

Abstract
Bacterial RNA polymerase (RNAP) interacts with conserved −10 and −35 promoter elements to recognize the promoter and to form an open complex in which DNA duplex around transcription start site melts. Using model DNA constructs (fork junction DNA) that mimic DNA structure found in the open complex we observed that the consequences of mutations in −10 promoter element for RNAP binding exhibited a striking dependence on the presence or absence of a functional −35 promoter element. A role of spacer DNA (a non-conserved DNA sequence connecting −10 and −35 promoter elements) in this phenomenon was probed with a series of fork junction DNA constructs containing perturbations to the spacer DNA. In the absence of a physical connection between the −10 and −35 DNA elements, or when −10 and −35 DNA elements were connected by a long flexible non-DNA linker, the dependence of RNAP interactions with −10 element on the strength of −35 element was lost. When these DNA elements were linked by a rigid DNA duplex or by a DNA duplex containing a short single-stranded gap, the coupling between the −10 and −35 binding activities was observed. These results indicated that promoter spacer DNA played an active role in integrating the functional consequences of RNA polymerase contacts with −10 and −35 promoter element. This role likely involves physical deformation of the spacer occurring in parallel with promoter melting as shown by Fluorescence Resonance Energy Transfer (FRET) experiments with the probes incorporated into spacer DNA.
Keywords: RNA polymerase, promoter, protein – DNA interactions
Graphical Abstract
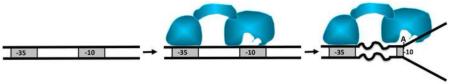
1. Introduction
Initiation of transcription, regardless of the origin and complexity of RNA polymerase (RNAP), involves a critical step of promoter melting which follows the initial recruitment of RNAP to the promoter [1, 2]. The mechanistic aspects of this step are still not completely understood. In bacteria a minimum of two of short-lived intermediates (designated I1 and I2) on the pathway to the open complex have been proposed [2-6]. The I1 intermediate corresponds to the closed complex in which promoter DNA remains double-stranded [3]. Conversion to I2 intermediate is rate limiting at least in some promoters [7] and recent data demonstrated that promoter DNA melting occurs at this step at λPR promoter [8]. Large conformational changes in promoter DNA and in the polymerase accompany promoter melting [2, 6]. Most notably, in addition to melting of DNA duplex, formation of the open complex involves a large-scale movement of downstream DNA duplex to position it in the main DNA binding channel of RNAP [6]. The order of these two events (i.e. if the placing of downstream DNA duplex in the RNAP channel precedes or follows duplex melting) may be promoter dependent [3-5, 9]. Sigma subunit of bacterial RNAP appears to play a crucial role in promoter melting [10].
Sigma subunit for a long time has been known to be responsible for the initial binding of RNAP to the promoter DNA [2, 11, 12]. It contains two DNA binding domains which can bind, in a sequence-specific manner, to the two conserved hexamers (−35 and −10 elements) which define a typical bacterial promoter [13]. This DNA binding activity of sigma is regulated by the protein-protein contact with the core enzyme [14-18]. Free sigma does not bind the promoter DNA sequences and it is believed that binding of sigma to the core enzyme unmasks DNA binding activity of sigma. This regulation of DNA binding activity involves a large scale domain movement which positions the DNA binding domains of sigma at a proper spacing compatible with simultaneous interactions with −10 and −35 elements [19]. It may also involve local conformational changes which could modulate intrinsic DNA binding activities of DNA binding domains of sigma [18] although recent data do not support this notion [20]. A clue to the active role of sigma in melting of promoter DNA has emerged from experiments which showed that sigma contained a strong single-stranded DNA binding site [21]. This site appeared to bind the nontemplate strand −10 element with high affinity and high sequence specificity [10, 21-23]. Promoter melting is believed to involve at least two phases: nucleation and bubble propagation [24]. Nucleation of melting occurs at the upstream boundary of −10 element [25] suggesting that ss binding activity of sigma may be important for the nucleation step.
Dissection of contributions of the conserved −10 nontemplate strand bases to the overall binding affinity revealed that two positions, adenine at position −11 and thymine at position −7 are the most important for the overall affinity [26]. Adenine at position −11 is of special interest as it is the most conserved base in −10 element and it is located at the boundary between duplex and single-stranded DNA in the open complex. This adenine is recognized specifically by RNA polymerase which targets N1 position of the adenine [26]. Thr429 has been suggested to be involved in N1 position recognition [27]. This is significant since N1 position is involved in hydrogen bonding in an A/T base pair. Thus, base pairing at this position and recognition of −11A are mutually exclusive further supporting a potential role of −11A interactions in initial nucleation of promoter melting. Recent data provided strong support for the role of nontemplate −11A in promoter melting nucleation [28, 29]. For example, stability of −11 base-pair has been shown to correlate with the rate of promoter melting [30]. Polymerase elements involved in binding the −10 element in single-stranded form constitute a very small fraction of the ~ 0.5 MDa RNAP holoenzyme. Functional studies confirmed that core of promoter melting activity is localized to a very small subset of all promoter-polymerase contacts [31, 32].
Interactions of RNAP with −35-promoter element are believed to be important primarily for initial promoter binding. The −35 promoter element is one of the first sites of the promoter to form a contact with RNAP during promoter binding by RNAP [5].
In the case of bacterial RNA polymerase, promoter melting is driven entirely by noncovalent interactions of the enzyme with nucleic acid template [1, 2]. Understanding of the mechanism of promoter melting by bacterial RNAP will thus require the analysis of how the favorable energy that is released by RNAP-promoter interactions is channeled by the enzyme into energetically unfavorable DNA melting step. RNAP contacts with −10 (in ds and ss form) and −35 (in ds form) promoter elements are the prime sources of the favorable free energy released upon RNAP-promoter complex formation. To make these contacts RNAP uses two conserved domains of σ subunit (region 2 and 4, respectively) that are far apart (~ 60Å) in the holoenzyme structure precluding a direct contact between them [33, 34]. This would suggest that energetic contributions of −10 and −35 contacts should be independent (i.e. should be additive). However, we describe here a striking dependence of the energetic effect of mutations in −10 promoter element on the presence or absence of a functional −35 element consistent with what we reported previously [31]. We show that spacer DNA connecting −10 and −35 promoter elements is responsible for integrating functional consequences of RNAP contacts with −10 and −35 promoter elements.
2. Materials and Methods
2.1 Materials
DTPA-AMCA-maleimide (diethylenetriaminepentaacetic acid-7-amino-4-methylcoumarin-maleimide) was prepared in our laboratory as described previously [35]. Cy™5 mono NHS was from Amersham Biosciences Corp. (Piscataway, NJ). Bodipy FL C1 iodoacetamide and tetramethylrhodamine iodoacetamide (TMRIA) were from Molecular Probes (Eugene, OR). The amine-VN phosphoramidate was obtained from Clontech (Palo Alto, CA). All other reagents were of highest purity commercially available. Core RNAP enzyme was purified from E. coli K12 cells (obtained from fermentation facility at University of Alabama) using the method of Burgess and Jendrisak [36] as modified by Polyakov et al. [37] and Hager et. al.[38]. Single-cysteine mutant of σ70 σ70) and wt σ70 were expressed and purified as described previously [17].[A59C]σ70 was labeled with DTPA(Eu3+)-AMCA-maleimide according to [35]. Labeled [A59C]σ70 was mixed with core polymerase in 1.5:1 ratio, incubated 30 minutes at 4°C and purified on a Superdex 200 10/30 GL sizing column (Pharmacia) in 50 mM HEPES (pH 7.9), 250 mM KCl, 10μM EDTA.
2.2 DNA constructs
The oligonucleotides were obtained from Integrated DNA Technologies Inc.(Coralville, IA) or from W.M. Keck Facility (Yale University, New Haven, CT). Fig. 1 summarizes DNA constructs that were used in this work. Details regarding the mutations introduced to the constructs are given in parenthesis next to the name of the construct. For example, C1(−10cons, −35cons) describes a C1 construct with both −10 and −35 elements having the consensus sequence (TATAAT and TTGACA), respectively. The “anticonsensus” sequences for these elements were CGCGGT and CCAGTG, respectively. The following oligonucleotides were prepared in order to obtain all the constructs illustrated in Fig. 1 (−10 and −35 elements are underlined):
Figure 1.

Schematic representation of DNA constructs used in this work. Sequences of the constructs are provided in Materials and Methods. C1 and C2 are fork junction DNA constructs with the last base-pair of ds portion at −11 and −12 position, respectively. C3 is analogous to C2 but contains a 1 bp gap in the nontemplate strand of the spacer region. C4 corresponds to a construct where short duplex containing −35 region is connected to a short hairpin contain −10 region via a long flexible linker. C5 and C6 are short hairpin and duplex constructs containing −10 and −35 sequences, respectively. R1 and R2 are Cy5-labeled constructs analogous to C2 and C5, respectively. C7 and C8 are 75 bp long promoter DNA constructs with fluorescence probes to monitor using FRET spacer DNA deformations (C7) and promoter melting (C8).
1. C1 was prepared by annealing the C1T and C1B oligonucleotides (C1T: GTG TTG ACA ATT TTA CCT CTG GCG TTT ATA ATG GT; C1B: TAA ACG CCA GAG GTA AAA TTG TCA ACA C).
2. C2 was prepared by annealing the C2T and C2B oligonucleotides (C2T: GGC ATC CTT GAC ATC ACG ACA GGT GAG CTG TAT AAT CAG; C2B: ACA GCT CAC CTG TCG TGA TGT CAA GGA TGC C). C2 containing 15 bp and 19 bp spacer DNA was prepared using oligonucleotides in which 17 bp spacer present in C2T and C2B oligonucleotides was replaced with TCA CGA CAT GAG CTG and TCA CGA CAG GCT GTA GCT G, respectively.
3. C3 was prepared by annealing C3T1, C3T2 and C3B oligonucleotides (C3T1: GGC ATC CTT GAC ATC; C3T2: CGA CAG GTG AGC TGT ATA ATC AG; C3B: ACA GCT CAC CTG TCG (Spacer9) GAT GTC AAG GAT GCC)
4. C4 was prepared by annealing C4B and C4T (C4T: ACA CGC (Spacer18) GCG TGT ATA ATG GT (Spacer18)6 TGA TGT CAA GGA TGC; C4B: GCA TCC TTG ACA TCA
5. C5: ACA CGC (Spacer9) GCG TGT ATA ATG GT.
6. C6 was prepared by annealing C6T and C6T (C6T: CGA TCC TTG ACA AAG CTG TTA C; C6B: GTA ACA GCT TTG TCA AGG ATC G).
7. R1 was prepared by annealing C1T and R1B oligonucleotides (R1B: TAA AC(amine-VN) CCA GAG GTA AAA TTG TCA ACA). The amino group of amine-VN residue was modified with Cy5-NHS and modified oligonucleotide was purified by HPLC.
7. R2: ACA CGC (Spacer9) GCG TGT ATA ATG GT- 3’amino. The amino group was modified with Cy5-NHS and modified oligonucleotide was purified by HPLC.
In addition to the above fork junction DNA constructs, the following oligonucleotides were used to prepare promoter DNA constructs for FRET-based detection of promoter melting and spacer deformation:
1. C7 was prepared by annealing C7T and C7B (C7T: AAT CCT CTA GAG TTG GAT AAA TAT CTA ACA CCG TGC GTG TTG ACA AT*T TTA CCT CTG GCG GTT ATA ATG GTT GCA; C7B: TGC AAC CAT TAT AAC CG*C CAG AGG TAA AAT TGT CAA CAC GCA CGG TGT TAG ATA TTT ATC CAA CTC TAG AGG ATT).
3. C8 was prepared by annealing C8T and C8B (C8T: AAT CCT CTA GAG TTG GAT AAA TAT CTA ACA CCG TGC GTG TTG ACA ATT TTA CCT CTG GCG GTT ATA ATG GTT GCA-3’dabcyl; C8B: 5′-Cy3-TGC AAC CAT TAT AAC CGC CAG AGG TAA AAT TGT CAA CAC GCA CGG TGT TAG ATA TTT ATC CAA CTC TAG AGG ATT). Hybridization of C7T with C7B (or C8T with C8B) produces DNA duplex (C7 or C8 (Fig. 1), espectively) corresponding to −74 to +1 fragment of λ PR promoter containing consensus −35 and −10 elements. The star denotes the positions where the phosphothio residues were incorporated into the DNA’s. C7T and C7B were labeled with fluorescence probes at the phoshothio residue by incubating the oligonucleotides with Bodipy FL C1 iodoacetamide (C7T, 13-fold molar excess of the dye) or with TMRIA (C7B, 14-fold molar excess of the dye) in 25 mM potassium phosphate buffer (pH 7.5) containing 20% DMF for 20hrs at 50 deg. Excess of unreacted dyes was removed by ethanol precipitation and labeled oligonucleotides were purified by reversed-phase HPLC.
2.3 Determination of relative binding affinity of DNA constructs
High affinity of DNA fork junction constructs for binding RNA polymerase makes the measurements of absolute RNAP binding affinity difficult. Thus, we employed previously described luminescence resonance energy transfer (LRET) based approach that allows site-specific determinations of the relative binding affinity of various DNA. Relative affinities of RNAP holoenzyme towards different unlabeled promoter DNA fragments were determined by measuring the ability of these molecules to compete with Cy5-labeled reference fork junction promoter fragment.
When the binding experiments are performed under conditions where the competitor and the Cy5-lableled reference binder are in excess of RNAP concentration and their equilibrium dissociation constants for binding to RNAP, the relative affinities of competitor DNA can be determined from the measurements of the relative degree of saturation of RNA polymerase with Cy5-labeled reference fork junction molecule and unlabeled competitor DNA as described previously (26) using following equations:
| (eq. 1) |
| (eq. 2) |
| (eq. 3) |
| (eq. 4) |
where νref and νmut are the degrees of saturation of RNAP with reference Cy5-labeled DNA and competitor unlabeled DNA, respectively; [DNAref]tot and [DNAmut]tot are the total concentrations of reference Cy5-labeled DNA and competitor unlabeled DNA, respectively; [RNAP]tot is the total concentration of RNAP; F0 and Fmut are sensitized acceptor emission in the absence and presence of unlabeled competitor DNA fragment, respectively.
Binding experiments were performed in a final volume of 20 μl at 25°C in binding buffer (50 mM HEPES (pH 7.9), 250 mM KCl, 10 mM EDTA, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA, 2.8% polyethylene glycol (8000)). The final concentrations of Cy5-labeled reference fork junction DNA (R1 or R2, Fig. 1) and RNAP holoenzyme containing [A59C]σ70 labeled with (Eu3+)DTPA-AMCA were 50 nM and 5 nM respectively. The concentration of unlabeled competitor promoter fragments was in nM to μM range depending on the relative affinity of the competitor for RNA polymerase holoenzyme. RNAP was added to the reaction mixture containing reference labeled DNA and unlabeled competitor DNA and after 30 min incubation at 25°C, sensitized acceptor emission at 668 nm and donor emission at 620 nm (excitation was at 360 nm) were determined in 384-well microplate using Analyst™ AD fluorescence microplate reader (BioSystems, Sunnyvale, CA). For each measurement signals resulting from 1000 lamp flashes were accumulated using 50 μsec delay and 1000 μsec gate.
2.4 FRET-based detection of promoter melting and spacer DNA conformational changes
Time course of fluorescence changes in C7 and C8 duplexes was monitored in 120 μl cuvette in the binding buffer on Aminco-Bowman AB2 spectrofluorometer. Fluorescence emission with excitation at 495 nm and emission at 520 nm of 10 nM solution of DNA construct was monitored as a function of time. At an indicated time, RNAP was added to a final concentration of 100 nM and recording of the time course of fluorescence emission was resumed.
3. Results
In this work we asked a question if the energetics of the contacts made by RNAP with −10 and −35 is independent from each other. The approach we took was to measure the energetic consequences on binding to RNAP of mutations in −10 conserved promoter element on the presence or absence of a consensus −35 element. If RNAP contacts −10 and −35 promoter elements independently, the energetic consequences of mutations in −10 promoter element should not depend on the presence or absence of functional −35 contact. On the other hand, if there were a coupling between these RNAP contacts, consequences of mutations in −10 promoter elements would be expected to be different depending on the presence or absence of strong −35 element.
Our binding measurements were performed using fork junction DNA constructs (such as, for example, C1 or C2, Fig. 1) which are well-established models to investigate interactions of RNAP holoenzyme important for the formation of the open complex [10, 26, 31, 39-41]. We utilized site-specific equilibrium competition assay to measure relative binding affinity DAN constructs [26, 31]. RNAP holoenzyme labeled with europium chelate at a unique cysteine residue (59) of sigma subunit was used in this assay. When a Cy5-labeled fork junction DNA (R1, Fig. 1) bound to the europium-labeled holoenzyme, luminescence energy transfer (LRET) [42] between the europium chelate and Cy5 was observed (Fig. 2, second bar) that could be used to detect RNAP-fork junction DNA complex formation. Since Cy5-labeld R1 was used in excess over RNAP concentration and the Kd of R1-RNAP complex (as verified by titrating RNAP with increasing concentrations of R1 (data not shown)), the signal depicted in Fig. 2 (second bar) corresponded to 100% of saturation of RNAP with R1. The LRET signal depicted in Fig. 2A corresponds to sensitized acceptor emission read with pulsed excitation of the donor with 50 μsec delay after excitation. This gated sensitized acceptor signal measurement allows elimination of any background of Cy5-labeled R1 that is not bound to RNAP (as illustrated by control experiment where europium chelate-labeled σ subunit alone was incubated with R1 (Fig. 2A, first bar). In the presence of unlabeled competitor fork junction DNA the observed LRET signal was reduced proportionally to the relative amounts of RNAP bound to R1 or the unlabeled competitor (Fig. 2A). The data illustrated in Fig. 2A could be then used to calculate the ratio of the binding constant of R1 and the competitor (using eq. 1-4) and from this the ratio of binding constant of unlabeled “wt” fork junction DNA (C1 in the case of data in Fig. 2) and a given construct can be calculated. This in turn provides the values of ΔΔG (the difference between binding free energies of wt and mutant ligands). The results of such calculations for the data in Fig. 2A are shown in Fig. 2B. LRET-based assay is site-specific i.e. the signal used to monitor the binding is generated by binding of the labeled fork junction DNA to the σ subunit. This is important since nucleic acids can bind nonspecifically at sites on other RNAP subunits. Using a site-specific assay eliminates potential artifacts due to such nonspecific interactions.
Figure 2.

LRET-based determination of relative binding affinity of fork junction DNA constructs. (A) Sensitized acceptor signal (LRET) in the absence of unlabeled competitor DNA (first two bars) and in the presence of competitor fork junction DNA’s containing indicated mutations. Data were normalized to the signal observed for the holoenzyme in the presence of Cy5 labeled R1 DNA construct (second bar). (B) Relative binding affinity of the fork junction DNA constructs calculated from the data shown in panel A using eq. 1 and 2.
The data in Fig. 2 are for the mutants derived from C1 fork junction that is based on λ PR promoter sequence. In agreement with our previous observations [31], the energetic consequences of mutations in −10 promoter element exhibited dramatic dependence on the status of −35 element. Mutations at three positions of −10 element were examined. Positions −11 and −7 are the most conserved in bacterial promoters and they have been previously shown to be the most important for binding short −10 element-only fork junction DNA’s [26]. In comparison, −8 position is much less conserved and mutating it in a context of short −10 element-only fork junction DNA’s had only relatively minor effect on RNAP binding affinity [26]. When the C1 fork junction DNA contained consensus −35 element, mutations at −11, −8 and −7 position had similar small effect on RNAP binding affinity. In contrast, when C1 fork junction DNA did not contain a strong −35 element, the effects of mutations at these three positions had very differential effect on RNAP binding affinity. Mutations at −11 and −7 had a large negative effect on binding affinity whereas mutation at position −8 had produced only a small decrease in the binding affinity, similar to the one observed with the C1 containing consensus −35 element. The last base-pair of the duplex part of the C1 fork junction DNA is at position −11. Very similar dependence of the energetics of −10 interactions on the presence of strong −35 element was observed in fork junction DNA that had a duplex terminating at position −12 (data not shown).
The dependence of −10 element interactions on the strength of −35 element interactions illustrated by Fig. 2 showed that −10 and −35 contacts are not energetically independent. A long-range coupling between these two interactions must take place since they involve two domains of σ subunit separated by ~60 Å. The most obvious structural element that connects sites of −10 and −35 element interactions with RNAP is spacer DNA between −10 and −35 elements. We have thus investigated a possible role of the spacer in coupling between −35 and −10 interactions with RNAP.
The first question that we asked was if the observed coupling is specific for the spacer sequence present in the λ PR promoter sequence. Fig. 3A shows the results of an experiment analogous to the one shown in Fig. 2 except that C2 fork junction DNA constructs were used in which λ PR spacer sequence was replaced with unrelated sequence. The results obtained with C2 fork junction DNA constructs were very similar to the results with C1 fork junction DNA constructs. In the presence of consensus −35 element, mutations at positions −11, −8, −7 had similar and very small effect on the affinity for RNAP, whereas in the absence of strong −35 element, mutations at −11 and −7 had large negative effect on RNAP affinity. We concluded that the observed coupling between −10 and −35 element binding is a general property of RNAP-fork junction DNA complexes and does not require a specific sequence of spacer DNA.
Figure 3.

The effect of −35 region on the energetic consequences of mutations in −10 region in a context of: (A) C2 fork junction DNA constructs; (B) C3 fork junction DNA constructs; (C) C5 short fork junction DNA (in the presence and absence of C6); (D) C4 construct.
The most common length of the spacer DNA in bacterial promoters is 17 bp (as in λ PR promoter). Spacer DNA lengths differing from consensus 17 bp are observed (in a range from 15-19 bp). We have thus tested if the dependence of the consequences of mutations in −10 element on the strength of −35 element required a specific spacer length (and thus specific spatial disposition of these elements). We compared the effect of −11 position mutation on fork junction DNA binding in the presence and absence of consensus −35 element in fork junction DNA constructs differing in spacer DNA length. A large dependence of the energetic consequences of −11 mutation on the presence of consensus −35 element was observed only with 17 bp spacer whereas much smaller effects were observed with 15 bp and 19 bp spacers (Fig. 4). These data further supported the role of spacer DNA in the coupling between −10 and −35 element binding by RNAP and demonstrated that this coupling required specific distance (positioning) between −10 and −35 promoter elements.
Figure 4.

Spacer DNA length dependence of the effect of −11A to G mutation on RNAP binding affinity. Black and grey bars correspond the data for fork junction DNA constructs containing and lacking consensus −35 element, respectively.
In principle the effect of −35 element on recognition of −10 element by RNAP could involve conformational changes of the protein induced by −35 element binding that would be transmitted to the −10 recognition domain or conformational changes within the spacer DNA itself (or a combination of both of these effects). The data depicted in Fig. 3B-D addressed this question. Fig. 3B shows the results of binding experiments using C3 fork junction DNA in which a single nick has been introduced into the nontemplate strand of spacer DNA. This small perturbation did not significantly alter the effect of −35 element on the energetics of −10 element recognition as the pattern of the effects of mutations in −10 element for C3 fork junction DNA was very similar to C2 fork junction DNA (Fig. 3A and 3B). In the next experiment (Fig. 3C) the connection between −10 and −35 elements was completely eliminated. The data shown in Fig. 3C is for a short hairpin fork junction DNA containing only −10 element (C5) in the presence and absence of a short duplex containing consensus −35 elements (C6). Removing entirely the connection between −10 and −35 promoter elements eliminated completely the dependence of −10 binding on the presence of consensus −35 element. This result argues against a mechanism in which spacer DNA plays only a passive role of positioning −35 element for interacting with RNAP to induce a conformational change in the protein that affects −10 element recognition.
Experiments illustrated in Fig. 3C could not be performed at high saturating concentrations of C6 duplex containing −35 element due to nonspecific direct competition of C6 duplexes with C5 fork junction DNA’s at very high concentrations of C6. We have thus performed an additional experiment to assure that the loss of the communication between −10 and −35 binding events shown in Fig. 3C is not due to insufficient binding of short −35 element duplex. We prepared a construct (C4, Fig. 1) in which the short hairpin fork junction DNA containing −10 element was connected with a short duplex containing −35 element by a non-DNA long flexible linker. In such construct the long flexible linker allows independent interactions of −35 and −10 elements but the local concentration of −35 element containing duplex is defined by the length of flexible linker and is expected to be in high micromolar range [43] assuring complete saturation of −35 binding site of RNAP. This was confirmed by experiments shown in Fig. 5 where the relative affinity of C1, C2 and C4 constructs was compared. While the affinity of C4 fork junction DNA was lower compared with C1 and C2 fork junction DNA’s (as expected), relative affinity of C4 construct containing the duplex with consensus −35 element was higher compared to the construct containing duplex with the anticonsensus −35 by roughly the same amount as observed with C1 or C2 fork junction DNA’s. This demonstrated that −35 element containing duplex was bound to its binding site on RNAP in RNAP-C4 complex.
Figure 5.

Relative affinity of the C1, C2 and C4 constructs for binding RNAP holoenzyme. The ratio of the binding constant for a given construct to a binding constant for C2(−10cons, −35cons) is plotted.
The data summarized in Fig. 3 indicated an active role of spacer promoter DNA in the coupling between −10 and −35 elements. The simplest mechanism by which spacer DNA could play such role would be if the binding of RNAP to fork junction DNA containing −10 and −35 elements produced conformational changes in the spacer DNA, the energetics of which would be either directly used to couple −10 and −35 recognition or indirectly used to drive conformational change in the protein. We therefore designed an experiment to test if deformations in the spacer DNA accompanying formation of the open complex could be indeed observed. To accomplish this we prepared two λ PR promoter constructs (−74 to +1) containing consensus −35 and −10 elements in which fluorescence probes were incorporated to allow real-time monitoring of the melting of promoter DNA (Fig. 6A) and the conformation of spacer DNA (Fig. 6B). In order to monitor promoter melting, Cy3 was incorporated at position +1 of the nontemplate strand whereas nonfluorescent quencher dabcyl was incorporated into position +1 of the template strand. When promoter DNA is in the duplex form, the quencher is in close proximity to the fluorophore resulting in low fluorescence emission intensity. Melting of the promoter DNA by RNAP would increase the distance between the fluorophore and the quencher resulting in increased fluorescence emission intensity. In agreement with these expectations, addition of RNAP to a solution of C8 duplex produced a rapid (within time of mixing) increase of fluorescence followed by slower phase of fluorescence increase. In control experiment with the C8 duplex containing only the donor label (Cy3), only the rapid phase of fluorescence increase was observed (data not shown) indicating that the fluorescence increase observed in the rapid phase resulted from the change of the environment of the label resulting from RNAP-promoter complex and the slow phase of fluorescence change was due to a decrease of FRET between the donor and acceptor probes. We interpret the fast phase of fluorescence increase as a result of the formation of initial closed complex followed by a slower phase corresponding to promoter melting. The time course of the slower phase could be reasonably well described by a single exponential although double exponential analysis produced a slightly better fit (Fig. 6A).
Figure 6.

Comparison of the kinetics of the melting of promoter DNA (A) and the deformations in the spacer DNA detected by FRET (B) between fluorescence probes placed in the constructs as schematically illustrated in panels A and B, respectively. Arrows depict time points of the addition of RNAP to a cuvette containing fluorescent promoter construct. Green and red lines correspond to nonlinear fits to single and double exponential kinetic models, respectively.
In order to monitor conformational changes in the spacer DNA in real-time, a donor-acceptor FRET pair of fluorochromes (Bodipy-FL and tetramethylrhodamine) was introduced into positions −26 and −15 of nontemplate and template strands, respectively. Addition of RNAP to a solution of C7 duplex produced a rapid (within the time of mixing) increase of fluorescence of the donor (BODIPY Fl) followed by slower phase of fluorescence increase (Fig. 6B). The pattern of changes observed using the probes incorporated into the spacer was very similar to those recorded by the probes monitoring promoter melting (Fig. 6 A&B). The rate constants describing the kinetics of the slow phase in the case of C7 duplex were very similar to those determined with C8 duplex. In control experiment with the C7 duplex containing only the donor label (BODIPY Fl), the rapid phase of fluorescence increase was also observed (data not shown) indicating that the fluorescence increase observed in this phase resulted from a change of the environment of the label resulting from RNAP-promoter complex formation rather then from a change of FRET between the donor-acceptor probes. While the slow phase was observed as well with donor-only C7 duplex, its amplitude was smaller indicating that at least some of the fluorescence increase in this phase was due to structural changes in the spacer resulting in a change of FRET between the donor and the acceptor. Taken together, we interpret the results shown in Fig. 6 as showing that conformational changes of spacer DNA occur concomitant with melting of promoter DNA containing consensus −10 and −35 elements.
4. Discussion
The data described here demonstrated that RNAP interactions with −10 and −35 promoter elements were not independent events and that spacer DNA played an active role in integrating the functional consequences of RNA polymerase contacts with −10 and −35 promoter elements. Nucleotide sequence of the spacer is not conserved (although some nonrandom distribution of bases in the spacer was noted [44]) indicating that whatever the function of the spacer might be, it is not likely to involve sequence specific protein-DNA interactions. It was observed that transcriptional activity of a promoter can be very significantly affected by changing the sequence of the spacer [45]. Fig. 7 schematically depicts possible roles that spacer DNA could play in the formation of the open complex. The simplest role (Fig. 7a) could be a passive role to correctly position the two conserved promoter regions so they can be efficiently recognized by RNAP. A variant of this role (Fig. 7b) could involve allosteric conformational changes in the σ subunit induced by −35 promoter element contact that could affect interactions at −10 element (and thus could affect steps beyond promoter recognition such as promoter melting). Fig. 7c illustrate a scenario in which initial binding of RNAP to the promoter induces spacer deformation that is relieved during promoter melting. The data reported here are most consistent with the spacer role illustrated in Fig. 7d in which spacer DNA undergoes a deformation during formation of the open complex, although scenarios depicted in Fig. 7 are not mutually exclusive.
Figure 7.

Potential roles of spacer DNA In “a” spacer DNA plays a passive role to position −10 and −35 element to allow recruitment of RNAP. In “b” the −35 contact induces an allosteric change in the protein that effects interactions of polymerase with −10 element. In “c” spacer DNA undergoes deformations triggered by initial RNAP interactions with −10 and −35 elements. In “d” spacer DNA undergoes deformations concomitant with promoter melting.
What could be the functional role of these spacer deformations? Spacer deformations have been previously proposed in so called “twist and melt” hypothesis in which an interaction of σ subunit with −10 and −35 elements was suggested to produce a twist (or other deformation) in the spacer DNA connecting −10 and −35 promoter elements [1, 46]. The energy stored in this DNA deformation would be then somehow used by RNAP to facilitate promoter melting (scenario “c” in Fig. 7). Our data indicate that spacer deformation occurs concomitant with promoter melting rather then preceding promoter melting in contrast what would be expected from “twist and melt” hypothesis. While our results do not preclude the role of spacer deformations in promoter melting, possible roles of spacer deformations in steps beyond promoter melting should be considered. For example, the energy stored in deformed spacer DNA upon promoter melting could be utilized in promoter escape step. In this step the favorable contacts of RNAP with promoter DNA that were used to drive promoter melting have to be broken to allow transition to processively elongating polymerase. While spacer deformations can not be absolutely essential for promoter melting (since short promoter fragments containing only −10 promoter element are melted by RNAP [31]), they could play role in increasing the rate of melting of promoter DNA since −10 only short DNA’s are melted by RNAP only very slowly. Experiments to dissect possible functional roles of spacer DNA deformations in promoter melting and(or) promoter escape are currently under way in our laboratory.
Spacer deformation in the complex of RNAP with fork junction DNA constructs could explain our paradoxical observations that, for example, a mutation of −11 adenine known to be important for promoter function had little effect on binding affinity of fork junction DNA when consensus −35 element is present. This could be explained by assuming that strong −35 and −10 elements are necessary to induce spacer DNA deformation. In such case a mutation at position −11 could on one hand decrease the binding affinity by removing a favorable direct contact and on the other hand if this contact is critical for the deformation of the spacer DNA, mutating −11A would also remove the energetic cost of such deformation. Thus, the net effect could be a very small decrease in affinity. When consensus −35 element is absent, region 4 domain of σ would bind DNA in nonspecific manner allowing sliding of the domain along DNA duplex. With such flexibility of positioning region 4 of σ there would be no deformation of the spacer (and the energetic costs associated with it) and thus mutation of −11A would produce a large decrease in binding affinity of fork junction DNA.
Acknowledgements
This work was supported by NIH Grant GM50514. This manuscript has been submitted in celebration of 25th Gibbs Conference that has been a continuous source of enlightenment and stimulation for applying thermodynamics to study biological mechanisms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFRENCES
- [1].Fiedler U, Timmers H.T. Marc. Peeling by binding or twisting by cranking: models for promoter opening and transcription initiation by RNA polymerase II. Bioessays. 2000;22:316–326. doi: 10.1002/(SICI)1521-1878(200004)22:4<316::AID-BIES2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- [2].Record. M.T. R, Jr, W.S., Craig ML, McQuade KL, Schlax PJ. In: Escherichia coli and Salmonella: cellular and molecular biology. Neidhardt FC, Curtis R III, Ingraham JL, Lin ECC, Low KR, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. ASM Press; Washington, DC: 1996. pp. 792–820. [Google Scholar]
- [3].Davis CA, Bingman CA, Landick R, Record MT, Jr., Saecker RM. Real-time footprinting of DNA in the first kinetically significant intermediate in open complex formation by Escherichia coli RNA polymerase. Proc Natl Acad Sci U S A. 2007;104:7833–7838. doi: 10.1073/pnas.0609888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rogozina A, Zaychikov E, Buckle M, Heumann H, Sclavi B. DNA melting by RNA polymerase at the T7A1 promoter precedes the rate-limiting step at 37 degrees C and results in the accumulation of an off-pathway intermediate. Nucleic Acids Res. 2009;37:5390–5404. doi: 10.1093/nar/gkp560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sclavi B, Zaychikov E, Rogozina A, Walther F, Buckle M, Heumann H. Real-time characterization of intermediates in the pathway to open complex formation by Escherichia coli RNA polymerase at the T7A1 promoter. Proc Natl Acad Sci U S A. 2005;102:4706–4711. doi: 10.1073/pnas.0408218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Haugen SP, Ross W, Gourse RL. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol. 2008;6:507–519. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saecker RM, Tsodikov OV, McQuade KL, Schlax PE, Jr., Capp MW, Record MT., Jr. Kinetic studies and structural models of the association of E. coli sigma(70) RNA polymerase with the lambdaP(R) promoter: large scale conformational changes in forming the kinetically significant intermediates. J Mol Biol. 2002;319:649–671. doi: 10.1016/S0022-2836(02)00293-0. [DOI] [PubMed] [Google Scholar]
- [8].Gries TJ, Kontur WS, Capp MW, Saecker RM, Record MT., Jr. One-step DNA melting in the RNA polymerase cleft opens the initiation bubble to form an unstable open complex. Proc Natl Acad Sci U S A. 2010;107:10418–10423. doi: 10.1073/pnas.1000967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen J, Darst SA, Thirumalai D. Promoter melting triggered by bacterial RNA polymerase occurs in three steps. Proc Natl Acad Sci U S A. 2010;107:12523–12528. doi: 10.1073/pnas.1003533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guo Y, Gralla JD. Promoter opening via a DNA fork junction binding activity. Proc Natl Acad Sci U S A. 1998;95:11655–11660. doi: 10.1073/pnas.95.20.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McClure WR. Mechanism and control of transcription initiation in prokaryotes. Annu Rev Biochem. 1985;54:171–204. doi: 10.1146/annurev.bi.54.070185.001131. [DOI] [PubMed] [Google Scholar]
- [12].deHaseth PL, Zupancic ML, Record MT., Jr. RNA polymerase-promoter interactions: the comings and goings of RNA polymerase. J Bacteriol. 1998;180:3019–3025. doi: 10.1128/jb.180.12.3019-3025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- [14].Dombroski AJ, Walter WA, Gross CA. Amino-terminal amino acids modulate sigma-factor DNA-binding activity. Genes Dev. 1993;7:2446–2455. doi: 10.1101/gad.7.12a.2446. [DOI] [PubMed] [Google Scholar]
- [15].Dombroski AJ, Walter WA, Gross CA. The role of the sigma subunit in promoter recognition by RNA polymerase. Cell Mol Biol Res. 1993;39:311–317. [PubMed] [Google Scholar]
- [16].Dombroski AJ, Walter WA, Record MT, Jr., Siegele DA, Gross CA. Polypeptides containing highly conserved regions of transcription initiation factor sigma 70 exhibit specificity of binding to promoter DNA. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- [17].Callaci S, Heyduk E, Heyduk T. Conformational changes of Escherichia coli RNA polymerase sigma70 factor induced by binding to the core enzyme. J Biol Chem. 1998;273:32995–33001. doi: 10.1074/jbc.273.49.32995. [DOI] [PubMed] [Google Scholar]
- [18].Young BA, Anthony LC, Gruber TM, Arthur TM, Heyduk E, Lu CZ, Sharp MM, Heyduk T, Burgess RR, Gross CA. A coiled-coil from the RNA polymerase beta’ subunit allosterically induces selective nontemplate strand binding by sigma(70) Cell. 2001;105:935–944. doi: 10.1016/s0092-8674(01)00398-1. [DOI] [PubMed] [Google Scholar]
- [19].Callaci S, Heyduk E, Heyduk T. Core RNA polymerase from E. coli induces a major change in the domain arrangement of the sigma 70 subunit. Mol Cell. 1999;3:229–238. doi: 10.1016/s1097-2765(00)80313-5. [DOI] [PubMed] [Google Scholar]
- [20].Mekler V, Pavlova O, Severinov K. Interaction of Escherichia coli RNA polymerase sigma70 subunit with promoter elements in the context of free sigma70, RNA polymerase holoenzyme, and the beta’-sigma70 complex. J Biol Chem. 2011;286:270–279. doi: 10.1074/jbc.M110.174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- [22].Huang X, Lopez de Saro FJ, Helmann JD. sigma factor mutations affecting the sequence-selective interaction of RNA polymerase with −10 region single-stranded DNA. Nucleic Acids Res. 1997;25:2603–2609. doi: 10.1093/nar/25.13.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Callaci S, Heyduk T. Conformation and DNA binding properties of a single-stranded DNA binding region of sigma 70 subunit from Escherichia coli RNA polymerase are modulated by an interaction with the core enzyme. Biochemistry. 1998;37:3312–3320. doi: 10.1021/bi972041m. [DOI] [PubMed] [Google Scholar]
- [24].Helmann JD, deHaseth PL. Protein-nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry. 1999;38:5959–5967. doi: 10.1021/bi990206g. [DOI] [PubMed] [Google Scholar]
- [25].Chen YF, Helmann JD. DNA-melting at the Bacillus subtilis flagellin promoter nucleates near −10 and expands unidirectionally. J Mol Biol. 1997;267:47–59. doi: 10.1006/jmbi.1996.0853. [DOI] [PubMed] [Google Scholar]
- [26].Matlock DL, Heyduk T. Sequence determinants for the recognition of the fork junction DNA containing the −10 region of promoter DNA by E. coli RNA polymerase. Biochemistry. 2000;39:12274–12283. doi: 10.1021/bi001433h. [DOI] [PubMed] [Google Scholar]
- [27].Schroeder LA, Karpen ME, deHaseth PL. Threonine 429 of Escherichia coli sigma 70 is a key participant in promoter DNA melting by RNA polymerase. J Mol Biol. 2008;376:153–165. doi: 10.1016/j.jmb.2007.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tsujikawa L, Strainic MG, Watrob H, Barkley MD, DeHaseth PL. RNA polymerase alters the mobility of an A-residue crucial to polymerase-induced melting of promoter DNA. Biochemistry. 2002;41:15334–15341. doi: 10.1021/bi026539m. [DOI] [PubMed] [Google Scholar]
- [29].Heyduk E, Kuznedelov K, Severinov K, Heyduk T. A consensus adenine at position −11 of the nontemplate strand of bacterial promoter is important for nucleation of promoter melting. J Biol Chem. 2006 doi: 10.1074/jbc.M601364200. [DOI] [PubMed] [Google Scholar]
- [30].Heyduk E, Kuznedelov K, Severinov K, Heyduk T. A consensus adenine at position −11 of the nontemplate strand of bacterial promoter is important for nucleation of promoter melting. J Biol Chem. 2006;281:12362–12369. doi: 10.1074/jbc.M601364200. [DOI] [PubMed] [Google Scholar]
- [31].Niedziela-Majka A, Heyduk T. Escherichia coli RNA polymerase contacts outside the −10 promoter element are not essential for promoter melting. J Biol Chem. 2005;280:38219–38227. doi: 10.1074/jbc.M507984200. [DOI] [PubMed] [Google Scholar]
- [32].Young BA, Gruber TM, Gross CA. Minimal machinery of RNA polymerase holoenzyme sufficient for promoter melting. Science. 2004;303:1382–1384. doi: 10.1126/science.1092462. [DOI] [PubMed] [Google Scholar]
- [33].Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- [34].Murakami KS, Masuda S, Darst SA. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 A resolution. Science. 2002;296:1280–1284. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
- [35].Heyduk E, Heyduk T. Architecture of a complex between the sigma70 subunit of Escherichia coli RNA polymerase and the nontemplate strand oligonucleotide. Luminescence resonance energy transfer study. J Biol Chem. 1999;274:3315–3322. doi: 10.1074/jbc.274.6.3315. [DOI] [PubMed] [Google Scholar]
- [36].Burgess RR, Jendrisak JJ. A procedure for the rapid, large-scall purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- [37].Polyakov A, Severinova E, Darst SA. Three-dimensional structure of E. coli core RNA polymerase: promoter binding and elongation conformations of the enzyme. Cell. 1995;83:365–373. doi: 10.1016/0092-8674(95)90114-0. [DOI] [PubMed] [Google Scholar]
- [38].Hager DA, Jin DJ, Burgess RR. Use of Mono Q high-resolution ion-exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- [39].Fenton MS, Gralla JD. Effect of DNA bases and backbone on sigma70 holoenzyme binding and isomerization using fork junction probes. Nucleic Acids Res. 2003;31:2745–2750. doi: 10.1093/nar/gkg400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fenton MS, Gralla JD. Roles for inhibitory interactions in the use of the −10 promoter element by sigma 70 holoenzyme. J Biol Chem. 2003;278:39669–39674. doi: 10.1074/jbc.M307412200. [DOI] [PubMed] [Google Scholar]
- [41].Fenton MS, Lee SJ, Gralla JD. Escherichia coli promoter opening and −10 recognition: mutational analysis of sigma70. Embo J. 2000;19:1130–1137. doi: 10.1093/emboj/19.5.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Selvin PR, Hearst JE. Luminescence energy transfer using a terbium chelate: improvements on fluorescence energy transfer. Proc Natl Acad Sci U S A. 1994;91:10024–10028. doi: 10.1073/pnas.91.21.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tian L, Heyduk T. Bivalent ligands with long nanometer-scale flexible linkers. Biochemistry. 2009;48:264–275. doi: 10.1021/bi801630b. [DOI] [PubMed] [Google Scholar]
- [44].Beutel BA, Record MT., Jr. E. coli promoter spacer regions contain nonrandom sequences which correlate to spacer length. Nucleic Acids Res. 1990;18:3597–3603. doi: 10.1093/nar/18.12.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu M, Tolstorukov M, Zhurkin V, Garges S, Adhya S. A mutant spacer sequence between −35 and −10 elements makes the Plac promoter hyperactive and cAMP receptor protein-independent. Proc Natl Acad Sci U S A. 2004;101:6911–6916. doi: 10.1073/pnas.0401929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].deHaseth PL, Helmann JD. Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA. Mol Microbiol. 1995;16:817–824. doi: 10.1111/j.1365-2958.1995.tb02309.x. [DOI] [PubMed] [Google Scholar]