

Abstract
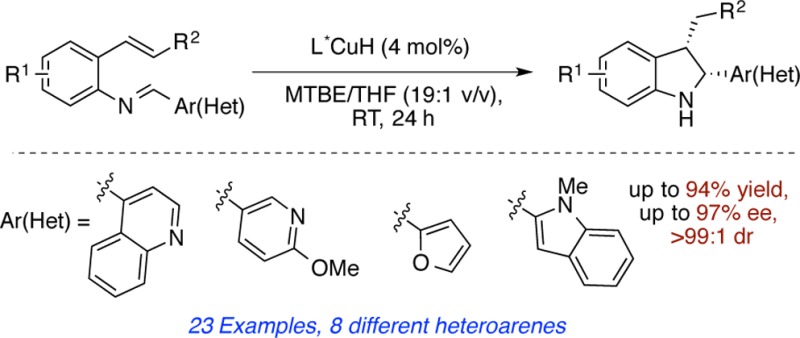
A diastereo- and enantioselective CuH-catalyzed method for the preparation of highly functionalized indolines is reported. The mild reaction conditions and high degree of functional group compatibility as demonstrated with substrates bearing heterocycles, olefins, and substituted aromatic groups, renders this technique highly valuable for the synthesis of a variety of cis-2,3-disubstituted indolines in high yield and enantioeselectivity.
Optically active 2,3-disubstituted indolines are of considerable interest in both synthetic and medicinal chemistry due to the numerous natural products and pharmaceuticals whose structures incorporate this heterocyclic framework.1 They also serve as key building blocks for drug discovery, as exemplified in various natural product-like chemical libraries based on the indoline framework.2
Consequently, a wide range of strategies have been developed for the asymmetric synthesis of these structures.3 Among these methods, the Brønsted acid-activated asymmetric hydrogenation of functionalized indoles is perhaps the most important strategy to their synthesis.4 Despite its attractiveness, the need to employ acidic reaction conditions, high pressure, and the lack of broad functional group compatibility manifested, have prevented the more widespread application of this approach. Other notable methods, such as the kinetic resolution of indolines,5 the Pd-catalyzed asymmetric intramolecular coupling reactions,6 tandem AN/SNAr processes,7 and asymmetric sparteine-mediated reactions,8 have also been developed. Despite these advances, the harsh reaction conditions,6,8 multistep synthesis,5 and modest to poor yields,6a,7 limits their synthetic applications. To date, no direct asymmetric approach is available to access 2,3-disubstituted indolines bearing a wide variety of functional groups. In particular, incorporation of pharmaceutically important heterocycles (e.g., indoles, quinolines, pyridines) with indolines in an asymmetric fashion is not well-known. Herein, we report a straightforward approach to the synthesis of 2,3-disubstituted indolines in high diastereo- and enantioselectivities through a CuH-based strategy. This protocol tolerates a broad range of functional groups, including heteroarenes and olefins.
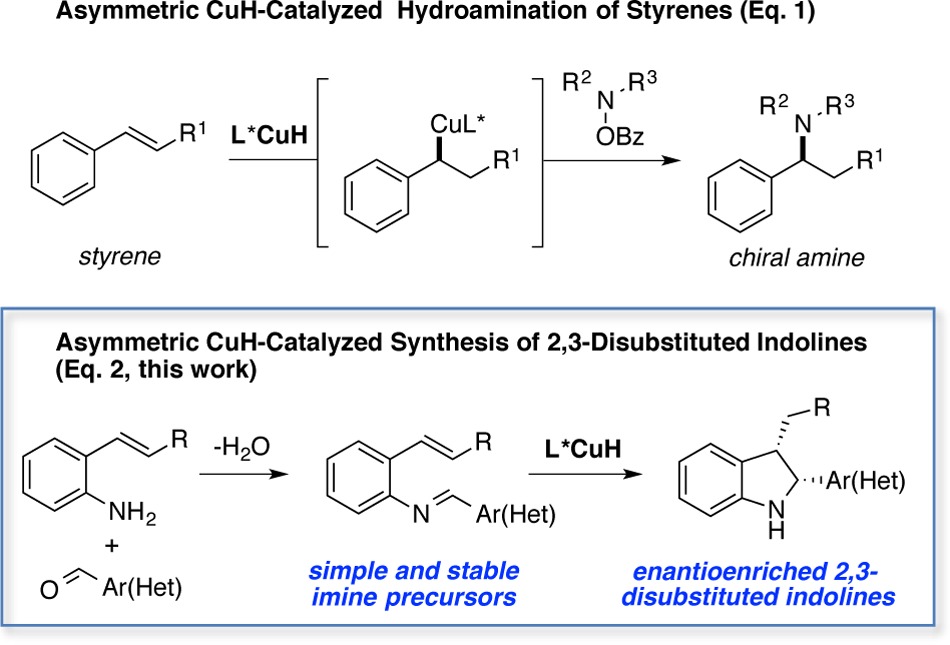
Recently, we and Miura and Hirano have independently reported the enantioselective CuH-catalyzed Markovnikov hydroamination of styrenes.9 Bearing in mind that these reactions proceed via organocopper intermediates that undergo amination with electrophilic hydroxylamine esters (eq 1), we questioned whether these species could be trapped intramolecularly with tethered imine electrophiles. In this manner a rapid and synthetically valuable approach to enantioenriched indolines with two contiguous chiral centers could be realized in two steps from readily available aminostyrene and aromatic aldehyde derivatives (eq 2).
 |
1 |
A scheme of our proposed strategy is illustrated in Figure 1. Based on literature precedent for related CuH-based systems,9−14 the active L*CuH species A could be formed by reacting Cu(OAc)2 with a chiral ligand and a stoichiometric amount of a hydrosilane.9a,10a,10b The olefinic moiety of 1a could then readily insert into Cu–H bond of A. Based on previous results we would expect this to occur in a highly enantioselective and in a Markovnikov fashion to form the alkylcopper intermediate B.9,10c−10f Intermediate B could then undergo an unprecedented cyclization with the adjacent imine to form C.11 Subsequent protonation of C with t-BuOH would then provide the desired indoline 2a, and generate [L*CuOt-Bu] species D, which is an ideal precursor for rapid regeneration of the active L*CuH A.12 In order for this approach to be successful, it is necessary that A react chemoselectively with the olefin over the imine.13 It is also necessary that the cyclization (B to C) is faster than the undesired protonation of B to 3.14
Figure 1.
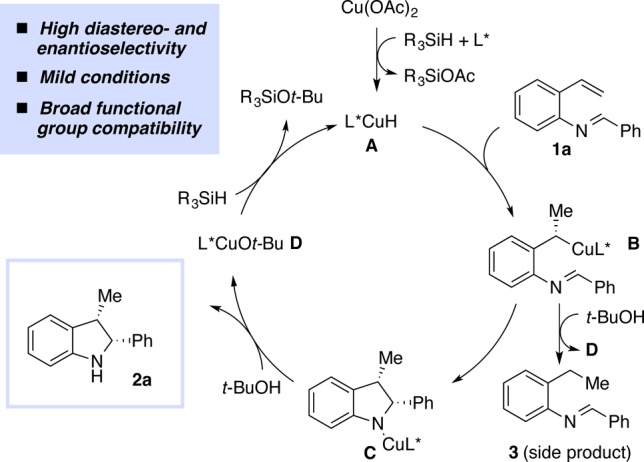
Proposed catalytic cycle for CuH-catalyzed synthesis of 2,3-disubstituted indolines.
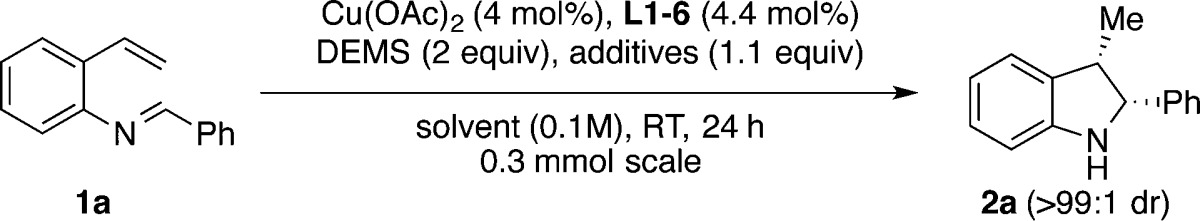
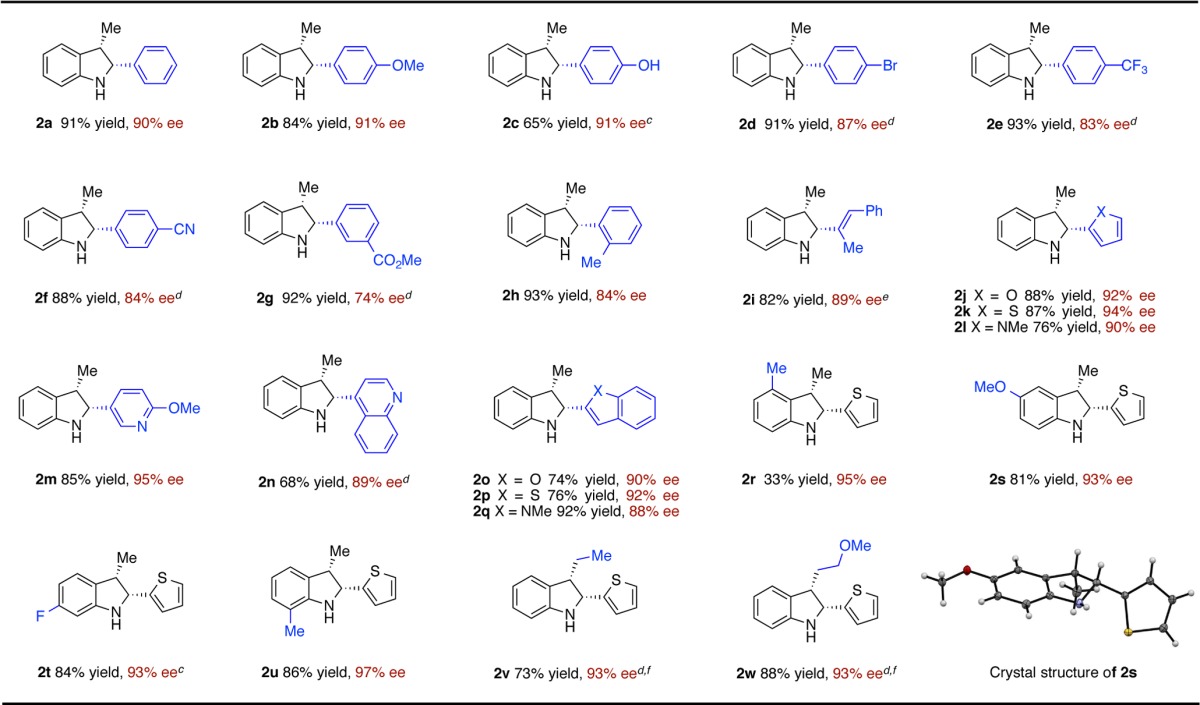
To test the viability of our proposed method, we examined styrene 1a as our model substrate. We begun using our reported conditions for the hydroamination of styrene9a (Table 1, entry 1). The reaction afforded the desired indoline 2a in 9% yield and 27% ee, as a single cis-diastereomer. However, when a stoichiometric amount of t-BuOH was added, the yield was improved to 67% yield, while the enantioselectivity was unchanged (entry 2). We attributed the improvement in catalyst turnover to the protonation of [L*Cu–N-indoline] C with t-BuOH (Figure 1), bypassing the slow transmetalation between C and R3Si–H needed for regeneration of L*CuH.15 We next examined several commercially available chiral bisphosphine ligands (entries 2–7), and we found that (S,S)-Ph-BPE proved to be best in terms of reactivity and enantioselectivity (84% yield, 84% ee, entry 7). Screening of solvents revealed that a mixture of MTBE/THF (19:1) was the best choice, leading to an enhanced yield and enantioselectivity (92% yield, 90% ee, entry 8).
Table 1. Reaction Optimization.
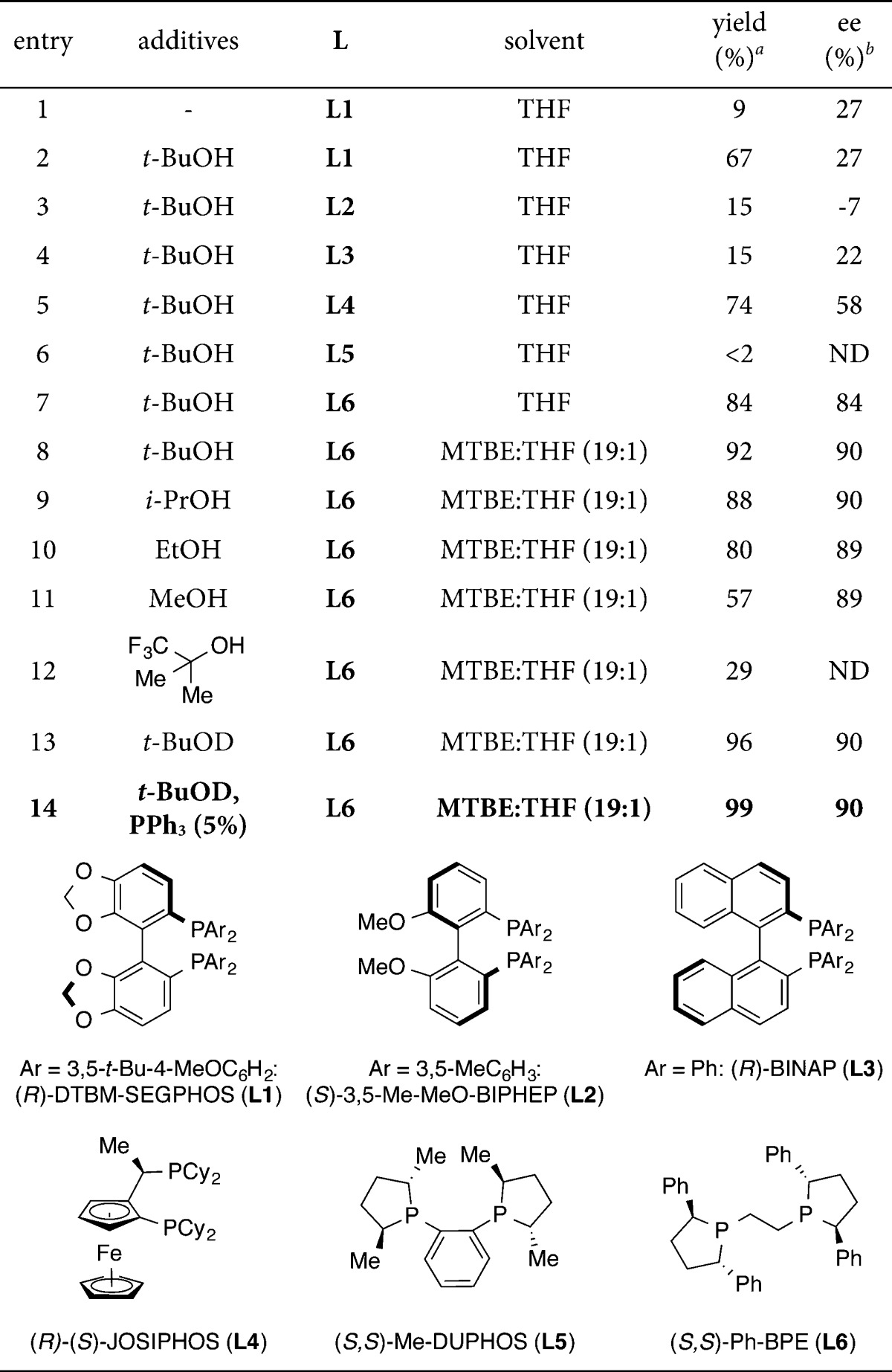
Yields were determined by 1H NMR analysis of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard.
Enantioselectivites were determined by chiral HPLC analysis. ND = not determined. DEMS = (EtO)2MeSiH.
We found that the choice of alcohol was important for the yield. For example, when using t-BuOH (entry 8) the reduced styrene side product 3 could be detected in 3% yield. Examining the use of i-PrOH, EtOH, MeOH, and 2-trifluoromethyl-2-propanol, afforded the desired product 2a in lower yields (29–88%, entries 9–12), while observing an increase in yield of the side product 3 in 5%, 15%, 36%, and 55%, respectively. When t-BuOD was used in lieu of t-BuOH, only a trace amount of 3 (<1%) could be detected, and a slight increase in yield of 2a was obtained (96%, entry 13) compared to t-BuOH (92%, entry 8). We attribute this to a deuterium isotope effect; i.e., the protonolysis of B to 3 is slower with t-BuOD than with t-BuOH.16
Lipshutz, in related CuH-catalyzed systems, has shown that addition of triphenylphosphine as a secondary ligand, significantly improves the catalyst turnover number.17 Applying this strategy to our method, further improved the yield that could be achieved without any erosion in enantioselectivity (entry 14).
With the optimized conditions in hand, we examined the reaction scope using a variety of stable 2-alkenylimine precursors 1a–w. These were obtained in good to excellent yields from readily available 2-alkenylanilines and aromatic aldehydes (Table 2). Generally, the reaction displayed good functional group tolerance, and in all cases (except for product 2i), the resulting indolines were obtained exclusively as the cis-diastereomers.18 The reason for the observed diastereoselectivitiy is not clear and remains to be elucidated.
Table 2. Substrate Scopea,b.
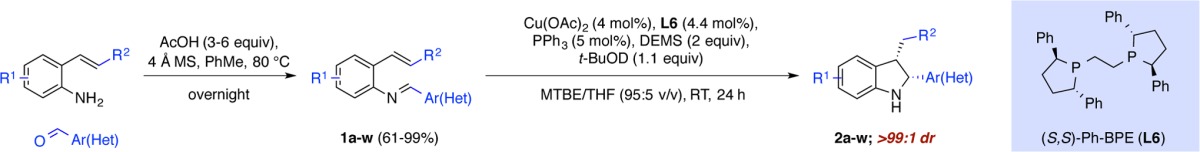
Isolated yield on 1 mmol scale (average of two runs).
Enantioselectivities were determined by chiral HPLC analysis.
3 equiv of DEMS was used.
Reaction performed in the absence of PPh3.
Isolated as a 11:1 diastereoselective ratio.
Cu(OAc)2 (8 mol%) and (S,S)-Ph-BPE (8.8 mol%) were used.
It was found that electron-neutral and -rich substitutents on the phenyl ring could be transformed, thus giving cis-indolines 2a–c,h in high yields and in good to excellent enantioselectivities. Electron-deficient substituents (2d–g) also gave satisfactory results but with slightly lower levels of enantioselectivity. For these examples, the yield was higher if the reaction was run in the absence of triphenylphosphine. The applicability of this method to access indolines bearing a phenol (2c), aryl bromide (2d), benzonitrile (2f), or methyl benzoate (2g) group is important, as these functional groups can be used as synthetic handles for further transformations. Moreover, the compatibility of a vinyl group, which is a valuable feature that can be problematic in methods using asymmetric hydrogenation,4 furnishes the resultant indoline 2i in good yield and enantioselectivity. This method also tolerates a variety of hetereoarenes. As illustrated in Table 2, five-membered (2j–l), six-membered (2m), and fused heterocycles (2n–q) could all be accommodated in good yields with excellent levels of enantioselectivity.
We next turned our attention to substrates bearing substituents on the aryl ring of styrene. We found that excellent enantioselectivites were obtained for indolines 2r–u.19 However, indoline 2r, with a methyl group at the 4-position displayed lower reactivity (33% yield), with 30% of the reduced imine could be also detected. We rationalize that the CuH insertion step is slower due to steric encumbrance of the methyl group in the 4-position.
We also briefly examined the extension of this method to β-substituted styrenes and found that indolines 2v,w could be obtained in high yield and excellent enantioselectivities. In these cases, 8 mol% catalyst loading was needed and best results were observed in the absence of triphenylphosphine.
In conclusion, we have developed a mild, Cu-catalyzed strategy for the highly diastereo- and enantioselective synthesis of a broad range of cis-2,3-disubstituted indolines. Due to the generality of the method and mild conditions employed, we believe this method will see considerable use in both academic and industrial settings.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award no. GM46059. E.A. thanks the Danish Council for Independent Research, Technology and Production Sciences for a postdoctoral fellowship. We thank Dr. Michael Pirnot and Dr. Yiming Wang for their advice on the preparation of this manuscript.
Supporting Information Available
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For selected examples, see:; a Crich D.; Banerjee A. Acc. Chem. Res. 2007, 40, 151. [DOI] [PubMed] [Google Scholar]; b Triggle D. J.; Mitchell J. M.; Filler R. CNS Drug Reviews 1998, 4, 87. [Google Scholar]; c Deutsch H. F.; Evenson M. A.; Drescher P.; Sparwasser C.; Madsen P. O. J. Pharm. Biomed. Anal. 1994, 12, 1283. [DOI] [PubMed] [Google Scholar]; d Silvestri R. J. Med. Chem. 2013, 56, 625. [DOI] [PubMed] [Google Scholar]; e Greig N. H.; Pei X.-F.; Soncrant T. T.; Ingram D. K.; Brossi A. Med. Res. Rev. 1995, 15, 3. [DOI] [PubMed] [Google Scholar]; f Neelamegam R.; Hellenbrand T.; Schroeder F. A.; Wang C.; Hooker J. M. J. Med. Chem. 2014, 57, 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Podoll J. D.; Liu Y.; Chang L.; Walls S.; Wang W.; Wang X. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 15573. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Poondra R. R.; Kumar N. N.; Nijian K.; Prakesch M.; Slater-Campagna V.; Reayi A.; Reddy T. P.; Choudhry A.; Barnes M. L.; Leek D. M.; Daroszewska M.; Lougheed C.; Xu B.; Schapira M.; Alaoui-Jamali M. A.; Arya P. J. Comb. Chem. 2009, 11, 303. [DOI] [PubMed] [Google Scholar]; c Gan Z. P.; Reddy T.; Quevillon S.; Couve-Bonnaire S.; Arya P. Angew. Chem., Int. Ed. 2005, 44, 1366. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Liu D.; Zhao G.; Xiang L. Eur. J. Org. Chem. 2010, 3975. [Google Scholar]; b Zhang D.; Song H.; Qin Y. Acc. Chem. Res. 2011, 44, 447. [DOI] [PubMed] [Google Scholar]
- a Wang D.-S.; Chen Q.-A.; Li W.; Yu C.-B.; Zhou Y.; Zhang X. J. Am. Chem. Soc. 2010, 132, 8909. [DOI] [PubMed] [Google Scholar]; b Duan Y.; Chen M.-W.; Chen Q.-A.; Yu C.-B.; Zhou Y.-G. Org. Biomol. Chem. 2012, 10, 1235. [DOI] [PubMed] [Google Scholar]; c Duan Y.; Chen M.-W.; Ye Z.-S.; Wang D.-S.; Chen Q.-A.; Zhou Y.-G. Chem.—Eur. J. 2011, 17, 7193. [DOI] [PubMed] [Google Scholar]; d Wang D.-S.; Tang J.; Zhou Y.-G.; Chen M.-W.; Yu C.-B.; Duan Y.; Jiang G.-F. Chem. Sci. 2011, 2, 803. [Google Scholar]; e Li C.; Chen J.; Fu G.; Liu D.; Liu Y.; Zhang W. Tetrahedron 2013, 69, 6839. [Google Scholar]; f Duan Y.; Li L.; Chen M.-W.; Yu C.-B.; Fan H.-J.; Zhou Y.-G. J. Am. Chem. Soc. 2014, 136, 7688. [DOI] [PubMed] [Google Scholar]; g Xiao Y.-C.; Wang C.; Yao Y.; Sun J.; Chen Y.-C. Angew, Chem. Int. Ed. 2011, 50, 10661. [DOI] [PubMed] [Google Scholar]
- a Arp F. O.; Fu G. C. J. Am. Chem. Soc. 2006, 128, 14264. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lopez-Iglesias M.; Busto E.; Gotor V.; Gotor-Fernández V. J. Org. Chem. 2012, 77, 8049. [DOI] [PubMed] [Google Scholar]; c Saito K.; Shibata Y.; Yamanaka M.; Akiyama T. J. Am. Chem. Soc. 2013, 135, 11740. [DOI] [PubMed] [Google Scholar]
- a Katayev D.; Nakanishi M.; Bürgi T.; Kündig P. E. Chem. Sci. 2012, 3, 1422. [Google Scholar]; b Saget T.; Lemouzy S. J.; Cramer N. Angew. Chem., Int. Ed. 2012, 51, 2238. [DOI] [PubMed] [Google Scholar]
- Ruano J. L. G.; Parra A.; Marcos V.; Pozo C. D.; Catalán S.; Monteagudo S.; Fustero S.; Poveda A. J. Am. Chem. Soc. 2009, 131, 9432. [DOI] [PubMed] [Google Scholar]
- Kang K. H.; Do J.; Park Y. S. J. Org. Chem. 2012, 77, 808. [DOI] [PubMed] [Google Scholar]
- a Zhu S.; Niljianskul N.; Buchwald S. L. J. Am. Chem. Soc. 2013, 135, 15746. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miki Y.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2013, 52, 10830. [DOI] [PubMed] [Google Scholar]
- a Zhu S.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 15913. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Niljianskul N.; Zhu S.; Buchwald S. L. Angew. Chem., Int. Ed. 2014, 54, 1638. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related asymmetric Cu-catalyzed Markovnikov hydroboration and aminoboration reactions of styrenes, see:; c Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 3160. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Matsuda N.; Hirano K.; Satoh T.; Miura M. J. Am. Chem. Soc. 2013, 135, 4934. [DOI] [PubMed] [Google Scholar]; e Grigg D. R.; Hoveln R. V.; Schomaker J. M. J. Am. Chem. Soc. 2012, 134, 16131. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062. [DOI] [PubMed] [Google Scholar]; For reviews dealing with CuH-catalyzed reactions, see:; g Deutsch C.; Krause N.; Lipshutz B. H. Chem. Rev. 2008, 108, 2916.and references therein. [DOI] [PubMed] [Google Scholar]; h Hesp K. D. Angew. Chem., Int. Ed. 2014, 53, 2034. [DOI] [PubMed] [Google Scholar]; i Fujihara T.; Semba K.; Terao J.; Tsuji Y. Catal. Sci. Technol. 2014, 4, 1699. [Google Scholar]
- Although no examples of an analogous intramolecular coupling reaction have been reported, an asymmetric intermolecular CuH-catalyzed coupling between vinylazaarenes and (hetero)aryl N-Boc imines has recently been developed;:Choi B.; Saxena A.; Smith J. J.; Churchill G. H.; Lam H. W. Synlett 2015, 26, 350. [Google Scholar]
- For examples of t-BuOH-accelerated CuH-catalyzed reactions, see:; a Chen J.-X.; Daeuble J. F.; Brestensky D. M.; Stryker J. M. Tetrahedron 2000, 56, 2153. [Google Scholar]; b Chen J.-X.; Daeuble J. F.; Stryker J. M. Tetrahedron 2000, 56, 2789. [Google Scholar]; c Hughes G.; Kimura M.; Buchwald S. L. J. Am. Chem. Soc. 2003, 11253. [DOI] [PubMed] [Google Scholar]; d Lipshutz B. H.; Servesko J. M.; Taft B. R. J. Am. Chem. Soc. 2004, 126, 8352. [DOI] [PubMed] [Google Scholar]; e Rainka M. P.; Aye Y.; Buchwald S. L. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5821. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lam H. W.; Saxena A.; Rupnicki L. J. Am. Chem. Soc. 2009, 131, 10386. [DOI] [PubMed] [Google Scholar]
- For CuH-catalyzed hydrosilylation of imines, see:Lipshutz B. H.; Shimizu H. Angew. Chem., Int. Ed. 2004, 43, 2228. [DOI] [PubMed] [Google Scholar]
- For protonation of carbon–copper bonds with alcohols in related CuH-catalyzed reactions, see:; a Semba K.; Fujihara T.; Xu T.; Terao J.; Tsuji Y. Adv. Synth. Catal. 2012, 354, 1542. [Google Scholar]; b Whittaker A. M.; Lalic G. Org. Lett. 2013, 15, 1112. [DOI] [PubMed] [Google Scholar]; c Shi S.-L.; Buchwald S. L. Nat. Chem. 2015, 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a conceptually related process, see:Verdaguer X.; Lange U. E. W.; Buchwald S. L. Angew. Chem., Int. Ed. 1998, 37, 1103. [DOI] [PubMed] [Google Scholar]
- Hoveyda, in related Cu-catalyzed processes, has shown that protonation of carbon–copper intermediates with MeOD-d4 is significantly slower than the reaction with MeOH. Thus, kH/kD = 2.7–3.6 have been measured. See:Jang W.; Zhugralin A. R.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 7859. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Noson K.; Chrisman W.; Lower A. J. Am. Chem. Soc. 2003, 125, 8779. [DOI] [PubMed] [Google Scholar]; See also ref (10a).
- This technique could also be applied on crude imines directly after solvent evaporation, as exemplified on 1a, although the yield was somewhat lower (74% isolated yield, 90% ee). Unfortunately, no product was observed on aliphatic imines, as demonstrated on (E)-2-methyl-N-(2-vinylphenyl)propan-1-imine.
- The absolute configuration of 2s was determined to be 2R,3S by X-ray analysis (see Table 2). Consequently, those of the other products were assigned by analogy.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.