15 to 25% higher than that of the wild type for the remainder of the time course (P ≤ 0.05 at each time point from 5 to 10 h, Student's t test) (Fig. 1a). In the wild type, the OdDHL concentration in the culture supernatant peaked after 4 h growth. The BHL concentration peaked slightly later, after 5 h growth. The concentrations of both AHLs declined thereafter (presumably due to alkalization of the growth medium [44]). No AHLs were detected in the supernatant of the lasI rhlI mutant culture. In parallel, cell-free supernatants from each strain were harvested throughout the growth curve and analyzed by 1H-NMR spectroscopy. Subtle differences in the profiles of NMR-visible metabolites in the culture supernatants of each strain were apparent, especially at the later time points (representative spectra taken after 1, 7, and 10 h of growth are shown in Fig. 1b). Given that QS is a transcriptional regulatory mechanism, we also assayed (in separate cultures, containing antibiotic selection) the promoter activity of lasB and rhlA, genes whose expression is known to be primarily dependent on OdDHL and BHL, respectively. This was done using the pQF50-based lacZ reporter constructs, pβ01 (to measure PlasB promoter activity) and pβ02 (to measure PrhlA activity). The β-galactosidase gene expression driven by PlasB and PrhlA was negligible in the lasI rhlI mutant background. In contrast, in the wild-type background, PlasB activity rose sharply between 4 and 6 h growth, and PrhlA activity increased between 5 and 7 h of growth (Fig. 1c), corresponding well with the accumulation of OdDHL and BHL, respectively (Fig. 1a).
To identify latent patterns in the 1H-NMR data, we used principal component analysis (PCA). This revealed that the “metabolic footprints” (45) of the wild-type and QS mutant were essentially indistinguishable until 5 h of growth (Fig. 2), corresponding to the point at which AHL concentrations peak in the wild type. Following this, the footprints diverged. This was especially noticeable with the variables contributing toward principal components 2 and 3 (Fig. 2b and c). We conclude that there is a good correlation between the onset of QS-dependent gene expression (Fig. 1c) and the metabolic divergence seen in the footprint data (Fig. 2).
The lasI rhlI mutant takes up alanine faster and excretes more acetate.
Inspection of the loadings underpinning the plot of PCA scores shown in Fig. 2 revealed two key variables driving divergence of the metabolic footprints between 5 and 7 h of growth in the lasI rhlI mutant compared to the wild type: (i) increased uptake of alanine (Fig. 3a) and (ii) increased levels of excreted acetate (Fig. 3b). In addition, and as noted above, the lasI rhlI mutant also grew to a higher overall cell density (Fig. 1a). These data suggest that the lasI rhlI mutant assimilated carbon more efficiently over this period. The reduced efficiency of carbon assimilation by the wild type could be explained by QS-dependent diversion of cellular resources toward secreted factors (46). However, while this would account for differences in output (re growth yield and acetogenesis) it does not explain differences in input (re uptake of alanine).
Perturbations to central metabolism revealed by cellular metabolic profiling.
We reasoned that the divergence in metabolic footprint of the wild-type and QS mutant after 5 h of growth was likely driven by differences in the intracellular metabolic activity of each strain. To investigate this, we analyzed the water-soluble fraction of cell extracts by GC-MS. Four sampling points were chosen to bracket the metabolic footprint divergence, which occurred at an OD of 3. These sampling points were ODs of 1.5, 2.5, 4.5, and 6.5. We selected sampling points based on optical density in order to account for differences in growth rates between the strains. Consistent with the footprinting data, the metabolic profiles of the wild-type and mutant strains were essentially identical at an OD of 1.5 and OD of 2.5 (Fig. 4). However, between an OD of 2.5 and OD of 4.5, the concentrations of about one-third of the >90 identified metabolites in the GC-MS analysis had become altered in the mutant (see Table S1 in the supplemental material). Key metabolites were affected in central carbon, amino acid, nucleic acid, fatty acid, and membrane metabolism, pointing to widespread metabolic retuning driven by AHL-dependent QS. For example, citrate and malate concentrations were both increased in the lasI rhlI mutant at an OD of 4.5, and by the time an OD of 6.5 was attained, other tricarboxylic acid (TCA) cycle intermediates, such as succinate and fumarate, were also more abundant. As the central hub of metabolism, the TCA cycle not only oxidizes carbon sources, it also provides anabolic precursors for biosynthesis. The reallocation of cellular resources toward the synthesis of AHL-induced secreted factors may explain the depletion of TCA intermediates in the wild type and may also help to account for its small but consistent lower growth yield relative to the lasI rhlI mutant.
AHL signaling induces fatty acid cyclopropanation and increased acyl chain length.
The GC-MS analysis revealed that in the lasI rhlI mutant, the concentrations of six out of the seven identified fatty acids were altered relative to the wild type at either an OD of 4.5 or an OD of 6.5: the levels of 2-hydroxypentanedioic acid were higher in the mutant, whereas the levels of 3-hydroxydecanoic acid, hexadecanoic acid, octadecanoic acid, nonanoic acid, and a monounsaturated fatty acid which we could not unambiguously identify were all lower in the mutant. To gain a better picture of the impact of QS on fatty acid metabolism, we used fatty acid methyl ester (FAME) GC-MS to analyze the total fatty acid content in the organic-phase extracts of the cells (Table 2). Consistent with our earlier observations, the wild-type and QS mutant fatty acid profiles were nearly identical before diverging at the onset of stationary phase. The fatty acid profile of wild-type cells (Fig. 5a) at later time points showed trends similar to those reported previously to be associated with stationary-phase adaptation, namely, increased fatty acid cyclopropanation (diamonds) and saturation (circles) with a concomitant reduction in the level of cis-unsaturated fatty acids (squares) (47–49).
TABLE 2.
Fatty acid profiles of bacterial cells as determined by FAME GC-MS analysis
| Characteristic and fatty acida | Mean value for characteristicb |
% complementationc |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD600 of 1.5 |
OD600 of 2.5 |
OD600 of 4.5 |
OD600 of 6.5 |
OD600 of 1.5 |
OD600 of 4.5 |
|||||||||
| WT | LIRI | WT | LIRI | WT | LIRI | WT | LIRI | BHL | OdDHL | BHL + OdDHL | BHL | OdDHL | BHL + OdDHL | |
| % of total for individual or types of fatty acids | ||||||||||||||
| 12:0 | 0 | 0 | 0 | 0.1 | 0.1 | 0 | 0.4 | 0.2** | NS | NS | NS | 97**↓ | 35**↓ | 109**↓ |
| 14:0 | 1.5 | 1.7** | 1.5 | 1.8** | 1.2 | 2.2** | 2.3 | 3.5** | NS | NS | 128*↓ | 76**↓ | 43**↓ | 102**↓ |
| 7Z-14:1 | 0.1 | 0.1 | 0 | 0 | 0 | 0 | 0.1 | 0.1** | NS | NS | NS | 84**↓ | 53**↓ | 92*↓ |
| 9Z-14:1 | 0.2 | 0.1 | 0.2 | 0.2 | 0.2 | 0.1 | 0.6 | 0.5 | NS | NS | NS | NS | NS | NS |
| 15:0 | 0.2 | 0.3 | 0.3 | 0.3 | 0.2 | 0.2 | 0.2 | 0.2 | NS | NS | −287*↑ | NS | 86*↑ | 203*↑ |
| 16:0 | 39 | 39 | 39 | 38.8 | 40.2 | 41.6** | 38.3 | 41.7** | NS | NS | NS | NS | NS | 135*↓ |
| 9Z-16:1 | 18 | 18.7** | 18 | 19** | 14.4 | 18.4** | 10.1 | 15.7** | NS | NS | NS | 69**↓ | 28**↓ | 82**↓ |
| 17:0 | 0.1 | 0.1 | 0.1 | 0.1** | 0.1 | 0.1** | 0.3 | 0.3 | NS | NS | 19,353*↑ | 46**↑ | 29**↑ | 126*↑ |
| 9cyc-17:0 | 0.3 | 0.3 | 0.3 | 0.3 | 1.6 | 1** | 3.5 | 2.1** | 46**↑ | NS | 116*↑ | 64**↑ | 24**↑ | 44**↑ |
| 18:0 | 5.2 | 5.1 | 5.6 | 5.2 | 4.9 | 4** | 14 | 10.7** | NS | NS | 132**↑ | 57**↑ | NS | 73*↑ |
| 11Z-18:1 | 35.2 | 34.5** | 34.6 | 34 | 35.6 | 31.9** | 26.7 | 24.1** | NS | NS | 172*↑ | 62**↑ | 34**↑ | 111**↑ |
| 11cyc-19:0 | 0.1 | 0.1** | 0.1 | 0.1 | 1.2 | 0.4** | 3.5 | 0.7** | 2**↑ | NS | 112*↑ | 54**↑ | 16**↑ | 52**↑ |
| 13Z-22:1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0** | 0.2 | 0.2 | NS | NS | NS | NS | NS | NS |
| Sum SFA | 46 | 46.2 | 46.6 | 46.3 | 46.8 | 48.1** | 55.5 | 56.6 | 67*↓ | NS | 245*↓ | 43**↓ | 36**↓ | 128**↓ |
| Sum CUFA | 53.5 | 53.4 | 53 | 53.3 | 50.4 | 50.5 | 37.6 | 40.5** | NS | NS | NS | NS | NS | 314*↑ |
| Sum CPFA | 0.5 | 0.3** | 0.4 | 0.4 | 2.9 | 1.4** | 7 | 2.8** | 27**↑ | NS | 115*↑ | 59**↑ | 20**↑ | 48**↑ |
| ACCL | 16.8 | 16.8** | 16.8 | 16.7 | 16.8 | 16.7** | 16.8 | 16.6** | NS | NS | 166*↑ | 62**↑ | 32**↑ | 101**↑ |
| C18/C16 ratio | ||||||||||||||
| All | 0.7 | 0.7** | 0.7 | 0.7 | 0.7 | 0.6** | 0.9 | 0.6** | NS | NS | 173**↑ | 57**↑ | 28**↑ | 102**↑ |
| SFA | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1** | 0.4 | 0.3** | NS | NS | 148**↑ | 52**↑ | NS | 80*↑ |
| CUFA | 2 | 1.8** | 1.9 | 1.8** | 2.5 | 1.7** | 2.7 | 1.5** | NS | NS | 144*↑ | 60**↑ | 26**↑ | 90**↑ |
| CPFA | 0.4 | 0.3** | 0.4 | 0.3 | 0.8 | 0.4** | 1 | 0.4** | NS | NS | NS | 56**↑ | 16**↑ | 80**↑ |
| CUFA + CPFA | 1.9 | 1.8** | 1.9 | 1.8** | 2.3 | 1.7** | 2.2 | 1.4** | NS | NS | 145*↑ | 61**↑ | 27**↑ | 95**↑ |
Individual fatty acids are indicated according to the carboxyl reference system; for example, “9Z-16:1” indicates a fatty acid with a 16-carbon acyl chain containing a cis C=C double bond at the ninth position from the carboxyl carbon (hexadec-9-enoic acid, or palmitoleic acid). Abbreviations: SFA, saturated fatty acid; CUFA, cis-unsaturated fatty acid; CPFA, cyclopropane fatty acid; ACCL, average carbon chain length.
Values for the wild type (WT) and the lasI rhlI mutant (LIRI) are shown. Values that are significantly different (P ≤ 0.01) from the WT value by two-tailed Student's t test are indicated (**).
Percent complementation was calculated as the percentage return toward the wild-type phenotype (values are shown only where P ≤ 0.05). Values that are significantly different from the value for the lasI rhlI mutant by two-tailed Student's t test are indicated by asterisks as follows: *, P ≤ 0.05; **, P ≤ 0.01. Values that are not significantly different (NS) are also indicated. Arrows pointing up and down indicate an increase or decrease in response to chemical complementation compared to the lasI rhlI mutant, respectively.
The lasI rhlI mutant showed a pattern of fatty acid saturation similar to that of the wild type. However, the degree of fatty acid cyclopropanation was less than half that of the wild type (Fig. 5b). Moreover, the average acyl chain length of fatty acids in the QS mutant decreased at higher optical densities, whereas they remained essentially unchanged in the wild type (Fig. 5c). This shortening of average acyl chain length was mainly driven by a lower 18:1/16:1 cis-unsaturated fatty acid ratio. Chemical complementation of the lasI rhlI mutant with synthetic AHLs restored the fatty acid profile toward wild-type levels (Table 2). Interestingly, cyclopropanation was more responsive to BHL than to OdDHL, suggesting that this may be more a consequence of Rhl signaling than Las signaling. These data suggest that AHL-dependent QS induces wild-type cell membranes to become less fluid and more chemically stable as the cells progress into stationary phase.
Polyamine concentrations are elevated in the lasI rhlI mutant.
Our GC-MS analysis of aqueous cell extracts indicated that amino acid and polyamine metabolism differ between the wild-type and mutant: 9 out of 22 amino acids or related metabolites were altered at an OD of 4.5. To investigate this further, we used a targeted LC-MS method to measure amino acid and polyamine metabolites in aqueous cell extracts harvested at an OD of 1.5 and an OD of 4.5 (see Table S2 in the supplemental material). One striking result was that the concentrations of putrescine and spermidine and their precursor ornithine were lower in wild-type cells than in lasI rhlI mutant cells during stationary phase (by 4-fold, 5-fold, and 1.8-fold, respectively): whereas the concentrations of these metabolites in the wild type decreased during the transition from exponential- to stationary-phase growth, concentrations in the lasI rhlI mutant remained more constant. Addition of BHL (and to a lesser extent also OdDHL) to lasI rhlI cultures decreased the stationary-phase concentrations of putrescine and spermidine toward wild-type concentrations (see Table S2). Interestingly, ornithine levels increased in the complemented lasI rhlI mutant.
DISCUSSION
Although there have been several proteomic and transcriptomic analyses of QS in P. aeruginosa (27–29), to our knowledge, no previous studies have examined the impact of QS on the small-molecule profile of this organism. In the current work, we redress this situation by showing that QS gives rise to substantial changes in most major domains of metabolism.
Several lines of evidence suggest that QS, a transcriptional regulatory system, may impact metabolic functions. For example, Schuster et al. (28) showed that QS positively controls glycogen phosphorylase (glgP), glycogen branching enzyme (glgB), and glycogen synthase (PA2165; glgA) transcription, perhaps indicating mobilization or remodeling of stored energy reserves. Consistent with this, the results of our GC-MS analysis revealed that a large number of sugars and sugar phosphates were perturbed in the lasI rhlI mutant (see Table S1 in the supplemental material), suggesting a general restructuring of carbohydrate metabolism. Schuster et al. also showed that glyceraldehyde 3-phosphate dehydrogenase (gapA) is positively QS regulated. Such a bottleneck at GapA in the lasI rhlI mutant could explain why we observed an accumulation of glycerophosphate in this mutant (glycerophosphate becomes converted to dihydroxyacetone phosphate and thence to glyceraldehyde 3-phosphate by GlpD and triose phosphate isomerase, respectively). Indeed, although there is substantial variation in the QS-controlled transcriptome depending on the growth conditions used, after excluding transcripts encoding secreted factors, transcripts encoding functions in central intermediary metabolism and fatty acid/phospholipid metabolism are by far the most extensively modulated functional class in the “core” (i.e., conserved across multiple studies) QS regulon (7). Interestingly, the Las signaling system appears to predominantly control the expression of genes involved in central intermediary metabolism, whereas the Rhl system regulates genes involved in fatty acid metabolism (7). Consistent with this, the data in Table 2 indicate that chemical complementation of lasI rhlI mutant cultures with BHL alone elicited larger changes in the cellular fatty acid pool than complementation with OdDHL alone. Moreover, we note that one of the most consistently upregulated QS-dependent genes across all microarray studies is fabH2, encoding 3-oxoacyl-ACP (acyl carrier protein) synthase III, involved in fatty acid synthesis. In addition, atoB (acetyl coenzyme A [acetyl-CoA] acetyltransferase), rhlA [3-(3-hydroxyalkanoyloxy) alkanoic acid synthase], and phaC2 [poly(3-hydroxyalkanoic acid synthase)]—all of which would be expected to influence the composition of the fatty acid/fatty acyl-CoA/fatty acyl-ACP pool—are QS regulated in most or all microarray analyses (18, 27, 28). Taken together, these data indicate that QS has a major impact on fatty acid metabolism and that at least some of this is likely driven by QS-dependent changes in the expression of the genes involved. However, beyond generalizations, it was difficult to correlate many of the metabolic changes that we observed with changes in the level of specific transcripts reported in earlier microarray studies. This may reflect the different growth conditions used in this work compared with previous microarray studies or the fact that as has been pointed out previously (7, 29), there is not always a good correlation between the transcriptional and posttranslational effects of QS. Moreover, metabolic control theory tells us that even relatively modest changes in enzyme activity (which may be influenced not only by the expression level of the enzyme but also by allosteric factors, etc.) can have a disproportionately large impact on metabolite concentrations.
Why should QS influence metabolism? Dandekar et al. have suggested that by bringing “private goods” such as intracellular metabolic functions under QS control, the emergence of “social cheats” may be suppressed (31). While this may indeed be the case for a select number of specific metabolic enzymes, we do not believe that this explanation accounts for the large-scale metabolic perturbations observed in the current study. A more prosaic explanation is that certain intracellular metabolic pathways are required for production of the biosynthetic building blocks that are needed for QS-controlled exoenzyme and secondary metabolite synthesis, so it makes sense to bring these under QS control. A related, but subtly different explanation is that the reallocation of existing resources—the “metabolic rewiring”—which accompanies QS-dependent upregulation of exoenzyme and secondary metabolite synthesis may itself lead to a global metabolic readjustment. One final contributory factor is that some QS-regulated metabolic genes might play a protective role; as noted by Schuster et al. (28), this may explain why, e.g., the glucose-6-phosphate dehydrogenase-encoding gene, PA3183, is regulated by QS. Our observation that the wild type displayed increased fatty acid saturation, chain length, and cyclopropanation (compared with the lasI rhlI mutant) upon entry into the stationary phase is consistent with QS playing a role in the response to membrane stress.
That QS-regulated products are metabolically costly and are likely to impose an energetic burden on the producer was nicely illustrated by Sandoz et al. (50), who cocultured wild-type P. aeruginosa and a lasR mutant under conditions in which QS-dependent extracellular proteases are required for growth. The lasR mutant (which could exploit the secreted proteases—“public goods” made by the wild type) grew approximately 60% faster than its wild-type counterpart (50). Consistent with the additional biosynthetic load imposed by QS, we found that wild-type cultures grew to a lower final optical density than the lasI rhlI mutant (especially following the period 4 to 6 h postinoculation, when AHL concentrations peaked). The redirection of metabolic resources away from central metabolism and toward QS-dependent biosynthesis may, in part, explain why the wild type was more depleted of TCA cycle intermediates compared with the lasI rhlI mutant. In addition, 1H-NMR spectroscopy footprinting revealed that the profile of extracellular low-molecular-weight compounds in the lasI rhlI mutant and wild-type cultures began to diverge after 4 to 6 h growth. This divergence was driven mainly by a decrease in the uptake of alanine by the wild type coupled with reduced (net) excretion of acetate (compared with the lasI rhlI mutant). Acetogenic overflow occurs when the influx of reduced carbon into cells exceeds the catabolic capacity of the TCA cycle and the electron transfer chain (51). Conversion of acetyl-CoA to acetate by phosphate acetyltransferase and acetate kinase yields ATP and recycles coenzyme A, thereby relieving CoASH bottlenecks (e.g., at 2-ketoglutarate dehydrogenase) and supporting higher growth rates (52). One explanation for the lower excreted acetate levels in wild-type culture supernatants is that acetogenic fermentation is depressed due to a QS-dependent increase in the capacity for cellular oxidative phosphorylation, perhaps mediated by AHL-regulated redox shuttles such as phenazines (53) and upregulated expression of the cytochrome oxidases coxAB and cyoAB, as well as genes in the Entner-Doudoroff pathway (18, 25, 27, 28). An alternative explanation is that since excreted acetate can be reassimilated once other carbon sources have been exhausted, the lasI rhlI mutant might exhibit impaired acetate uptake. Interestingly, in Vibrio fischeri, QS has been previously shown to directly control this “acetate switch” by stimulating the expression of acetyl-CoA synthetase (ACS) (encoded by acsA), required for acetate uptake and assimilation (54). However, in P. aeruginosa, acsA is one of the few genes that is QS repressed (28, 29), so we favor a model in which acetogenesis is depressed by QS in the wild type.
Bacteria tune the properties of their membranes by regulating fatty acid biosynthesis and by directly modifying existing membrane lipids (55). cis C=C double bonds disrupt acyl chain packing in membrane bilayers and are vulnerable to peroxidation (56). By increasing the length and saturation of lipid acyl chains, bacteria adapt their membranes to higher temperatures, nutrient deprivation, and resistance to membrane stressors such as lipophilic solvents, acids, and reactive oxygen species (57–61). Like other Gram-negative bacteria, P. aeruginosa can cyclopropanate cis-monounsaturated lipid acyl chains by inserting methylene groups across their C=C double bonds; this allows rapid adaptation of membrane properties without metabolically costly de novo fatty acid synthesis (62, 63). Cyclopropane acyl chains are chemically stable and appear to have effects similar to those of cis-monounsaturated fatty acids on membrane fluidity (64). Cyclopropanation of unsaturated fatty acyl chains occurs in response to nutrient deprivation (48, 64, 65) and can increase resistance to membrane stressors, including reactive oxygen species and low pH (56, 62, 66). Consistent with previous studies, we observed increased fatty acid saturation, chain length, and cyclopropanation as the wild-type PAO1 strain entered stationary phase (48, 63). However, in the lasI rhlI mutant, the average acyl chain length decreased upon entry to stationary phase, and the degree of cyclopropanation was half that of the wild type. These phenotypes were complemented by the addition of exogenous AHLs. Our data therefore indicate that QS contributes toward the development of a less fluid and more chemically stable membrane as the culture exits exponential-phase growth. We do not yet know whether this is a direct or indirect consequence of QS.
We also observed that polyamine levels were decreased by AHL-dependent QS. Polyamines are multifunctional molecules associated with a variety of stress responses, notably against oxidative stress (67, 68). Polyamines execute these functions by diverse means, including modulating gene expression, modifying the activities of cell membrane components, and by scavenging free radicals. By sequestering free radicals, polyamines can protect cell components (especially GC-rich DNA) against reactive oxygen species (ROS)-induced stress. In the wild type, QS is known to elicit increased expression of enzymes involved in ROS detoxification, such as superoxide dismutases (Mn-SOD and Fe-SOD), the major catalase KatA (69), and NADPH-generating glucose-6-phosphate dehydrogenase (NADPH is required for the synthesis of glutathione). Indeed, it has been suggested recently that since they are “private goods,” the expression of these detoxification enzymes is another key factor suppressing the appearance of “social cheats” in the population (70). Since these enzymes would no longer be upregulated in the lasI rhlI mutant, we speculate that the elevated levels of polyamines reflect an alternative metabolic response to increased ROS-induced stress as the culture enters stationary phase.
In summary, we have shown here that QS has a major impact on metabolism in P. aeruginosa. Almost every major domain of metabolism was affected, with particularly notable changes associated with carbohydrate, polyamine, and fatty acid/lipid metabolism. The changes that we observed seem to be consistent with a variety of drivers, ranging from altered QS-dependent transcription of “metabolic genes” to the effect(s) of global “metabolic readjustment” as a consequence of QS-dependent exoproduct synthesis and a general stress response, among others. However, most of the metabolic changes that we observed could not be easily inferred from inspection of available microarray data sets. This reinforces the notion that multiple levels of regulatory control impact on cellular physiology and that extreme caution should be used when attempting to extrapolate from the transcriptome to the metabolome (or vice versa).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a BBSRC studentship to P.W.D.
Baljit Ubhi is thanked for assistance with the LC-MS analysis. James Hodgkinson and David Spring are thanked for kindly providing OdDHL and BHL.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02557-14.
REFERENCES
- 1.Ramos J-L. 2004. Pseudomonas, vol 1 Genomics, life style and molecular architecture. Kluwer Academic Publishers, New York, NY. [Google Scholar]
- 2.Römling U, Kader A, Sriramulu DD, Simm R, Kronvall G. 2005. Worldwide distribution of Pseudomonas aeruginosa clone C strains in the aquatic environment and cystic fibrosis patients. Environ Microbiol 7:1029–1038. doi: 10.1111/j.1462-2920.2005.00780.x. [DOI] [PubMed] [Google Scholar]
- 3.Kerr KG, Snelling AM. 2009. Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect 73:338–344. doi: 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 4.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 5.Gaspar MC, Couet W, Olivier J-C, Pais AACC, Sousa JJS. 2013. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: a review. Eur J Clin Microbiol Infect Dis 32:1231–1252. doi: 10.1007/s10096-013-1876-y. [DOI] [PubMed] [Google Scholar]
- 6.Ramos J-L. 2004. Pseudomonas, vol 3 Biosynthesis of macromolecules and molecular metabolism. Kluwer Academic Publishers, New York, NY. [Google Scholar]
- 7.Schuster M, Greenberg EP. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 8.Palmer KL, Aye LM, Whiteley M. 2007. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J Bacteriol 189:8079–8087. doi: 10.1128/JB.01138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos J-L. 2004. Pseudomonas, vol 2 Virulence and gene regulation. Kluwer Academic Publishers, New York, NY. [Google Scholar]
- 10.Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH. 1993. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260:1127–1130. doi: 10.1126/science.8493556. [DOI] [PubMed] [Google Scholar]
- 11.Smith RS, Iglewski BH. 2003. P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol 6:56–60. doi: 10.1016/S1369-5274(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 12.Smith D, Wang J-H, Swatton JE, Davenport P, Price B, Mikkelsen H, Stickland H, Nishikawa K, Gardiol N, Spring DR, Welch M. 2006. Variations on a theme: diverse N-acyl homoserine lactone-mediated quorum sensing mechanisms in gram-negative bacteria. Sci Prog 89:167–211. doi: 10.3184/003685006783238335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winson MK, Camara M, Latifi A, Foglino M, Chhabra SR, Daykin M, Bally M, Chapon V, Salmond GP, Bycroft BW. 1995. Multiple N-acyl-l-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 92:9427–9431. doi: 10.1073/pnas.92.20.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. 1996. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol 21:1137–1146. doi: 10.1046/j.1365-2958.1996.00063.x. [DOI] [PubMed] [Google Scholar]
- 15.Pesci E, Pearson J, Seed P, Iglewski B. 1997. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 179:3127–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner VE, Li L-L, Isabella VM, Iglewski BH. 2007. Analysis of the hierarchy of quorum-sensing regulation in Pseudomonas aeruginosa. Anal Bioanal Chem 387:469–479. doi: 10.1007/s00216-006-0964-6. [DOI] [PubMed] [Google Scholar]
- 17.Dekimpe V, Déziel E. 2009. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology 155:712–723. doi: 10.1099/mic.0.022764-0. [DOI] [PubMed] [Google Scholar]
- 18.Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Høiby N, Givskov M. 2003. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J 22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imperi F, Massai F, Pillai CR, Longo F, Zennaro E, Rampioni G, Visca P, Leoni L. 2013. New life for an old drug: the anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob Agents Chemother 57:996–1005. doi: 10.1128/AAC.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobsen TH, Bjarnsholt T, Jensen PØ, Givskov M, Høiby N. 2013. Targeting quorum sensing in Pseudomonas aeruginosa biofilms: current and emerging inhibitors. Future Microbiol 8:901–921. doi: 10.2217/fmb.13.57. [DOI] [PubMed] [Google Scholar]
- 21.O'Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. 2013. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A 110:17981–17986. doi: 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raina S, De Vizio D, Odell M, Clements M, Vanhulle S, Keshavarz T. 2009. Microbial quorum sensing: a tool or a target for antimicrobial therapy? Biotechnol Appl Biochem 54:65–84. doi: 10.1042/BA20090072. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen TB, Givskov M. 2006. Quorum sensing inhibitors: a bargain of effects. Microbiology 152:895–904. doi: 10.1099/mic.0.28601-0. [DOI] [PubMed] [Google Scholar]
- 24.Hassett DJ, Cuppoletti J, Trapnell B, Lymar SV, Rowe JJ, Sun Yoon S, Hilliard GM, Parvatiyar K, Kamani MC, Wozniak DJ, Hwang S-H, McDermott TR, Ochsner UA. 2002. Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv Drug Deliv Rev 54:1425–1443. doi: 10.1016/S0169-409X(02)00152-7. [DOI] [PubMed] [Google Scholar]
- 25.Heurlier K, Dénervaud V, Haas D. 2006. Impact of quorum sensing on fitness of Pseudomonas aeruginosa. Int J Med Microbiol 296:93–102. doi: 10.1016/j.ijmm.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 26.Chugani S, Kim BS, Phattarasukol S, Brittnacher MJ, Choi SH, Harwood CS, Greenberg EP. 2012. Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc Natl Acad Sci U S A 109:E2823–E2831. doi: 10.1073/pnas.1214128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. 2003. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol 185:2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuster M, Lostroh CP, Ogi T, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol 185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arevalo-Ferro C, Hentzer M, Reil G, Görg A, Kjelleberg S, Givskov M, Riedel K, Eberl L. 2003. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ Microbiol 5:1350–1369. doi: 10.1046/j.1462-2920.2003.00532.x. [DOI] [PubMed] [Google Scholar]
- 30.Goo E, Majerczyk CD, An JH, Chandler JR, Seo Y-S, Ham H, Lim JY, Kim H, Lee B, Jang MS, Greenberg EP, Hwang I. 2012. Bacterial quorum sensing, cooperativity, and anticipation of stationary-phase stress. Proc Natl Acad Sci U S A 109:19775–19780. doi: 10.1073/pnas.1218092109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dandekar AA, Chugani S, Greenberg EP. 2012. Bacterial quorum sensing and metabolic incentives to cooperate. Science 338:264–266. doi: 10.1126/science.1227289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beatson SA, Whitchurch CB, Semmler ABT, Mattick JS. 2002. Quorum sensing is not required for twitching motility in Pseudomonas aeruginosa. J Bacteriol 184:3598–3604. doi: 10.1128/JB.184.13.3598-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monson R, Foulds I, Foweraker J, Welch M, Salmond GPC. 2011. The Pseudomonas aeruginosa generalized transducing phage phiPA3 is a new member of the phiKZ-like group of “jumbo” phages, and infects model laboratory strains and clinical isolates from cystic fibrosis patients. Microbiology 157:859–867. doi: 10.1099/mic.0.044701-0. [DOI] [PubMed] [Google Scholar]
- 34.R Development Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 35.Choi K-H, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Ramsay J. 2013. High-throughput β-galactosidase and β-glucuronidase assays using fluorogenic substrates. Bio-protocol 3:e827. [Google Scholar]
- 37.Swift S, Karlyshev AV, Fish L, Durant EL, Winson MK, Chhabra SR, Williams P, Macintyre S, Stewart GS. 1997. Quorum sensing in Aeromonas hydrophila and Aeromonas salmonicida: identification of the LuxRI homologs AhyRI and AsaRI and their cognate N-acylhomoserine lactone signal molecules. J Bacteriol 179:5271–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winson MK, Swift S, Fish L, Throup JP, Jørgensen F, Chhabra SR, Bycroft BW, Williams P, Stewart GSAB. 1998. Construction and analysis of luxCDABE-based plasmid sensors for investigating N-acyl homoserine lactone-mediated quorum sensing. FEMS Microbiol Lett 163:185–192. doi: 10.1111/j.1574-6968.1998.tb13044.x. [DOI] [PubMed] [Google Scholar]
- 39.Atherton HJ, Bailey NJ, Zhang W, Taylor J, Major H, Shockcor J, Clarke K, Griffin JL. 2006. A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR-α null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiol Genomics 27:178–186. doi: 10.1152/physiolgenomics.00060.2006. [DOI] [PubMed] [Google Scholar]
- 40.Morrison WR, Smith LM. 1964. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J Lipid Res 5:600–608. [PubMed] [Google Scholar]
- 41.Ubhi BK, Davenport PW, Welch M, Riley J, Griffin JL, Connor SC. 2013. Analysis of chloroformate-derivatised amino acids, dipeptides and polyamines by LC–MS/MS. J Chromatogr B 934:79–88. doi: 10.1016/j.jchromb.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol 57:289–300. [Google Scholar]
- 43.Mikkelsen H, Duck Z, Lilley KS, Welch M. 2007. Interrelationships between colonies, biofilms, and planktonic cells of Pseudomonas aeruginosa. J Bacteriol 189:2411–2416. doi: 10.1128/JB.01687-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yates EA, Philipp B, Buckley C, Atkinson S, Chhabra SR, Sockett RE, Goldner M, Dessaux Y, Cámara M, Smith H, Williams P. 2002. N-acylhomoserine lactones undergo lactonolysis in a pH-, temperature-, and acyl chain length-dependent manner during growth of Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Infect Immun 70:5635–5646. doi: 10.1128/IAI.70.10.5635-5646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kell DB, Brown M, Davey HM, Dunn WB, Spasic I, Oliver SG. 2005. Metabolic footprinting and systems biology: the medium is the message. Nat Rev Microbiol 3:557–565. doi: 10.1038/nrmicro1177. [DOI] [PubMed] [Google Scholar]
- 46.Nouwens AS, Beatson SA, Whitchurch CB, Walsh BJ, Schweizer HP, Mattick JS, Cordwell SJ. 2003. Proteome analysis of extracellular proteins regulated by the las and rhl quorum sensing systems in Pseudomonas aeruginosa PAO1. Microbiology 149:1311–1322. doi: 10.1099/mic.0.25967-0. [DOI] [PubMed] [Google Scholar]
- 47.Dubois-Brissonnet F, Malgrange C, Guérin-Méchin L, Heyd B, Leveau J. 2000. Effect of temperature and physiological state on the fatty acid composition of Pseudomonas aeruginosa. Int J Food Microbiol 55:79–81. doi: 10.1016/S0168-1605(00)00198-7. [DOI] [PubMed] [Google Scholar]
- 48.Dubois-Brissonnet F, Malgrange C, Guérin-Méchin L, Heyd B, Leveau JY. 2001. Changes in fatty acid composition of Pseudomonas aeruginosa ATCC 15442 induced by growth conditions: consequences of resistance to quaternary ammonium compounds. Microbios 106(414):97–110. [PubMed] [Google Scholar]
- 49.Härtig C, Loffhagen N, Harms H. 2005. Formation of trans fatty acids is not involved in growth-linked membrane adaptation of Pseudomonas putida. Appl Environ Microbiol 71:1915–1922. doi: 10.1128/AEM.71.4.1915-1922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandoz KM, Mitzimberg SM, Schuster M. 2007. Social cheating in Pseudomonas aeruginosa quorum sensing. Proc Natl Acad Sci U S A 104:15876–15881. doi: 10.1073/pnas.0705653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolfe AJ. 2005. The acetate switch. Microbiol Mol Biol Rev 69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Mansi M. 2004. Flux to acetate and lactate excretions in industrial fermentations: physiological and biochemical implications. J Ind Microbiol Biotechnol 31:295–300. doi: 10.1007/s10295-004-0149-2. [DOI] [PubMed] [Google Scholar]
- 53.Price-Whelan A, Dietrich LEP, Newman DK. 2007. Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 189:6372–6381. doi: 10.1128/JB.00505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Studer SV, Mandel MJ, Ruby EG. 2008. AinS quorum sensing regulates the Vibrio fischeri acetate switch. J Bacteriol 190:5915–5923. doi: 10.1128/JB.00148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y-M, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 56.Pradenas GA, Díaz-Vásquez WA, Pérez-Donoso JM, Vásquez CC. 2013. Monounsaturated fatty acids are substrates for aldehyde generation in tellurite-exposed Escherichia coli. Biomed Res Int 2013:563756. doi: 10.1155/2013/563756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denich TJ, Beaudette LA, Lee H, Trevors JT. 2003. Effect of selected environmental and physico-chemical factors on bacterial cytoplasmic membranes. J Microbiol Methods 52:149–182. doi: 10.1016/S0167-7012(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 58.Keweloh H, Diefenbach R, Rehm H-J. 1991. Increase of phenol tolerance of Escherichia coli by alterations of the fatty acid composition of the membrane lipids. Arch Microbiol 157:49–53. [DOI] [PubMed] [Google Scholar]
- 59.Massa EM, Vińals AL, Farías RN. 1988. Influence of unsaturated fatty acid membrane component on sensitivity of an Escherichia coli fatty acid auxotroph to conditions of nutrient depletion. Appl Environ Microbiol 54:2107–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pesakhov S, Benisty R, Sikron N, Cohen Z, Gomelsky P, Khozin-Goldberg I, Dagan R, Porat N. 2007. Effect of hydrogen peroxide production and the Fenton reaction on membrane composition of Streptococcus pneumoniae. Biochim Biophys Acta 1768:590–597. doi: 10.1016/j.bbamem.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Sikkema J, de Bont JA, Poolman B. 1995. Mechanisms of membrane toxicity of hydrocarbons. Microbiol Rev 59:201–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grogan D, Cronan J. 1997. Cyclopropane ring formation in membrane lipids of bacteria. Microbiol Mol Biol Rev 61:429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hancock IC, Meadow PM. 1969. The extractable lipids of Pseudomonas aeruginosa. Biochim Biophys Acta 187:366–379. doi: 10.1016/0005-2760(69)90010-1. [DOI] [PubMed] [Google Scholar]
- 64.Loffhagen N, Härtig C, Geyer W, Voyevoda M, Harms H. 2007. Competition between cis, trans and cyclopropane fatty acid formation and its impact on membrane fluidity. Eng Life Sci 7:67–74. doi: 10.1002/elsc.200620168. [DOI] [Google Scholar]
- 65.Guckert JB, Hood MA, White DC. 1986. Phospholipid ester-linked fatty acid profile changes during nutrient deprivation of Vibrio cholerae: increases in the trans/cis ratio and proportions of cyclopropyl fatty acids. Appl Environ Microbiol 52:794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang YY, Cronan JE. 1999. Membrane cyclopropane fatty acid content is a major factor in acid resistance of Escherichia coli. Mol Microbiol 33:249–259. doi: 10.1046/j.1365-2958.1999.01456.x. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida M, Kashiwagi K, Shigemasa A, Taniguchi S, Yamamoto K, Makinoshima H, Ishihama A, Igarashi K. 2004. A unifying model for the role of polyamines in bacterial cell growth, the polyamine modulon. J Biol Chem 279:46008–46013. doi: 10.1074/jbc.M404393200. [DOI] [PubMed] [Google Scholar]
- 68.Rhee HJ, Kim E-J, Lee JK. 2007. Physiological polyamines: simple primordial stress molecules. J Cell Mol Med 11:685–703. doi: 10.1111/j.1582-4934.2007.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hassett DJ, Ma J-F, Elkins JG, McDermott TR, Ochsner UA, West SEH, Huang C-T, Fredericks J, Burnett S, Stewart PS, McFeters G, Passador L, Iglewski BH. 1999. Quorum sensing in Pseudomonas aeruginosa controls expression of catalase and superoxide dismutase genes and mediates biofilm susceptibility to hydrogen peroxide. Mol Microbiol 34:1082–1093. doi: 10.1046/j.1365-2958.1999.01672.x. [DOI] [PubMed] [Google Scholar]
- 70.García-Contreras R, Nuñez-López L, Jasso-Chávez R, Kwan BW, Belmont JA, Rangel-Vega A, Maeda T, Wood TK. 2015. Quorum sensing enhancement of the stress response promotes resistance to quorum quenching and prevents social cheating. ISME J 9:115–125. doi: 10.1038/ismej.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 72.Farinha MA, Kropinski AM. 1990. Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol 172:3496–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikeda T, Kajiyama K, Kita T, Takiguchi N, Kuroda A, Kato J, Ohtake H. 2001. The synthesis of optically pure enantiomers of N-acyl-homoserine lactone autoinducers and their analogues. Chem Lett 30:314–315. doi: 10.1246/cl.2001.314. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.