

Abstract
We investigated diverse genomic selections using high-density single nucleotide polymorphism data of five distinct cattle breeds. Based on allele frequency differences, we detected hundreds of candidate regions under positive selection across Holstein, Angus, Charolais, Brahman, and N'Dama. In addition to well-known genes such as KIT, MC1R, ASIP, GHR, LCORL, NCAPG, WIF1, and ABCA12, we found evidence for a variety of novel and less-known genes under selection in cattle, such as LAP3, SAR1B, LRIG3, FGF5, and NUDCD3. Selective sweeps near LAP3 were then validated by next-generation sequencing. Genome-wide association analysis involving 26,362 Holsteins confirmed that LAP3 and SAR1B were related to milk production traits, suggesting that our candidate regions were likely functional. In addition, haplotype network analyses further revealed distinct selective pressures and evolution patterns across these five cattle breeds. Our results provided a glimpse into diverse genomic selection during cattle domestication, breed formation, and recent genetic improvement. These findings will facilitate genome-assisted breeding to improve animal production and health.
Keywords: cattle genome, population structure, positive selection, haplotype network
Introduction
As one of most important farm animals, cattle are used for a variety of purposes including dairy, beef, hide production, and labor. The majority of the global cattle population can be classified into one of two groups: Humpless (taurine) and humped (indicine or zebu) cattle with dramatic phenotypic differences (Bradley et al. 1996; Bovine HapMap Consortium 2009). Earlier studies indicated that these two subspecies diverted from the last common ancestor between 275,000 and 850,000 years ago (MacHugh et al. 1997; Hiendleder et al. 2008). Each subspecies appeared to be separately domesticated with taurine cattle in the Fertile Crescent approximately 10,000–8,000 years ago and indicine cattle in the Indus Valley approximately 6,000–8,000 years ago (Loftus et al. 1994; Larson et al. 2014). A third independent domestication was proposed in Africa (Troy et al. 2001); however, a recent result argued against it (Decker et al. 2014). After these domestication events, continuous human selective breeding for desirable traits like easy management and high production has been more documented in taurine cattle than in indicine cattle. Since the early 1800’s, breed development was based on phenotype selection on coat color and polled phenotypes, and included the imposition of severe bottlenecks followed by breed expansion via artificial insemination. During the last 50 years, animal breeding based on quantitative genetics has resulted in a remarkable progress in improving production traits for milk and meat (Andersson and Georges 2004). Therefore, selection (natural and human-imposed) and nonselective forces (the demographic events and introgression) drove changes within the cattle genome. Their combined effects have created exceptional phenotypic diversity and genetic adaptation to local environment across the globe within the modern cattle breeds.
It is generally accepted that there are four mechanisms of evolutionary change: Mutation, genetic drift, gene flow or migration (demographic history), and selection. However, only selection is locus specific, while the first three forces work uniformly across the whole genome. Selection can be divided into three modes: Positive, purifying (or negative selection, eliminating a deleterious mutation), and balancing selection (including heterozygote advantage and frequency-dependent selection). Positive selection is a mode of natural selection that drives the increase in prevalence of advantageous alleles due to their favorable effects on fitness (Biswas and Akey 2006; Kelley and Swanson 2008; Oleksyk et al. 2010). Genetic hitchhiking refers changes in the frequency of an allele because of linkage with a positively selected or neutral allele at another locus. The availability of genomic data has spurred many approaches for mapping positive selection, mainly based on reduced local variability, deviations in the marker frequency, increased linkage disequilibrium (LD), and extended haplotype structure. These methods such as CLR, CMS, FST, EHH, iHS, and hapFLK (Tajima 1989; Fay and Wu 2000; Sabeti et al. 2002; Nielsen et al. 2005; Voight et al. 2006; Grossman et al. 2010; Fariello et al. 2013) have been widely used in human, mouse, rat, and domesticated animals like dogs, cattle, sheep, pigs, horses, and chickens (Waterston et al. 2002; Gibbs et al. 2004; Rubin et al. 2010, 2012; Kijas et al. 2012; Petersen et al. 2013). One method (di) was recently developed to identify genomic regions indicative of selection with a high degree of genetic differentiation between dog breeds (Akey et al. 2010). Distinct from FST, which measures the fraction of total genetic variation between two populations, the di value is defined as a function of unbiased estimates of all pairwise FST between one breed and the remaining breeds within a population. It is suited for detecting selection specific to a particular breed, or subset of breeds, and isolating the direction of change. It was utilized to track lineage-specific signatures of selection in the dog and horse genomes, revealing its power to detect selection acting on both newly arisen and preexisting variations (Akey et al. 2010; Petersen et al. 2013).
Selection mapping is a powerful approach, together with genome-wide association studies, to detect candidate genes associated with quantitative traits. Selection mapping in cattle has been previously investigated using a lower density markers like BovineSNP50 array (Flori et al. 2009; Hayes et al. 2009; Qanbari et al. 2010, 2011; Stella et al. 2010; Rothammer et al. 2013). Only recently similar studies were reported based on a higher density markers like BovineHD array (Porto-Neto et al. 2013; Utsunomiya et al. 2013; Kemper et al. 2014; Perez et al. 2014). More recently, sequence-based signatures were reported in Fleckvieh (Qanbari et al. 2014). However, these studies focused on limited breeds with specific traits. Therefore, it is possible many breed-specific selection signatures remain undetected due to lack of comparison across breeds. There are a few of targeted studies of the haplotype pattern and evolution on selected gene families like Toll-like receptors in cattle (Seabury et al. 2010). However, to our knowledge, no systematic effort has been reported to investigate the haplotype pattern and evolution of positively selected genes in the cattle genome.
In this study, we investigated diverse genomic selection using high-density single nucleotide polymorphism (SNP) data of five distinct cattle breeds, including Holstein (HOL), Angus (ANG), Charolais (CHL), Brahman (BRM), and N'Dama (NDA). HOL, ANG, and CHL are taurine breeds from Europe. HOLs represent the highest-production dairy animals, originally from the Netherlands and northern Germany. Their black-and-white color was due to artificial selection by the breeders. ANG cattle, first developed in Scotland, are used in beef production. They are naturally polled (do not have horns) and solid black or red in color. CHL is a dual purpose breed (both milk and beef) originated in France, which is known for its large body size, bone structure, and white to cream coat. NDA is an indigenous local taurine breed from West Africa. With a small size and fawn coat, NDA is well known for its trypanotolerant and shows superior resistance to ticks and other parasites (http://www.ansi.okstate.edu/breeds/cattle/, last accessed December 2, 2014). BRM is a composite of several zebu breeds imported from India (Guzerat, Kankrej, Gir, and others), and was first bred in America in the 1880s for beef production with a minor taurine contribution (Decker et al. 2014). The BRM is known for its gray coat, heat tolerance, and disease resistance. We performed a genome-wide scan with the BovineHD SNP genotypes to map selection signatures among these five diverse cattle breeds. Selected regions were further independently validated by next-generation sequencing. We also conducted a genome-wide association analysis of milk production traits for 26,362 HOLs and found that regions under selection were likely functional. In addition, we investigated haplotype pattern and evolution of positively selected genes in the cattle genome.
Results
Genetic Diversity, Breed Relationship, and LD
We chose five genetically diverse and geographically distinct breeds to systematically investigate the selection signatures in cattle (supplementary table S1, Supplementary Material online), including HOL, ANG, CHL, BRM, and NDA. Several quality control steps were used to assure SNP data quality. These filters included missing data per individual, missing data per marker, and allele frequency. A total of 710,681 SNPs were kept after these QC steps. We estimated the average heterozygosity using the filtered SNPs within each breed, and results indicated that significant genetic diversities exist among five breeds. The average heterozygosities for the three European taurine breeds were just over 0.3 for HOL, ANG, and CHL whereas they were lower for the indicine breed BRM (0.25) and the African taurine breed NDA (0.22) (supplementary table S2, Supplementary Material online). The inbreeding coefficient, F, was also estimated for the five breeds. The average values of F were less than 0.1 for the three European taurine breeds, just over 0.25 for BRM and NDA (supplementary table S2, Supplementary Material online).
Multidimensional scaling (MDS) analysis revealed a typical triangular relationship among the five breeds from three continents, with the first dimension separating the taurine and indicine breeds, and the second dimension separating the European taurine from African taurine (supplementary fig. S1A, Supplementary Material online). The well-separated clusters indicated that the chosen samples were powerful to explore the genomic characteristics of these five breeds. Admixture analysis was also performed using 72,945 LD-filtered SNPs to identify ancestry and admixture level among the five breeds when the number of clusters, K, varied from 2 to 5. When K = 2, the clustering pattern reflects the primary, predomestication division of taurine from indicine cattle. When K = 3, NDA separates from the European breeds, suggesting a early divergence. When K = 5, the five breeds can be unequivocally separated into five closed endogamous breeding units with expected levels of admixture (supplementary fig. S1B, Supplementary Material online). We also used PLINK to construct a neighbor-joining tree based on pairwise nucleotide genetic distances as shown in supplementary fig. S1C, Supplementary Material online (Saitou and Nei 1987; Purcell et al. 2007). Two main features are obvious: 1) cattle from the same breed always cluster together and 2) little structure is observed in the internal branches within a breed. The patterns were consistent with the fact that modern breeds were derived from population bottlenecks with closed gene pools. Modern breed formation happened within a short period of time, and a certain amount of genetic variations exist within a breed. We also analyzed LD decay patterns in the five breeds. We found that LD in cattle genome generally decays rapidly and the decay rates could vary greatly among breeds and chromosomes, as reported previously (Bovine HapMap Consortium 2009; Espigolan et al. 2013; Qanbari et al. 2014).
Candidate Regions and Genes under Positive Selection
We used selection mapping to explore the differences among breeds selected for different purposes (milk or beef). We calculated average di values in a nonoverlapping sliding window way for all 11,651 widows of 50 neighboring SNPs. The average length and standard deviation (SD) of the 50-SNP sliding windows was 212 ± 110 kb. The genome-wide distribution of the window di values is shown in figure 1. For each breed, we defined candidate selection regions using two thresholds: The top 1% or 5% windows with highest average di values in the empirical distribution (Voight et al. 2006; Pickrell et al. 2009). Thus, a total of 117 or 583 windows were identified under these two criteria for each breed (supplementary table S3, Supplementary Material online). To keep a high specificity, our results were mainly based on the top 1%, that is, 117 windows unless stated otherwise.
Fig. 1.

Genomic distribution of selection regions in five cattle breeds. The distribution of di for each 50-SNP windows across all auto chromosomes is shown for each breed. Alternating color indicate di values from adjacent chromosomes. Breeds are abbreviated as described in supplementary table S1, Supplementary Material online.
To investigate the shared or breed-specific selection regions, we plotted a Venn diagram (fig. 2) and a Circos plot (supplementary fig. S3, Supplementary Material online) (Zhang et al. 2013), and counted the numbers of overlapping windows among the five breeds. Out of 470 merged nonredundant windows, 383 windows (∼81.5%) were unique to only one breed. On the other hand, 87 windows (∼21.2%) were shared by two or more breeds, and within them 24 windows (∼5.1%) were shared by three or more breeds.
Fig. 2.

Venn diagram for shared versus breed-specific selection events among five cattle breeds. Top 1% (117) windows of the average di values were compared across five breeds.
Based on the bovine RefSeq gene annotation, there were 44, 38, 44, 39, and 23 windows, which did not overlap with any gene in HOL, ANG, CHL, BRM, and NDA, respectively (supplementary table S3, Supplementary Material online). We then focused on the left windows which overlap with genes. Out of a total of 13,775 genes, we detected 1,133 and 3,480 genes (8.23% and 25.26%) within the top 1% and 5% windows, respectively (table 1 and details in supplementary table S3, Supplementary Material online). And both of them were significantly enriched for gene content as determined in our 10,000 permutation tests. We further performed DAVID analyses on breed-specific loci for each breed separately, with genes in shared windows excluded (supplementary table S4, Supplementary Material online). Interestingly, we found a number of functional clusters that were significantly enriched among these breeds. In ANG, the top two enriched clusters were defense response and the voltage-gated potassium channel complex. In HOL, enrichments were found for lymphocyte activation, protein kinase activity, ATP binding, ubiquitin conjugation, and mitochondrial lumen and matrix. In CHL, transcription coactivator activity, calcium binding, and protein ubiquitination were overrepresented. In NDA, significantly enriched clusters were for T-cell activation, hematopoiesis, carbohydrate catabolic process, and bone, skeletal, and embryo development. Finally, BRM showed distinct, extensive and strong enrichments for 12 clusters, including cytoskeleton, adherens junction, intracellular organelle lumen, activities for arylesterase, peptidase, ligase, protein dimerization and adenyl nucleotide binding, and response to DNA damage stimulus and stress.
Table 1.
Selected Genes under Positive Selections.
| Gene | Long Name | Breed | Genome Position | Traits | Cattle Studies | Studies in Other Species |
|---|---|---|---|---|---|---|
| Known genes in cattle | ||||||
| LAP3 | Leucine aminopeptidase 3 | CHL | chr6:38,574,590–38,600,027 | Milk production traits | Flori et al. 2009 | |
| LCORL | Ligand dependent nuclear receptor corepressor-like | CHL | chr6:38,840,864–38,992,112 | Body size | Flori et al. 2009 | Rubin et al. 2012; Petersen et al. 2013 |
| KIT | V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | CHL; HOL | chr6:71,796,318–71,917,430 | Coat color | Stella et al. 2010 | Kijas et al. 2012; Rubin et al. 2012 |
| MC1R | Melanocortin 1 receptor | ANG; BRM; CHL; HOL | chr18:14,757,332–14,759,082 | Coat color | Flori et al. 2009 | Petersen et al. 2013 |
| NUDCD3 | NudC domain-containing protein 3 | HOL | chr4:77,598,538–77,670,088 | Mitosis | Flori et al. 2009 | |
| WIF1 | WNT inhibitory factor 1 | ANG; BRM; CHL | chr5:48,917,722–49,009,466 | Mammalian mesoderm segmentation | Bovine HapMap Consortium et al. 2009 | |
| SAR1B | SAR1 homolog B | HOL | chr7:47,717,605–47,747,828 | Milk production traits | Larkin et al. 2012 | |
| ASIP | Agouti signaling protein | ANG; HOL | chr13:64,213,312–64,239,962 | Coat color | Norris and Whan 2008 | |
| LRIG3 | Leucine-rich repeats and immunoglobulin-like domains protein 3 | CHL | chr5:54,884,394–54,936,361 | Elongated body axis | Akey et al. 2010 | |
| Known genes in other domesticated animals: | ||||||
| FGF5 | Fibroblast growth factor 5 | BRM | chr6:96,723,382–96,746,176 | Coat color | Cadieu et al. 2009 | |
| OSTN | Osteocrin | CHL; HOL | chr1:76,685,699–76,721,920 | An inhibitor of osteoblast | Rubin et al. 2012 | |
| NCAPG | Non-SMC condensin I complex, subunit G | CHL | chr6:38,765,969–38,812,055 | Body size | Petersen et al. 2013 | |
| ABCA12 | ATP-binding cassette, subfamily A (ABC1), member 12 | BRM | chr2:103,520,024–103,720,887 | Ichthyosis fetalis; skin keratinization, calf birth weight | Charlier et al. 2008; Cole et al. 2014 | Tennessen and Akey 2011 |
| ACTN3 | Actinin, alpha 3 | BRM | chr29:45,230,630–45,242,406 | Skeletal and cardiac muscle | Gu et al. 2009; MacArthur et al. 2007 | |
| NPR2 | Atrial natriuretic peptide receptor 2 precursor | BRM | chr8:60,368,097–60,379,016 | Skeletal morphology and body size | ||
| Kijas et al. 2012 | ||||||
| chr8:60,381,937–60,385,961 | ||||||
Genes under Positive Selection
We identified genes from positive selection regions and then compared them with published literature. We categorized them into three classes. Class 1, genes reported before in cattle: We detected many genes which have been previously identified under positive selection in cattle (table 1; Bovine HapMap Consortium 2009; Flori et al. 2009; Hayes et al. 2009; Stella et al. 2010; Larkin et al. 2012). We found two well-known genes related to coat color, that is, KIT in CHL and HOL and MC1R in ANG, BRM, CHL, and HOL. We also identified NUDCD3 in HOL and WIF1 in ANG, BRM, and CHL, which may be involved in cell division and developmental pathways (Zhou et al. 2006; Cai et al. 2009). In addition, LAP3 (leucine aminopeptidase 3) and LCORL (ligand-dependent nuclear receptor corepressor-like) were detected in CHL, which are associated with milk production traits and body size, respectively (Flori et al. 2009; Pryce et al. 2011). We further detected gene SAR1B (SAR1 homolog B) under selection in HOL, which was briefly reported to be associated with milk production and disease resistance in dairy cattle (Larkin et al. 2012). Class 2, genes reported before in other domesticated animals: We discovered some genes which were previously reported to be under positive selection in other domesticated species but not in cattle. These included coat color related genes (ASIP and FGF5), body size-related genes (NPR2, NCAPG, and LRIG3), and skeletal muscle-related gene (OSTN). Class 3, genes reported bin human: We further detected that the following bovine genes, whose human ortholog genes were previously reported to before under selection in human populations. For example, signals of selective sweeps of NTRK2, CNGA3, ETFA, ISL2, KCND2, and SYN3 were found in human populations, according to geographic locations (Pickrell et al. 2009). Evidence for positive selection of IRAK3, LLPH, TMBIM4, BTBD11, GCK, AKAP3, and CISH were reported in African populations (Granka et al. 2012; Jarvis et al. 2012). Four blood pressure related genes (CLCN6, KIAA2013, FBXO2, and FAM117A) were identified in a human genome-wide association study (Newton-Cheh et al. 2009). Promoter regions of many neural- and nutrition-related genes (RPL37A and GLMN) also experienced positive selection during human evolution (Haygood et al. 2007).
When we relaxed the threshold from 1% to 5%, we obtained more previously reported bovine genes. For example, GHR (growth hormone receptor) on chr20 has effects on milk yield and composition in dairy cow (Blott et al. 2003; Viitala et al. 2006) and body size in dog (Rimbault et al. 2013, Wiener and Wilkinson 2011). Other additional genes are related to muscle formation and fatty (ACTC1, FABP3, and HBEGF) (Gerbens et al. 1997; Rahman et al. 2010; Stella et al. 2010; Qanbari et al. 2012); mammary gland and daily production (ITFG1, ITGB3, RTN4, SULT1E1, and TMCC3) (Lemay et al. 2009; Barreiro and Quintana-Murci 2010; Stella et al. 2010; Larkin et al. 2012); and immunity and nervous system (CD79A, NEUROD6, and SPOCK1) (Kosiol et al. 2008; Bovine HapMap Consortium 2009; Gautier et al. 2009). If we include published results from other species, we found more genes related to coat color (KRT71 and RSPO2) (Cadieu et al. 2009), body size (BMP2, IGF1) (Sutter et al. 2007; Kijas et al. 2012; Petersen et al. 2013), nervous system (MBP, SMO, and TLX3) (Axelsson et al. 2013), and immune process (CCR2) (Kijas et al. 2012).
Validation by Next-Generation Sequencing
To validate high-density SNP array results, we carried out additional analyses based on next-generation sequencing. Because LAP3 was reported before to be under positive selection in HOL (Flori et al. 2009) and we also identified LAP3 in CHL and CHL is a dual purpose breed (both milk and beef), we chose to investigate selection signature in HOL near the LAP3 gene. Using SNP calls generated from next-generation sequencing (NGS) data, we checked the 24.1-kb region (92 SNPs) flanking the LAP3 locus in HOL and ANG. We plotted MAF and calculated FST between HOL and ANG to look for high degrees of breed differentiation. Their trends agreed well with our di results derived from the BovineHD array. As expected, there was an excess of high FST values among the 92 SNPs, although FST values varied to a certain degree (fig. 3). The elevated FST was predominantly due to SNPs located near LAP3, with allele frequencies that were generally higher in HOL than in ANG. This elevated FST revealed and confirmed the signature of selection near the LAP3 gene.
Fig. 3.
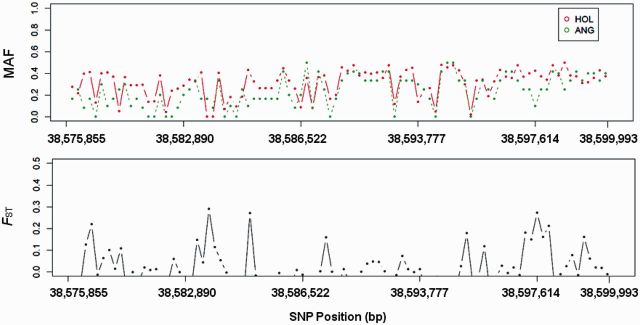
MAF and FST plot for LAP3 genes between HOL and ANG using 92 SNPs extracted from next-generation sequencing.
Association Tests
To further validate the genes involved in selection for milk production, we performed association tests using 26,362 BovineSNP50 array data from HOL to test whether LAP3 and SAB1B were related to five milk production traits (milk yield [MY], fat yield [FY], protein yield [PY], fat percentage [FP], and protein percentage [PP]). Within a 652-kb region containing 12 SNPs from the BovineSNP50 array, LAP3 gene located between the sixth (Hapmap26308-BTC-057761) and seventh SNPs (Hapmap43470-BTA-114677) (fig. 4). We identified 5, 5, 3, 5, and 1 significant SNPs associated with MY, FY, PY, FP, and PP traits, respectively (-log10 P value ≥ 7 as the threshold). Based on the high-density HOL BovineHD data, we observed two large haploblocks in strong LD with the same 652-kb region. Among them, one 155 kb block from BovineHD0600010649 to BovineHD0600010693 surrounding LAP3 also contains the sixth (Hapmap26308-BTC-057761) and seventh SNPs (Hapmap43470-BTA-114677), which were significantly associated with FP (fig. 4). Similar haplotype LD patterns were found in the neighboring region of 600 kb (12 SNPs) around SAR1B (supplementary fig. S4, Supplementary Material online). We identified 2, 2, 2, 1, and 1 associated SNPs for MY, FY, PY, FP, and PP, respectively. There were three long and tightly linked neighboring haploblocks in the proximity of SAR1B. The sixth SNP (BTB-01846474) was also significantly associated with FP (supplementary fig. S4, Supplementary Material online).
Fig. 4.
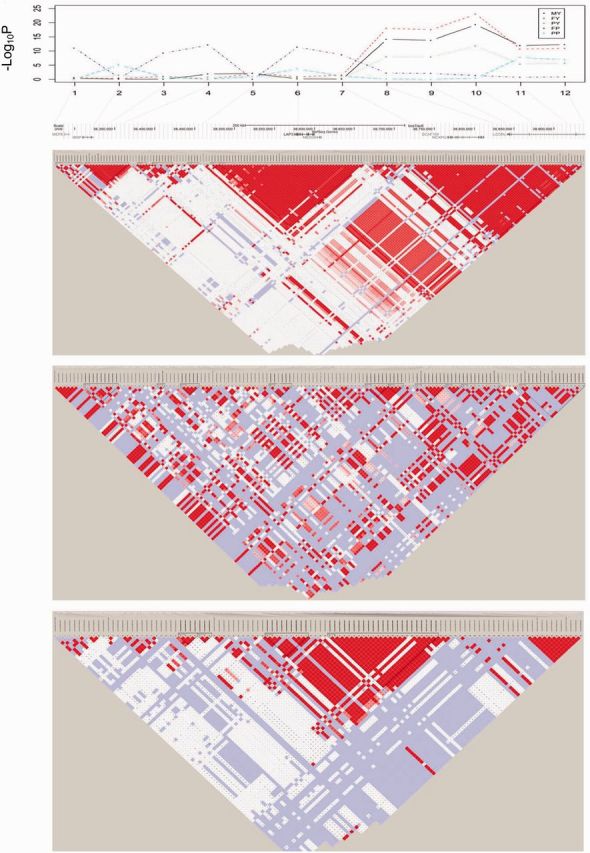
Association test of LAP3 with HOL milk production traits and haplotype LD analyses in HOL, BRM, and NDA.
Haplotype Homozygosity
One genetic signature of an incomplete selective sweep is a region of extensive LD (termed extended haplotype homozygosity or EHH) and low variation on high-frequency chromosomes carrying the derived beneficial mutation relative to chromosomes with the ancestral allele. Therefore, we compared the haploblock LD patterns around LAP3 and SAR1B and found dramatic differences among the five breeds. We calculated D′ and r2 between SNPs to evaluate the LD pattern in these regions. Pairwise comparisons show that strong LD within sites over short distance. For the locus near LAP3, we detected an extensive high LD level in HOL as compared with BRM and NDA (fig. 4). It is worthwhile to note that NDA display an intermediate LD level between HOL and BRM. BRM’s haploblocks seem short and not tightly linked in these regions. A similar LD pattern was found in the locus around SAR1B (supplementary fig. S4, Supplementary Material online).
The recombination rate is another important parameter, which influences the LD level and the strength and frequency of positive selection. We estimated fine-scale recombination rates based on approximate conditionals model near these genes. Although we did detect dramatic differences in recombination rates across five breeds near genes like GHR and ABCA12 (supplementary fig. S5, Supplementary Material online), all breeds showed little recombination near LAP3 and SAR1B. In summary, although the differences between HOL and BRA could be related to ancient events like subspeciation, the comparison between HOL and NDA agree well with the hypothesis that the selected allele and its haplotype could rise rapidly in frequency through genetic hitchhiking in the process of a strong selective sweep.
Haplotype Network Analysis
We further investigated haplotype pattern and evolution to further characterize selective pressures near selected genes including LAP3 and SAR1B. We obtained 131 haplotypes within the 175.7 kb haploblock region near LAP3. We detected the top four haplotypes: H1, H2, H3, and H4 with a frequency of 13.41%, 7.33%, 7.04%, and 6.12%, respectively (fig. 5). Among them, top haplotype H1 contained CHL (53.13%), HOL (1.41%), and ANG (8.84%). H2 contained only NDA (59.03%), but not any of the other four breeds. H3 contained HOL (17.18%), ANG (6.31%), and CHL (5.37%). We also observed three exclusive haplotypes for NDA. Fourteen haplotypes clustered together only in the BRM samples. Altogether, this pattern indicated separate haplotypes were clustered only for BRM or NDA, while overlapping haplotypes were identified for HOL, ANG, and CHL. It is interesting to note that most BRM haplotypes cluster exclusively together in one individual branch.
Fig. 5.
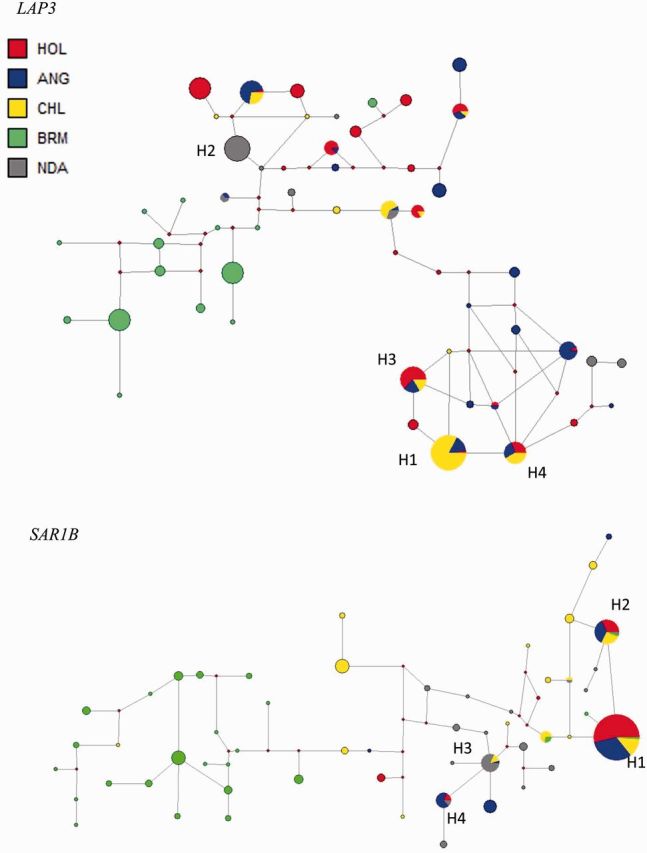
Haplotype networks of two loci. (A) LAP3 and (B) SAR1B. Each node represents a different haplotype, with the size of the circle proportional to frequency. Branch lengths are proportional to the number of nucleotide differences. Circles are color coded according to population (red: HOL, blue: ANG, yellow: CHL, green: BRM, and gray: NDA).
We obtained 95 haplotypes within the 404.0 kb haploblock region near SAR1B. The top haplotype, H1 (with frequency of 37.32%), was found in HOL (77.29%), ANG (52.54%), CHL (23.15%), and BRM (1.67%) (fig. 5). H2 (with frequency of 10.90%) included HOL (13.62%), ANG (16.74%), CHL (14.73%), BRM (1.67%), and NDA (1.14%). H3 (5.99%) had a large proportion of NDA (37.83%), ANG (1.25%), and CHL (4.82%). A similar general trend was found: Separate haplotypes were clustered only for NDA or BRM, while overlapping haplotypes were identified for HOL, ANG, and CHL.
We also studied genes related to growth and body size (LCORL, LRIG3, FGF5, and NCAPG), coat color (KIT and MC1R), and other functions, obtaining similar results (supplementary fig. S6, Supplementary Material online). Combined with LD pattern results, the haplotype network analyses indicated 1) common overlapping haplotypes were often identified for HOL, ANG, and CHL, while separate distinct haplotypes were clustered only for NDA or BRM; 2) the high SNP diversity and differential LD patterns near some genes, suggesting that they have been under different evolution pressure in these five breeds.
Discussion
Diverse geographic adaptation and selection have contributed to the shared and population-specific phenotypes in many species. For example, environment contributed to the process of selection and evolution of bovine genes in the cattle breeds of West Africa (Gautier et al. 2009) and the Senepol breed of Caribbean (Flori et al. 2012). Previous studies have shown that purifying selection is the most dominant mode of selection on genes of the innate immune system in human and cattle (Mukherjee et al. 2009; Seabury et al. 2010). To explore the cattle breed diversity and breed-specific selection signatures, we compared the significant candidate regions among five breeds. Shared selection regions across populations were found in five cattle breeds. One possibility is that different breeds have been under similar artificial selection intention. It is interesting to note that many of these genes have also been reported in other species. One possibility is that these genes carried out similar functions and went through similar adaptations in different species. On the other hand, we also found the unique loci, which likely contain genes and variations that confer breed-specific phenotypes. For example, DAVID analysis for the genes specific to individual breeds revealed differentially enriched molecular functions, suggesting that each breed was under different selections for unique phenotypes. We detected 12 DAVID clusters showing positive selection only in BRM. These genes may be related to the fact that founding zebu breeds for BRM were developed with inadequate food supplies, insect pests, parasites, and diseases in a tropical climate. Extensive breed-specific regions agreed well with the idea that differential selection often sorts individuals into separate breeds with distinct phenotypes.
The diversity of selection signatures across the genome could provide critical insights for the understanding of gene function and evolution. Our results have revealed a series of genes involved in coat color, growth, and milk production under selection. Coat color variation in domestic animals is evidence of phenotypic adaptation in response to selection (Hubbard et al. 2010). Genes underlying such variation are of considerable interest in breed formation and tracing molecular evolution in domestic animals. For example, in the domestic pig the evolutionary pattern of MC1R demonstrated that coat color phenotypes were likely a direct result of artificial selection (Fang et al. 2009). KIT is known to play roles in coat color in many species including cattle (Hayes et al. 2010; Stella et al. 2010; Kemper et al. 2014; Qanbari et al. 2014), pig (Amaral et al. 2011; Rubin et al. 2012), horse (Haase et al. 2007; McCue et al. 2012), and sheep (Kijas et al. 2012). This gene was also reported to be related to reproduction (Koch et al. 2009), and expressed in the lactating bovine mammary gland (Flori et al. 2009; Lemay et al. 2009). Segregating quantitative trait locus around GHR affected milk composition and yield (Blott et al. 2003; Khatkar et al. 2004; Viitala et al. 2006). In this study, we found differential recombination rates near GHR in five breeds, which correlated to the distinct LD patterns in taurine (as represented by HOL) and indicine breeds (BRM).
LAP3 catalyses the removal of N-terminal amino acids and is involved in protein maturation and degradation. LAP3 (chr6:38,574,590–38,600,027) is only 544 kb downstream of to ABCG2 (chr6:37,959,536–38,030,586), which affects milk production (Cohen-Zinder et al. 2005; Olsen et al. 2005). LAP3 and nearby genes NCAPG and LCORL have also been found to be associated with direct calving ease in Piedmontese cattle (Bongiorni et al. 2012) and feed intake and growth in beef cattle (Lindholm-Perry et al. 2011). Positive selection of LAP3 has been reported in European HOLs, Chinese HOLs, and five Italian cattle breeds (Gautier et al. 2009; Pan et al. 2013; Mancini et al. 2014). The haplotype diversity near LAP3 in the haplotype network analysis reflected diverse evolutionary patterns across breeds, and no significant recombination event was observed in this gene, so we conclude that differential selection pressures are responsible for the diverse haplotype distributions across breeds. SAR1B belongs to the Sar1-ADP ribosylation factor family of small GTPases, which govern the intracellular trafficking of proteins in coat protein-coated vesicles (Schekman and Orci 1996). It is annotated by the GO term of secretary pathway as it is associated with the milk fat globule membrane in dairy cattle (Lemay et al. 2009). It was first reported under selection in HOL in a whole-genome resequencing and haplotype-phasing effort (Larkin et al. 2012). In this study, we provided additional evidence that this gene is under positive selection as it is included in a couple of major haplotypes. Our haplotype network results shed light into the evolution of the SAR1B haplotypes across breeds. For instance, we found dominant haplotypes were common across multiple breeds, and the most plausible explanation is that positive selection in multiple breeds resulted in their increased frequencies. On the other hand, breed-specific haplotypes and changes in gene frequency for particular haplotypes may represent the imprint of diverse selections for an individual breed.
Our data generally agree well with the common beliefs that NDA and the ancestral founding breeds for BRM have a long history of local geographical adaptation while the three European taurine breeds (HOL, ANG, and CHL) went through recent strong artificial selection for milk and meat production. We found that LD patterns estimated using high-density SNP array offer detailed LD characteristics across breeds. The diverse LD patterns may indicate changes in genomic regions in different populations after selection for a particular allele, the various timescales of cattle domestication and breed formation, or specific characteristics of traits under selection.
Potential Pitfalls
Our study had detected many genes previously reported to be under positive selection. However, some genes were not detected by the di statistics, and this situation was reported before (Petersen et al. 2013). One example was the RXFP2 gene (ENSBTAG00000015132, chr12:29234959–29280832), which was found in polled sheep and cattle (Kijas et al. 2012; Allais-Bonnet et al. 2013) but not detected in our studies. Another example was the 547 kb polled locus in the 1.5- to 2.0-Mb region on chromosome 1 in cattle (Drogemuller et al. 2005; Medugorac et al. 2012; Seichter et al. 2012). We did find some marginally significant differences in terms of pairwise FST value and minor alleles frequency among five breeds (supplementary figs. S7 and S8, Supplementary Material online) near these loci, but they did not achieve significance due to different study designs, samples, and statistical methods. Window size and target locus length could also affect the detection. For example, genes with limited numbers of SNPs (such as ASIP) need further analysis. Another example is LAP3, which only contains 13 SNPs from BovineHD array. Luckily, with FST test using 92 NGS-based SNPs and LAP3 haplotype network analysis, our results revealed direct evidence of positive selection signals involved in milk production trait in HOL.
This study applied the di statistic to detect signatures of positive selection using common, moderate-frequency SNP data. It is noted that LD as measured by r2 depends on allele frequencies and the SNPs included on the Illumina Bovine arrays, which were selected based on their allele frequency and genome coverage (Pritchard et al. 2000; Matukumalli et al. 2009). With sequencing costs decreasing rapidly, high-quality, high-coverage whole-genome sequence information will make it feasible to study positive selection free of ascertainment bias.
While this study was being completed, Kemper et al. reported that selection for complex traits leaves little or no classic signatures of selection (Kemper et al. 2014). The authors attributed this to the fact that “selection response is caused by weak selection at many sites across the genome, probably for previously segregating variants”. Several lines of evidences (multiple independent studies, different samples and methods, NGS data, and haplotype network analyses) suggest our results are probably not analytical artifacts. It is well known, even in human population genomics studies, that different samples and methods often make it difficult to compare the results from different selection mapping efforts (Kelley et al. 2006; Akey 2009; Hohenlohe et al. 2010).
Conclusion
We carried out a genome-wide scan of selection signature using high-density SNP array across five diverse cattle breeds. Our analysis identified multiple genes under positive selection that are related to coat color, milk production, growth and body size, and other functions. Even though some genes have been previously reported, we confirmed the positive selections using high-density SNP arrays and more diverse samples. We further validated them (LAP3 and SAR1B) with next-generation sequencing and association tests. Haplotype diversity and network analyses revealed distinct selective pressures and evolution patterns across these five cattle breeds. Our results are unique in indicating that diverse genomic selections during speciation, domestication, breed formation, and recent genetic improvement could contribute to the cattle breed diversity.
Materials and Methods
Samples and SNP Data Quality Control
We retrieved a total of 169 samples genotyped on Illumina BovineHD BeadChip (Illumina Inc., San Diego, CA) from five cattle breeds, including HOL, ANG, CHL, BRM, and NDA. On the BovineHD Genotyping BeadChip, SNP markers were uniformly distributed throughout the cattle genome with a median interval less than 3 kb. We used PLINK 1.7 (Purcell et al. 2007) and custom in-house R scripts to convert file formats, manage data, and control quality. To minimize close relationships and genome sharing among samples, we removed closely related individuals when the pi-hat value (an identity-by-descent or IBD estimation) was more than 0.4. All chosen samples showed a genotyping success rate of more than 99%. The SNPs were filtered with MAF ≥ 0.05 and geno ≥ 0.1 (i.e., only SNPs with a 90% genotyping rate or higher). A total of 710,681 autosomal SNPs passed these filters. We then used 581,820 informative autosomal SNPs across breeds to calculate the di values.
Population Structure and Phylogenetic Analysis
MDS analysis was conducted using a total of 72,945 SNPs after LD-based pruning (>0.2). Pairwise genetic distances (4 dimensions) were used to identify the relationship between populations with PLINK (command line arguments: -mds -plot 4). Population structure was examined using Structure 2.3 (Pritchard et al. 2000; Falush et al. 2003). Each analysis was performed using 5,000 replicates and 2,000 burn-in cycles under admixture and correlated allele frequencies models. Genetic distance (D) between pairwise combination of individuals was calculated using PLINK (Purcell et al. 2007), where D = 1-(IBS2+0.5IBS1)/N: IBS1 and IBS2 are the number of loci that share either one or two alleles identical by state (IBS), respectively and the N is the number of loci (Stevens et al. 2011). Neighbor-joining phylogenetic trees were built based on pairwise genetics distance using PHYLIP 3.69 (http://www.phylip.com/, last accessed December 2, 2014). The phylogenetic trees were visualized with Figtree 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/, last accessed December 2, 2014).
Genome-Wide LD Estimation
To estimate genome-wide levels of LD in five breeds, pairwise LD measures for all retained SNPs were performed using the PLINK “-ld” option. We used the default window size of 1 Mb, and set “–ld-window-r2” to 0 in order to get all pairs reported. The LD decay along genomic distance was fitted by smooth.spline function in R (version 2.15.1).
Selection Mapping
We measured the locus-specific divergence in allele frequencies for each breed based on unbiased estimates of pairwise FST, that is, di, as described previously (Akey et al. 2010). Briefly, for each of 581,820 SNPs, we calculated the expected value and SD of FST between breeds i and j. For each breed, di was averaged over the SNPs contained within nonoverlapping windows of 50 SNPs. There were a total of 11,651 windows in the cattle genome. The top 1% (117) or 5% (583) regions with highest average di scores were used in the downstream analyses. Custom Perl scripts were used to annotate regions with RefSeq, Ensemble, and human-mapped genes. We performed enrichment analysis using DAVID Functional Annotation Tools (Huang et al. 2009). Only clusters with enrichment scores more than 1 (P value < 0.10) were considered.
Validation with Next-Generation Sequencing
Individual HOL and ANG samples were sequenced using the Illumina HiSeq2000 platform, with library preparation and sequence generation according to the manufacturer's protocols. Sequences were aligned against the UMD 3.1 reference genome and read-pairing was performed with the Burrows-Wheeler Aligner (BWA) v. 0.7.3a-r367 (Li and Durbin 2010). The SAM files were converted to BAM files and unmapped reads were removed with SAMtools v. 0.1.18 (Li et al. 2009). File sorting, marking and removal of duplicates, and indexing were performed with Picard v. 1.88 (http://broadinstitute.github.io/picard/, last accessed December 2, 2014). The resulting files were realigned and recalibrated with the Genome Analysis Toolkit v. 2.3-3 (GATK) (McKenna et al. 2010; DePristo et al. 2011) using a test data set containing known variant sites on LD, BovineHD and BovineSNP50 arrays as well as validated variant sites obtained from dbSNP (http://www.ncbi.nlm.nih.gov/SNP/, last accessed December 2, 2014). Default settings were used for BWA, Samtools, Picard, and GATK. SNPs and indels were called separately using the GATK Unified Genotyper within a 200 bp window surrounding the targeted intervals. Multisample calling was employed to detect SNPs in all samples. Variants were selected using the variant quality score recalibration threshold of 2.30 (99% of known high-quality SNPs identified). SNPs were filtered if their call rates were less than 80% across all HOL and ANG samples. FST and MAF frequency plot for LAP3 genes across HOL and ANG used 92 SNPs extracted from the GATK calls. FST between two breeds were calculated by using weighted analysis of variance in Genepop (Rousset 2008).
Association Tests for Milk Production Traits
We chose to confirm two genes, LAP3 and SAR1B, under positive selection in a genome-wide association analysis involving milk production traits for HOLs. We retrieved and analyzed BovineSNP50 BeadChip (Illumina Inc.) data for 26,362 HOLs. Predicted transmitting abilities (PTAs) for five milk production traits (MY, FY, PY, FP, and PP) were used in association tests (Xu et al. 2014a). PTAs are estimates of an animal’s additive genetic merit for a trait. Target regions were detected using line regression after FDR correction to estimate the significant associated SNPs as described previously (Xu et al. 2014b).
LD, Recombination rate, and Haplotype Network Analysis
Genotypes for the SNPs in each gene were extracted as unphased information. LD parameters (D′ and r2) and LD blocks were computed using the Haploview 4.2 program (Barrett et al. 2005). Recombination rates were estimated using PHASE 2.1 under the approximate conditionals model for varying recombination rate (Li and Stephens 2003; Crawford et al. 2004). To explore the diversity of haplotypes and evolutionary relationships cross populations, haplotypes and their frequencies were estimated separately for each breed using PHASE 2.1 (Stephens et al. 2001; Crawford et al. 2004). To obtain reliable results, we employs an iterative scheme to perform inference with 10,000 iterations and 10,000 burn-ins, also we increased the number of iterations of the final run of the algorithm using option -X 100, for detailed see http://stephenslab.uchicago.edu/instruct2.1.pdf, last accessed December 2, 2014. We constructed haplotype networks for functional genes such as LAP3, SAR1B, LCORL, KIT, MC1R, and MIF1. For LAP3, SAR1B, and MC1R, we extended to include the full LD block length. Phylogenetic relationships among the identified haplotypes were inferred through a median-joining network analysis by Network 4.6.12 (http://www.fluxus-engineering.com/, last accessed December 2, 2014).
Availability of Data
SNP genotype data and population genetics and evolutionary analysis results are available upon request for research purposes.
Supplementary Material
Supplementary figures S1–S8 and tables S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank members of the Bovine HapMap Consortium for sharing their samples. The authors also thank Reuben Anderson and Alexandre Dimitriv for technical assistance. This work was supported in part by Agriculture and Food Research Initiative (AFRI) grant No. 2011-67015-30183 from U.S. Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA). Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. The USDA is an equal opportunity provider and employer.
References
- Akey JM. Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 19. 2009:711–722. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107:1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allais-Bonnet A, Grohs C, Medugorac I, Krebs S, Djari A, Graf A, Fritz S, Seichter D, Baur A, Russ I, et al. Novel insights into the bovine polled phenotype and horn ontogenesis in Bovidae. PLoS One. 2013;8:e63512. doi: 10.1371/journal.pone.0063512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral AJ, Ferretti L, Megens HJ, Crooijmans RP, Nie H, Ramos-Onsins SE, Perez-Enciso M, Schook LB, Groenen MA. Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PLoS One. 2011;6:e14782. doi: 10.1371/journal.pone.0014782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson L, Georges M. Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet. 2004;5:202–212. doi: 10.1038/nrg1294. [DOI] [PubMed] [Google Scholar]
- Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar A, Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Blott S, Kim J, Moisio S, Schmidt-Kuntzel A, Cornet A, Berzi P, Cambisano N, Ford C, Grisart B, Johnson D, et al. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics. 2003;163:253–266. doi: 10.1093/genetics/163.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongiorni S, Mancini G, Chillemi G, Pariset L, Valentini A. Identification of a short region on chromosome 6 affecting direct calving ease in Piedmontese cattle breed. PLoS One. 2012;7:e50137. doi: 10.1371/journal.pone.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovine HapMap Consortium. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324:528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley DG, MacHugh DE, Cunningham P, Loftus RT. Mitochondrial diversity and the origins of African and European cattle. Proc Natl Acad Sci U S A. 1996;93:5131–5135. doi: 10.1073/pnas.93.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, Vonholdt BM, Rhue A, Boyko A, Byers A, et al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009;326:150–153. doi: 10.1126/science.1177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Yang Y, Shen M, Zhou T. Inhibition of cytokinesis by overexpression of NudCL that is localized to the centrosome and midbody. Cell Res. 2009;19:1305–1308. doi: 10.1038/cr.2009.118. [DOI] [PubMed] [Google Scholar]
- Charlier C, Coppieters W, Rollin F, Desmecht D, Agerholm JS, Cambisano N, Carta E, Dardano S, Dive M, Fasquelle C, et al. Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat Genet. 2008;40:449–454. doi: 10.1038/ng.96. [DOI] [PubMed] [Google Scholar]
- Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ, Everts-van der Wind A, Lee JH, Drackley JK, Band MR, Hernandez AG, Shani M, et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–944. doi: 10.1101/gr.3806705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JB, Waurich B, Wensch-Dorendorf M, Bickhart DM, Swalve HH. A genome-wide association study of calf birth weight in Holstein cattle using single nucleotide polymorphisms and phenotypes predicted from auxiliary traits. J Dairy Sci. 2014;97:3156–3172. doi: 10.3168/jds.2013-7409. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet. 2004;36:700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- Decker JE, McKay SD, Rolf MM, Kim J, Molina AA, Sonstegard TS, Hanotte O, Gotherstrom A, Seabury CM, Praharani L, et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014;10:e1004254. doi: 10.1371/journal.pgen.1004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogemuller C, Wohlke A, Momke S, Distl O. Fine mapping of the polled locus to a 1-Mb region on bovine chromosome 1q12. Mamm Genome. 2005;16:613–620. doi: 10.1007/s00335-005-0016-0. [DOI] [PubMed] [Google Scholar]
- Espigolan R, Baldi F, Boligon AA, Souza FR, Gordo DG, Tonussi RL, Cardoso DF, Oliveira HN, Tonhati H, Sargolzaei M, et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genomics. 2013;14:305. doi: 10.1186/1471-2164-14-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Larson G, Ribeiro HS, Li N, Andersson L. Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genet. 2009;5:e1000341. doi: 10.1371/journal.pgen.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics. 2013;193:929–941. doi: 10.1534/genetics.112.147231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M. The genome response to artificial selection: a case study in dairy cattle. PLoS One. 2009;4:e6595. doi: 10.1371/journal.pone.0006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flori L, Gonzatti MI, Thevenon S, Chantal I, Pinto J, Berthier D, Aso PM, Gautier M. A quasi-exclusive European ancestry in the Senepol tropical cattle breed highlights the importance of the slick locus in tropical adaptation. PLoS One. 2012;7:e36133. doi: 10.1371/journal.pone.0036133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Flori L, Riebler A, Jaffrezic F, Laloe D, Gut I, Moazami-Goudarzi K, Foulley JL. A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics. 2009;10:550. doi: 10.1186/1471-2164-10-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbens F, Rettenberger G, Lenstra J, Veerkamp J, Pas M. Characterization, chromosomal localization, and genetic variation of the porcine heart fatty acid-binding protein gene. Mamm Genome. 1997;8:328–332. doi: 10.1007/s003359900433. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–391. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- Granka JM, Henn BM, Gignoux CR, Kidd JM, Bustamante CD, Feldman MW. Limited evidence for classic selective sweeps in African populations. Genetics. 2012;192:1049–1064. doi: 10.1534/genetics.112.144071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883–886. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- Gu J, Orr N, Park SD, Katz LM, Sulimova G, MacHugh DE, Hill EW. A Genome Scan for Positive Selection in Thoroughbred Horses. PLoS ONE. 2009;4(6):e5767. doi: 10.1371/journal.pone.0005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase B, Brooks SA, Schlumbaum A, Azor PJ, Bailey E, Alaeddine F, Mevissen M, Burger D, Poncet PA, Rieder S, et al. Allelic heterogeneity at the equine KIT locus in dominant white (W) horses. PLoS Genet. 2007;3:e195. doi: 10.1371/journal.pgen.0030195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes BJ, Chamberlain AJ, MacEachern S, Savin K, McPartlan H, MacLeod I, Sethuraman L, Goddard ME. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim Genet. 2009;40:176–184. doi: 10.1111/j.1365-2052.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME. Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010;6:e1001139. doi: 10.1371/journal.pgen.1001139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Wray GA. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat Genet. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- Hiendleder S, Lewalski H, Janke A. Complete mitochondrial genomes of Bos taurus and Bos indicus provide new insights into intra-species variation, taxonomy and domestication. Cytogenet Genome Res. 2008;120:150–156. doi: 10.1159/000118756. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Phillips PC, Cresko WA. Using population genomics to detect selection in natural populations: key concepts and methodological considerations. Int J Plant Sci. 2010;171:1059–1071. doi: 10.1086/656306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hubbard JK, Uy JA, Hauber ME, Hoekstra HE, Safran RJ. Vertebrate pigmentation: from underlying genes to adaptive function. Trends Genet. 2010;26:231–239. doi: 10.1016/j.tig.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Jarvis JP, Scheinfeldt LB, Soi S, Lambert C, Omberg L, Ferwerda B, Froment A, Bodo JM, Beggs W, Hoffman G, et al. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet. 2012;8:e1002641. doi: 10.1371/journal.pgen.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Res. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JL, Swanson WJ. Positive selection in the human genome: from genome scans to biological significance. Annu Rev Genomics Hum Genet. 2008;9:143–160. doi: 10.1146/annurev.genom.9.081307.164411. [DOI] [PubMed] [Google Scholar]
- Kemper KE, Saxton SJ, Bolormaa S, Hayes BJ, Goddard ME. Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics. 2014;15:246. doi: 10.1186/1471-2164-15-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatkar MS, Thomson PC, Tammen I, Raadsma HW. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet Sel Evol. 2004;36:163–190. doi: 10.1186/1297-9686-36-2-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, et al. Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10:e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch D, Sakurai M, Hummitzsch K, Hermsdorf T, Erdmann S, Schwalbe S, Stolzenburg JU, Spanel-Borowski K, Ricken AM. KIT variants in bovine ovarian cells and corpus luteum. Growth Factors. 2009;27:100–113. doi: 10.1080/08977190802707571. [DOI] [PubMed] [Google Scholar]
- Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin DM, Daetwyler HD, Hernandez AG, Wright CL, Hetrick LA, Boucek L, Bachman SL, Band MR, Akraiko TV, Cohen-Zinder M, et al. Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc Natl Acad Sci U S A. 2012;109:7693–7698. doi: 10.1073/pnas.1114546109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson G, Piperno DR, Allaby RG, Purugganan MD, Andersson L, Arroyo-Kalin M, Barton L, Climer VC, Denham T, Dobney K, et al. Current perspectives and the future of domestication studies. Proc Natl Acad Sci U S A. 2014;111:6139–6146. doi: 10.1073/pnas.1323964111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay DG, Lynn DJ, Martin WF, Neville MC, Casey TM, Rincon G, Kriventseva EV, Barris WC, Hinrichs AS, Molenaar AJ, et al. The bovine lactation genome: insights into the evolution of mammalian milk. Genome Biol. 2009;10:R43. doi: 10.1186/gb-2009-10-4-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, Wheeler TL, Ferrell CL, Jenkins TG, Snelling WM, et al. Association, effects and validation of polymorphisms within the NCAPG—LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011;12:103. doi: 10.1186/1471-2156-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus RT, MacHugh DE, Bradley DG, Sharp PM, Cunningham P. Evidence for two independent domestications of cattle. Proc Natl Acad Sci U S A. 1994;91:2757–2761. doi: 10.1073/pnas.91.7.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Seto JT, Raftery JM, Quinlan KG, Huttley GA, Hook JW, Lemckert FA, Kee AJ, Edwards MR, Berman Y, et al. Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nat Genet. 2007;39:1261–1265. doi: 10.1038/ng2122. [DOI] [PubMed] [Google Scholar]
- MacHugh DE, Shriver MD, Loftus RT, Cunningham P, Bradley DG. Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus) Genetics. 1997;146:1071–1086. doi: 10.1093/genetics/146.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini G, Gargani M, Chillemi G, Nicolazzi EL, Marsan PA, Valentini A, Pariset L. Signatures of selection in five Italian cattle breeds detected by a 54K SNP panel. Mol Biol Rep. 2014;41:957–965. doi: 10.1007/s11033-013-2940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O'Connell J, Moore SS, Smith TP, Sonstegard TS, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4:e5350. doi: 10.1371/journal.pone.0005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue ME, Bannasch DL, Petersen JL, Gurr J, Bailey E, Binns MM, Distl O, Guerin G, Hasegawa T, Hill EW, et al. A high density SNP array for the domestic horse and extant Perissodactyla: utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012;8:e1002451. doi: 10.1371/journal.pgen.1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medugorac I, Seichter D, Graf A, Russ I, Blum H, Gopel KH, Rothammer S, Forster M, Krebs S. Bovine polledness—an autosomal dominant trait with allelic heterogeneity. PLoS One. 2012;7:e39477. doi: 10.1371/journal.pone.0039477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. Proc Natl Acad Sci U S A. 2009;106:7073–7078. doi: 10.1073/pnas.0811357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris BJ, Whan VA. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008;18:1282–1293. doi: 10.1101/gr.072090.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksyk TK, Smith MW, O'Brien SJ. Genome-wide scans for footprints of natural selection. Philos Trans R Soc Lond B Biol Sci. 2010;365:185–205. doi: 10.1098/rstb.2009.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen HG, Lien S, Gautier M, Nilsen H, Roseth A, Berg PR, Sundsaasen KK, Svendsen M, Meuwissen TH. Mapping of a milk production quantitative trait locus to a 420-kb region on bovine chromosome 6. Genetics. 2005;169:275–283. doi: 10.1534/genetics.104.031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Zhang S, Jiang J, Jiang L, Zhang Q, Liu J. Genome-wide detection of selective signature in Chinese Holstein. PLoS One. 2013;8:e60440. doi: 10.1371/journal.pone.0060440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez OBA, Utsunomiya YT, Meszaros G, Bickhart DM, Liu GE, Van Tassell CP, Sonstegard TS, Da Silva MV, Garcia JF, Solkner J. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46:19. doi: 10.1186/1297-9686-46-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, Bailey E, Bannasch D, Binns MM, Borges AA, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9:e1003211. doi: 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto-Neto LR, Sonstegard TS, Liu GE, Bickhart DM, Da Silva MV, Machado MA, Utsunomiya YT, Garcia JF, Gondro C, Van Tassell CP. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics. 2013;14:876. doi: 10.1186/1471-2164-14-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce JE, Hayes BJ, Bolormaa S, Goddard ME. Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011;187:981–984. doi: 10.1534/genetics.110.123943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, Thaller G, Simianer H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12:318. doi: 10.1186/1471-2164-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10:e1004148. doi: 10.1371/journal.pgen.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Pimentel EC, Tetens J, Thaller G, Lichtner P, Sharifi AR, Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41:377–389. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- Qanbari S, Strom TM, Haberer G, Weigend S, Gheyas AA, Turner F, Burt DW, Preisinger R, Gianola D, Simianer H. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens. PLoS One. 2012;7:e49525. doi: 10.1371/journal.pone.0049525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman FB, Kadowaki Y, Ishihara S, Tobita H, Imaoka H, Fukuhara H, Aziz MM, Furuta K, Amano Y, Kinoshita Y. Fibroblast-derived HB-EGF promotes Cdx2 expression in esophageal squamous cells. Lab Invest. 2010;90:1033–1048. doi: 10.1038/labinvest.2010.71. [DOI] [PubMed] [Google Scholar]
- Rimbault M, Beale HC, Schoenebeck JJ, Hoopes BC, Allen JJ, Kilroy-Glynn P, Wayne RK, Sutter NB, Ostrander EA. Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res. 2013;23:1985–1995. doi: 10.1101/gr.157339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothammer S, Seichter D, Forster M, Medugorac I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genomics. 2013;14:908. doi: 10.1186/1471-2164-14-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F. genepop'007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Martinez BA, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci U S A. 2012;109:19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Seabury CM, Seabury PM, Decker JE, Schnabel RD, Taylor JF, Womack JE. Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle. Proc Natl Acad Sci U S A. 2010;107:151–156. doi: 10.1073/pnas.0913006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seichter D, Russ I, Rothammer S, Eder J, Forster M, Medugorac I. SNP-based association mapping of the polled gene in divergent cattle breeds. Anim Genet. 2012;43:595–598. doi: 10.1111/j.1365-2052.2011.02302.x. [DOI] [PubMed] [Google Scholar]
- Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P. Identification of selection signatures in cattle breeds selected for dairy production. Genetics. 2010;185:1451–1461. doi: 10.1534/genetics.110.116111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens EL, Heckenberg G, Roberson ED, Baugher JD, Downey TJ, Pevsner J. Inference of relationships in population data using identity-by-descent and identity-by-state. PLoS Genet. 2011;7:e1002287. doi: 10.1371/journal.pgen.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, et al. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–115. doi: 10.1126/science.1137045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Akey JM. Parallel adaptive divergence among geographically diverse human populations. PLoS Genet. 2011;7:e1002127. doi: 10.1371/journal.pgen.1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AC, Cullup T, Norgett EE, Hill T, Barton S, Dale BA, Sprecher E, Sheridan E, Taylor AE, Wilroy RS, et al. ABCA12 is the major harlequin ichthyosis gene. J. Invest Dermatol. 2006;126:2408–2413. doi: 10.1038/sj.jid.5700455. [DOI] [PubMed] [Google Scholar]
- Troy CS, MacHugh DE, Bailey JF, Magee DA, Loftus RT, Cunningham P, Chamberlain AT, Sykes BC, Bradley DG. Genetic evidence for Near-Eastern origins of European cattle. Nature. 2001;410:1088–1091. doi: 10.1038/35074088. [DOI] [PubMed] [Google Scholar]
- Utsunomiya YT, do Carmo AS, Carvalheiro R, Neves HH, Matos MC, Zavarez LB, Perez O'Brien AM, Solkner J, McEwan JC, Cole JB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52. doi: 10.1186/1471-2156-14-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitala S, Szyda J, Blott S, Schulman N, Lidauer M, Maki-Tanila A, Georges M, Vilkki J. The role of the bovine growth hormone receptor and prolactin receptor genes in milk, fat and protein production in Finnish Ayrshire dairy cattle. Genetics. 2006;173:2151–2164. doi: 10.1534/genetics.105.046730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wiener P, Wilkinson S. Deciphering the genetic basis of animal domestication. Proc Biol Sci. 2011;278:3161–3170. doi: 10.1098/rspb.2011.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Cole JB, Bickhart DM, Hou Y, Song J, VanRaden PM, Sonstegard TS, Van Tassell CP, Liu GE. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genomics. 2014a;15:683. doi: 10.1186/1471-2164-15-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Hou Y, Bickhart DM, Song J, Van Tassell CP, Sonstegard TS, Liu GE. A genome-wide survey reveals a deletion polymorphism associated with resistance to gastrointestinal nematodes in Angus cattle. Funct Integr Genomics. 2014b;14:333–339. doi: 10.1007/s10142-014-0371-6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Meltzer P, Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics. 2013;14:244. doi: 10.1186/1471-2105-14-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Zimmerman W, Liu X, Erikson RL. A mammalian NudC-like protein essential for dynein stability and cell viability. Proc Natl Acad Sci U S A. 2006;103:9039–9044. doi: 10.1073/pnas.0602916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.