

Abstract
Rhodium and molybdenum catalyzed allenic [2 + 2 + 1] cycloaddition reactions give 4-alkylidene and α-alkylidene cylopentenones, respectively. The selective reaction of one double bond of the allene over another is controlled by the transition metal and not the substrate structure. Calculations were performed to explain this unique control element using the B3LYP functional as implemented in Gaussian 03. The 6-31G(d) basis set was applied to all elements except rhodium, which is described with the LANL2 effective core potential and the LANL2DZ basis set. The product-determining step for both reaction pathways is oxidative addition of the metal to the alkynyl allene to form the corresponding metallocycles B and B′. The transition state calculations strongly suggest that geometry constraints imposed by the metal in the transition state are the key controlling factor of the double bond selectivity. The transition state structure of rhodium-catalyzed oxidative addition has a distorted square planar geometry that affords a lower transition state energy when coordinated to the distal double bond of the allene. In turn, the distorted trigonal bipyramidal geometry of molybdenum in the transition state structure imposes conformational constraints upon binding to the distal double on the allene and thus leads to the energetically preferred complexation and reaction with the proximal double bond.
Transition metal catalyzed cyclocarbonylation reactions of allenes provide rapid entry into molecular skeletons that may otherwise be difficult to synthesize. However, advantages of incorporating an allene moiety into a synthetic sequence can be negated by selectivity issues associated with this underutilized functional group.1 Recently, we observed that during a cyclocarbonylation reaction of an allene, only one of the double bonds reacted, depending upon the metal catalyst employed.2 The selective reaction of one double bond over the other is precedented,3 but in most cases the substrate structure, not the reagent, is the controlling factor. We have subsequently taken advantage of the new transition metal catalyzed control element by applying this to the first synthetically feasible [2 + 2 + 1] approach to bicyclo[5.3.0]decanes.4 In addition, we have also shown that this regiocontrol element can be applied to other types of transition metal catalyzed cyclocarbonylation reactions.5
Since this regioselectivity trend appears to be general we were eager to understand the nature of this transition metal directed control element. As a first step in our approach, we sought to understand the selectivity via theoretical investigations,6 since the information gained from these calculations could also guide us in designing additional laboratory experiments. The mechanism outlined in Figure 1 provides the basis for our analysis. Initially, some assumptions were made concerning the generally accepted mechanism of this transition metal catalyzed [2 + 2 + 1] cycloaddition. It was assumed that the first step in the cycloaddition reaction involves complexation of the alkyne and the allene to the catalyst giving either intermediate A or A′. Next, oxidative addition affords the metallocyles B or B′, which undergo carbon monoxide insertion to yield the products C or C′, respectively. Finally, reductive elimination provides either the α-alkylidene cyclopentenone D or 4-alkylidene cyclopentenone D′, depending upon the metal catalyst employed in the reaction.
Figure 1.
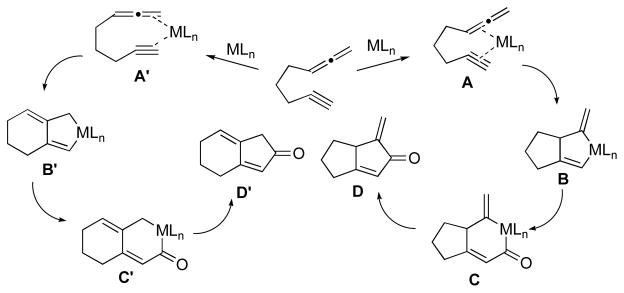
Proposed mechanism of allenic [2 + 2 + 1] cyclocarbonylation
Potential energy pathways for the proposed mechanisms were mapped out using density functional theory with the B3LYP functional implemented in Gaussian 03 to establish the most likely rate-determining step. The composition of the rhodium complex was chosen based upon experiment whereby a variety of rhodium(I) catalysts were screened and all gave similar results. Thus, it was reasoned that the additional complexity of introducing ligands other than carbon monoxide was not beneficial or necessary. For the rhodium-based catalyst it was determined that the oxidative addition step had the highest energy of activation and was the least reversible. Thus, product formation is determined early on along the reaction coordinate and our attention was focused on the transition states for the oxidative addition step of the rhodium catalyzed reaction.
The free energy landscape for the rhodium-catalyzed reaction is shown in Figure 2.7 The first step of the reaction is the oxidative addition of the alkynyl allene to the rhodium catalyst leading to metallocycle 1a. This step is rate-limiting with a free energy barrier of 16.8 kcal/mol. The relative free energies were calculated at 90 °C, the temperature at which the reactions were performed. Carbonyl insertion and reductive elimination steps giving 2 and 3 have calculated free energy barriers of 1.0 and 11.5 kcal/mol, respectively. These last two steps are energetically more feasible if an additional CO ligand is added to the metal complex.
Figure 2.

Energy landscape for rhodium catalyzed reaction.8 Parts of the pathway where there is an extra CO present are shown in blue.
The free energy landscape for the molybdenum catalyzed process is shown in Figure 3. The composition of the molybdenum complex is based upon experiment, where molybdenum aryl tricarbonyl complexes are used to catalyze the same CO insertion reaction. The rate determining step was found to be the insertion of CO into the metallocycle 6. This differs from the rhodium catalyzed process where the oxidative addition step was rate-determining. Interestingly, the free energy of the transition state for the CO insertion is still 2.5 kcal/mol lower than that for the oxidative addition of the metal to the distal double bond of the allene. Moreover, all low energy transition states for the oxidative addition step involve the formation of a metallocycle via a selective reaction with the proximal double bond of the allene. Finally, the reductive elimination step is energetically a very favorable process.
Figure 3.

Energy landscape for molybdenum catalyzed reaction.8 Parts of the pathway where there is an extra CO present are shown in blue.
Extensive searches were performed on the oxidative addition step of the rhodium and molybdenum catalyzed reactions and the results of these calculations are depicted in Figures 4 and 5, respectively. Interestingly, for the molybdenum process all low energy complexation species involved the proximal double bond of the allene (compare 7a and 11, Figure 5). The same was true for the transition states (compare ts7a-6 and ts11-13, figure 5). For the rhodium case, all low energy complexes involved the distal double bond of the allene (compare 4a and 12a, Figure 4) and likewise for the transition states (compare ts4a-1a and ts12a-14, figure 4). For both rhodium and molybdenum metallocycles, the larger sized ring is preferred over the smaller ring. (For molybdenum compare 13 and 6, Figure 5 and for rhodium compare 1a and 14, Figure 4.) As a consequence, the energy difference of the molybdenum and rhodium catalyzed reactions is reflected in the transition states of oxidative addition step. For example, in the rhodium case the conversion of the complex to the metallocycle is an exothermic process by ~15.6 kcal/mol and proceeds via an early transition state. Thus, transition structure ts4a-1a possesses a slightly distorted square planar geometry, nearly the same as the starting complex 4a. The geometry constraints imposed by the metal in the transition state preclude complexation of the proximal double bond of the allene, leading to an energetically preferred complexation with the distal double bond, which in turn leads to the thermodynamically favored metallocycle 1a. Alternatively, conversion of the complexed molybdenum species 7a to the molybdenum metallocycle 6 is a slightly endothermic process with a later transition state. The trigonal bipyramidal geometry of metal in this transition state imposes conformational constraints upon binding to the distal double on the allene and thus leads to the energetically preferred complexation with the proximal double bond, leading to the kinetically preferred 6. It is important to point out that experimentally iridum gives very similar results to rhodium and tungsten gives the same regioselectivities as molybdenum, further supporting the conclusion that it is the transition state geometry of the metal that drives the selectivity of this reaction.
Figure 4.

Rhodium catalyzed oxidative addition.8
Figure 5.

Molybdenum catalyzed oxidative addition.8
In conclusion, computational evidence is provided to explain an unusual selective reaction of either of the double bonds of the allene in an intramolecular allenic [2 + 2 + 1] cycloaddition.
Supplementary Material
Acknowledgments
We thank the NSF (CHE0518253) and the NIH (GM54161) for financial support and KMB thanks Johnson & Johnson for a Focused Giving Award and the University of Pittsburgh for a Chancellor’s Distinguished Research Award. Further thanks go to Professor Tara Meyer for helpful discussions. The calculations were carried out on computers in the University of Pittsburgh’s Center for Molecular and Materials Simulations.
Footnotes
Supporting Information Available. Computational details and references, including structures, energies and vibrational frequencies of all stationary points are provided in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org.
Contributor Information
Kay M. Brummond, Email: kbrummon@pitt.edu.
Kenneth D. Jordan, Email: jordan@pitt.edu.
References
- 1.Hashmi ASK. Angew Chem, Int Ed Eng. 2000;39:3590. [PubMed] [Google Scholar]
- 2.Brummond KM, Chen H, Fisher KD, Kerekes AD, Rickards B, Sill PC, Geib SJ. Org Lett. 2002;4:1931. doi: 10.1021/ol025955w. [DOI] [PubMed] [Google Scholar]
- 3.For a catalyst controlled [4 + 2] diene-allene cycloaddition, see: Wender PA, Jenkins TE, Suzuki S. J Am Chem Soc. 1995;117:1843.
- 4.Brummond KM, Gao D. Organic Lett. 2003;5:3491. doi: 10.1021/ol035322x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Brummond KM, Chen H, Sill PC, You L. J Am Chem Soc. 2002;124:15186. doi: 10.1021/ja027588p. [DOI] [PubMed] [Google Scholar]; (b) Brummond KM, Chen H, Mitasev B, Casarez AD. Organic Lett. 2004;6:2161. doi: 10.1021/ol049390a. [DOI] [PubMed] [Google Scholar]; (c) Brummond KM, Mitasev M. Organic Lett. 2004;6:2245. doi: 10.1021/ol0492391. [DOI] [PubMed] [Google Scholar]
- 6.For an application of DFT calculations to: an intermolecular [5 + 2] reaction, see Yu ZX, Wender PA, Houk KN. J Am Chem Soc. 2004;126 doi: 10.1021/ja048739m.91254 and for an application to an intermolecular [4 + 2 + 2] reaction, see Baik MH, Baum EW, Burland MC, Evans PA. J Am Chem Soc. 2005;127:1602. doi: 10.1021/ja043521l.
- 7.All stationary points were located and vibrational analysis were calculated using the B3LYP functional as implemented in Gaussian 03. The 6-31G(d) basis set was applied to all elements except Rh, which was described with the LANL2 effective core potential and the LANL2DZ basis set. Relative free energies were calculated at a temperature of 90 °C which was used in the experiments. Citations of the computational methods and references are given in Supporting Information.
- 8.All energies used in figures are Gibbs free energies in kcal/mol and were calculated in the absence of solvent. Analogous calculations on an azepine systemin refluxing THF were performed using COSMO9 and the solvent did not have a significant effect on the outcome of these calculations. Unpublished results from our laboratory.
- 9.Klamt A, Schüürmann G. J Chem Soc Perkin Trans. 1993;2:799. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.