

Specific recognition of centromere-specific histone variant CENP-A-containing chromatin by CENP-N is an essential process in the assembly of the kinetochore complex at centromeres prior to mammalian cell division. Fang et al. show that a CENP-A-specific RG loop (Arg80/Gly81) plays an essential and dual regulatory role in this process, providing insights into the mechanisms by which the recruitment of CENP-N is regulated by the structural transitions between compaction and relaxation of centromeric chromatin during the cell cycle.
Keywords: RG loop, CENP-A, higher-order chromatin structure, CENP-N, chromosome congression, cell cycle
Abstract
Specific recognition of centromere-specific histone variant CENP-A-containing chromatin by CENP-N is an essential process in the assembly of the kinetochore complex at centromeres prior to mammalian cell division. However, the mechanisms of CENP-N recruitment to centromeres/kinetochores remain unknown. Here, we show that a CENP-A-specific RG loop (Arg80/Gly81) plays an essential and dual regulatory role in this process. The RG loop assists the formation of a compact “ladder-like” structure of CENP-A chromatin, concealing the loop and thus impairing its role in recruiting CENP-N. Upon G1/S-phase transition, however, centromeric chromatin switches from the compact to an open state, enabling the now exposed RG loop to recruit CENP-N prior to cell division. Our results provide the first insights into the mechanisms by which the recruitment of CENP-N is regulated by the structural transitions between compaction and relaxation of centromeric chromatin during the cell cycle.
The centromere is a specialized chromatin domain that provides a platform for kinetochore assembly and microtubule attachment, thus playing a crucial role in chromosome segregation during mitosis (Allshire and Karpen 2008). A centromere-specific histone H3 variant (CenH3; termed CENP-A in humans) (Earnshaw and Cleveland 2013; Talbert and Henikoff 2013) functions as an epigenetic mark for the formation and maintenance of centromeres (Falk and Black 2012; Gurard-Levin et al. 2014; Müller and Almouzni 2014). In human cells, CENP-A is specifically recognized and deposited into centromeres by its chaperone, HJURP (Dunleavy et al. 2009; Foltz et al. 2009). CENP-A is a highly divergent member in the H3 family, with only ∼60% similarity within the histone folding domain (HFD), and contains distinct C-terminal and N-terminal tails as compared with canonical histone H3. In addition, the overall crystal structure of the CENP-A-containing nucleosome (CENP-A nucleosome) is similar to that of the canonical H3-containing nucleosome (H3 nucleosome), with two main differences (Tachiwana et al. 2011). First, the shorter αN helix of CENP-A contributes to the flexible nature of DNA at the entry/exit site of CENP-A nucleosome. Second, the RG loop of CENP-A (the two CENP-A-specific residues—Arg80 and Gly81—in loop1 [L1]), which is located at the lateral surface of the CENP-A nucleosome, is largely exposed in the mononucleosome structure. Moreover, centromeric chromatin is considered to have a distinct higher-order organization, existing in a more condensed structure than bulk chromatin (Gilbert and Allan 2001; Panchenko et al. 2011; Geiss et al. 2014).
In addition to CENP-A, human centromeres/kinetochores contain at least 16 nonhistone proteins that collectively belong to the constitutive centromere-associated network (CCAN), which is assembled onto the centromeric chromatin (Perpelescu and Fukagawa 2011). CENP-N, an indispensible member of CCAN, was identified as the first “reader” of epigenetic marks present in the CENP-A-containing chromatin (CENP-A chromatin) and was shown to directly bind to the centromere targeting domain (CATD) of CENP-A (Carroll et al. 2009). In addition, CENP-N, together with another member of CCAN (CENP-C), recruits several other CCAN proteins and outer kinetochore proteins to attach microtubules and ensure faithful segregation of sister chromatids (Foltz et al. 2006; Carroll et al. 2009, 2010). Importantly, down-regulation of CENP-N components by RNAi results in mitotic defects of chromosome congression (Foltz et al. 2006) and reduces the loading of nascent CENP-A onto centromeres (Carroll et al. 2009). Thus, CENP-N appears to be an essential factor for proper functioning of both the kinetochore complex and the centromere.
It has been proposed that the CCAN complex functions as a structural scaffold stably associated with centromeric chromatin, whereas CENP-N is dynamically recruited to centromeres/kinetochores during the cell cycle (McClelland et al. 2007; Hellwig et al. 2011). However, the molecular mechanisms by which CENP-N is recruited to centromeres/kinetochores in a cell cycle-dependent manner remain unknown. In this study, using biochemical, biophysical, and cell-based assays, we revealed the dual function of the RG loop of CENP-A in the recruitment of CENP-N and the folding of centromeric chromatin. Our results show for the first time that structural transitions of the higher-order structure of centromeric chromatin orchestrate the temporal loading of CENP-N to centromeres/kinetochores via regulating the accessibility of the RG loop in CENP-A chromatin during the cell cycle.
Results
The RG loop of CENP-A plays a key role in the recruitment of CENP-N to centromeres/kinetochores
In order to define the regions of CENP-A that are responsible for the recruitment of CENP-N to CENP-A chromatin, a series of human CENP-A/H3 (H3.1) chimeras were constructed (Fig. 1A). To monitor the interaction between CENP-A and CENP-N, we used a cell-based approach using the LacO/I targeting system in A03_1 cells (Supplemental Fig. S1A; Liu et al. 2012). Initially, to test whether the LacI-CENP-A or LacI-H3 is incorporated into chromatin in our system, the retention ratio of LacI dots was analyzed following IPTG treatment. The results showed that >90% of LacI dots for both LacI-CENP-A and LacI-H3 could still be detected, whereas LacI protein alone was detected at very low levels following IPTG treatment (Supplemental Fig. S1B,C), indicating that histones can be efficiently incorporated into chromatin at the LacO locus. Our results are in agreement with two recent reports that demonstrated that LacI-CENP-A is assembled into chromatin at or near LacO arrays using a similar system (Logsdon et al. 2015; Tachiwana et al. 2015). Therefore, using this LacO/I targeting system, the interaction between CENP-N and CENP-A chromatin can be analyzed. Interestingly, we found that CENP-N was efficiently targeted to CENP-A chromatin but not to H3-containing chromatin (H3 chromatin), demonstrating the validity of our system. Moreover, CENP-N was found to interact with chromatin containing either CENP-AH3CTD, H3NTDCENP-A, or H3CATD but not with CANTDH3CACTD (Fig. 1B,C). Our results revealed that both the N-terminal and C-terminal regions of CENP-A are dispensable for CENP-N binding, and the CATD domain is sufficient for the interaction between CENP-N and CENP-A chromatin, which is consistent with previously published data (Carroll et al. 2009).
Figure 1.

The RG loop plays a key role in the recruitment of CENP-N (see also Supplemental Figs. S1, S2). (A) A schematic depiction of the junctions of the CENP-A (red) and H3 (blue) chimeras. CENP-A domains, R80G81 residues, and amino acid positions are also indicated. (B) CENP-N targeting assay. Representative images of A03_1 cells transfected with mCherry-CENP-N (red) along with eGFP-LacI-CENP-A, H3, or the indicated chimeras and mutants (green) and stained with DAPI (blue). Bar, 10 µm. (C) Statistical analysis of the proportion of cells with colocalization as shown in B. (D) Side view of CENP-A nucleosome, with the CENP-A molecule shown in magenta, and superimposition of CENP-A (magenta) and H3 (orange) L1 regions. Arrows indicate the tip of the CENP-A L1 containing Arg80 and Gly81 residues (green). (E) ClustalX2 alignment of the CATD domain in CENP-A orthologs from Homo sapiens (Hs), Pongo abelii (Pa), Bos taurus (Bt), and Mus musculus (Mm) and the corresponding region in human histone H3. Residues R80G81 are highlighted in red. (F) HJURP-tethering CCAN experiments. Representative images of A03_1 cells transfected with mCherry-LacI-HJURP (red) along with HA-CENP-A or HA-CENP-AR80A/G81A (magenta) and eGFP-CENP-N or CENP-C (green) and stained with DAPI (blue). HA-tagged proteins were immunoblotted with an anti-HA antibody. Bar, 10 µm. (G) Statistical analysis of the proportion of cells with colocalization as shown in F. (H) Mononucleosome immunoprecipitation experiments. 293T cells were transfected with constructs or siRNA oligos as indicated. Mononucleosomes were generated and immunoprecipitated with anti-Flag beads and immunoblotted with the indicated antibodies. (I) Overexpression of mCherry-CENP-N in stable HeLa cells expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A and colocalization analysis. SNAP-CENP-As was labeled with Oregon Green (green). Bar, 10 µm. (J) Statistical analysis of the proportion of cells with colocalization as shown in I.
Next, we set out to identify the precise residues of CENP-A that are responsible for CENP-N binding. The CATD domain contains L1 and helix2 (α2). As described previously, the RG loop protrudes from the core of the CENP-A mononucleosome (Fig. 1D; Tachiwana et al. 2011) and can serve as a potential binding site for the CCAN proteins. The R80G81 residues are conserved across higher mammal species and therefore are prime candidates for interrogation (Fig. 1E). We constructed CENP-A and H3CATD mutants in which the R80G81 residues were substituted by A80A81 (named CENP-AR80A/G81A and H3-CATDR80A/G81A). Using the LacO/I targeting assay, we showed that these two mutants greatly impaired their interaction with CENP-N (Fig. 1B,C). In addition, in the presence of IPTG, the disruption of the RG loop of CENP-A also impaired the binding of CENP-N (Supplemental Fig. S1D,E), indicating that the RG loop is crucial for the recruitment of CENP-N by CENP-A nucleosomes.
Previously, CENP-C was reported as another protein directly interacting with CENP-A nucleosomes (Kato et al. 2013); thus, we assessed whether the impaired recruitment of CENP-N to CENP-AR80A/G81A chromatin is because of the impact of the mutation of the RG loop of CENP-A on the bindings to CENP-C. Using the same LacO/I targeting assay, we showed that CENP-AR80A/G81A had little effect on the interaction with CENP-C in comparison with wild-type CENP-A (Supplemental Fig. S1F,G). These results suggested that CENP-AR80A/G81A specifically impaired the binding of CENP-N. To investigate the interaction between CENP-A and CENP-N in a more physiological context, we performed HJURP-tethering experiments as previously reported (Barnhart et al. 2011). We found that the mutants CENP-AR80A/G81A and CENP-AR80A did not affect their recruitment by HJURP (Supplemental Fig. S1H–J). Next, we analyzed the effects of CENP-A mutants on the binding of CENP-N to the HJURP-CENP-A-tethering arrays. Consistent with the above results, our analysis showed that the binding of CENP-N is inhibited in the HJURP-CENP-AR80A/G81A arrays (Fig. 1F,G). In contrast, CENP-AR80A/G81A showed little effect on the interaction with CENP-C (Fig. 1F,G).
Because the LacI–LacO-tethering assay is nonphysiological to some extent, to confirm the above results, we performed mononucleosome immunoprecipitation experiments to assess the interaction of CENP-N with CENP-A mononucleosomes (Foltz et al. 2006). Our results demonstrated that the mutation of the RG loop in CENP-A impaired the recruitment of CENP-N to CENP-A nucleosomes, whereas the recruitment of CENP-C to CENP-A nucleosomes remained unaffected (Fig. 1H; Supplemental Fig. S1K), which demonstrates that the effect of CENP-AR80A/G81A is specific for CENP-N. Interestingly, we also observed that the binding of CENP-L to CENP-AR80A/G81A nucleosomes was greatly reduced. Since CENP-L has been reported to be a protein downstream from CENP-N (Carroll et al. 2009; Hellwig et al. 2011), we hypothesized that the reduction of CENP-L may be a consequence of impaired binding of CENP-N. In addition, we analyzed the binding of CENP-N to CENP-A nucleosomes upon knockdown of CENP-L using RNAi. Interestingly, we found that the knockdown of CENP-L did not affect the specific binding of CENP-N to wild-type CENP-A nucleosomes (Fig. 1H; Supplemental Fig. S1K,L). Taken together, our results suggested that the RG loop of CENP-A is critical for CENP-N binding. This binding is independent of CENP-C and is not affected by the interaction between CENP-N and CENP-L.
In order to determine whether these mutations affect the deposition of CENP-N onto centromeres/kinetochores, we generated stable HeLa cell lines expressing either SNAP-CENP-A or SNAP-CENP-AR80A/G81A. As previously reported, endogenous CENP-A was found to be down-regulated in these cell lines (Supplemental Fig. S1M), possibly by cell-intrinsic mechanisms that ensure that CENP-A protein levels are within a tightly regulated range (Tomonaga et al. 2003; Lacoste et al. 2014). Analysis of fluorescent imaging showed that the recruitment of CENP-N to the centromeres/kinetochores was greatly impaired in cells expressing SNAP-CENP-AR80A/G81A (Fig. 1I,J), suggesting that the RG loop is important for CENP-N recruitment. In comparison, the levels of CENP-C recruitment to centromeres/kinetochores showed no differences between cells expressing either wild-type or mutant CENP-A (Supplemental Fig. S1N). Together, these results indicated that the conserved RG loop is specifically required for the recruitment of CENP-N, but not CENP-C, to centromeric chromatin. Because Arg80 is a positively charged residue, we tested whether this characteristic contributes to the interaction between CENP-N and CENP-A chromatin. A series of mutants were generated and tested for binding ability using the LacO/I targeting assay. We found that only CENP-AR80K and CENP-AG81A displayed levels of CENP-N binding that were similar to those observed for wild-type CENP-A. In contrast, CENP-AR80E, CENP-AR80M, CENP-AR80A, CENP-AG81E, and CENP-AG81Y could not interact with CENP-N (Supplemental Fig. S2). The results indicated that both the positive charge property of Arg80 and the size of Gly81 are critical for the binding of CENP-N.
Compaction of CENP-A chromatin impairs the binding of CENP-N
To confirm that the RG loop of CENP-A is essential for the direct binding of CENP-N, an in vitro gel-shift assay was employed. A His-tagged truncated form of CENP-N (His-CENP-N1–289, CENP-N for short in the following) was used in the following in vitro biochemical experiments. Previously, it had been shown that CENP-N1–289 was sufficient for the interaction between CENP-N and CENP-A nucleosomes (Carroll et al. 2009). It is of note that CENP-N was found to be present in its polymeric form (pentamer or hexamer), as shown by gel filtration analysis (Supplemental Fig. S3A). The results showed that CENP-N directly and specifically binds to mononucleosomes containing wild-type CENP-A but not to mononucleosomes containing either mutant CENP-AR80A/G81A or canonical H3 (Fig. 2A), which is consistent with our cell-based results described above.
Figure 2.

CENP-N specifically binds to open CENP-A chromatin but not to compact CENP-A chromatin (see also Supplemental Fig. S3). (A) In vitro mononucleosome-binding assay. Mononucleosomes (0.15 µM) were incubated with His-CENP-N1–289mono in different amounts and separated by 6% native PAGE gel. (B) In vitro polynucleosome-binding assay. Polynucleosomes (0.15 µM) were incubated with His-CENP-N1–289 in different amounts and analyzed by 1.0% agarose gel. (C) Statistical analysis of the proportion of nucleosome binding as shown in B. (D) In vitro polynucleosome-binding assay. Polynucleosomes (0.15 µM) were incubated with His-CENP-C426–943 in different amounts and analyzed by 1.0% agarose gel. (E) Statistical analysis for the proportion of nucleosome binding as shown in D. (F) In vitro mononucleosome-binding assay. Mononucleosomes (0.15 µM) were incubated with His-CENP-N1–289 in different amounts in the absence or presence of MgCl2 and analyzed by 6% native PAGE gel. (G) In vitro polynucleosome-binding and sucrose gradient assay. Chromatin was incubated with His-CENP-N1–289 in a molar ratio of 1:2 and analyzed by sucrose gradient sedimentation. Each sample was fractionated by sucrose gradient centrifugation, and DNA collected from individual fractions was sized on agarose gel.
In addition, we assessed whether CENP-N interacts with CENP-A-containing polynucleosomes (also as CENP-A chromatin). Thus, regularly spaced polynucleosomal arrays were reconstituted using DNA templates containing 12 tandem repeats of 177-base-pair (bp) “601 nucleosome positioning” sequence (12mer-177-bp-601 DNA for short) and recombinant octamers (Song et al. 2014). EM (electron microscopy) imaging confirmed that both the H3 and CENP-A chromatin formed similar “beads on a string” 12mer arrays (Supplemental Fig. S3B). Surprisingly, the binding of CENP-N to either CENP-A or H3 chromatin was undetectable in the presence of MgCl2 (Supplemental Fig. S3B). It has been reported that nucleosomal arrays fold into compact chromatin fibers in the presence of MgCl2 (Fan et al. 2002; Chen et al. 2013), and CENP-A chromatin exhibits a more condensed structure than bulk chromatin in vitro (Panchenko et al. 2011; Geiss et al. 2014) and in vivo (Gilbert and Allan 2001). Therefore, we hypothesized that, in the presence of MgCl2, CENP-A nucleosomal arrays fold into a compact chromatin, resulting in concealment of the RG loop and thus preventing the binding of CENP-N to CENP-A chromatin. To test this hypothesis, we measured the binding ability of CENP-N to CENP-A chromatin using in vitro gel-shift assay combined with changes in MgCl2 concentration. When MgCl2 was removed from the binding buffer, binding of CENP-N to CENP-A chromatin was observed as expected (Fig. 2B,C). Moreover, we found that CENP-C could specifically bind with CENP-A chromatin but not with H3 chromatin, and the addition of MgCl2 showed little effect on the interaction between CENP-C and CENP-A chromatin (Fig. 2D,E; Supplemental Fig. S3C). In order to rule out the possibility that MgCl2 itself affected the binding of CENP-N to CENP-A mononucleosomes, we compared the binding affinity of CENP-N to CENP-A mononucleosomes in the absence or presence of MgCl2. The results showed that the addition of MgCl2 had almost no effect on the binding of CENP-N to CENP-A mononucleosomes (Fig. 2F). Our results indicated that the addition of MgCl2 to CENP-A chromatin specifically prevents the binding of CENP-N to CENP-A chromatin.
To verify these results, we used sucrose gradient sedimentation to test the interaction between CENP-N and CENP-A chromatin. In the absence of MgCl2, a distinct shift of the CENP-A chromatin band was observed upon the addition of CENP-N at a molar ratio of 1:2, while no such shift was visible in the presence of MgCl2 (Fig. 2G), thus confirming the results described above.
CENP-A chromatin adopts a compact ‘ladder-like’ structure
To characterize the precise role of CENP-A on the folding of chromatin fibers, the folding of reconstituted nucleosomal arrays containing H3 or CENP-A was measured by both analytical ultracentrifugation (AUC) and EM analysis as described previously (Supplemental Fig. S4A; Chen et al. 2013). EM imaging confirmed that, in the absence of MgCl2, both the H3-containing and CENP-A-containing nucleosomal arrays displayed similar nucleosome occupancy, with >90% saturation, and adopted a typical extended “beads on a string” conformation (Fig. 3A). The compact structures of H3 or CENP-A arrays formed in the presence of 1.0 mM MgCl2 were visualized by EM analysis of negative-stained samples. In the presence of 1.0 mM MgCl2, >80% of the CENP-A arrays folded into a compact “ladder-like” structure, while little “ladder-like” structure was observed in H3 arrays (Fig. 3A,B). In addition, the folding behavior of H3 or CENP-A arrays was also measured by AUC, and the two arrays displayed similar average sedimentation coefficients (Save) of 36S in the absence of MgCl2 (Fig. 3C,D). As MgCl2 concentration increased, nucleosomal arrays started to fold more tightly. In the presence of 1.0 mM MgCl2, the Save for the H3 nucleosomal arrays was 44S (Fig. 3C,E). In contrast, CENP-A nucleosomal arrays compacted more readily and formed a more condensed structure, with a Save of 56S at 1.0 mM MgCl2 (Fig. 3D,E).
Figure 3.

Effects of CENP-A on the folding of chromatin arrays (see also Supplemental Figs. S4, S5). (A) EM images of the canonical H3-containing, CENP-A-containing, CENP-AR80A/G81A-containing, and H3RG-containing nucleosomal arrays (by metal-shadowing method; bar, 50 nm) and their related compact states in 1.0 mM MgCl2 (by negatively stained method; bar, 100 nm). (B) Statistical analysis of the proportion of chromatin in “ladder-like” conformation as shown in A. (C,D) Sedimentation coefficient distribution plots for canonical H3-containing and CENP-A-containing nucleosomal arrays in 0, 0.5, and 1.0 mM MgCl2. (E) The Save values of the nucleosomal arrays containing CENP-A and H4Δ1–23, CENP-AR80A/G81A, and H3RG compared with the ones containing wild-type CENP-A or H3. (F–H) Sedimentation coefficient distribution plots for CENP-AR80A/G81A-containing, H3RG-containing, and CENP-A/HAΔ1–23-containing nucleosomal arrays in 0, 0.5, and 1.0 mM MgCl2. (I) EM images of the CENP-A/HAΔ1–23-containing nucleosomal arrays (by metal-shadowing method; bar, 50 nm) and their related compact states in 1.0 mM MgCl2 (by negatively stained method; bar, 100 nm). (J) In vitro polynucleosome-binding assay. Polynucleosomes (0.15 µM) containing CENP-A or CENP-A/HAΔ1–23 were incubated with His-CENP-N1–289 in different amounts in the absence or presence of 1.0 mM MgCl2 and analyzed by 1.0% agarose gel. (K) Statistical analysis of the proportion of nucleosome binding as shown in J.
Human centromeres consist of megabase-long stretches of α-satellite DNA, which are repetitive sequences that contain ∼171-bp monomers (Rudd and Willard 2004). Recently, the nucleosome repeat length (NRL) was reported as a key factor for the degree of folding of chromatin fiber (Routh et al. 2008; Correll et al. 2012). To investigate whether NRL affects the specific compaction of CENP-A chromatin, we reconstituted H3 and CENP-A nucleosomal arrays on 12mer-171-bp-601 DNA. The AUC results showed that CENP-A chromatin with short NRL also folded into a more compact structure than H3 chromatin did, similar to the results with 12mer-177-bp-601 DNA (Supplemental Fig. S4B–D). In order to study the effects of chromatin folding on the native centromeric chromatin, we reconstituted nucleosomal arrays using 12mer-171-bp-α-satellite DNA. The AUC results revealed that these CENP-A arrays folded into a more compact structure than H3 arrays did (Supplemental Fig. S4E–G). Taken together, our results suggest that CENP-A enhances the folding of chromatin regardless of the underlying DNA sequences or NRL present and facilitates the formation of a compact “ladder-like” chromatin structure in the presence of MgCl2.
The RG loop of CENP-A is critical for the formation of a ‘ladder-like’ chromatin structure
To define the regions of CENP-A that are responsible for the formation of a “ladder-like” chromatin structure, the folding of nucleosomal arrays containing H3/CENP-A chimeras (Supplemental Fig. S5A) was analyzed using AUC and EM. Interestingly, the H3CATD and H3NTDCENP-A chimeras (both containing the CATD domain) showed a degree of chromatin compaction similar to that of wild-type CENP-A, whereas CANTDH3 (without the CATD domain) displayed a degree of chromatin folding similar to that of H3 (Supplemental Fig. S5B–E). Together, our results indicate that the CATD domain of CENP-A is important for enhancing the folding of CENP-A chromatin.
As described above, the RG loop of CENP-A is located at the side surface of the CENP-A mononucleosome (Fig. 1D), where it may impact chromatin structure. To test this hypothesis, CENP-A mutants CENP-AR80A/G81A and CENP-AV82A/D83A (mutations of V82D83 to A82A83) were generated. The ability to promote chromatin compaction was greatly impaired in the CENP-AR80A/G81A mutant (Fig. 3E,F). Furthermore, in the presence of MgCl2, hardly any compact “ladder-like” structures were observed in these mutant nucleosomal arrays (Fig. 3A,B). In contrast, the CENP-AV82A/D83A mutant had effects on chromatin compaction similar to that observed for wild-type CENP-A (Supplemental Fig. S5F,G). When inserting the residues RG into canonical H3 (H3RG), we found that this mutant caused chromatin compaction similar to that observed for wild-type CENP-A, with most of the H3RG chromatin adopting a “ladder-like” structure (Fig. 3A,B,E,G). These results clearly showed that the RG loop of CENP-A plays a key role in enhancing chromatin compaction and that it is essential for the formation of a “ladder-like” chromatin structure.
According to the crystal structure of the CENP-A mononucleosome, the RG loop is located at the side face of the nucleosome and is largely exposed in mononucleosome. We therefore speculated that the RG loop facilitates the formation of the “ladder-like” structure of CENP-A chromatin. Once the formation of the “ladder-like” structure is initiated, the RG loop becomes concealed, ultimately resulting in impaired recruitment of CENP-N.
It was previously reported that the amino acids 14–19 and R23 of H4 were critical for H3 chromatin folding (Dorigo et al. 2003; Song et al. 2014); thus, we assessed whether the effect of histone H4 tail on the folding/compaction of CENP-A chromatin could modulate the binding of CENP-N to CENP-A chromatin. Our AUC and EM results showed that incorporation of tailless H4 (H4Δ1–23) could indeed impair the folding and formation of a “ladder-like” structure of CENP-A chromatin induced by MgCl2 (Fig. 3E,H,I). Interestingly, we found that CENP-N bound equally to the CENP-A/HAΔ1–23 chromatin in the absence and presence of MgCl2 (Fig. 3J,K), which suggested that the “ladder-like” conformation of CENP-A chromatin hinders the accessibility of the RG loop for the CENP-N binding. Next, we investigated whether prebound CENP-N affects the compaction of CENP-A chromatin induced by MgCl2. To this end, we preincubated CENP-N with CENP-A chromatin in the absence of MgCl2 and then measured the compaction of CENP-A and monitored the binding of CENP-N to CENP-A chromatin upon addition of MgCl2. Intriguingly, we found that most CENP-A chromatin folded into a compact “ladder-like” structure after addition of MgCl2. In the meanwhile, following addition of MgCl2, the prebound CENP-N was found to be largely dissociated/unloaded from CENP-A chromatin (Supplemental Fig. S5H,I). These results suggested that the nucleosome–nucleosome interaction (which is mediated by the RG loop) within the compacted CENP-A chromatin fiber is stronger than the affinity between CENP-N and the RG loop of CENP-A nucleosome.
CENP-N is recruited onto centromeres/kinetochores during middle/late S phase
The above results demonstrated that in vitro, CENP-N bound only to open CENP-A chromatin but not to compact CENP-A chromatin, indicating that the dynamic states of higher-order structure of centromeric chromatin play a critical role in regulating the recruitment of CENP-N. Indeed, previous fluorescence and biophysical experiments reported that CENP-N bound to centromeres/kinetochores in a cell cycle-dependent manner (McClelland et al. 2007; Hellwig et al. 2011). To test this in our system, we generated a stable HeLa cell line expressing SNAP-CENP-N and quantified the binding levels of SNAP-CENP-N to centromeres/kinetochores at different phases during the cell cycle. The location of SNAP-CENP-N was detected and showed in colocalization with centromeres, revealing that SNAP tag does not affect the deposition behavior of SNAP-CENP-N (Supplemental Fig. S6A). Our quantitative analysis of the fluorescence measurements showed that CENP-N bound to centromeres/kinetochores increased at early S phase, peaked at middle/late S and G2 phase, and decreased during mitosis and G1 phase (Fig. 4A,B). In addition, we analyzed the cellular abundance of SNAP-CENP-N protein during the cell cycle and found that the protein levels of SNAP-CENP-N did not change across the cell cycle (Supplemental Fig. S6B–D). These results indicate that cellular abundance of CENP-N protein was not the cause for the observed enrichment of CENP-N protein on centromeres/kinetochores during S and G2 phase.
Figure 4.

Specific loading of CENP-N in middle/late S phase (see also Supplemental Fig. S6). (A) Representative images of HeLa cells expressing SNAP-CENP-N in different cell cycle phases. SNAP-CENP-N was labeled with TMR-Star (red), and cell cycle phases were determined by combined staining with CENP-F antibody (gray), PCNA antibody (green), and DAPI (blue). Bar, 10 µm. (B) Statistical analysis of the relative fluorescence intensity of SNAP-CENP-N during the cell cycle as shown in A. (C) Outline of cell synchronization and the labeling regimen for CENP-N loading. (D) HeLa cells expressing SNAP-CENP-N (red) were synchronized and labeled as depicted in C, and cell cycle phases were determined by combined staining with CENP-F antibody (gray), PCNA antibody (green), and DAPI (blue). Bar, 10 µm. (E) Statistical analysis of relative fluorescence intensity of SNAP-CENP-N newly synthesized and deposited onto centromeres as shown in D.
To analyze the temporal deposition of CENP-N onto centromeres/kinetochores, we used HeLa cells stably expressing SNAP-CENP-N in combination with a classical SNAP quench pulse chase assay. Cells were arrested in the G2/M boundary by incubation with 25 ng/mL nocodazole for 16 h. Subsequently, cellular old SNAP-CENP-N was quenched with BG block for 30 min, and the cells were then released to re-enter the cell cycle. Once the majority of cells entered into late G1/early S phase ∼12 h later, the newly synthesized SNAP-CENP-N was labeled with TMR-Star for 30 min (Fig. 4C). After TMR-Star labeling, cells were collected every 4 h, and the abundance of CENP-N recruited to centromeres/kinetochores was determined. The results showed that newly synthesized SNAP-CENP-N was deposited into centromeres (Supplemental Fig. S6E,F), and this deposition was clearly observed from the middle S phase, reached its peak level in late S phase, and decreased to very low levels during mitosis and the following G1 phase (Fig. 4D,E). It was previously reported that CENP-N underwent a low recovery in middle/late S phase using FRAP experiments (Hellwig et al. 2011). Taken together, we conclude that CENP-N can stably bind with centromeric chromatin when it is loaded during middle/late S phase and may dissociate from centromeres/kinetochores during mitosis and G1 phase.
Structural transitions of centromeric chromatin during the cell cycle in vivo
The mechanisms by which the loading of CENP-N is dynamically regulated during the cell cycle remain unclear. Recently, it had been described that the timing of CENP-A deposition is uncoupled from the replication of centromeric DNA; i.e., centromeric DNA replicates in S phase, while CENP-A protein is synthesized in G2 phase and replenished in the subsequent early G1 phase (Jansen et al. 2007). In that case, the concentration of CENP-A nucleosomes at centromeres is diluted to half after DNA replication and regained in the next G1 phase, indicating that the chromatin state at centromeres will alter after centromeric DNA replication. To test the effect of the CENP-A concentration on the compaction of chromatin arrays, nucleosomal arrays were reconstituted by titrating CENP-A octamers with H3 octamers before analysis of the folding behavior by AUC and EM. We found that the chromatin assembled with mixed CENP-A and H3 octamers was homogenous in our AUC analysis, while the mixture of two homotypic chromatins (mixture of preassembled CENP-A and H3 chromatin) was clearly shown as two components (Supplemental Fig. S7A). The results indicated that the mixed CENP-A/H3 arrays were heterotypic (which represented mixed CENP-A/H3 nucleosomes on a single DNA molecule) rather than the mixture of homotypic CENP-A and H3 chromatin. Using a CENP-A titrating assay, we found that the lower the ratio of CENP-A to H3, the less compact the state of the chromatin arrays (Supplemental Fig. S7B–G). In addition, our EM data showed that the decreased concentration of CENP-A also impaired the formation of the compact “ladder-like” structure (Supplemental Fig. S7H). Thus, dilution of CENP-A results in a less condensed state of chromatin compared with CENP-A-saturated chromatin, suggesting that CENP-A chromatin will change from a compact state in G1 phase to an open state after DNA replication in S phase. In addition, we analyzed the interaction between CENP-N and these polynucleosomes containing different proportions of CENP-A. The results showed that in the absence of MgCl2, the polynucleosomes interacted with CENP-N even in the 25% proportion of CENP-A as long as the polynucleosomes contained CENP-A. In addition, the ability to bind CENP-N directly correlated with an increase in CENP-A in the polynucleosomes (Supplemental Fig. S7I,J). Moreover, the addition of MgCl2 reduced the binding of CENP-N to chromatin containing 50% CENP-A but did not completely impair the binding (Supplemental Fig. S7K,L).
Our biochemical results showed that the compaction of CENP-A chromatin greatly impaired the binding of CENP-N due to decreased accessibility of the RG loop. We therefore hypothesized that structural transitions of centromeric chromatin coordinate the cell cycle-dependent recruitment of CENP-N. To verify the hypothesis, we employed an AB-FRET (acceptor photobleaching-based FRET) to monitor the structural transitions of centromeric chromatin during the cell cycle. Because the FRET value is correlated with the distance of fluorescently labeled adjacent nucleosomes (Fig. 5A), this technique can be used to detect chromatin compaction in living cells (Llères et al. 2009). AB-FRET assays were performed in HeLa cells expressing SNAP-CENP-A. Each SNAP-tagged CENP-A was covalently labeled with only one fluorescent chromophore; thus, the same centromere can be simultaneously labeled with both Oregon Green (donor, 490|514) and TMR-Star (acceptor, 554|580) chromophores (Fig. 5B). In AB-FRET, the acceptor chromophore is destroyed by photobleaching, thereby preventing energy transfer occurring from donor to acceptor, resulting in an observable increase in donor fluorescence (Fig. 5C, spot 1).
Figure 5.

Centromeric chromatin structure changes in a cell cycle-dependent manner (see also Supplemental Figs. S7, S8). (A) Schematic diagram of FRET relative to chromatin structure. The closer the distance between donor and acceptor, the higher the FRET signals that are obtained. The distance between donor and acceptor could reflect the chromatin structure. (B) Representative images of HeLa cells expressing SNAP-CENP-A colabeled with Oregon Green (green) and TMR-Star (red). Bar, 10 µm. (C) Schematic diagram of AB-FRET. Two locations in centromeres/kinetochores, 1 and 2, were selected for fluorescence intensity analysis before and after acceptor bleaching (see enlargement below). At spot 1, the acceptor fluorophore was bleached, and the fluorescence intensity of donor emission increased, indicating that FRET occurred between TMR-labeled CENP-A and Oregon Green-labeled CENP-A. Spot 2 was not photobleached and served as an intrinsic control for non-FRET effect. (D) FRET measurements between CENP-A nucleosomes in HeLa cells expressing SNAP-CENP-A throughout the cell cycle. (E) FRET measurements between CENP-AR80A/G81A nucleosomes in HeLa cells expressing SNAP-CENP-AR80A/G81A compared with that between CENP-A nucleosomes in HeLa cells expressing SNAP-CENP-A. (F) FRET measurements between CENP-A or CENP-AR80A/G81A nucleosomes in HeLa cells expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A throughout the cell cycle.
Using the AB-FRET assay, we analyzed the chromatin dynamics in centromeres across the cell cycle (Supplemental Fig. S8A). Interestingly, a significant FRET signal was detected in mid-G1 phase and remained high over late G1 and the G1/S boundary (Fig. 5D). However, the FRET signal rapidly declined to the background levels as soon as cells entered into S phase and then remained at a low level throughout the subsequent G2 and M phases (Fig. 5D). To test whether the observed FRET signals reflected levels of chromatin folding, we measured the FRET signals before and after disrupting the higher-order chromatin organization by micrococcal nuclease (MNase) treatment. Interestingly, in comparison with the high FRET signals in the untreated G1-phase nuclei, FRET signals were undetectable in MNase-treated G1-phase nuclei (Supplemental Tables S1, S2; Supplemental Fig. S8B,C). Our results confirmed that the FRET value faithfully reflected higher-order chromatin organization. Taken together, we conclude that the centromeric chromatin converts to an open state as cells enter into S phase, which is accompanied with the increased loading of CENP-N onto centromeres/kinetochores during middle/late S phase.
Our above in vitro biophysical studies showed that the RG loop was important for the compaction of CENP-A chromatin. We also examined whether mutations of the RG loop affected the higher-order structure of centromeric chromatin in vivo by AB-FRET. Interestingly, we found that FRET signals were significantly lower in unsynchronized cells expressing SNAP-CENP-AR80A/G81A relative to cells expressing its wild-type form, indicating that the centromeric chromatin structure might be less compact in mutant cells than in wild-type cells (Fig. 5E). To clarify in which cell cycle phase centromeric chromatin became less compact, we synchronized the mutant cells and released them into G1, S, G2, or M phase. Similar to the results in wild-type cells, notable FRET signals were detected in the G1/S boundary, and the signals reduced to background levels when the mutant cells entered into S phase. Surprisingly, during G1 phase, particularly in early G1 phase, the FRET signals were much lower in mutant cells than that in wild-type cells (Fig. 5F). It is known that CENP-A at centromeres is halved during DNA replication in S phase, and newly synthesized CENP-A is complemented during late telophase and early G1 phase (Jansen et al. 2007). Therefore, the much lower FRET signal observed during early/middle G1 (6 h) might result from the delayed deposition of newly synthesized CENP-AR80A/G81A in mutant cells. To test this, we analyzed the timing of deposition of newly synthesized CENP-A in both wild-type and mutant cells using a SNAP quench pulse chase assay. Indeed, our results showed that the loading of a newly synthesized CENP-AR80A/G81A mutant was found to be delayed (Supplemental Fig. S8D–F). Very little mutant CENP-A (CENP-AR80A/G81A) was deposited into centromeres in early G1 phase when compared with wild-type CENP-A, which might be the reason why a much lower FRET signal was observed in mutant cells than in wild-type cells at early/middle G1 phase (6 h). It is of note that our aforementioned results demonstrated that the mutation of RG loop in CENP-A did not affect the HJURP recognition with CENP-A (Supplemental Fig. S1H–J). Therefore, we proposed that the mutation of the RG loop might cause the deficiency of cell cycle progression or affect the timing of CENP-A loading through unknown mechanisms, either of which might account for the delayed deposition of CENP-AR80A/G81A.
The RG loop of CENP-A ensures cell proliferation and chromosome congression
Our results showed that the RG loop of CENP-A was responsible for the binding of CENP-N in vitro and in vivo, indicating that the RG loop plays an important role in kinetochore formation and chromosome segregation. In order to investigate the biological role of the RG loop in vivo, we examined the effects of mutations in the RG loop on cell proliferation and mitotic progress (Fig. 6A). In HeLa cell lines expressing either SNAP-CENP-A or SNAP-CENP-AR80A/G81A, the locations of exogenous SNAP-CENP-As were detected by TMR-Star labeling, which showed that SNAP-CENP-As colocalized with ACA (human anti-centromere antibody)-labeled centromeres (Fig. 6B). Protein levels of endogenous and exogenous CENP-As were analyzed by Western blot, which showed that both forms were present at levels that were similar between the two cell lines (Supplemental Fig. S1M). Importantly, we found that mutations in the RG loop resulted in inhibition of cell proliferation (Fig. 6C) concomitant with an increase in the percentage of cells in G2/M phase (Fig. 6D), suggesting that a part of cells undergoes abnormal mitosis. Moreover, immunofluorescence imaging of cells in M phase demonstrated that, in contrast to wild-type cells, mutant cells displayed mitotic defects in chromosome congression, such as the formation of three or four spindle poles in one single mitotic cell instead of two spindle poles (Fig. 6E; Supplemental Fig. S9). Similar defects were observed in the CENP-N-depleted cells, as reported previously (Foltz et al. 2006; McClelland et al. 2007). The proportion of cells with mitotic defects was striking, with up to 35% of the mutant cells defective, but with wild-type cells remaining mostly intact (Fig. 6F). Moreover, some multinucleated cells containing three or four nuclei were observed in the mutant cells, indicating that at least some of the mutant cells eventually exited from mitosis, although not cytokinesis (Supplemental Fig. S9). In addition, mitotic defects in cells expressing CENP-AR80A/G81A might be responsible for the delayed deposition of CENP-AR80A/G81A at very early G1 phase as observed above (Supplemental Fig. S8D–F).
Figure 6.

Functions of the RG loop in ensuring cell proliferation and chromosome congression (see also Supplemental Fig. S9). (A) Schemes for construction of stable HeLa cell lines expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A and depletion of endogenous CENP-A. (B) Centromere localization of exogenous SNAP-CENP-As. SNAP tag was labeled with TMR-Star (red), and the centromere was detected with ACA (green). Bar, 10 µm. (C–E) Cell growth analysis (C), FACS analysis (D), and representative M-phase immunofluorescence images (E) of HeLa cells expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A. Cells were stained with TMR-Star (for CENP-A; red), tubulin (for microtubule; green), and DAPI (for DNA; blue). Bar, 10 µm. (F) Statistical analysis of the proportion of cells with M-phase defects in chromosome congression as shown in E. (G) Western blot assay to detect the amounts of endogenous and exogenous CENP-As before and after depletion of endogenous CENP-A in stable HeLa cells expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A, respectively. (H) Colony outgrowth assay in the stable HeLa cells expressing SNAP-CENP-A or SNAP-CENP-AR80A/G81A after depletion of endogenous CENP-A. The numbers at the left indicate the numbers of initial cells, while the numbers in the image indicate the numbers of surviving clones.
Despite the significant mitotic defects and delay in cell proliferation in the mutant cells, there was still a large portion of mutant cells without any obvious defects. To eliminate the possibility that remaining endogenous wild-type CENP-A could rescue these cells, we further knocked down the endogenous CENP-A by shRNA-mediated RNAi. As shown by Western blot analysis, endogenous CENP-A was greatly deleted after RNAi in both wild-type and mutant cells (Fig. 6G). We found that this depletion of endogenous CENP-A resulted in fewer surviving mutant cells, less than half the number of wild-type cells (Fig. 6H). Taken together, our results suggest that the RG loop of CENP-A plays a key role in ensuring cell proliferation and chromosome congression.
In summary, the results from our in vitro and in vivo studies demonstrate that the RG loop of CENP-A plays an important role in cell proliferation and chromosome congression by coordinately regulating structural transitions of centromeric chromatin and cell cycle-dependent recruitment of CENP-N. In the G1 phase, centromeric chromatin is present in a compact state in which the RG loop is concealed, and CENP-N fails to be loaded onto centromeres/kinetochores. However, when cells enter into S phase, centromeric DNA is replicated, and the occupancy of CENP-A is decreased. The centromeric chromatin is subsequently converted to a rather open state, and the RG loop is exposed to promote the stable loading of CENP-N onto centromeres/kinetochores. When cells reached the next G1 phase, the newly synthesized CENP-A was incorporated into centromeres, leading to the conversion of chromatin to a compact state, and, with the RG loop concealed again, the CENP-N was released from centromeres (Fig. 7).
Figure 7.
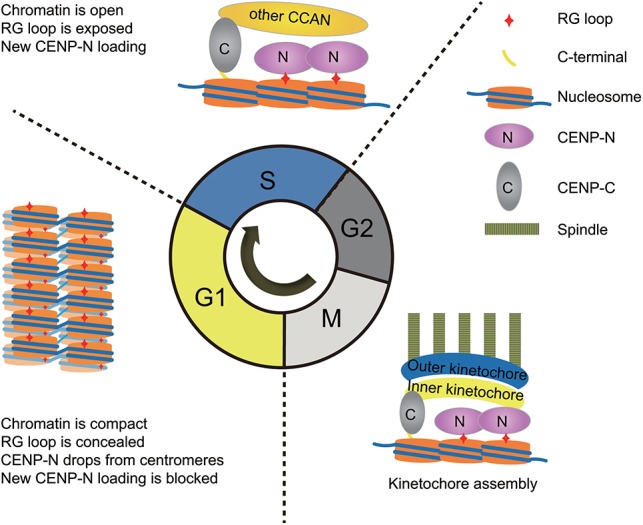
Model for structural transitions of centromeric chromatin regulating the cell cycle-dependent recruitment of CENP-N via modulating the accessibility of the RG loop of CENP-A.
Discussion
CENP-A chromatin displays a distinct higher-order organization
The structural features of centromeric chromatin, including the nucleosome composition and higher-order organization, are likely to provide epigenetic information that physically distinguishes centromeric chromatin from the rest of the chromosome (Black and Cleveland 2011). Based on several earlier studies, a number of contradictory models have been proposed for the composition and stoichiometry of CENP-A nucleosomes (Furuyama and Henikoff 2009; Black and Cleveland 2011; Bui et al. 2012; Miell et al. 2013, 2014; Padeganeh et al. 2013; Codomo et al. 2014; Walkiewicz et al. 2014). However, several more recent reports supported the idea that, throughout the entire cell cycle, CENP-A chromatin mainly consists of octameric nucleosomes rather than nonoctameric forms (for example, tetrasomes, hemisomes, and hexasomes) (Hasson et al. 2013; Padeganeh et al. 2013).
In our study, we found for the first time that the incorporation of CENP-A facilitated the formation of a compact “ladder-like” structure in the presence of MgCl2, as revealed by thorough AUC and EM analysis. Two recent crystallographic studies of CENP-A homotypic and CENP-A/H3.3 heterotypic nucleosomes revealed that, in comparison with H3.1 and H3.3, the αN helix of CENP-A is shorter, which results in a weaker interaction between DNA at the entry/exit site of CENP-A nucleosomes and histone cores and leads to an open conformation of the linker DNA (Luger et al. 1997; Tachiwana et al. 2011; Arimura et al. 2014). In addition, a lysine in place of H3R49 in CENP-A probably results in a less protected αN helix of CENP-A, which consequently increases the flexibility of the linker DNA at the entry/exit sites of CENP-A nucleosomes (Conde e Silva et al. 2007; Panchenko et al. 2011). The distinct arrangements of the linker DNA might account for a criss-crossed “zig-zag” conformation for H3 nucleosomes and a parallel “ladder-like” conformation for CENP-A nucleosomes, as proposed recently (Lyubchenko 2014). Interestingly, a recent study using cryo-EM tomography showed that CENP-A arrays are more condensed, with a closer proximity between spatially adjacent nucleosomes (N and N + 2), which consequently promotes the face-to-face stacking of the adjacent nucleosomes under physiological salt conditions (Geiss et al. 2014). These two hypotheses are consistent with our findings of a compact “ladder-like” structure of CENP-A chromatin.
In addition, numerous studies have shown that the internucleosomal interactions between histones are important for the folding of nucleosomal arrays into a 30-nm chromatin fiber (Schalch et al. 2005; Song et al. 2014). For example, the internucleosomal interactions between the positively charged residues of the H4 N-terminal tail (residues 16–23) and the acidic patch of the H2A–H2B heterodimer are responsible for the twist between the tetranucleosomal units in the 30-nm chromatin fiber (Song et al. 2014). Apart from the flexible entry/exit DNA in the CENP-A nucleosomes, one crystallographic study also showed that the RG loop is located at the lateral surfaces of the CENP-A nucleosomes, rendering L1 longer and protruding from the core of the mononucleosome (Tachiwana et al. 2011). Altogether, we proposed that the open conformation of the linker DNAs and the nucleosome–nucleosome interactions mediated by the RG loops are critical for the formation of the straight “ladder-like” structure of CENP-A chromatin fiber.
The structure of centromeric chromatin undergoes cell cycle-dependent transitions
Recently, a number of studies demonstrated that the assembly of CENP-A is temporally regulated by phosphorylation/dephosphorylation of CENP-A at Ser68 (Yu et al. 2015), chaperone HJURP (Müller et al. 2014), and the “priming” factor Mis18 complex (McKinley and Cheeseman 2014). The tight regulation results in a change of CENP-A in a strictly cell cycle-dependent manner and ensures proper centromere function during mitosis after temporal recruitment of specific kinetochore proteins. Centromeric DNA is replicated during early S phase (Weidtkamp-Peters et al. 2006; Koren et al. 2010), while CENP-A is replenished in the next early G1 phase prior to the next round of centromeric DNA replication (Shelby et al. 2000; Jansen et al. 2007). Thus, the CENP-A concentration is halved after centromeric DNA replication. Accordingly, two main alternations may occur on the structures of centromeric chromatin after DNA replication during S phase: (1) composition of the nucleosomes and (2) higher-order chromatin organization. A recent study showed that CENP-A nucleosomes undergo transitions in nucleosome composition during the cell cycle (Bui et al. 2012). However, other studies support the notion that CENP-A nucleosomes are octameric throughout the cell cycle (Hasson et al. 2013; Padeganeh et al. 2013).
Employing an AB-FRET assay, we demonstrated that the higher-order organization of centromeric chromatin underwent a structural transition from a compact state in G1 phase to an open state in S phase. Our in vitro AUC and EM analyses demonstrated that the dilution of CENP-A impairs the folding ability of chromatin in vitro, which may result in the structural transitions of centromeric chromatin in vivo from a compact state in the G1 phase to an open state in the S phase. In addition, it has been demonstrated that both H3.1 and H3.3 are deposited onto centromeres in S phase, but only H3.3 acts as a placeholder for newly assembled CENP-A in G1 phase (Dunleavy et al. 2011). It has been documented that CENP-A can form a heterotypic particle with H3.3 (Lacoste et al. 2014). Furthermore, it had been shown recently that the heterotypic CENP-A/H3.3 nucleosome forms an unexpectedly stable structure as compared with the CENP-A nucleosome (Arimura et al. 2014). Previously, we reported that incorporation of H3.3 can prevent the compaction of the chromatin fiber (Chen et al. 2013); thus, we hypothesized that the dynamics of H3.3 at centromeres play important roles in the regulation of the higher-order organization of centromeric chromatin during the cell cycle. Furthermore, the high FRET signals that we observed during G1 phase are consistent with the previously described dynamics of CENP-A assembly, which starts in late telophase and peaks in G1 phase (Jansen et al. 2007). We therefore propose that the increased occupancy of CENP-A at centromeres during G1 phase results in a compact chromatin fiber.
Coordination of the transitions of the higher-order structure of centromeric chromatin with the cell cycle-dependent recruitment of CENP-N
Numerous studies have demonstrated that the structure and function of the centromere are epigenetically defined by the presence of CENP-A (Black et al. 2007; Guse et al. 2011; Panchenko et al. 2011; Fachinetti et al. 2013). Similar to other chromatin marks, it is important to understand how the epigenetic information encoded in CENP-A-containing nucleosomes is read and transmitted. A recent study reported that the higher hydrophobicity of the C-terminal tail of CENP-A is a primary determinant for the recognition of the CENP-A nucleosome by CENP-C (Kato et al. 2013).
In this study, we demonstrated that the RG loop of CENP-A can serve as a key determinant for the recognition of the CENP-A nucleosome by the “reader” CENP-N. Interestingly, we found that the RG loop not only provides the recognition site for the binding of CENP-N to CENP-A nucleosome but also facilitates the folding of CENP-A arrays into a compact “ladder-like” chromatin structure. The CATD domain has been found to be required for the deposition of CENP-A into centromeres by its specific chaperone, HJURP (Foltz et al. 2009), suggesting that the RG loop may have a potential role in this process as well. However, we show that mutations of the RG loop have little effect on the deposition of CENP-A into centromeres, which is consistent with one previous report (Bassett et al. 2012). Frangakis and colleagues (Geiss et al. 2014) proposed that the compact structure of CENP-A arrays may enable the binding of centromere- and kinetochore-related proteins. However, our results showed that the compact “ladder-like” CENP-A chromatin greatly impaired the binding of CENP-N via concealing the RG loop. As mentioned above, we demonstrated that higher-order chromatin organization at centromeres undergoes a structural transition from a compact chromatin in G1 phase to an open chromatin in S phase. Using a classical SNAP quench pulse chase assay, we verified that CENP-N was stably loaded in middle/late S phase. Interestingly, this structural transition of chromatin is consistent with the dynamic loading of CENP-N onto centromeres during the cell cycle. Thus, we conclude that the cell cycle-dependent loading of CENP-N is highly coordinated with structural transitions of the higher-order structure of centromeric chromatin.
A recent study showed that CENP-A nucleosomes were converted from tetrameric to octameric nucleosomes at the transition from G1 to S phase (Bui et al. 2012). Thus, it is possible that the increased binding of CENP-N to centromeres observed in S phase may be a result of transitional compositions of CENP-A nucleosomes from the tetrameric to the octameric state. However, it was demonstrated earlier that tetrameric CENP-A nucleosomes possess an affinity to CENP-N similar to that of octameric CENP-A nucleosomes (Carroll et al. 2009), supporting our conclusion that the structural transitions of centromeric chromatin, rather than the transitional nucleosome compositions at centromeres, coordinate the temporal loading of CENP-N during the cell cycle. Our finding that the RG loop of CENP-A functions as a switch for the cell cycle-dependent loading of CENP-N onto centromeres/kinetochores via structural transitions of higher-order chromatin organization at centromeres should facilitate further mechanistic understanding of the structure and function of centromeric chromatin.
Materials and methods
Plasmids, proteins, and antibodies
All plasmids used in this study were constructed following standard molecular biology techniques. The expression and purification of proteins and the antibodies used are described in detail in the Supplemental Material.
Cell culture, synchronization, and immunofluorescence
HeLa cells and A03_1 cells were cultured under standard conditions. For cell cycle-dependent analysis, cells were blocked by either hydroxyurea or nocodazole treatment for G1/S and G2/M arrest, respectively. Immunofluorescence assays were performed under standard conditions. Additional experimental details are provided in the Supplemental Material.
LacO/I targeting assays
LacO/I targeting assays were performed as previously described with some modifications (Liu et al. 2012). In brief, plasmids were transfected into cells with LipoD293 (SignaGen) according to the manufacturer's instructions. At 48 h after transfection, cells were collected and stained with DAPI followed by observation with fluorescence microscopy. Additional experimental details are provided in the Supplemental Material.
Mononucleosome immunoprecipitation
The mononucleosome immunoprecipitation assay was performed as previously described with some modifications (Foltz et al. 2006). In brief, native mononucleosomes were prepared by sonication and MNase digestion of 293T cells and immunoprecipitated with the Flag beads. Additional experimental details are provided in the Supplemental Material.
SNAP quench and pulse labeling
Cells were seeded onto glass coverslips in a 24-well plate to minimize the required incubation volumes. SNAP tag activity was pulse-labeled with 3 µM TMR-Star (New England Biolabs, S9105S) or quenched with 2 µM BG block (New England Biolabs, S9106S) in complete growth medium for 30 min at 37°C. Additional experimental details are provided in the Supplemental Material.
SNAP-labeling AB-FRET measurements
FRET measurements were performed as previously described with some modifications (Hellwig et al. 2009; Bui et al. 2012). Cells were labeled with 0.6 µM TMR-Star and 3 µM Oregon Green using the methods described above. Additional experimental details are provided in the Supplemental Material.
DNA, histone, and octamer purification and chromatin assembly
DNA, histone, and octamer purification was performed as previously described (Chen et al. 2013). Additional details are provided in the Supplemental Material.
EM and sedimentation velocity AUC analysis
EM and AUC analyses were performed as previously described with some modifications (Chen et al. 2013). The sedimentation experiments were performed at 18,000 rpm. Additional experimental details are provided in the Supplemental Material.
Nucleosome-binding assay
Nucleosomes were prepared and mixed with His-CENP-N1–289 or His-CENP-C426–943 in different amounts for 1.5 h at room temperature. After incubation, the complexes were analyzed by 6.0% native PAGE gel electrophoresis or 1.0% native agarose gel electrophoresis. Additional experimental details are provided in the Supplemental Material.
Sucrose density gradient sedimentation
Sucrose density gradient sedimentation was performed as previously described with some modifications (Margueron et al. 2008). Additional experimental details are provided in the Supplemental Material.
Graphics and statistics
Multiple sequence alignments were calculated with ClustalX2. Figures were composed with Adobe Illustrator CS5. Error bars indicate ±SD from at least three independent experiments or samples. Student's t-test was used for comparisons of two independent treatments. For all tests, a P-value <0.05 (*) was considered significant, while a P-value <0.01 (**) was considered highly significant.
Supplementary Material
Acknowledgments
We are grateful to Dr. Genevieve Almouzni, Dr. Torsten Juelich, and Dr. Bing Zhu for critical reading and discussion of our manuscript. This work was supported by grants to G.L. from the National Natural Science Foundation of China (91219202), the Ministry of Science and Technology of China (2015CB856200 and 2011CB966300), and the Chinese Academy of Sciences (CAS) Strategic Priority Research Program (XDA01010304); to P.C. from National Natural Science Foundation of China (31471218) and the Key Research Program (KJZD-EW-L05); and to R.-M.X. from the National Natural Science Foundation of China (31210103914). All fluorescence and EM imaging data were collected at the Center for Bioimaging, Core Facility for Protein Sciences, Institute of Biophysics, Chinese Academy of Sciences.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.259432.115.
References
- Allshire RC, Karpen GH. 2008. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9: 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Shirayama K, Horikoshi N, Fujita R, Taguchi H, Kagawa W, Fukagawa T, Almouzni G, Kurumizaka H. 2014. Crystal structure and stable property of the cancer-associated heterotypic nucleosome containing CENP-A and H3.3. Sci Rep 4: 7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MC, Kuich PHJL, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR. 2011. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol 194: 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, DeNizio J, Barnhart-Dailey MC, Panchenko T, Sekulic N, Rogers DJ, Foltz DR, Black BE. 2012. HJURP uses distinct CENP-A surfaces to recognize and to stabilize CENP-A/histone H4 for centromere assembly. Dev Cell 22: 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Cleveland DW. 2011. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell 144: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Jansen LET, Maddox PS, Foltz DR, Desai AB, Shah JV, Cleveland DW. 2007. Centromere identity maintained by nucleosomes assembled with histone H3 containing the CENP-A targeting domain. Mol Cell 25: 309–322. [DOI] [PubMed] [Google Scholar]
- Bui M, Dimitriadis Emilios K, Hoischen C, An E, Quénet D, Giebe S, Nita-Lazar A, Diekmann S, Dalal Y. 2012. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell 150: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Silva MCC, Godek KM, Jansen LET, Straight AF. 2009. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nat Cell Biol 11: 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. 2010. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhao J, Wang Y, Wang M, Long H, Liang D, Huang L, Wen Z, Li W, Li X, et al. 2013. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev 27: 2109–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codomo CA, Furuyama T, Henikoff S. 2014. CENP-A octamers do not confer a reduction in nucleosome height by AFM. Nat Struct Mol Biol 21: 4–5. [DOI] [PubMed] [Google Scholar]
- Conde e Silva N, Black BE, Sivolob A, Filipski J, Cleveland DW, Prunell A. 2007. CENP-A-containing nucleosomes: easier disassembly versus exclusive centromeric localization. J Mol Biol 370: 555–573. [DOI] [PubMed] [Google Scholar]
- Correll SJ, Schubert MH, Grigoryev SA. 2012. Short nucleosome repeats impose rotational modulations on chromatin fibre folding. EMBO J 31: 2416–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Bystricky K, Richmond TJ. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol 327: 85–96. [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. 2009. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137: 485–497. [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Almouzni G, Karpen GH. 2011. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G1 phase. Nucleus 2: 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Cleveland DW. 2013. CENP-A and the CENP nomenclature: response to Talbert and Henikoff. Trends Genet 29: 500–502. [DOI] [PubMed] [Google Scholar]
- Fachinetti D, Folco HD, Nechemia-Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LET, et al. 2013. A two-step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol 15: 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Black BE. 2012. Centromeric chromatin and the pathway that drives its propagation. Biochim Biophys Acta 1819: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JY, Gordon F, Luger K, Hansen JC, Tremethick DJ. 2002. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat Struct Biol 9: 172–176. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Black BE, Bailey AO, Yates JR, Cleveland DW. 2006. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol 8: 458–469. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. 2009. Centromere-specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell 137: 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama T, Henikoff S. 2009. Centromeric nucleosomes induce positive DNA supercoils. Cell 138: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss CP, Keramisanou D, Sekulic N, Scheffer MP, Black BE, Frangakis AS. 2014. CENP-A arrays are more condensed than canonical arrays at low ionic strength. Biophys J 106: 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Allan J. 2001. Distinctive higher-order chromatin structure at mammalian centromeres. Proc Natl Acad Sci 98: 11949–11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Quivy J-P, Almouzni G. 2014. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 83: 487–517. [DOI] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. 2011. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson D, Panchenko T, Salimian KJ, Salman MU, Sekulic N, Alonso A, Warburton PE, Black BE. 2013. The octamer is the major form of CENP-A nucleosomes at human centromeres. Nat Struct Mol Biol 20: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig D, Hoischen C, Ulbricht T, Diekmann S. 2009. Acceptor-photobleaching FRET analysis of core kinetochore and NAC proteins in living human cells. Eur Biophys J 38: 781–791. [DOI] [PubMed] [Google Scholar]
- Hellwig D, Emmerth S, Ulbricht T, Döring V, Hoischen C, Martin R, Samora CP, McAinsh AD, Carroll CW, Straight AF, et al. 2011. Dynamics of CENP-N kinetochore binding during the cell cycle. J Cell Sci 124: 3871–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LET, Black BE, Foltz DR, Cleveland DW. 2007. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 176: 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou B-R, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. 2013. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science 340: 1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Tsai H-J, Tirosh I, Burrack LS, Barkai N, Berman J. 2010. Epigenetically-inherited centromere and neocentromere DNA replicates earliest in S-phase. PLoS Genet 6: e1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste N, Woolfe A, Tachiwana H, Garea Ana V, Barth T, Cantaloube S, Kurumizaka H, Imhof A, Almouzni G. 2014. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol Cell 53: 631–644. [DOI] [PubMed] [Google Scholar]
- Liu C-P, Xiong C, Wang M, Yu Z, Yang N, Chen P, Zhang Z, Li G, Xu R-M. 2012. Structure of the variant histone H3.3–H4 heterodimer in complex with its chaperone DAXX. Nat Struct Mol Biol 19: 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llères D, James J, Swift S, Norman DG, Lamond AI. 2009. Quantitative analysis of chromatin compaction in living cells using FLIM–FRET. J Cell Biol 187: 481–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, Black BE. 2015. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol 208: 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Lyubchenko YL. 2014. Centromere chromatin: a loose grip on the nucleosome? Nat Struct Mol Biol 21: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. 2008. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32: 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland SE, Borusu S, Amaro AC, Winter JR, Belwal M, McAinsh AD, Meraldi P. 2007. The CENP-A NAC/CAD kinetochore complex controls chromosome congression and spindle bipolarity. EMBO J 26: 5033–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM. 2014. Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell 158: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miell MDD, Fuller CJ, Guse A, Barysz HM, Downes A, Owen-Hughes T, Rappsilber J, Straight AF, Allshire RC. 2013. CENP-A confers a reduction in height on octameric nucleosomes. Nat Struct Mol Biol 20: 763–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miell MDD, Straight AF, Allshire RC. 2014. Reply to ‘CENP-A octamers do not confer a reduction in nucleosome height by AFM’. Nat Struct Mol Biol 21: 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S, Almouzni G. 2014. A network of players in H3 histone variant deposition and maintenance at centromeres. Biochim Biophys Acta 1839: 241–250. [DOI] [PubMed] [Google Scholar]
- Müller S, Montes de Oca R, Lacoste N, Dingli F, Loew D, Almouzni G. 2014. Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3CENP-A loading. Cell Rep 8: 190–203. [DOI] [PubMed] [Google Scholar]
- Padeganeh A, Ryan J, Boisvert J, Ladouceur A-M, Dorn JF, Maddox PS. 2013. Octameric CENP-A nucleosomes are present at human centromeres throughout the cell cycle. Curr Biol 23: 764–769. [DOI] [PubMed] [Google Scholar]
- Panchenko T, Sorensen TC, Woodcock CL, Kan Z-Y, Wood S, Resch MG, Luger K, Englander SW, Hansen JC, Black BE. 2011. Replacement of histone H3 with CENP-A directs global nucleosome array condensation and loosening of nucleosome superhelical termini. Proc Natl Acad Sci 108: 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perpelescu M, Fukagawa T. 2011. The ABCs of CENPs. Chromosoma 120: 425–446. [DOI] [PubMed] [Google Scholar]
- Routh A, Sandin S, Rhodes D. 2008. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc Natl Acad Sci 105: 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd MK, Willard HF. 2004. Analysis of the centromeric regions of the human genome assembly. Trends Genet 20: 529–533. [DOI] [PubMed] [Google Scholar]
- Schalch T, Duda S, Sargent DF, Richmond TJ. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436: 138–141. [DOI] [PubMed] [Google Scholar]
- Shelby RD, Monier K, Sullivan KF. 2000. Chromatin assembly at kinetochores is uncoupled from DNA replication. J Cell Biol 151: 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R-M, Zhu P, Li G. 2014. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344: 376–380. [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi-Takanaka Y, Oda T, Sato M, Park S-Y, et al. 2011. Crystal structure of the human centromeric nucleosome containing CENP-A. Nature 476: 232–235. [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Müller S, Blümer J, Klare K, Musacchio A, Almouzni G. 2015. HJURP involvement in de novo CenH3CENP-A and CENP-C recruitment. Cell Rep 11: 22–32. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2013. Phylogeny as the basis for naming histones. Trends Genet 29: 499–500. [DOI] [PubMed] [Google Scholar]
- Tomonaga T, Matsushita K, Yamaguchi S, Oohashi T, Shimada H, Ochiai T, Yoda K, Nomura F. 2003. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res 63: 3511–3516. [PubMed] [Google Scholar]
- Walkiewicz MP, Dimitriadis EK, Dalal Y. 2014. CENP-A octamers do not confer a reduction in nucleosome height by AFM. Nat Struct Mol Biol 21: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidtkamp-Peters S, Rahn H-P, Cardoso M, Hemmerich P. 2006. Replication of centromeric heterochromatin in mouse fibroblasts takes place in early, middle, and late S phase. Histochem Cell Biol 125: 91–102. [DOI] [PubMed] [Google Scholar]
- Yu Z, Zhou X, Wang W, Deng W, Fang J, Hu H, Wang Z, Li S, Cui L, Shen J, et al. 2015. Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Dev Cell 32: 68–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.