

Abstract
Novel therapies are needed for treating the increasing prevalence of hepatic steatosis in Western populations. In this regard, dipeptidyl peptidase-4 (DPP-4) inhibitors have recently been reported to attenuate the development of hepatic steatosis, but the potential mechanisms remain poorly defined. In the current study, 4-week-old C57Bl/6 mice were fed a high-fat/high-fructose Western diet (WD) or a WD containing the DPP-4 inhibitor, MK0626, for 16 weeks. The DPP-4 inhibitor prevented WD-induced hepatic steatosis and reduced hepatic insulin resistance by enhancing insulin suppression of hepatic glucose output. WD-induced accumulation of hepatic triacylglycerol (TAG) and diacylglycerol (DAG) content was significantly attenuated with DPP-4 inhibitor treatment. In addition, MK0626 significantly reduced mitochondrial incomplete palmitate oxidation and increased indices of pyruvate dehydrogenase activity, TCA cycle flux, and hepatic TAG secretion. Furthermore, DPP-4 inhibition rescued WD-induced decreases in hepatic PGC-1α and CPT-1 mRNA expression and hepatic Sirt1 protein content. Moreover, plasma uric acid levels in mice fed the WD were decreased after MK0626 treatment. These studies suggest that DPP-4 inhibition ameliorates hepatic steatosis and insulin resistance by suppressing hepatic TAG and DAG accumulation through enhanced mitochondrial carbohydrate utilization and hepatic TAG secretion/export with a concomitant reduction of uric acid production.
Introduction
Obesity is becoming an epidemic disease in Western cultures, affecting more than one-third of the U.S. adult population (1). Nonalcoholic fatty liver disease (NAFLD) progressing to steatohepatitis and cirrhosis is also increasing in epidemic proportions concurrent with the obesity epidemic (2,3). The dramatic rise in obesity and NAFLD appears to be partly due to consumption of a Western diet (WD) containing high amounts of fat and fructose, and fructose consumption in the U.S. has more than doubled in the last 3 decades (2,4). Hepatic insulin resistance that develops with consumption of high-fat and high-fructose diets is closely linked to NAFLD and increases the risk for the development of type 2 diabetes (5,6). Therefore, novel strategies targeting hepatic steatosis and insulin resistance have received considerable attention in recent years (7).
The gut-derived incretin hormones, glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), play important roles in both postprandial and long-term glucose homeostasis by enhancing glucose-stimulated insulin secretion and suppressing glucagon release (8). The exopeptidase dipeptidyl peptidase-4 (DPP-4), a serine protease found in the plasma and on the surface of diverse cells, rapidly degrades circulating GLP-1 and GIP, limiting the half-life of these hormones. Plasma DPP-4 activity and expression of DPP-4 on various inflammation-promoting immune cells is increased in obesity and diabetes, raising the possibility that DPP-4 inhibition may reduce systemic and tissue inflammation (9,10). In this regard, there is emerging evidence that DPP-4 inhibition may be a novel therapeutic strategy to prevent the development of hepatic insulin resistance and hepatic steatosis (11–13). However, the precise mechanisms and mediators involved in this hepatic protection are not well understood.
Accumulating evidence suggests that hepatic insulin resistance is caused by dysfunction in three pathways of energy metabolism (14,15). First, excess carbohydrate flux (glucose, fructose) is associated with resistance to the suppressive effect of insulin on hepatic glucose production and excess disposal of carbons via de novo lipogenesis (14,16). Second, elevation in lipid synthesis (or reduced lipid secretion/export) leads to accumulation of hepatic triacylglycerols (TAG), which are inert but often track with increased levels of bioactive lipid intermediates diacylglycerols (DAGs) and ceramides that putatively lead to hepatic insulin resistance (17,18). Third, the hepatic steatosis linked to insulin resistance is associated with mitochondrial dysfunction and altered hepatic fatty acid oxidation (15,19,20). The effect of DPP-4 inhibition on these metabolic processes has not been previously examined.
MK0626 is a DPP-4 inhibitor closely related to sitagliptin with pharmacokinetics suited to investigation in a rodent model (21,22). Here, we test the hypothesis that DPP-4 inhibition with MK0626 will attenuate WD-induced hepatic steatosis and insulin resistance by reducing hepatic lipid intermediate (DAGs and ceramides) accumulation with consequent improvement in hepatic mitochondrial function and metabolism.
Research Design and Methods
The animals used in this study were cared for in accordance with National Institutes of Health guidelines. All procedures were approved in advance by the University of Missouri Institutional Animal Care and Use Committee.
Animals and Experimental Design
C57Bl/6 mice were purchased from Charles River, Inc. MK0626 was added to mouse chow to a final concentration of 33 mg/kg chow to achieve a dose and plasma level of ∼10 mg/kg/day and 300 nmol/L, respectively (≥80% inhibition of plasma DPP-4; Merck) based on previous pharmacology studies in rodents (21) and as previously published by our group and others (22–24). Male mice were divided into four groups (n = 10–12 per group) to include C57Bl/6 control diet (CD), C57Bl/6 treated with MK0626 (CD-MK), Western diet (WD), and WD treated with MK0626 (WD-MK). Diets were initiated at 4 weeks of age for 16 weeks, and mice were killed at 20 weeks of age. Mice fed the CD consumed product #58Y2 (TestDiet, St. Louis, MO), providing 18.0% of energy as protein (16.9 g/100 g), 10.2% of energy as fat (4.3 g/100 g), and 71.8% of energy as carbohydrate (67.4 g/100 g). Mice fed the WD consumed product #58Y1 (TestDiet), providing 17.6% of energy as protein (20.5 g/100 g), 46.4% of energy as fat (24 g/100 g), and 36.0% of energy as carbohydrate (41.8 g/100 g) with 17.5% fructose and 17.5% sucrose. The primary sources of fat in both diets were corn oil and lard.
Mice were anesthetized with sodium pentobarbital (100 mg · kg–1) after a 5-h fast and killed by exsanguination by removal of the heart. Retroperitoneal and epididymal adipose tissue fat pads were removed from exsanguinated animals and weighed. For acute insulin stimulation studies (additional n = 6–7 per group), food was removed 5 h before mice were given an intraperitoneal injection of insulin (Humulin; 2.5 units/kg), and tissues were harvested under anesthesia 20 min after injection.
Biochemical Parameters and DPP-4 Activity
Plasma alanine aminotransferase (ALT) activity, cholesterol, nonesterified fatty acids (NEFA), uric acid, and TAG concentrations were determined by automated analyzer. Plasma and liver DPP-4 activity was fluorometrically assessed, as previously described by our group (22,23).
Body Composition
The percent body fat was measured by a nuclear magnetic resonance imaging whole-body composition analyzer (EchoMRI 4in1/1100; Echo Medical Systems, Houston, TX). This noninvasive measure was performed on conscious mice.
Hepatic Histology and TAG, DAG, and Ceramide Content
After animals were killed, the liver was immediately removed, rinsed in chilled PBS, blotted dry, weighed, and flash frozen in liquid nitrogen for storage at −80°C or placed in 10% neutral buffered formalin for formalin-fixation. Hematoxylin and eosin staining was used for evaluation of steatosis by light microscopy, as previously described by our group (19). Liver tissue was homogenized, and lipids were isolated by extraction into chloroform with appropriate internal standards included for each protocol. Extracted lipids were resuspended and diluted in methanol/chloroform (4:1 by volume) before analysis by electrospray ionization-mass spectrometry using a Thermo Electron TSQ Quantrum Ultra instrument (San Jose, CA). DAG molecular species were quantified as sodiated adducts using selected reaction monitoring, as previously described, with the intensity of each species normalized to that of the internal standard Di-20:0 DAG (25). TAG aliphatic groups were quantified by TAG fingerprinting techniques, with neutral loss scanning for the loss of each fatty acid from the TAG species and comparisons with that of the neutral loss 268, which is derived from the internal standard Tri-17:1 TAG (26). Individual ceramide molecular species were quantified in negative ion mode using neutral loss 256 by comparing the ion intensity of individual molecular species with that of the internal standard (17:0 ceramide) after corrections for type I and type II 13C isotope effects.
Western Blot Analyses
Western blots were performed as described earlier for oxidative phosphorylation (OXPHOS) electron transport chain complexes I through V (MitoProfile Total OXPHOS Rodent WB Antibody Cocktail; Abcam, Cambridge, MA), NAD-dependent deacetylase sirtuin-1 (Sirt1; Santa Cruz Biotechnology, Santa Cruz, CA), NAD-dependent deacetylase sirtuin-3 (Sirt3; Cell Signaling, Beverly, MA), microsomal triglyceride transfer protein (MTTP; Santa Cruz Biotechnology), apolipoprotein B100 (apoB100; Abcam), fatty acid synthase (FAS, Cell Signaling), acetyl-CoA carboxylase (ACC; Cell Signaling), sterol regulatory element binding protein (SREBP-1c; Santa Cruz Biotechnology), protein kinase B (Akt; Cell Signaling), and phospho-Akt Ser473 (Cell Signaling) (19,27). Membranes stained with 0.1% amido-black (Sigma-Aldrich) were quantified to control for differences in protein loading or transfer of band densities, as previously described (19).
Mitochondrial Studies
Palmitate and Pyruvate Oxidation
Complete and incomplete oxidation of [1-14C]-labeled palmitate (American Radiochemicals, St. Louis, MO), [1-14C]pyruvate (PerkinElmer, Boston, MA), and [2-14C]pyruvate (PerkinElmer) were measured in fresh isolated hepatic mitochondria preparations, as previously described (27). Pyruvate ([1-14C] and [2-14C]) were oxidized to 14CO2 by isolated hepatic mitochondria in the appropriate reaction buffer. [1-14C]pyruvate oxidation was used as an index of pyruvate dehydrogenase activity (PDH) and [2-14C]pyruvate oxidation as an index of tricarboxylic acid (TCA) cycle flux (28).
Mitochondrial Respiration
Mitochondrial respiration was assessed using high-resolution respirometry (Oroboros Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria), as previously described (27). Briefly, oxygen flux was measured by addition of glutamate (5 mmol/L) and malate (2 mmol/L) to the chambers in the absence of ADP (GM-state 2) for assessment of state 2 respiration. OXPHOS with electron flux through complex I was then quantified by titration of ADP (25–125 μmol/L) (GM+ADP: state 3-complex I) for assessment of state 3 respiration. Maximal ADP respiration with electron flux through complex I and complex II was assessed by the addition of succinate (10 mmol/L) (succinate: state 3-complex I+II). Finally, maximal capacity of the electron transport system was assessed by uncoupling with the addition of 0.25 μmol/L FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; uncoupled).
TAG Secretion Assay
To assess hepatic TAG secretion, mice fasted for 5 h were injected intraperitoneally with the lipase inhibitor Poloxamer 407 (1 g/kg body weight) as a 75 mg/mL solution in saline, as previously described (29). Blood samples were drawn into heparinized tubes at 0, 1, 2, and 3 h after injection, and plasma was separated and assayed for triglycerides, as described above. Hepatic triglyceride production rates were calculated from the slope of the curve and expressed as mg/dL/h.
mRNA Expression
Total RNA was extracted from frozen liver using RNeasy kit and used for cDNA preparation and quantitative real-time PCR with commercially available primers (19). Results were quantified by the ddCT method relative to the housekeeping gene cyclophilin b, which relative gene expression did not differ among groups (P = 0.6).
Hyperinsulinemic-Euglycemic Clamp
The Vanderbilt University Animal Care and Use Committee approved all procedures required for the hyperinsulinemic-euglycemic clamp. Catheters were implanted into a carotid artery and a jugular vein of mice for sampling and infusions, respectively, 5 days before the study, as described by Berglund et al. (30). Hyperinsulinemic-euglycemic clamps were performed on mice fasted for 5 h using a modification of the method described by Ayala et al. (31). [3-3H]glucose was primed (2.4 μCi) and continuously infused for 90-min equilibration and basal sampling periods (0.04 μCi/min). [3-3H]glucose was mixed with the nonradioactive glucose infusate (infusate specific activity of 0.4 μCi/mg) during the 2-h clamp period. Arterial glucose was clamped using a variable rate of glucose (plus trace [3-3H]glucose) infusion, which was adjusted based on the measurement of blood glucose at 10-min intervals. Mixing radioactive glucose with the nonradioactive glucose infused during a clamp minimized deviations in arterial glucose specific activity, and steady-state conditions are achieved. Baseline blood or plasma variables were calculated as the mean of values obtained in blood samples collected at −15 and −5 min. At time 0, insulin infusion (2.5 mU/kg of body weight per min) was started and continued for 120 min. Mice received heparinized saline-washed erythrocytes from donors at 5 μL/min to prevent a fall in hematocrit. Insulin clamps were validated by assessment of blood glucose over time. Blood was taken at 80–120 min for the determination of [3-3H]glucose.
At the end of the clamps, animals were anesthetized, and the liver was removed and immediately frozen. Rates of whole-body glucose appearance and uptake were determined as the ratio of the [3H]glucose infusion rate to the specific activity of the plasma glucose during the final 40 min of the clamps. Hepatic glucose production during the clamps was determined by subtracting the glucose infusion rate from the whole-body glucose appearance. The glucose infusion rates across time have been previously reported by our group (22).
Statistical Analysis
Differences in outcomes among groups were determined using two-way ANOVA, and main effects (diet, drug) and interactions were considered significant when P < 0.05. IBM SPSS Statistics 22 software was used for the analysis. Significant diet-by-drug interactions were followed up with Fisher least significant differences post hoc analyses.
Results
Effects of WD and MK0626 on Body Weight, Percent Body Fat, Liver Weight, and Biochemical Parameters
WD-induced increases in body weight (+60% compared with CD, P < 0.001; Table 1), adiposity (+2.5-fold compared with the CD and CD-MK, P < 0.0001; Table 1), and food intake (Table 1) were not affected by MK0626. However, MK0626 administration suppressed liver weights in the WD-fed mice (P < 0.001; Table 1). WD feeding increased serum ALT levels by 40% (main effect, P = 0.01), but MK0626 treatment did not affect serum ALT in either diet group. In addition, MK0626 increased serum TAGs (main effect, P = 0.04), and the WD increased serum cholesterol (main effect, P = 0.03), but serum NEFA was not affected by diet or drug treatment (Table 1). We recently reported increased levels of plasma DPP-4 activity in WD-fed mice and suppression of DPP-4 activity by MK0626 (22,23). In the current study, DPP-4 activity was not increased in liver homogenates of WD-fed (nondrug-treated) mice, but MK0626 treatment caused significant (P < 0.01) inhibition of DPP-4 activity in the CD-MK and WD-MK groups (Table 1). In addition, WD significantly increased plasma uric acid levels (main effect, P = 0.003; Table 1), with MK0626 treatment significantly (P = 0.03) lowering uric acid levels in the CD-MK and WD-MK groups, similar to what we previously reported (22).
Table 1.
Animal, liver, and plasma characteristics
| CD | CD-MK | WD | WD-MK | |
|---|---|---|---|---|
| Body weight (g) | 27.5 ± 0.6 | 27.7 ± 0.7 | 39.0 ± 1.4* | 37.2 ± 1.4* |
| Liver weight (g) | 1.14 ± 0.04 | 1.02 ± 0.05# | 1.46 ± 0.08* | 1.16 ± 0.04*# |
| Fat pad mass (g) | 0.76 ± 0.09 | 0.84 ± 0.11 | 2.80 ± 0.14* | 2.52 ± 0.21* |
| Body fat (%) | 14.5 ± 2.2 | 17.7 ± 0.6 | 35.1 ± 0.8* | 36.7 ± 1.7* |
| Food intake (kcal/week) | 84.0 ± 3.4 | 81.7 ± 4.9 | 89.7 ± 3.6* | 95.2 ± 5.1* |
| Plasma ALT (units/L) | 12.3 ± 1.0 | 14.2 ± 0.8 | 22.4 ± 2.8* | 18.3 ± 1.9* |
| Plasma total cholesterol (mg/dL) | 112.9 ± 7.4 | 111.9 ± 7.0 | 137.7 ± 8.1* | 126.1 ± 7.8* |
| Plasma triglycerides (mg/dL) | 54.1 ± 3.1 | 59.6 ± 4.4# | 61.1 ± 5.3 | 67.9 ± 3.8# |
| Plasma NEFA (nmol/L) | 0.70 ± 0.06 | 0.70 ± 0.08 | 0.83 ± 0.08 | 0.76 ± 0.07 |
| Plasma uric acid (mg/dL) | 0.45 ± 0.04 | 0.40 ± 0.03# | 0.72 ± 0.08* | 0.49 ± 0.04*# |
| Liver DPP-4 activity (RLUs) | 28,391 ± 1,551 | 9,729 ± 1,362# | 27,567 ± 1,931 | 7,336 ± 679# |
Values are means ± SE. n = 8–12 per group except n = 6–7 per group for percent body fat and n = 6–8 per group for food intake. Fat pad mass is the sum of epididymal and retroperitoneal fat pads. RLUs, relative light units.
*P < 0.05, main effect of diet.
#P < 0.05, main effect of MK compound.
MK0626 Ameliorates WD-Induced Hepatic Insulin Resistance
Euglycemia was maintained in all groups during the 2-h clamp procedure and did not differ significantly among the groups (Fig. 1A). In addition, as we previously showed (22), WD-fed mice displayed significant whole-body insulin resistance, with ∼50% lower glucose infusion rates during the hyperinsulinemic clamp compared with CD mice; whole-body insulin sensitivity was not improved by MK0626 treatment (Fig. 1A and B). Not surprising, plasma insulin during basal and insulin clamp conditions was significantly higher in WD versus CD mice (P < 0.05; Fig. 1C), but did not differ within each diet between drug- and nondrug-treated groups. More importantly, MK0626 treatment rescued hepatic insulin resistance induced by WD feeding, as assessed by insulin-mediated suppression of hepatic glucose production during the hyperinsulinemic-euglycemic clamp (Fig. 1D). Given the chronic nature of the hyperinsulinemia during the clamp and the activation of multiple kinases and phosphatases, hepatic insulin signaling was assessed after acute insulin-stimulation studies. The blunted insulin suppression of hepatic glucose output in WD-fed animals was associated with impaired hepatic insulin signaling at the phosphorylation of Akt (Ser473), which was increased with MK0626 treatment (main effect for diet and drug, P < 0.05 for each; Fig. 1E).
Figure 1.

DPP-4 inhibition attenuated WD-induced hepatic insulin resistance. Values are means ± SE. Blood glucose levels and glucose infusion rate to maintain euglycemia (A), glucose infusion rate (GIR) during the final 40 min (steady-state) of a hyperinsulinemic-euglycemic clamp (B), plasma insulin during basal and steady-state insulin clamp conditions (C), hepatic glucose production during the basal and insulin-stimulated condition and percent insulin suppression of hepatic glucose output during the clamp (D), and hepatic insulin signaling at the level of Akt (Ser473 phosphorylation) from acute insulin stimulation studies (E). n = 5 per group for clamp data and n = 6 per group for acute insulin stimulation studies. *P < 0.05, main effect of diet. #P < 0.05, main effect of MK compound.
Effects of WD and MK0626 on Hepatic TAG, DAG, and Ceramide Content
MK0626 treatment largely ameliorated the marked accumulation of lipid droplets induced by the WD (representative hematoxylin and eosin staining shown in Fig. 2A). This observation was confirmed with mass spectrometric analysis of hepatic TAG content (Fig. 2B), with total hepatic TAG (insert) and each major fatty acid species of TAG dramatically elevated in WD-fed mice. Normalization to the CD values was observed with MK0626 treatment. Analysis of the fatty acid composition (mol%) revealed that feeding the diet rich in corn oil resulted in the expected increase in 18:2 in WD-fed animals, whereas MK0626 reduced the 18:2 percentage in this group. Analysis of hepatic liver DAG concentrations (Fig. 3A) revealed that in WD-fed mice, DAG fatty acids were more unsaturated, similar to the TAG composition. All fatty acid species assessed (34:3, 34:2, 34:1, 36:3, 36:2, 36:1, 38:6, and 38:4) were significantly elevated by the WD (main effect, P < 0.01; Fig. 3A). More importantly, MK0626 treatment significantly reduced the content of these DAG species and total hepatic DAG content in WD-fed mice (diet-by-drug interaction, P < 0.01). In contrast, total hepatic ceramide content did not differ among the groups (Fig. 3C), but 16:0, 18:0, and 20:0 were significantly (P < 0.05; Fig. 3B) increased by WD feeding, and 24:1 and total unsaturated ceramides were significantly (P < 0.05; Fig. 3B and 3C) decreased by WD feeding.
Figure 2.

DPP-4 inhibition reduces WD-induced hepatic steatosis and hepatic TAG accumulation. Values are means ± SE. A: Hematoxylin and eosin staining of hepatocytes for lipid droplets. B: Hepatic lipids were extracted and analyzed by liquid chromatography–mass spectrometry, and hepatic TAG content was determined as fatty acid content of TAG. C: Distribution of fatty acid species in TAG. n = 5 per group. *P < 0.01, main effect of diet. #P < 0.01, diet-by-drug interaction, WD-MK vs. WD.
Figure 3.

DPP-4 inhibition lowers WD-induced hepatic DAG accumulation. Hepatic lipids were extracted and analyzed by liquid chromatography–mass spectrometry. Values are means ± SE. Fatty acid species and total fatty acid content in DAG (A), and fatty acid species (B) and total fatty acid (C) content in ceramides. n = 5 per group. *P < 0.01, main effect of diet. $P < 0.01, main effect for drug; #P < 0.01, diet-by-drug interaction, WD-MK vs. WD.
Effects of MK0626 on Hepatic TAG Synthesis and Secretion Markers
To determine whether increased accumulation of hepatic TAG was due to elevations in de novo lipogenesis, we determined hepatic mRNA levels and protein content of ACC, FAS, and SREBF (SREBP-1c). WD feeding significantly reduced ACC and FAS mRNA and protein levels and SREBF mRNA expression (P < 0.05; Fig. 4A and B). In addition, MK0626 significantly lowered ACC and FAS mRNA expression in CD mice (diet and drug interaction, P < 0.05; Fig. 4A). Hepatic TAG secretion was assessed after the administration of the lipase inhibitor Poloxamer 407. Hepatic TAG secretion was reduced by 40% in WD-fed mice (P < 0.001; Fig. 4C and D), a reduction that was partially rescued by MK0626 treatment (diet-by-drug interaction, P = 0.02; Fig. 4C and D). Hepatic TAG secretion rates corresponded to WD feeding–induced reductions in apoB mRNA expression (Fig. 4E) but not directly to apoB100 protein content (Fig. 4F). In addition, MK0626 significantly increased hepatic MTTP mRNA expression (Fig. 4E), but MTTP content did not differ among the groups (Fig. 4F).
Figure 4.

Effects of DPP-4 inhibition on hepatic de novo lipogenesis markers and hepatic TAG secretion. Values are means ± SE. Hepatic mRNA expression for ACC, FAS and SREBF (A), protein content for ACC, FAS and SREBP-1c (B), hepatic TAG secretion time course (C) and rate (D), mRNA expression for MTTP and apoB (E), and protein content for MTTP and apoB100 (F). n = 8–10 per group for gene expression and protein content. n = 6–7 per group for TAG secretion studies. *P < 0.01, main effect of diet. &P < 0.05 for interaction (significantly different than CD). $P < 0.05 for interaction (significantly different than WD).
MK0626 Improves Indices of Hepatic Mitochondrial PDH Activity and TCA Cycle Flux and Decreases Incomplete Fatty Acid Oxidation
To evaluate the effects of DPP-4 inhibition on hepatic mitochondrial function, we assessed several indices of carbohydrate and fatty acid oxidation and mitochondrial respiration. The mitochondrial oxidation of [1-14C]pyruvate and [2-14C]pyruvate, indices of PDH activity and TCA cycle flux, respectively, did not differ between CD and WD mice (Fig. 5A and B). However, MK0626 significantly increased [1-14C]pyruvate oxidation and [2-14C]pyruvate oxidation in CD-fed and WD-fed mice (main effect for drug for each, P < 0.05; Fig. 5A and B). Examination of mitochondrial fatty acid oxidation revealed a dramatic suppression of complete [1-14C]palmitate oxidation to CO2 in WD-fed animals, a suppression not corrected by MK0626 administration (Fig. 5C). However, MK0626 treatment significantly decreased incomplete [1-14C]palmitate oxidation captured as acid soluble metabolites (P < 0.05; Fig. 5D). Hepatic mitochondrial state 2, state 3, and maximal uncoupled respiration was not affected by WD feeding or MK0626 administration (Fig. 5E).
Figure 5.

Effects of DPP-4 inhibition on hepatic [1-14C]pyruvate oxidation to CO2 (A), [2-14C]pyruvate oxidation to CO2 (B), [1-14C]palmitate oxidation to CO2 (C), incomplete [1-14C]palmitate oxidation (D), and mitochondrial respiration (E) in isolated mitochondria. n = 6–10 per group. Values are means ± SE. *P < 0.05, significant main effect for diet. #P < 0.05, significant main effect for MK compound.
MK0626 Partially Rescues WD-Induced Downregulation in Genes and Proteins Regulating Hepatic Mitochondrial Function
We examined gene expression and protein content of several markers of hepatic mitochondrial biogenesis and content (Fig. 6). Hepatic PGC-1α and CPT-1 were significantly reduced in WD versus CD mice, reductions of which were completely prevented by MK0626 (diet-by-drug interaction, P < 0.05; Fig. 6A). In addition, although mRNA levels of TFAM and PPAR-α were not decreased in WD-fed mice, the levels were significantly increased after MK0626 treatment (P < 0.05; Fig. 6A). We also evaluated hepatic protein levels of OXPHOS subunits, Sirt1, and Sirt3 by Western blot. As shown in Fig. 6B, hepatic protein levels of OXPHOS subunits I-V remained unaltered in WD-fed mice and in WD-fed mice treated with MK0626. However, the WD-induced reduction in protein deacetylase Sirt1 was completely prevented in the WD-MK treatment group (Fig. 6C). Moreover, MK0626 treatment increased hepatic Sirt3 protein content in isolated mitochondria in WD-fed mice (diet-by-drug interaction, P = 0.03; Fig. 6D).
Figure 6.

Effects of DPP-4 inhibition on hepatic mitochondrial genes PGC-1α, TFAM, CPT-1, and PPAR-α (A), OXPHOS complex I-V protein content (B), Sirt1 protein content (C), and Sirt3 protein content (D) measured in isolated mitochondria. n = 6–10 per group for Western blot analyses and n = 8–10 per group for gene expression. Values are means ± SE. *P < 0.05, significant main effect for diet; #P < 0.05, significant main effect for MK compound; &P < 0.05 for interaction (significantly different than CD); $P < 0.05 for interaction (significantly different than WD).
Discussion
In this investigation, we provide mechanistic insight by which DPP-4 inhibition protects against development of hepatic insulin resistance and steatosis in a mouse model fed a high-fat and high-fructose WD. These potential mechanisms are physiologically interrelated, and as shown in Fig. 7, independent lines of evidence demonstrated that the MK0626 compound resulted in a multifaceted improvement in liver insulin sensitivity and fatty acid metabolism. Specifically, DPP-4 inhibition ameliorated hepatic DAG accumulation independent of changes in body weight or adiposity. It also increased hepatic TAG export/secretion, enhanced indices of mitochondrial carbohydrate utilization, and decreased incomplete hepatic mitochondrial fatty acid oxidation, with a concomitant reduction of uric acid production.
Figure 7.
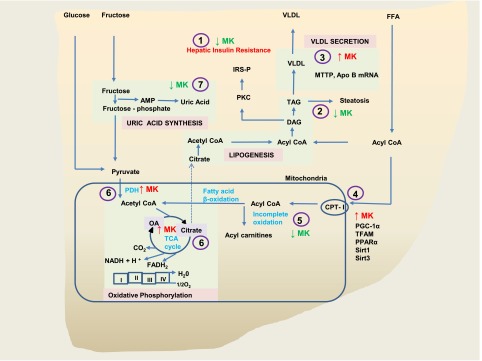
Mechanistic schematic of DPP-4 inhibition with MK0626 on hepatic metabolism and hepatic insulin resistance. Improvement in hepatic insulin resistance by DPP-4 inhibitor MK0626 (1) resulted from decreased levels of DAG and TAG (2), increasing hepatic TAG secretion (3), enhancing select genes and proteins involved in mitochondrial content and function (4), suppressing incomplete oxidation of fatty acids (5), enhancing utilization of metabolites through enhanced PDH activity (6) and TCA cycle flux (6), presumably decreasing carbon intermediates available of lipogenesis and lowering uric acid levels (7) and thereby suppressing the lipogenic effects of uric acid.
NAFLD is now considered an integral component of obesity-related metabolic syndrome, and this condition is a risk factor for the progression of cardiovascular and renal disease (6). Hepatic steatosis is strongly linked to the development of hepatic insulin resistance (14,19). Emerging evidence suggests that DPP-4 inhibition or augmentation of GLP-1 using GLP-1 receptor agonists may be useful in suppressing hepatic insulin resistance and/or steatosis (11–13,32,33). Interestingly, GLP-1 receptor activation with exendin-4 reversed hepatic steatosis (32,33), in part by decreasing hepatic lipogenesis but not through enhancing hepatic VLDL production (33) as we have shown with DPP-4 inhibition in the current study. The strategy of DPP-4 inhibition is especially intriguing, given the observation that T-helper cell surface expression of DPP-4 and serum levels of DPP-4 are elevated in obesity, insulin resistance, and diabetes (9,10). In addition, hepatic DPP-4 expression is elevated in patients with NAFLD, and serum DPP-4 and hepatic expression are both related to NAFLD severity (34,35). Our current findings are in support of the DPP-4 inhibition strategy in the management of liver disease, because administration of MK0626 dramatically suppressed the development of WD-induced hepatic steatosis in concert with enhanced insulin suppression of hepatic glucose production. Previous in vitro hepatocyte studies demonstrate direct effects of GLP-1 and GLP-1 receptor activation on increasing Akt phosphorylation, induction of genes controlling fatty acid oxidation (36,37), and direct in vitro effects of DPP-4 inhibition on hepatic stellate cell activation (38). Therefore, the reported improvements with MK0626 shown in this study are likely due to changes in both GLP-1 and GLP-1 receptor activation as well as to direct effects of DPP-4 inhibition on hepatocyte metabolism.
The mechanisms underlying obesity-related hepatic insulin resistance and fatty liver disease are under intense investigation. In addition, the hepatic protective effects of DPP-4 inhibitors are not well understood but likely go beyond mere improvement of glycemic control through enhanced glucose-stimulated insulin secretion (10). Hepatic insulin action to regulate hepatic glucose output is mediated through activation of the insulin receptor, insulin receptor substrates (IRS-1 and -2), phosphatidylinositol 3-kinase, and Akt pathway (14). Under normal insulin-sensitive conditions, insulin inhibits glycogenolysis and gluconeogenesis, suppressing glucose production (39). However, in the insulin-resistant state, defects in hepatic insulin signaling impair insulin-suppression of hepatic glucose production, leading to hyperglycemia and compensatory hyperinsulinemia (40). Increased accumulation of lipid metabolites/intermediates, such as DAGs and ceramides, are thought to be initiators in the development of insulin resistance (14,41). Numerous studies have implicated hepatic DAGs in potentially causing hepatic insulin resistance (14,19), although recent studies did not support the importance of hepatic accumulation of DAGs (42). In addition, polyunsaturated fatty acids in DAG are thought to be responsible for PKC activation (43). Lipidomic analysis revealed significant accumulation of polyunsaturated fatty acids in DAG in WD-fed mice, and DPP-4 inhibition with MK0626 completely abrogated this accumulation. Previous studies have also implicated hepatic ceramides in hepatic insulin resistance (44); however, in this investigation, total hepatic ceramide content was unchanged with WD feeding or MK0626 administration. This is in agreement with recent reports showing a lack of correlation between hepatic insulin resistance and ceramide accumulation (44,45), and collectively, these data support a stronger role for hepatic DAG accumulation in WD-induced hepatic insulin resistance in the present model.
Our group previously demonstrated a significant role for de novo fatty acid synthesis in liver TAG accumulation in NAFLD patients (20). In addition, increased fat and sucrose consumption can induce lipogenic genes, and this phenomenon is suppressed in rodents by DPP-4 inhibitors (12,46). In the current study, however, hepatic mRNA expression and protein content for lipogenic markers ACC, FAS, and SREBP were downregulated with the WD, with no further reduction seen with MK0626. Direct suppression of these lipogenic genes is a likely consequence of the high saturated fatty acid content in the WD diet (47). This notion is further supported by recent observations that lipogenic genes are suppressed with a high-fat and high-fructose combination diet but not with high fat or high fructose alone (46). Our findings suggest that the protective effects of DPP-4 inhibition in this model may not be mediated through direct suppression of hepatic de novo lipogenesis.
The export of hepatic TAG is dependent on VLDL-TAG packaging, a process requiring MTTP and apoB (48). Hepatic MTTP expression is decreased in the setting of insulin resistance and NAFLD (49), and marked accumulation of hepatic TAG is not always accompanied by increased secretion of VLDL (44). Here we demonstrate a dramatic reduction in hepatic TAG secretion in WD-fed mice, a finding that likely is contributing to the hepatic steatosis in the model. In addition, we demonstrate for the first time that DPP-4 inhibition partially rescued hepatic TAG secretion in WD mice. Circulating plasma TAGs were also increased in the MK0626-treated mice. Interestingly, hepatic TAG secretion was actually blunted with MK0626 in the CD-fed mice, a finding likely related to the minimal hepatic TAG present in the low fat–fed mice. We also observed WD-induced reductions in hepatic apoB mRNA expression, and MK0626-induced increases in MTTP and apoB100 mRNA expression in WD-fed mice. These data indicate that DPP-4 inhibition with MK0626 may be alleviating hepatic TAG accumulation, in part through increased hepatic TAG secretion/export.
Recent reports suggest that hepatic mitochondrial dysfunction may be an initial event in liver lipid accumulation (15,19) and intimately linked to the development of hepatic insulin resistance (50). Sirt1 is involved in mitochondrial biogenesis and mitochondrial metabolism and regulates the mRNA levels of PPAR-α, PGC-1α, CPT-1, and TFAM (51,52). Studies have shown that fructose can suppress Sirt1 in hepatocytes (53) and that GLP-1 agonism can increase Sirt1 levels in high fat–fed mice (37). Results of the current investigation demonstrate that WD-induced suppression of the Sirt1 level was completely prevented with DPP-4 inhibition. Furthermore, although not significantly decreased with WD-feeding, Sirt3, a mitochondrial protein deacetylase known to regulate mitochondrial function (52), was increased by 25% with DPP-4 inhibition. The WD-induced suppression in Sirt1 expression was accompanied by WD-induced impairment in complete mitochondrial palmitate oxidation that was not rescued by MK0626 treatment. This could be due to the effects of fructose, because fructose administration is associated with inhibition of fatty acid oxidation (54), raising the possibility of a fructose-mediated inhibition of fatty acid oxidation that is not entirely relieved by MK0626. However, MK0626 administration prevented WD-induced suppression of hepatic PGC-1α and CPT-1 mRNA expression and significantly increased TFAM and PPAR-α mRNA expression in the WD-fed mice, suggesting potentially better maintenance of mitochondrial biogenesis with DPP-4 inhibition. The examination of the direct role of DPP-4 inhibition on mitochondrial biogenesis is warranted in future investigations.
If β-oxidation is not matched with enhanced activity of the TCA cycle, this will lead to the incomplete oxidation of lipids and the accumulation of acetyl-CoA metabolites (acetyl-carnitine) (55). Lipid intermediates from incomplete oxidation of fatty acids are also linked to hepatic insulin resistance and mitochondrial dysfunction (56). These metabolites may also be directed to fatty acid biosynthesis pathways and are implicated in tissue insulin insensitivity and mitochondrial oxidative stress (55). To our knowledge, we report for the first time that DPP-4 inhibition decreased incomplete palmitate oxidation in WD-fed mice, thereby suggesting the possible role of suppressed incomplete fatty acid oxidation by MK0626 in improving hepatic insulin resistance.
DPP-4 inhibition with MK0626 also enhanced indices of mitochondrial carbohydrate oxidation, including PDH activity and TCA cycle flux, which was not accompanied by significant alterations in hepatic mitochondrial respiration or in changes in electron transport chain protein expression. These novel results suggest that DPP-4 inhibition enhances the oxidation and disposal of glucose/fructose carbon intermediates, thereby potentially reducing the concentrations of these intermediates for lipogenesis. The direct examination of fructose metabolism is warranted in future investigations.
High fructose consumption is known to increase liver uric acid production, deplete high-energy phosphates, and elevate glucose flux (57). In addition, uric acid has been shown to cause hepatic steatosis in cultured hepatocytes by enhancing lipogenesis; whereas, suppression of uric acid production has been shown to ameliorate hepatic steatosis (4,58,59). MK0626 seems to be unique among DPP-4 inhibitors, because it decreased plasma uric acid in WD mice, suggesting additional hepatic protective effects of MK0626 through enhancing fructose metabolism and shunting away from the uric acid pathway.
In summary, the current investigation highlights newly described pleiotropic protective effects of DPP-4 inhibition on hepatic metabolism and lipid accumulation. Our findings indicate that DPP-4 inhibition with MK0626 ameliorated high-fat/high-fructose–induced hepatic insulin resistance, hepatic steatosis, and hepatic DAG accumulation independent of changes in body weight or adiposity. DPP-4 inhibition also increased hepatic TAG secretion, enhanced indices of mitochondrial carbohydrate utilization with concomitant reduction of uric acid production, and decreased incomplete hepatic mitochondrial fatty acid oxidation. These findings collectively demonstrate a strong potential clinical utility for DPP-4 inhibition in the prevention of hepatic insulin resistance and development of hepatic steatosis.
Article Information
Acknowledgments. The authors thank Brenda Hunter (University of Missouri) for editorial assistance; Dr. Matthew E. Morris, Grace Meers, Nathan Rehmer, Dongqing Chen, Mona Garro, and Alex Meuth (University of Missouri) for technical assistance; and Vanderbilt University School of Medicine Mouse Metabolic Phenotyping Center (DK-059637) (Nashville, TN) for performing the hyperinsulinemic-euglycemic clamps.
Funding. This work was supported with resources and the use of facilities at the Harry S. Truman Memorial Veterans' Hospital in Columbia, MO. This work was supported by HL-074214 (D.A.F.), HL-111906 (D.A.F.), DK-088940 (J.P.T.), HL-73101-07 (J.R.S.), HL-107910-03 (J.R.S.), VA-Merit System 0018 (J.R.S.), and VHA-CDA2 1299 (R.S.R.).
Duality of Interest. Research support from Merck Pharmaceuticals was provided to J.H. and R.N. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. A.R.A., J.H., G.L., C.M., J.P.T., J.R.S., and R.S.R. contributed to the study concept and design. A.R.A., D.A.F., G.L., C.M., M.M.D., K.D.F., J.P.T., E.J.P., J.R.S., and R.S.R. acquired data. A.R.A., D.A.F., J.P.T., E.J.P., J.R.S., and R.S.R. analyzed and interpreted the data. A.R.A., E.J.P., J.R.S., and R.S.R. drafted the manuscript. A.R.A., J.H., D.A.F., R.N., G.L., C.M., M.M.D., K.D.F., J.P.T., E.J.P., J.R.S., and R.S.R. critically revised the manuscript for important intellectual content. A.R.A., E.J.P., and R.S.R. performed the statistical analysis. J.H., D.A.F., R.N., J.P.T., J.R.S., and R.S.R. obtained funding. R.S.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA 2010;303:235–241 [DOI] [PubMed] [Google Scholar]
- 2.Sowers JR, Whaley-Connell A, Hayden MR. The role of overweight and obesity in the cardiorenal syndrome. Cardiorenal Med 2011;1:5–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrera F, George J. The role of diet and nutritional intervention for the management of patients with NAFLD. Clin Liver Dis 2014;18:91–112 [DOI] [PubMed] [Google Scholar]
- 4.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013;62:3307–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013;57:2525–2531 [DOI] [PubMed] [Google Scholar]
- 6.Bray GA. Fructose and risk of cardiometabolic disease. Curr Atheroscler Rep 2012;14:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fruci B, Giuliano S, Mazza A, Malaguarnera R, Belfiore A. Nonalcoholic fatty liver: a possible new target for type 2 diabetes prevention and treatment. Int J Mol Sci 2013;14:22933–22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest 2007;117:24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Firneisz G, Varga T, Lengyel G, et al. Serum dipeptidyl peptidase-4 activity in insulin resistant patients with non-alcoholic fatty liver disease: a novel liver disease biomarker. PLoS One 2010;5:e12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckerle Mize DL, Salehi M. The place of GLP-1-based therapy in diabetes management: differences between DPP-4 inhibitors and GLP-1 receptor agonists. Curr Diab Rep 2013;13:307–318 [DOI] [PubMed] [Google Scholar]
- 11.Samson SL, Bajaj M. Potential of incretin-based therapies for non-alcoholic fatty liver disease. J Diabetes Complications 2013;27:401–406 [DOI] [PubMed] [Google Scholar]
- 12.Kern M, Klöting N, Niessen HG, et al. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS One 2012;7:e38744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirakawa J, Fujii H, Ohnuma K, et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 2011;60:1246–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rector RS, Thyfault JP, Uptergrove GM, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol 2010;52:727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longato L. Non-alcoholic fatty liver disease (NAFLD): a tale of fat and sugar? Fibrogenesis Tissue Repair 2013;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011;108:16381–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorden DL, Ivanova PT, Myers DS, et al. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One 2011;6:e22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rector RS, Morris EM, Ridenhour S, et al. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 2013;57:2213–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 2011;14:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edmondson SD, Mastracchio A, Mathvink RJ, et al. (2S,3S)-3-Amino-4-(3,3-difluoropyrrolidin-1-yl)-N,N-dimethyl-4-oxo-2-(4-[1,2,4]triazolo[1,5-a]-pyridin-6-ylphenyl)butanamide: a selective alpha-amino amide dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem 2006;49:3614–3627 [DOI] [PubMed] [Google Scholar]
- 22.Nistala R, Habibi J, Lastra G, et al. Prevention of obesity-induced renal injury in male mice by DPP4 inhibition. Endocrinology 2014;155:2266–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bostik B Habibi J, Ma L, et al. . Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of Western diet induced obesity. Metabolism 2014;63:1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Xiao J, Tian H, et al. The DPP-4 inhibitor MK0626 and exercise protect islet function in early pre-diabetic kkay mice. Peptides 2013;49:91–99 [DOI] [PubMed] [Google Scholar]
- 25.Demarco VG, Ford DA, Henriksen EJ, et al. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology 2013;154:159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han X, Gross RW. Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem 2001;295:88–100 [DOI] [PubMed] [Google Scholar]
- 27.Fletcher JA, Meers GM, Linden MA, et al. Impact of various exercise modalities on hepatic mitochondrial function. Med Sci Sports Exerc 2014;46:1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noland RC, Koves TR, Seiler SE, et al. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem 2009;284:22840–22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Millar JS, Cromley DA, McCoy MG, Rader DJ, Billheimer JT. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. J Lipid Res 2005;46:2023–2028 [DOI] [PubMed] [Google Scholar]
- 30.Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 2008;57:1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 32.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006;43:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parlevliet ET, Wang Y, Geerling JJ, et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS One 2012;7:e49152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balaban YH, Korkusuz P, Simsek H, et al. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann Hepatol 2007;6:242–250 [PubMed] [Google Scholar]
- 35.Miyazaki M, Kato M, Tanaka K, et al. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol Med Rep 2012;5:729–733 [DOI] [PubMed] [Google Scholar]
- 36.Gupta NA, Mells J, Dunham RM, et al. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010;51:1584–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J, Hong SW, Chae SW, et al. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PLoS One 2012;7:e31394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaji K, Yoshiji H, Ikenaka Y, et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J Gastroenterol 2014;49:481–491 [DOI] [PubMed] [Google Scholar]
- 39.Dentin R, Liu Y, Koo SH, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 2007;449:366–369 [DOI] [PubMed] [Google Scholar]
- 40.Marchesini G, Brizi M, Morselli-Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 1999;107:450–455 [DOI] [PubMed] [Google Scholar]
- 41.Jornayvaz FR, Shulman GI. Diacylglycerol activation of protein kinase Cε and hepatic insulin resistance. Cell Metab 2012;15:574–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farese RV Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 2012;15:570–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marignani PA, Epand RM, Sebaldt RJ. Acyl chain dependence of diacylglycerol activation of protein kinase C activity in vitro. Biochem Biophys Res Commun 1996;225:469–473 [DOI] [PubMed] [Google Scholar]
- 44.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010;51:679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magkos F, Su X, Bradley D, et al. . Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012;142:1444–1446 e1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renaud HJ, Cui JY, Lu H, Klaassen CD. Effect of diet on expression of genes involved in lipid metabolism, oxidative stress, and inflammation in mouse liver-insights into mechanisms of hepatic steatosis. PLoS One 2014;9:e88584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ou J, Tu H, Shan B, et al. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A 2001;98:6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol 2013;48:434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higuchi N, Kato M, Tanaka M, et al. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp Ther Med 2011;2:1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 2010;31:364–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Price NL, Gomes AP, Ling AJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 2012;15:675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fletcher JA, Meers GM, Linden MA, et al. Impact of Various Exercise Modalities on Hepatic Mitochondrial Function. Med Sci Sports Exerc 2014;46:1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rebollo A, Roglans N, Baena M, et al. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim Biophys Acta 2014;1841:514–524 [DOI] [PubMed]
- 54.Prager GN, Ontko JA. Direct effects of fructose metabolism on fatty acid oxidation in a recombined rat liver mitochondria-hish speed supernatant system. Biochim Biophys Acta 1976;424:386–395 [DOI] [PubMed] [Google Scholar]
- 55.Koves TR, Li P, An J, et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 2005;280:33588–33598 [DOI] [PubMed] [Google Scholar]
- 56.Adams SH, Hoppel CL, Lok KH, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 2009;139:1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arden C, Petrie JL, Tudhope SJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes 2011;60:3110–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishimoto T, Lanaspa MA, Rivard CJ, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013;58:1632–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–40744 [DOI] [PMC free article] [PubMed] [Google Scholar]