

Abstract
Background
Whole genome sequences (WGS) have proliferated as sequencing technology continues to improve and costs decline. While many WGS of model or domestic organisms have been produced, a growing number of non-model species are also being sequenced. In the absence of a reference, construction of a genome sequence necessitates de novo assembly which may be beyond the ability of many labs due to the large volumes of raw sequence data and extensive bioinformatics required. In contrast, the presence of a reference WGS allows for alignment which is more tractable than assembly. Recent work has highlighted that the reference need not come from the same species, potentially enabling a wide array of species WGS to be constructed using cross-species alignment. Here we report on the creation a draft WGS from a single bighorn sheep (Ovis canadensis) using alignment to the closely related domestic sheep (Ovis aries).
Results
Two sequencing libraries on SOLiD platforms yielded over 865 million reads, and combined alignment to the domestic sheep reference resulted in a nearly complete sequence (95% coverage of the reference) at an average of 12x read depth (104 SD). From this we discovered over 15 million variants and annotated them relative to the domestic sheep reference. We then conducted an enrichment analysis of those SNPs showing fixed differences between the reference and sequenced individual and found significant differences in a number of gene ontology (GO) terms, including those associated with reproduction, muscle properties, and bone deposition.
Conclusion
Our results demonstrate that cross-species alignment enables the creation of novel WGS for non-model organisms. The bighorn sheep WGS will provide a resource for future resequencing studies or comparative genomics.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1618-x) contains supplementary material, which is available to authorized users.
Keywords: Cross-species alignment, Comparative genomics, Domestication
Background
Widespread use of high-throughput sequencers has allowed an ever increasing number of species to have a whole genome sequence (WGS) prepared. While many of these have been model or domestic organisms, a wide array of taxa continue to be sequenced (as reviewed in [1]). WGS opens the door for a multitude of subsequent analyses including: 1) creation of phylogenies and assessment of broader evolutionary patterns and innovations [2, 3]. 2) Annotation of genes [4] and identification of rearrangements or gene expansions [5, 6]. 3) Discovery of large sets of markers [7, 8]. 4) Resequencing studies, including those that are genome-wide yet not full coverage (e.g. transcriptomics or reduced representation sequencing) but benefit from the presence of a reference genome [9]. Resequencing at any scale also allows for ‘population genomics’ including investigations of local adaptation or population differentiation [10, 11], demographic history [12, 13], and the genetic basis of phenotypic traits [14].
In the absence of a reference, construction of a WGS necessitates de novo methodologies [15]. These methods require large volumes of raw sequence data which are arranged into contigs and then joined to scaffolds by either computational methods [16], anchoring with outside information (e.g. a linkage map, BACs, or FISH), or continued sequencing [17]. Such an endeavor is still relatively expensive and challenging in terms of the bioinformatics involved, making it beyond the capability of many research programs. However, the presence of a reference sequence enables reads to be aligned to the reference which is much faster and allows for lower sequence depths than de novo assembly [17, 18]. Recent work has highlighted that the reference need not come from the same species the reads are from [19–22] opening these methods to a wide array of ‘genome-enabled’ taxa [23].
There are a number of reasons why we are motivated to produce a bighorn sheep (Ovis canadensis) WGS. First, this species has a complex demographic history in North America that has been profoundly influenced by anthropogenic activity, having experienced intense hunting, local extirpations and disease-related die-offs, as well as translocations and reintroductions throughout its range [24–29]. These events are expected to have significant genetic/genomic consequences [26, 28, 30] that merit further study. Second, there are several long-term study populations in which individual based questions such as the genetic basis of complex traits [31, 32] and linkages between individual genetic variation and differences in fitness [33, 34] can be addressed using genomic data. Finally, bighorn sheep are an excellent candidate for cross-species approaches since genomic resources for domestic sheep (Ovis aries, [35, 36]) can be easily applied to bighorn sheep as they are a close relative (~3 million years divergent; [37]) and are expected to share a high degree of genomic synteny [38]. Genomic resources have been recently developed for bighorn sheep [38–42], but future resequencing efforts would be aided by species specific genomic sequence data.
Here we use cross-species alignment to create a draft genome from a single ram sequenced using ABI SOLiD technology. The pros and cons of different high-throughput sequencers have been discussed at length elsewhere [43–46]. Choice of a specific platform balances read length, the amount of sequence data output, error profiles, and cost. SOLiD technology is well-suited for resequencing studies as it returns high volumes of data and the sequence-by-ligation strategy is able to distinguish sequencing errors from true nucleotide variants during alignment [47, 48]. Based on our alignment we called variants, annotated SNPs relative to domestic sheep, and conducted enrichment analysis of those SNPs showing fixed differences.
Results
SOLiD sequencing and alignment
Whole-genome sequencing of a single bighorn sheep ram was performed using two libraries and ABI SOLiD platforms. Prior to trimming the 50 × 50 bp mate-paired library contained 311,847,628 reads, while the 75 bp fragment library contained 555,575,794 reads. Filtering and alignment were then conducted on both libraries in CLC Genomics Workbench (version 5.0). Post-trimming, read count was 218,239,459 (70% retained) and 506,697,724 (91% retained) for the mate-paired and fragment libraries respectively.
The resulting reads from each library were then independently aligned to domestic sheep chromosomes version 3.1 [36]. When aligned on its own the mate-paired library had 174,894,731 reads map to the reference, of which 115,727,618 were in pairs with an average distance of 1108 nucleotides between pairs, while the fragment library had 377,008,050 reads map to the reference. Once merged, the two libraries covered 95% of the reference genome with an average read depth of 12 (104 SD).
Variant calling
In total, 15,622,884 variants (14,583,355 SNPs and 1,039,529 indels) passed our filtering thresholds (quality ≥30, read depth between 6 and 200) and were called compared to the domestic sheep reference using SAMtools version 0.1.17 [49]. Of the putatively bi-allelic SNPs relative to the domestic sheep reference, 9,831,700 were transitions and 4,320,985 were transversions (ti/tv = 2.275; which is similar to the 2.1 ratio observed for genomic data in many mammalian studies [50]). Insertions were slightly more common than deletions (Additional File 1). To assess SNP calling accuracy, genotypes from the aligned genome were compared to those generated for the same individual on the Ovine Infinium®HD SNP BeadChip [35]. Of the 606,006 loci present on the array 422,975 loci were successfully genotyped in our bighorn sheep sample. Note that a decrease in amplification success is expected from cross-species application of SNP chips [51, 52]. 407,465 (~96%) of these chip loci were present in the list of variants identified by sequencing, and over 93% of the loci showed agreement (Table 1). To assess the effects of filtering on these results an additional set of filtering criteria was applied to the sequence-derived SNPs. Increasing our stringency thresholds for SNPs in the WGS decreased the number chip loci that were present in the list of SNPs identified by sequencing (n = 329,690; ~78%), but increased concordance to ~95%. In both cases the major source of discordance was loci called heterozygous in the WGS but homozygous from the chip data (Table 1).
Table 1.
Number of loci showing concordance or discordance between the genome and the Ovine Infinium®HD SNP BeadChip
| Original filter | Stringent filter | |
|---|---|---|
| Same genotype | 377129 | 314734 |
| Heterozygous on chip, Homozygous in sequence | 456 | 130 |
| Homozygous on chip, Heterozygous in sequence | 22837 | 9565 |
| Alternate homozygotes | 7169 | 5261 |
Annotation and enrichment analysis
SnpEff [53] assigned 18,176,092 functional classes to our SNPs based on annotation of the domestic sheep genome. Note that the number of classes assigned is larger than the number of loci due to the fact that the categories are not mutually exclusive. The vast majority of the SNPs were predicted to be intronic or intergenic and 102,231 were assigned to coding regions or have predicted functional effects (Fig. 1, Additional File 2). Of those 102,231 loci, 52,381 SNPs were found to have fixed differences between our bighorn sheep and the domestic sheep reference, from which 25,472 were identified as non-synonymous and 27,198 were identified as synonymous. Note that sum of the number of synonymous and non-synonymous SNPs is larger than the total number of fixed differences because a locus may be classified as both synonymous and non-synonymous if a gene has more than one annotated transcript. Gene Ontology (GO) terms were available for 26,629 of the SNPs with fixed differences (9,752 non-synonymous and 16,877 synonymous) representing 6,963 genes (3,948 non-synonymous and 5,932 synonymous). We looked for functional enrichment between non-synonymous and synonymous SNPs using BLAST2GO [54]. When reduced to the most specific GO terms, we found 11 biological process GO terms to be over represented and 29 to be underrepresented in the non-synonymous set compared to the synonymous set (Additional File 3). Note that gene length was positively correlated to the number of annotated loci for both the non-synonymous and synonymous sets (r = 0.43 and 0.61 respectively). But given that this association was constant between both non-synonymous and synonymous gene sets we do not think it biases our results. However, one gene, titin, was ~3 times larger than all other genes considered so we repeated the GO enrichment analysis dropping titin, which reduced the level of correlation (r = 0.37 and 0.51 respectively). When titin is removed from the datasets the number of overrepresented and underrepresented terms decrease to 9 and 15 respectively; all of which were common to the set including titin, except for one underrepresented term (cellular protein metabolic process; GO 0044267) that was unique to the second analysis (Additional File 3).
Fig. 1.
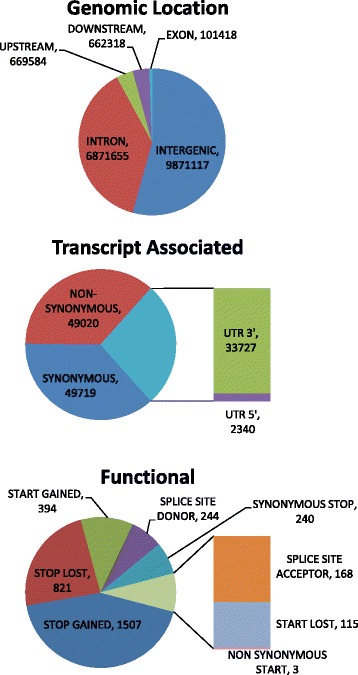
Distribution of SNP annotations and effect predictions
Discussion
Here we present a draft bighorn sheep WGS created by cross-species alignment to a domestic sheep reference sequence. Other studies have attempted de novo assembly with SOLiD sequencing data [55–57], but this was not an option in our case due to the high read depth required by such methods for a mammalian sized genome. Our work more closely resembles that of Canavez et al. and [22] Wang et al. [19]. Canavez et al. created a draft genome for an indicine bull (Bos indicus) through alignment of SOLiD reads to a taurine cow (Bos taurus) reference genome (divergence ~250 kya) [22]. While Wang et al. used SOLiD sequencing in a reference guided assembly of a black grouse (Tetrao tetrix) draft genome. However, Wang et al. [19] used a combination of de novo and alignment methods as the large divergence time between black grouse and domestic chicken (Gallus gallus) used as a reference (~30-40 mya) may hinder sequences from aligning properly. In contrast, bighorn and domestic are much less divergent which allows for successful direct alignment of reads: over 76% of our quality filtered reads mapped to the reference genome. Once merged, our two sequencing libraries provided 95% coverage of the reference and average 12x (104 SD) sequence depth.
Our alignment produced a large database of SNP markers for future studies. Approximately 6% of genotypes from a high-density SNP chip were discordant with those from the genome alignment, and increasing the quality thresholds for loci discovered in the genome only marginally decreased mismatches to ~4%. In both cases the major source of discordance was loci called heterozygous in the genome alignment but homozygous from the SNP chip. This source of discordance could be caused by incorrect joining of paralogous regions due to our procedure of randomly mapping ambiguous alignments. However, given the overall high concordance between the genome aligned SNPs and those on the SNP chip we are confident that the majority of our genotypes represent real SNPs. These markers add to the set of SNPs already available for this species [39, 42].
Genome scans of domestic sheep breeds have shown a number of regions that have been differentiated due to domestication [36, 58]. Therefore, we expect alleles associated with production traits to have been swept to or near fixation relative to a wild ancestor as well. Our GO term analysis of fixed SNP differences compared to the domestic sheep reference highlighted 40 biological process GO terms with significantly different representation in SNPs tagged as non-synonymous versus synonymous. Two of the gene ontologies that were associated with amino-acid changes relative to the domestic sheep reference involved reproduction: spermatogenesis (GO:0007283), and negative regulation of mammary gland epithelial cell proliferation (GO:0033600). This mirrors recent work has highlighted the genetic effects domestication had on reproductive traits of several sheep breeds [58, 59]. Another term that was over-represented in the non-synonymous gene set was ossification involved in bone maturation (GO:0043931). This term is noteworthy given the relationship of bones to horns which are bony projections covered by a keratinous sheath [60]. Horns are a major determinant of reproductive success in bighorn sheep, where larger males with bigger horns win antagonistic encounters and gain access to females [61, 62]; however, in many breeds of domestic sheep horns have been selected against leading to gene-level consequences [58]. All but two of the overrepresented biological process terms (skeletal muscle adaptation (GO:0043501) and maintenance of fidelity involved in DNA-dependent DNA replication (GO:0045005)) remained significant when titin (the largest gene in the dataset) was removed from the analysis.
For genes less likely to have amino acid changes, 14 of the 29 GO terms were related to muscles or muscle fibers, particularly cardiac muscles, e.g.: cardiac muscle hypertrophy (GO:0003300), cardiac myofibril assembly (GO:0055003), cardiac muscle fiber development (GO:0048739), adult heart development (GO:0007512), regulation of relaxation of cardiac muscle (GO:1901897), sarcomerogenesis (GO:0048769). It is interesting to note these differences associated with muscle properties, given that the domestic sheep reference genome was built from a meat-producing breed, the Texel [36, 63]. As mentioned above, body size is an important life history characteristic for male bighorn sheep as it relates to access to females, while larger females have been found to have longer lifespans [64]. Selective breeding for meat production in domestic sheep could favor conservation of the genes or developmental pathways that produce large muscles in bighorn sheep. However, analysis with REVIGO [65] indicated that there was overlap in these GO terms with 10 terms falling into two more representative terms: cardiac muscle hypertrophy (GO:0003300; containing two other terms) and cardiac muscle tissue morphogenesis (GO:0055008; containing eight other terms). In addition, nine of these terms become non-significant when titin (which has known associations with muscle properties, including body size, in cattle Bos taurus; [66, 67] and pigs Sus scrofa; [68]) is removed from the datasets.
Two factors are important to keep in mind when interpreting the results of our GO analysis. The first is that though it is tempting attribute the majority of differences we observed here to domestication and selective breeding, there are likely to be additional factors at play. In particular, natural selection as bighorn sheep and the progenitor to domestic sheep diverged, as well as genetic drift. Second, we are only comparing SNP sites from one individual’s genome to a reference sequence. This likely results in missing polymorphisms within either species, leading to incorrect annotation of fixed differences. However, we present the results only as a preliminary analysis to highlight candidate ontologies that may contribute to differentiation between the species. Such results will need to be confirmed by additional sequencing, alternate analyses (e.g. genome scans), and perhaps functional characterization [69].
While our draft genome represents a step forward in the genomic resources available for bighorn sheep this single genome is representative of a specific demographic history, an example of the ‘n = 1 constraint’ [70]. Future population genomic studies using additional individuals from Ram Mountain or other populations can confirm the variants we describe here, discover additional variants, and more fully examine the demographic history of bighorn sheep [71]. Expanded sequencing efforts would also allow for comparative genomics to further identify ancestral states and regions of selection relative to domestic sheep. In addition, our bighorn sheep genome can aid reference guided genome assembly [20–22] of other Ovis species using a genome that has not been subject to strong selective breeding.
Conclusion
In this study, we created a WGS for bighorn sheep using the closely related domestic sheep as a reference for alignment. This procedure was highly successful, covering 95% of the reference with an average read depth of 12 (104 SD). From this sequence we were able to call 15,622,848 variants and found 40 GO terms with significantly different representation in fixed SNPs tagged as non-synonymous versus synonymous. We hypothesize that these differences may largely be a result of selection during domestication. Our results demonstrate that cross-species alignment enables the creation of novel WGS for non-model organisms. The bighorn sheep WGS will provide a resource for future resequencing studies or comparative genomics both for other populations of bighorn sheep or species within the Ovis genus.
Methods
Sample collection & sequencing
Total genomic DNA was extracted from tissue of a single bighorn sheep from Ram Mountain (Alberta, Canada), using standard phenol–chloroform extraction protocols [72]. From this, two libraries were constructed and sequenced. The first was a mate-paired library the details of which are provided in [40]. Briefly, preparation used the reagents and protocols provided by Applied Biosystems with and an expected insert size of ~1.5 kb. Emulsion PCR was performed using the SOLiD EZ bead system (Life Technologies Corporation). Both forward and reverse tags were sequenced to 50 bases using an Applied Biosystems SOLiD 4 sequencer (Life Technologies Corporation). The second library was a fragment library sequenced to 75 bases using a SOLiD 5500xl sequencer (Life Technologies Corporation). The resulting xsq files were converted to csfasta and qual scores format using XSQ Tool (Life Technologies Corporation).
Alignment & variant calling
Sequence quality assessment and alignment were conducted with CLC Genomics Workbench (version 5.0; CLC bio, Cambridge, MA, USA). For each library, sequences were quality trimmed allowing for 1 ambiguous nucleotide, a quality score limit of 0.05, and minimum read length of 15 nucleotides. The resulting reads from each library were then independently aligned to domestic sheep chromosomes (version 3.1; [36]). Alignment parameters were set with no reference masking, mismatch cost of 2, insertion/deletion cost of 3, length fraction of 0.5, and similarity fraction of 0.8. Meaning at least 50% of a read must have at least 90% identity to the reference to be aligned. Non-specific matches were mapped randomly. Once mapped, PCR duplicates were removed from the alignment. We then merged the mate-paired and fragment mappings and removed PCR duplicates from the merged file. The merged alignment was then exported both as consensus fasta sequences as well as a BAM file for use in subsequent analyses. When generating the consensus fasta sequences we allowed for ambiguities (e.g. IUPAC codes W, R, etc.) and inserted N’s proportional to the length of the domestic sheep reference for regions of zero coverage. We elected to leave differences between our bighorn sheep sequence and domestic sheep reference as ambiguities in case additional sequencing reveals those sites to represent unobserved shared polymorphisms.
Variants were called from the consensus alignment BAM files using the mpileup command in SAMtools version 0.1.17 [49] and filtered in bcftools. Specifically, variants were required to have a minimum quality of 30 and a read depth between 6 and 200. VCFfilter version 0.1.11 [73] was then used to assess indel length distribution and calculate transition transversion (ti/tv) ratio using 100 basepair windows. As a quality check, genotypes from the aligned genome were compared to those generated for the same individual on the Ovine Infinium®HD SNP BeadChip, a newly developed SNP array for domestic sheep that contains 606,006 loci [35]. For this analysis raw intensity data were converted into genotype calls using GenomeStudio (Illumina) and SNP cluster information based on domestic sheep reference samples provided by the International Sheep Genomics Consortium. All genotype calls with GenCall scores less than 0.6, or GenTrain scores lower than 0.8, were removed from the data set. When assessing concordance between genotypes from the SNP array and the draft WGS we first positioned SNPs from the array in the reference assembly by comparing 50 nucleotides on either side of the locus position using BLAST with an E value of 1e−9. Loci with more than one match were excluded from analysis. In total this procedure excluded 45,979 loci. To assess the effects of filtering on the recovery of chip SNPs by sequencing and on concordance between the chip and the sequence genotypes an additional set of filtering criteria was applied to the sequence-derived SNPs. In this case we increased stringency, requiring read depths greater than 5 but less than the mean plus 3 SD, at least one forward or reverse alternative allele read (where applicable), no other variants within 5 bp, and genotype quality greater than 10.
Annotation and enrichment analysis
SnpEff version 3.1 [53] was used to predict functional classes (e.g. intergenic or intronic) and effect types (e.g. synonymous or non-synonymous) of the loci by comparing our SNPs to annotations from the domestic sheep genome (database oar3.1, downloaded Sept 2013). Note that within functional classes and effect types, categories are not mutually exclusive, for example a SNP can be classified as both intronic and in the 5’-UTR.
For enrichment analysis we first filtered SNPs to only those that were fixed between our bighorn sheep alignment and the domestic sheep reference using SNPsift [74]. We then split the resulting loci into two categories: 1) those with likely functional consequences (i.e. non-synonymous coding, start gained, start lost, stop gained, stop lost) and 2) those showing synonymous effects (i.e. synonymous coding, synonymous start). GO terms were added to the SNPs in these lists from the Ovis aries gene set (Oar v3.1) using BioMart [75] and Ensembl version 77 [76]. The two groups were then compared using BLAST2GO [54] which employs a Fisher’s Exact Test via the Gossip package [77]. Specifically, we used a two tailed test with false discovery correction of Benjamini and Hochberg [78] set at 0.0001. Evaluation of GO enrichment among candidate genes was restricted to terms within the biological process category.
Data accessibility
Raw reads have been deposited on NCBI SRA as SRR1752652 (mate-paired library) and SRR1752652 (fragment library), and genome fasta have been deposited at DDBJ/EMBL/GenBank under the accession JZLK00000000.
Additional files
Histogram of insertion/deletion lengths in the bighorn draft genome relative to the domestic sheep reference.
Summary of predicted effects of each SNP by chromosome as assigned by SnpEff.
GO enrichment summary between loci predicted to be non-synonymous and those predicted to be synonymous.
Acknowledgements
The tissue sample used in this study was previously collected under research protocols that were approved by the University of Alberta Animal Use and Care Committee, affiliated with the Canadian Council for Animal Care (Certificate 610901). The library preparation and sequencing were performed in the Agricultural Genomics and Proteomics Unit at the University of Alberta. JMM’s graduate research was funded by an Alberta Innovates graduate scholarship, a Natural Sciences and Engineering Research Council of Canada (NSERC) Vanier scholarship, the University of Alberta, and the Killam Foundation. DWC is supported by an NSERC Discovery Grant. We would like to thank Catherine I Cullingham, Corey S. Davis, and Rene M Malenfant for helpful discussions and guidance during the execution of this study; as well as two anonymous reviewers who helped to improve the manuscript.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Sequence data was provided by SSM. JMM and XL conducted bioinformatic analyses with analytical guidance from PS and DWC. JMM drafted the original manuscript which all authors provided input on throughout its preparation. All authors approved the final manuscript.
Contributor Information
Joshua M Miller, Email: jmm1@ualberta.ca.
Stephen S Moore, Email: s.moore3@uq.edu.au.
Paul Stothard, Email: stothard@ualberta.ca.
Xiaoping Liao, Email: liao_xp@tib.cas.cn.
David W Coltman, Email: david.coltman@ualberta.ca.
References
- 1.Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends in Ecology & Evolution. 2014;29(1):51–63. doi: 10.1016/j.tree.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, et al. Great ape genetic diversity and population history. Nature. 2013;499(7459):471–5. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Telford MJ, Copley RR. Improving animal phylogenies with genomic data. Trends in Genetics. 2011;27(5):186–95. doi: 10.1016/j.tig.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet. 2012;13(5):329–42. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
- 5.Bourque G, Zdobnov E, Bork P, Pevzner P, Tesler G. Comparative architectures of mammalian and chicken genomes reveal highly variable rates of genomic rearrangements across different lineages. Genome Research. 2005;15(1):98–110. doi: 10.1101/gr.3002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao H, Bourque G. Recovering genome rearrangements in the mammalian phylogeny. Genome Research. 2009;19(5):934–42. doi: 10.1101/gr.086009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg L, et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol. 2010;8(9). [DOI] [PMC free article] [PubMed]
- 8.Fang X, Zhang Y, Zhang R, Yang L, Li M, Ye K, et al. Genome sequence and global sequence variation map with 5.5 million SNPs in Chinese rhesus macaque. Genome Biol. 2011;12(7). doi:10.1186/gb-2011-1112-1187-r1163. [DOI] [PMC free article] [PubMed]
- 9.Vijay N, Poelstra JW, Künstner A, Wolf JBW. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Molecular Ecology. 2013;22(3):620–34. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- 10.Angeloni F, Wagemaker N, Vergeer P, Ouborg J. Genomic toolboxes for conservation biologists. Evolutionary Applications. 2012;5(2):130–43. doi: 10.1111/j.1752-4571.2011.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funk WC, McKay JK, Hohenlohe PA, Allendorf FW. Harnessing genomics for delineating conservation units. TREE. 2012;27(9):489–96. doi: 10.1016/j.tree.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–6. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheehan S, Harris K, Song YS. Estimating variable effective population sizes from multiple genomes: a sequentially Markov conditional sampling distribution approach. Genetics. 2013;194(3):647–62. doi: 10.1534/genetics.112.149096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brondum RF, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46(8):858–65. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 15.Miller J, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95(6):315–27. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunt M, Newbold C, Berriman M, Otto T. A comprehensive evaluation of assembly scaffolding tools. Genome Biology. 2014;15(3):R42. doi: 10.1186/gb-2014-15-3-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekblom R, Wolf JBW. A field guide to whole-genome sequencing, assembly and annotation. Evol Appl. 2014;7(9):1026-1042. [DOI] [PMC free article] [PubMed]
- 18.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–82. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 19.Wang B, Ekblom R, Bunikis I, Siitari H, Hoglund J. Whole genome sequencing of the black grouse (Tetrao tetrix): reference guided assembly suggests faster-Z and MHC evolution. BMC Genomics. 2014;15(1):180. doi: 10.1186/1471-2164-15-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnerre S, Lander E, Lindblad-Toh K, Jaffe D. Assisted assembly: how to improve a de novo genome assembly by using related species. Genome Biol. 2009;10(8). [DOI] [PMC free article] [PubMed]
- 21.Kim J, Larkin D, Cai Q, Asan, Zhang Y, Ge R, et al. Reference-assisted chromosome assembly. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(5):1785–90. doi: 10.1073/pnas.1220349110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canavez FC, Luche DD, Stothard P, Leite KRM, Sousa-Canavez JM, Plastow G, et al. Genome sequence and assembly of Bos indicus. Journal of Heredity. 2012;103(3):342–8. doi: 10.1093/jhered/esr153. [DOI] [PubMed] [Google Scholar]
- 23.Kohn MH, Murphy WJ, Ostrander EA, Wayne RK. Genomics and conservation genetics. Trends in Ecology & Evolution. 2006;21(11):629–37. doi: 10.1016/j.tree.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Berger J. Persistence of different-sized populations: an empirical assessment of rapid extinctions in bighorn sheep. Conservation Biology. 1990;4(1):91–8. doi: 10.1111/j.1523-1739.1990.tb00271.x. [DOI] [Google Scholar]
- 25.Festa-Bianchet M, Pelletier F, Jorgenson JT, Feder C, Hubbs A. Decrease in horn size and increase in age of trophy sheep in Alberta over 37 years. The Journal of Wildlife Management. 2014;78(1):133–41. doi: 10.1002/jwmg.644. [DOI] [Google Scholar]
- 26.Hedrick PW. Conservation genetics and the persistence and translocation of small populations: bighorn sheep populations as examples. Animal Conservation. 2014;17(2):106–14. doi: 10.1111/acv.12064. [DOI] [Google Scholar]
- 27.Johnson HE, Mills LS, Wehausen JD, Stephenson TR, Luikart G. Translating effects of inbreeding depression on component vital rates to overall population growth in endangered bighorn sheep. Conservation Biology. 2011;25(6):1240–9. doi: 10.1111/j.1523-1739.2011.01739.x. [DOI] [PubMed] [Google Scholar]
- 28.Olson ZH, Whittaker DG, Rhodes OE. Translocation history and genetic diversity in reintroduced bighorn sheep. The Journal of Wildlife Management. 2013;77(8):1553–63. doi: 10.1002/jwmg.624. [DOI] [Google Scholar]
- 29.Shackleton DM, Shank CC, Wikeem B. Natural history of rock mountain and California bighorn sheep. In: Valdez R, Krausman PR, editors. Mountain sheep of North America. Tuscon: The University of Arizona Press; 1999. pp. 78–138. [Google Scholar]
- 30.Coltman DW, O’Donoghue P, Jorgenson JT, Hogg JT, Strobeck C, Festa-Bianchet M. Undesirable evolutionary consequences of trophy hunting. Nature. 2003;426(6967):655–8. doi: 10.1038/nature02177. [DOI] [PubMed] [Google Scholar]
- 31.Réale D, Martin J, Coltman DW, Poissant J, Festa-Bianchet M. Male personality, life-history strategies and reproductive success in a promiscuous mammal. Journal of Evolutionary Biology. 2009;22(8):1599–607. doi: 10.1111/j.1420-9101.2009.01781.x. [DOI] [PubMed] [Google Scholar]
- 32.Poissant J, Davis CS, Malenfant RM, Hogg JT, Coltman DW. QTL mapping for sexually dimorphic fitness-related traits in wild bighorn sheep. Heredity. 2012;108:256–63. doi: 10.1038/hdy.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JM, Poissant J, Hogg JT, Coltman DW. Genomic consequences of genetic rescue in an insular population of bighorn sheep (Ovis canadensis) Molecular Ecology. 2012;21(7):1583–96. doi: 10.1111/j.1365-294X.2011.05427.x. [DOI] [PubMed] [Google Scholar]
- 34.Miller JM, Malenfant RM, David P, Davis CS, Poissant J, Hogg JT, et al. Estimating genome-wide heterozygosity: effects of demographic history and marker type. Heredity. 2014;112:240–7. doi: 10.1038/hdy.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kijas JW, Porto-Neto L, Dominik S, Reverter A, Bunch R, McCulloch R, et al. Linkage disequilibrium over short physical distances measured in sheep using a high-density SNP chip. Animal Genetics. 2014;45(5):754–7. doi: 10.1111/age.12197. [DOI] [PubMed] [Google Scholar]
- 36.Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014;344(6188):1168–73. doi: 10.1126/science.1252806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunch T, Wu C, Zhang Y, Wang S. Phylogenetic analysis of snow sheep (Ovis nivicola) and closely related taxa. Journal of Heredity. 2006;97(1):21–30. doi: 10.1093/jhered/esi127. [DOI] [PubMed] [Google Scholar]
- 38.Poissant J, Hogg JT, Davis CS, Miller JM, Maddox JF, Coltman D. Genetic linkage map of a wild genome: genomic structure, recombination and sexual dimorphism in bighorn sheep. BMC Genomics. 2010;11(524). doi:10.1186/1471-2164-1111-1524. [DOI] [PMC free article] [PubMed]
- 39.Miller JM, Poissant J, Kijas J, Coltman DW, TISGC A genome-wide set of SNPs detects population substructure and long range linkage disequilibrium in wild sheep. Molecular Ecology Resources. 2011;11(2):314–22. doi: 10.1111/j.1755-0998.2010.02918.x. [DOI] [PubMed] [Google Scholar]
- 40.Miller JM, Malenfant RM, Moore SS, Coltman DW. Short reads, circular genome: skimming SOLiD sequence to construct the bighorn sheep mitochondrial genome. Journal of Heredity. 2012;103(1):140–6. doi: 10.1093/jhered/esr104. [DOI] [PubMed] [Google Scholar]
- 41.Poissant J, Shafer ABA, Davis CS, Mainguy J, Hogg JT, CÔTÉ ST, et al. Genome-wide cross-amplification of domestic sheep microsatellites in bighorn sheep and mountain goats. Molecular Ecology Resources. 2009;9(4):1121–6. doi: 10.1111/j.1755-0998.2009.02575.x. [DOI] [PubMed] [Google Scholar]
- 42.Genomic Resources Development Consortium. Coltman DW, Hogg JT, Miller JM. Genomic resources notes accepted 1 April 2013–31 May 2013. Molecular Ecology Resources. 2013;13(5):965. doi: 10.1111/1755-0998.12142. [DOI] [PubMed] [Google Scholar]
- 43.Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107(1):1. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glenn TC. Field guide to next-generation DNA sequencers. Molecular Ecology Resources. 2011;11(5):759–69. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 45.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, et al. The potential and challenges of nanopore sequencing. Nat Biotech. 2008;28(10):1146–53. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 47.Ondov B, Varadarajan A, Passalacqua K, Bergman N. Efficient mapping of applied biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics. 2008;24(23):2776–7. doi: 10.1093/bioinformatics/btn512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu YT, Tsung EF, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Research. 2009;19(9):1527–41. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wakeley J. The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscore its significance. Trends in Ecology & Evolution. 1996;11(4):158–62. doi: 10.1016/0169-5347(96)10009-4. [DOI] [PubMed] [Google Scholar]
- 51.Miller JM, Kijas JW, Heaton MP, McEwan JC, Coltman DW. Consistent divergence times and allele sharing measured from cross-species application of SNP chips developed for three domestic species. Molecular Ecology Resources. 2012;12(6):1145–50. doi: 10.1111/1755-0998.12017. [DOI] [PubMed] [Google Scholar]
- 52.Sechi T, Coltman DW, Kijas JW. Evaluation of 16 loci to examine the cross-species utility of single nucleotide polymorphism arrays. Animal Genetics. 2010;41(2):199–202. doi: 10.1111/j.1365-2052.2009.01972.x. [DOI] [PubMed] [Google Scholar]
- 53.Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 55.Cerdeira LT, Carneiro AR, Ramos RTJ, de Almeida SS, D´Afonseca V, Schneider MPC, et al. Rapid hybrid de novo assembly of a microbial genome using only short reads: Corynebacterium pseudotuberculosis I19 as a case study. Journal of Microbiological Methods. 2011;86(2):218–23. doi: 10.1016/j.mimet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Umemura M, Koyama Y, Takeda I, Hagiwara H, Ikegami T, Koike H, et al. Fine de novo sequencing of a fungal genome using only solid short read data: verification on aspergillus oryzae RIB40. PLoS One. 2013;8(5). [DOI] [PMC free article] [PubMed]
- 57.Genomic Resources Dev Consortium. Bensch S, Coltman D, Davis C, Hellgren O, Johansson T, et al. Genomic resources notes accepted 1 June 2013-31 July 2013. Molecular Ecology Resources. 2014;14(1):218. doi: 10.1111/1755-0998.12166. [DOI] [PubMed] [Google Scholar]
- 58.Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lv F-H, Agha S, Kantanen J, Colli L, Stucki S, Kijas JW, et al. Adaptations to climate-mediated selective pressures in sheep. Molecular Biology and Evolution. 2014;31(12):3324–43. doi: 10.1093/molbev/msu264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis EB, Brakora KA, Lee AH. Evolution of ruminant headgear: a review. Proceedings of the Royal Society B: Biological Sciences. 2011;278(1720):2857–65. doi: 10.1098/rspb.2011.0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coltman DW, Festa-Bianchet M, Jorgenson JT, Strobeck C. Age-dependent sexual selection in bighorn rams. Proceedings of the Royal Society B-Biological Sciences. 2002;269(1487):165–72. doi: 10.1098/rspb.2001.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pelletier F, Festa-Bianchet M. Sexual selection and social rank in bighorn rams. Animal Behaviour. 2006;71(3):649–55. doi: 10.1016/j.anbehav.2005.07.008. [DOI] [Google Scholar]
- 63.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38(7):813–8. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 64.Gaillard JM, Festa-Bianchet M, Delorme D, Jorgenson JT. Body mass and individual fitness in female ungulates: bigger is not always better. Proceedings of the Royal Society B: Biological Sciences. 2000;267(1442):471–7. doi: 10.1098/rspb.2000.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6(7). [DOI] [PMC free article] [PubMed]
- 66.Lee K-T, Chung W-H, Lee S-Y, Choi J-W, Kim J, Lim D, et al. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics. 2013;519(14). [DOI] [PMC free article] [PubMed]
- 67.Sasaki Y, Nagai K, Nagata Y, Doronbekov K, Nishimura S, Yoshioka S, et al. Exploration of genes showing intramuscular fat deposition-associated expression changes in musculus longissimus muscle. Animal Genetics. 2006;37(1):40–6. doi: 10.1111/j.1365-2052.2005.01380.x. [DOI] [PubMed] [Google Scholar]
- 68.Braglia S, Davoli R, Zappavigna A, Zambonelli P, Buttazzoni L, Gallo M, et al. SNPs of MYPN and TTN genes are associated to meat and carcass traits in Italian large white and Italian duroc pigs. Molecular Biology Reports. 2013;40(12):6927–33. doi: 10.1007/s11033-013-2812-z. [DOI] [PubMed] [Google Scholar]
- 69.Stinchcombe JR, Hoekstra HE. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity. 2008;100(2):158–70. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- 70.Buerkle C, Gompert Z, Parchman T. The n = 1 constraint in population genomics. Molecular Ecology. 2011;20(8):1575–81. doi: 10.1111/j.1365-294X.2011.05046.x. [DOI] [PubMed] [Google Scholar]
- 71.Bolormaa S, Kijas J, Coltman DW, Daetwyler HD, MacLeod IM. 10th world congress of genetics applied to livestock production: 2014 2014. Vancouver, British Columbia, Canada: Asas; 2014. Inferring ancestral demography of domestic and wild sheep using whole-genome sequence. [Google Scholar]
- 72.Sambrook J, Russell D. Molecular cloning: a laboratory manual. Cold Spring Harbor Press (NY): 2001.
- 73.Danecek P, Auton A, Abecasis G, Albers C, Banks E, DePristo M, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program. Genetics: SnpSift. Frontiers in; 2012. p. 3(35). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kinsella R, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database-the Journal of Biological Databases and Curation. 2011:bar030. [DOI] [PMC free article] [PubMed]
- 76.Flicek P, Amode M, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Research. 2014;42(D1):D749–55. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blüthgen N, Brand K, Čajavec B, Swat M, Herzel H, Beule D. Biological profiling of gene groups utilizing gene ontology. Genome Informatics. 2005;16(1):106–15. [PubMed] [Google Scholar]
- 78.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histogram of insertion/deletion lengths in the bighorn draft genome relative to the domestic sheep reference.
Summary of predicted effects of each SNP by chromosome as assigned by SnpEff.
GO enrichment summary between loci predicted to be non-synonymous and those predicted to be synonymous.
Data Availability Statement
Raw reads have been deposited on NCBI SRA as SRR1752652 (mate-paired library) and SRR1752652 (fragment library), and genome fasta have been deposited at DDBJ/EMBL/GenBank under the accession JZLK00000000.