

Abstract
Diazeniumdiolate-based aspirin prodrugs have previously been shown to retain the anti-inflammatory properties of aspirin while protecting against the common side effect of stomach ulceration. Initial analysis of two new prodrugs of aspirin that also release either nitroxyl (HNO) or nitric oxide (NO) demonstrated increased cytotoxicity toward human lung carcinoma cells compared to either aspirin or the parent nitrogen oxide donor. In addition, cytotoxicity was significantly lower in endothelial cells, suggesting cancer-specific sensitivity. To assess the chemotherapeutic potential of these new prodrugs in breast cancer, we studied their effect both in cultured cells and in a nude mouse model. Both prodrugs reduced growth of breast adenocarcinoma cells more effectively than the parent compounds while not being appreciably cytotoxic in a related non-tumorigenic cell line (MCF-10A). The HNO donor also was more cytotoxic than the related NO donor. The basis for the observed specificity was investigated in terms of impact on metabolism, DNA damage and repair, apoptosis, angiogenesis and metastasis. The results suggest a significant pharmacological potential for treatment of breast cancer.
Keywords: nitroxyl, nitric oxide, NONOate, diazeniumdiolate, IPA/NO, DEA/NO, NSAID, aspirin, anti-inflammatory, anticancer
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are used extensively for treatment of pain, inflammation and fever [1]. Daily intake of aspirin also reduces the risk of cardiovascular disease [2, 3], however with concomitant increases in the incidence of a variety of adverse side effects including gastrointestinal bleeding and ulceration [3–6]. Diverse structural modifications have been employed to improve the benefit versus risk profile of NSAIDs. For instance, increased steric bulk has been shown to provide protection against the gastrointestinal irritation that is common to use of traditional NSAIDs [7–10] by improving specificity towards cyclooxygenase (COX)-2 over COX-1 [11]. However, the cardioprotection observed for aspirin is not replicated by other NSAIDs, as chronic use increases the risk of myocardial infarction and stroke [12].
Another synthetic strategy that has been employed to improve the NSAID safety profile is to couple other moieties to NSAIDs to produce prodrugs with multiple functionalities. For example, recent attention has been given to nitric oxide (NO) derivatives (NO-NSAIDs) [13–15], which are designed to harness the beneficial cardiovascular and gastrointestinal effects of NO (e.g., regulation of blood pressure, inhibition of platelet aggregation, repair of mucosal injury) [16–19]. Such adducts do in fact retain the anti-inflammatory and analgesic properties of the NSAID while reducing gastrointestinal, cardiovascular and renal side effects in animal models (see [15]). A significantly lower degree of gastric injury has also been apparent in clinical trials [15, 20].
NO-NSAIDs are formulated by covalently attaching NSAIDs to nitrate esters [21], diazeniumdiolates (also called as NONOates) [22, 23] and other NO donors [24]. Interestingly, the covalent linker itself has been suggested to reduce NSAID toxicity by increasing stability and absorption rate of the prodrug in the gastrointestinal tract [25]. The linker can also augment localized delivery of NO [26, 27], which is a critical feature in NO donor-based treatment of conditions other than hypertension. Cleavage of the linker may also produce bioactive agents.
The prodrug strategy with respect to NO donors was initially developed by Keefer and colleagues who derivatized diazeniumdiolates in the early 1990s [28]. Esterase-sensitive derivatives were later found to significantly decrease proliferation of leukemia cells compared to the parent donor [29]. Incorporation by design of a thiol-sensitive functionality to produce a construct named JS-K significantly enhanced antiproliferative activity [30] and opened up a new research area in the expansive arena of diazeniumdiolates as drug candidates.
The role of NO in cancer is complicated in that NO production can be mutagenic yet can affect apoptosis, proliferation, migration, adhesion, angiogenesis and vascular permeability [31–33]. Often, low levels of NO are suggested to be pro-tumorigenic, while production of higher, sustained levels of NO can have cytostatic and cytotoxic effects on cancer cells [34, 35]. However, expression of the inducible isoform of NO synthase (iNOS) has been reported in malignancies of the breast, prostate, lung, brain and colon [36–45]. Detection of increased iNOS levels also predicts poor survival in estrogen receptor α-negative (ER(−)) breast cancer patients [46]. Thus, inhibition of iNOS within cancer cells may be therapeutic [32] while delivery of exogenous NO may initiate tumor regression in simulation of the immune system. NO donors have in fact been shown to lead to chemo- [47, 48] and radiosensitization [49–51] and to overcome drug resistance by tumor cells [52].
COX-2 expression [53] predicts poor outcome similarly to iNOS, indicating that inflammation is a major driving force in both oncogenesis and resistance to conventional therapeutic regimes [54]. Epidemiological studies show that daily intake of NSAIDs, primarily aspirin, reduces risk of colon, lung, prostate, esophageal, stomach, ovarian and breast cancer [55–60]. Initial studies of malignant melanoma, Hodgkin’s disease, and adult leukemia also found NSAIDs to be protective [61–63]. Early analysis also indicates that NO-NSAIDs inhibit cancer growth [64, 65].
Recently nitroxyl (HNO), which is one electron more reduced than NO, has emerged as a biologically relevant nitrogen oxide [66], with promise for the treatment of cardiovascular diseases such as heart failure and ischemic reperfusion injury [67–69]. HNO donors have also been used clinically in the treatment of alcoholism [70, 71]. Although HNO has yet to be demonstrated to be endogenously produced, a role in inhibition of tumor development by HNO donors is emerging [72]. In particular, the thiophilicity of HNO [73–75] leads to irreversible thiol modification of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [76, 77]. This in turn results in inhibition of glycolysis, which occurs at a substantially accelerated rate in hypoxic, solid tumors compared to normal cells [78].
Other critical thiol-containing enzymes that facilitate cancer progression may also be targets of HNO-donating anti-cancer agents. Consistent with this idea, HNO has been shown to inhibit breast cancer [72] and neuroblastoma [79] proliferation in mouse xenografts as well as in culture, through increased apoptosis [72]. In addition to the direct effects of HNO on cancer cells, HNO donors may be useful adjuvant agents to cancer chemotherapy. HNO has also been shown to inhibit poly(ADP-ribose) polymerases (PARP) in a breast cancer cell line [80]. As an important component of the DNA repair machinery, PARP is a major target in the design of anti-cancer agents. Since a number of chemotherapies and radiation therapy are based on inducing DNA damage in cancer cells, inhibition of PARP by HNO donors may increase the efficacy of these treatments.
One attractive attribute of diazeniumdiolates in terms of NO donation is the ability to tune the rate of decomposition based on amine identity [81]. In addition, diazeniumdiolates can be tuned to function as HNO donors, by utilizing a primary rather than secondary amine as the scaffold [82–85]. Recently, we prepared two new diazeniumdiolate-NSAID adducts (NONO-NSAIDs) by derivatizing both a primary and secondary amine-based diazeniumdiolate with aspirin to produce O2-(acetylsalicyloyloxymethyl)-1-(N-isopropylamino)-diazen-1-ium-1,2-diolate (IPA/NO-aspirin) and O2-(acetylsalicyloyloxymethyl)-1-(N,N-diethylamino)-diazen-1-ium-1,2-diolate (DEA/NO-aspirin) [26]. A comparison of their chemical and biological properties demonstrated the expected retention of the analgesic and anti-inflammatory properties of aspirin as well as known effects of HNO or NO donors. In addition, these conjugates showed enhanced cytotoxicity toward human lung epithelial carcinoma cells (A549) while not being appreciably toxic toward human umbilical vascular endothelial cells (HUVECs). Here, we sought to further elucidate the mechanism behind this observed selectivity, with breast cancer as the model.
Materials and Methods
Reagents
Unless otherwise noted, chemicals were purchased from Sigma-Aldrich and used without further purification. IPA/NO, DEA/NO, IPA/NO-aspirin and DEA/NO-aspirin were synthesized and purified according to previously published procedures [26, 86]. Stock solutions of NONO-aspirin prodrugs (100–1000×) were prepared in DMSO and stored at −20°C. The final concentration in media or calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (PBS) was adjusted to 0.1% DMSO. Stock solutions of IPA/NO or DEA/NO (1000×) were prepared similarly but in 10 mM NaOH. Concentrations were determined directly prior to use from the extinction coefficient at 250 nm (ε of 8,000 M−1 cm−1) [87].
Instrumentation
UV-visible spectroscopy was performed with an Agilent Hewlett-Packard 8453 diode-array spectrophotometer equipped with an Agilent 89090A thermostat. Absorbance and fluorescence measurements of 96 well plates were accomplished with a Perkin-Elmer HTS 7000 Bio Assay reader. Cells were counted using a Beckman Coulter Z-2 cell and particle counter. Cell viability and angiogenesis was monitored using a Nikon eclipse TS-100 inverted microscope. An Eppendorf thermocycler was used to prepare cDNA. Sequence detection was then carried out using 7300 Real-Time PCR System from Applied Biosystems. An iBlot® gel transfer device from Invitrogen, Carlsbad, CA was used for dry transfer in Western blots. DNA damage was assessed using a Comet Assay® Electrophoresis System (Trevigen Inc. Gaithersburg, MD).
Cell Culture
Human breast adenocarcinoma cells (MDA-MB-231, MCF-7 and MDA-MB-468; American Type Culture Collection, Manassas, VA) were grown as monolayers in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hyclone, Thermo Fisher Scientific Inc., Waltham, MA), penicillin (50 units/mL) and streptomycin (50 mg/mL; Life Technologies, Inc., Grand Island, NY). Non-tumorigenic, human breast epithelial cells (MCF-10A; American Type Culture Collection) and HUVECs (Cell Applications, Inc., San Diego, CA) were grown as monolayers in endothelial growth media (Lonza Inc., US-Allendale, NJ or Cell Applications, Inc., San Diego, CA). Cells were seeded at a density of 1 × 106 cells per 100 cm2 culture dish and were incubated at 37 °C in 5% CO2 at 80% relative humidity. Single-cell suspensions were obtained by trypsinization (0.05% trypsin/EDTA, Invitrogen, Carlsbad, CA). MDA-MB-231 cells stably transfected with green fluorescent protein (GFP) and G418 antibiotic resistance gene were a generous gift from Dr. Aldona Karaczyn from the Maine Medical Center Research Institute, Scarborough, ME. MDA-MB-231-GFP cells were grown as described for MDA-MB-231 cells with the addition of 418 antibiotics (100 mg/mL) to ensure vector presence.
Cell viability as determined by the colorimetric MTT assay
Cells were plated at 8,000–10,000 cells per well in a 96-well plate and grown overnight as above. The cells were then treated with different concentrations of the prodrugs (10–200 μM) or precursors (50–200 μM) for 48 h at 37 °C. After addition of 10 μL of a solution of 2 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to each well, the plate was incubated for 1 h at 37 °C. The media was then removed, 100 μL DMSO was added to each well, and absorbance was recorded at 550 nm.
Cell viability as determined by colonogenic analysis
MDA-MB-231 cells were plated at 400,000 cells per 60 mm dish and grown for 48 h as above. The cells were treated in growth media containing different concentrations (1–1000 μM) of NONO-aspirin prodrugs for 24 h at 37 °C. After treatment, the cells were washed twice with PBS, trypsinized, counted and plated at a density of 100, 1000 or 10,000 per 60 mm plate. For each dose determination, cells were plated in triplicate, and each experiment was repeated at least twice. After 10–12 d, the colonies were stained with crystal violet (0.5% w/v) and counted using a Stemi microscope.
Mouse tumor model
Analysis of tumor growth was carried out using protocols approved by the Committee on the Ethics of Animal Experiments of the National Cancer Institute (RBB-144) and adhered with the recommendations in the United States National Research Council’s “Guide for the Care and Use of Laboratory Animals,” the United States Public Health Service’s “Guide for the Care and Use of Laboratory Animals” and the “Policy on Humane Care and Use of Laboratory Animals”. The vivarium was maintained at 23 °C on a 12 h light/12 h dark cycle with ad libitum access to food and water. Nude mice (n = 40) under general anesthesia were implanted with 7.5 × 105 MDA-MB-231 cells transfected with GFP by injection underneath the fourth left mammary gland. Prior to implantation, pedal withdrawal and eyelid reflexes were examined to ensure that mice were under stage III of anesthesia. At 14 d post-inoculation, the mice were randomly divided into four groups and treated by daily injection of equimolar doses (10 μL of 100 mM stock) of aspirin (9.00 mg/kg), IPA/NO-aspirin (15.8 mg/kg) or DEA/NO-aspirin (16.3 mg/kg) or with vehicle (DMSO). After five weeks, the tumor size was measured using in vivo fluorescent imaging for quantification of the GFP tag. In brief, mice were under general anesthesia throughout the whole body imaging process, and GFP signals were captured and quantified in an Xenogen IVIS 100 Imaging System. To assess metastasis in the brain, the animals were subsequently sacrificed following the approved method and guidelines.
To assess the stability of GFP in proliferating cells as well as its sensitivity to exposure to NO or HNO, MB-231-GFP cells were grown to 60% confluence in 200 μL media in a 96 well plate (5,000 cells per well) for 24 h. After washing once with PBS and addition of fresh media, the cells were exposed to 2 μL of 10 mM NaOH or to sublethal doses of IPA/NO (50 μM) or DEA/NO (75 μM) at 37 °C. Fluorescence intensity was then measured (λem 509 nm, λex 435 nm) at 0, 1, 2, 4, 6, 24 and 48 h in a Perkin Elmer Victor X fluorescence plate reader.
Caspase-3 activity
Caspase-3 activity was measured using a fluorescence assay kit (Cat No. 10009135, Cayman Chemical). Cells were plated at a density of 50,000 per well in a 96 well plate and grown overnight. The cells were treated with different concentrations of NONO-aspirin prodrugs (25–100 μM) or DMSO (0.1%) for 24 h. The plate was then centrifuged at 3000 rpm, and the media was aspirated. Lysis buffer (100 μL) was added to each well, and the plate was incubated for 30 min at room temperature. After addition of caspase-3 substrate solution (100 μL) to each well, the plate was and incubated for 30 min, after which fluorescence was measured at excitation of 485 nm and emission of 535 nm.
Alkaline Comet assay
Cells were plated at a density of 50,000 per well in 12 well plates and grown overnight. They were then treated with sublethal doses of IPA/NO (50 μM) or DEA/NO (75 μM) for 12 h, and the assay was conducted using a Comet assay kit (Cat No. 4250-050-K, Trevigen, MD) as described in the manufacture’s protocol.
GAPDH activity
GAPDH activity was measured using an assay kit (Cat No. AM1639, Applied Biosystems). Cells were plated at a density of 30,000 per well and grown overnight. They were then treated with 25–100 μM IPA/NO-aspirin or DEA/NO-aspirin for 1 h, after which 200 μL of KDalert lysis buffer was added to each well. The plate was incubated at 4 °C for 20 min to lyse the cells, and 10 μL of cell lysate was transferred to a clean 96 well plate. After addition of 90 μL of KDalert Master Mix, fluorescence was measured at excitation of 540 nm and emission of 570 nm.
Measurement of oxidative species
Cells were plated at a density of 30,000 cells per well in a 96 cell plate and grown overnight in RPMI 1640 media containing 10% FBS and 1% penicillin-streptomycin (100×). 4-Amino-5-methylamino-2′,7′-dichlorofluorescein diacetate (DCF-2DA, Sigma Aldrich) in DMSO (1000×) was diluted to a final concentration of 10 μM in PBS. The media was aspirated from each well and was replaced by 100 μL of the DCF-2DA solution. The plate was incubated for 30 min at 37 °C. Each well was then washed three times with PBS (pH 7.4) to remove excess dye. NONO-aspirin prodrugs dissolved in DMSO (1000×) were then added to achieve a final concentration of 100 μM. The increase in fluorescence intensity with time was measured at an excitation of 485 nm and emission of 535 nm.
Measurement of angiogenesis
Matrigel (50 μL) was added to each well in a 96 well plate and incubated for 2 h at 37 °C to allow the gel to solidify. Then, a 100 μL cell suspension of 2 × 105 cells/mL with varied concentrations (0, 1, 10 μM) of NONO-aspirin prodrugs or controls was added to each well. Pictures of different wells were captured after 12 h using an inverted microscope.
Measurement of mRNA levels using RT-PCR
Cells were plated at a density of 1 × 106 per plate on 100 mm plates, were grown overnight and then were treated with 100 μM of IPA/NO-aspirin, DEA/NO-aspirin, IPA/NO, DEA/NO or aspirin for 1 to 24 h. RNA was harvested in TRIzol reagent following the manufacture’s protocol. cDNA was prepared and real-time PCR was performed as described. A high capacity cDNA Kit (Applied Biosystems Cat #4322171) was used to produce cDNA. RNA was heated at 60 °C for 10 min prior to cDNA synthesis, and the concentration was measured using a NanoDrop 1000 spectrophotometer. Samples were prepared such that each tube contained 25 μL of total RNA at a final concentration of 0.1 μg/μL. To each tube reverse transcription buffer (5.0 μL, 10×), dNTPs (2.0 μL, 25×), random hexamers (5.0 μL, 10×), MultiScribe reverse transcriptase (50 U/μL, 2.5 μL) and nuclease-free water (10.5 μL) were added such that the final volume was 50 μL. A thermocycler was then used to produce cDNA. Then, 40 ng/μL (1.6 μL cDNA and 2.4 μL H2O) was aliquoted to a PCR tube. To each tube 4 μL of forward and reverse primer, 72 μL of H2O, 80 μL of Taq Master Mix was added to obtain a total volume of 160 μL. Hypoxanthine phosphoribosyltransferase (HPRT)-1 was used as a house keeping gene. Aliquots of 40 μL/well were dispensed in triplicate, and expression was recorded.
Measurement of protein levels via Western blot
Cells were plated at a concentration of 1 × 106 and grown for 24 h. After overnight serum starvation, the cells were treated with sublethal doses of IPA/NO (50 μM) or DEA/NO (75 μM) for different time intervals (1–44 h). Protein cell extracts were prepared by washing the cells twice with cold PBS, scraping, centrifuging at 150 g for 5 min, removing the supernatant and then resuspending in lysis buffer (Cell Signaling Technology, Inc., Danvers, MA) containing a protease inhibitor mixture (Calbiochem, Gibbstown, NJ). After a 30-min incubation on ice, the samples were centrifuged at 14,000 × g, and the supernatant protein concentration was determined by the bichoncinic acid method (Pierce, Thermo Fischer Scientific Inc., Rockford, IL) by recording absorbance at 560 nm. Protein samples (20 μg) were subjected to polyacrylamide gel electrophoresis (120 V) on 15% Tris-glycine acrylamide gels (NOVEX–Invitrogen, Carlsbad, CA). After transfer to polyvinylidene fluoride Immobilon-P membranes (Millipore), samples were probed with rabbit polyclonal or mouse monoclonal antibodies (Cell Signaling Technology, Inc., Danvers, MA). Bands were visualized with horseradish peroxidase-conjugated secondary antibodies (1:2,000–10,000; Cell Signaling Technology, Inc., Danvers, MA) and chemiluminescent substrate (ECL plus Western blotting kit; GE, Piscataway, NJ). HPRT protein-loading controls were run for each gel. Gel images were scanned and quantified by using a FluoroChem SP imaging system using AlphaEase FC software (Alpha Innotech, San Leandro, CA, USA).
Data analysis
Data are presented as mean ± SD or SEM, as indicated in the figure legends. Statistical analysis was performed with Prism 6.02 (GraphPad Software, San Diego, CA) using Student’s t-test.
Results and Discussion
Breast cancer is only second to lung cancer in prevalence among women [88]. In 2013 in the US alone, more than 200,000 new cases of breast cancer were reported (breastcancer.org). Estrogen is essential for growth and development of breast tissue but has also been associated with breast cancer [89, 90]. An increase in estrogen levels is a common biomarker for many types of breast cancer. Patients with both early and metastatic estrogen ER(+) breast cancer (70–80% of cases) [91] have the option to receive hormonal based therapies including tamoxifen [92] and aromatase inhibitors [93], which decrease mortality. Tamoxifen, which is a non-steroidal selective estrogen receptor modulator (SERM), is a pioneering medicine used worldwide in treatment of all stages of ER(+) breast cancer. Tamoxifen is also prescribed prophylactically for women at high risk of developing breast cancer. Despite being highly effective against breast cancer, tamoxifen may induce serious side effects, including blood clots, stroke and endometrial cancer [94, 95]. Also as is common with chemotherapy, cancer cells often develop resistance against tamoxifen treatment [93].
Both overexpression of COX-2 and increased prostaglandin biosynthesis show a strong correlation of carcinogenesis and metastasis in breast cancer [96]. This suggests that inflammation leads to drug resistance in this specific patient population. Furthermore, ER(−) cancer is not dependent on estrogen for proliferation and is comparatively more difficult to treat, leading to relatively poor prognosis. Thus, new chemotherapeutics are necessary for treatment of breast cancer, and evidence points to investigating the efficacy of NONO-NSAIDs in this context.
IPA/NO-aspirin and DEA/NO-aspirin (Scheme 1) offer the benefits of being of the same compound class, thus facilitating comparison of the impact of respective HNO or NO donation. Furthermore, the mechanisms of decomposition have been previously elucidated [26], and it is clear that these compounds decompose intracellularly. Here, we examine in detail the impacts of these prodrugs on breast cancer progression.
Scheme 1.
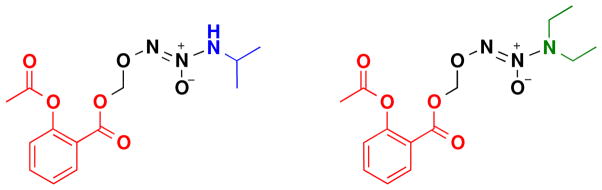
Structures of IPA/NO-aspirin and DEA/NO-aspirin
Effect of NONO-aspirin prodrugs on cell viability
While the cytotoxicity of both aspirin and ionic diazeniumdiolates in various cancer cells lines is low (IC50 > 1 mM) [72, 83, 97], NO-NSAIDs and related constructs significantly inhibit growth of cultured cancer cells as well as xenografts in mouse models [13, 26, 30, 64, 97–102]. Keefer and colleagues have previously shown that the derivatized diazeniumdiolate JS-K was significantly cytotoxic toward a variety of breast cancer cell lines (>50% reduction in viability at 1 μM JS-K) [102]. The impact was more modest for non-malignant epithelial cells up to 10 μM JS-K. We have also previously observed that IPA/NO-aspirin and DEA/NO-aspirin selectively reduced proliferation of lung carcinoma cells (IC50 values of ~100 μM) compared to normal endothelial cells [26].
Here, we assessed the impact of IPA/NO-aspirin and DEA/NO-aspirin compared to aspirin or the parent diazeniumdiolate on proliferation of four human breast cell lines: MDA-MB-231 (ER(−) adenocarcinoma), MDA-MB-468 (ER(−) adenocarcinoma), MCF-7 (ER(+) adenocarcinoma) and MCF-10A (non-tumorigenic, immortalized human breast epithelial cells). Our prior analysis indicated that these NONO-aspirin prodrugs were metabolized in cultured cells in less than 10 h [26]. After 48 h, the reduction in viability by each prodrug was comparable in all three breast adenocarcinoma lines, regardless of sensitivity to estrogen (Figure 1A–C, Table 1), and was similar to that observed previously in lung carcinoma cells and by clonogenic assay (data not shown). Cancer cells appear to generally be more sensitive to IPA/NO-aspirin compared to the NO-donating analogue. The lack of an effect on nonmalignant epithelial cells at up to 200 μM (Figure 1D) supports a cancer-dependent response. As expected the precursors did not impact viability at concentrations below 200 μM. This suggests that the prodrug itself may be involved or that concomitant delivery of NO or HNO may lead to chemosensitization.
Figure 1.


Effect of NONO-aspirin prodrugs (10–200 μM), aspirin (50–200 μM) or parent diazeniumdiolates (200 μM) on the viability of A) MDA-MB-231, B) MDA-MB-468, C) MCF-7 or D) MCF-10A cells. Cells were treated for 48 h at 37 °C, and cell survival was determined using the MTT assay. Figures are representative of three trials with data plotted as mean percentage versus untreated cells ± SD of the four replicates per plate). Vehicle (DMSO or 10 mM NaOH) was added as a control to otherwise untreated cells.
Table 1.
IC50 values (μM) for IPA/NO-aspirin or DEA/NO-aspirin in human breast cells
| cell line | IPA/NO-aspirin | DEA/NO-aspirin |
|---|---|---|
| MDA-MB-231 | 92 ± 1 | 102 ± 10 |
| MDA-MB-468 | 81 ± 2 | 90 ± 4 |
| MCF-7 | 93 ± 2 | 122 ± 3 |
| MCF-10A | >200 | >200 |
Values are mean ± SD for three trials, except for MCF-10A cells, which did not reach a 50% reduction at 200 μM.
Effect of NONO-aspirin prodrugs on growth of MDA-MB-231 xenografts
We primarily focused further studies on the effects of the NONO-aspirin prodrugs on one of the ER(−) lines, MDA-MB-231 cells, due to the more aggressive nature of this type of breast cancer. The chemotherapeutic effect of IPA/NO-aspirin and DEA/NO-aspirin was investigated in xenografts of GFP-transfected MDA-MB-231 cells, which allow noninvasive monitoring of tumor size. At the conclusion of two weeks of tumor growth and five weeks of treatment, aspirin did not significantly decrease the size of the primary tumor compared to the control (Figure 2A,B). A substantial, although not statistically significant reduction in fluorescence intensity was observed for DEA/NO-aspirin. Although masses were visible, upon preliminary examination they appeared to be primarily fibrotic tissue. Treatment with IPA/NO-aspirin led to both a significant decrease in fluorescence intensity and tumor mass. This suggests that the HNO-donating derivative not only effective inhibits tumor progression but also is tumoricidal.
Figure 2.
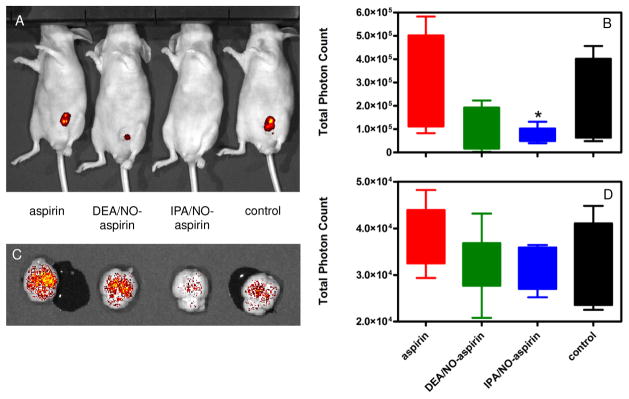
Effect of NONO-aspirin prodrugs or aspirin in nude mice implanted with 7.5 × 105 MDA-MB-231 cells stably transfected with GFP. The cells were allowed to grow for 14 d, and the animals (40) were randomly divided into four groups (control, aspirin, IPA/NO-aspirin or DEA/NO-aspirin). Treated groups were injected daily with equimolar doses of DEA/NO-aspirin or IPA/NO-aspirin (16 mg/kg) or aspirin (9 mg/kg) or with DMSO for the next five weeks. Tumor size was then measured using in vivo fluorescent imaging for quantification of the GFP tag: A) qualitative image of individual animals, B) quantitative analysis of fluorescence intensity at the primary tumor site (n = 5), C) qualitative image of individual brains, D) quantitative analysis of fluorescence intensity due to metastasis to the brain (n = 7). *p < 0.001 vs. control.
Fukuto and coworkers previously performed a similar study on the impact of HNO on MDA-MB-231 xenograft growth in SCID mice [72]. The HNO donor was Angeli’s salt (Na2N2O3), which is the most commonly used HNO donor in biological studies and is of the diazeniumdiolate class. Like IPA/NO, Angeli’s salt decomposes spontaneously with a short half-life (~4 min) under physiological conditions [87, 103, 104]. Treatment with Angeli’s salt led to some inhibition of tumor growth at both 17 and 50 mg/kg compared to control. A similar result was obtained with pheochromocytoma cells [79]. These studies are consistent with our finding that extracellular decomposition of ionic diazeniumdiolates is less effective than intracellular delivery by derivatized analogues [26].
Since MDA-MB-231 cells have a propensity to form metastatic tumors, the effect of the prodrugs was also studied on metastasis to the brains of the mice. While IPA/NO-aspirin and DEA/NO-aspirin both decreased metastasis compared to aspirin and untreated controls, the results were not statistically significant (Figure 2C,D).
Since nitration of GFP is known to reduce fluorescence intensity [105], the impact of HNO and NO on the GFP signal in MDA-MB-231-GFP cells during 48 h of growth was investigated to ensure that the reduction in signal in Figure 2 was not due to a chemical modification or modulation of GFP transcription. Over this period, fluorescence intensity increased, suggesting that cells were proliferating, and fluorescence intensity was not significantly impacted by the presence of 50 μM IPA/NO or 75 μM DEA/NO compared to NaOH control (data not shown). This indicates that the reduction in signal in Figure 2 was a result of tumor regression as a function of exposure to the prodrugs.
Mechanism of cytotoxicity
The role of NO in cancer has been investigated extensively [33]. We sought to more fully understand the underlying mechanism for the observed cancer-dependent response and the higher sensitivity of cancer cells to the HNO-donating adduct. A first consideration when comparing the effects of HNO to those of NO is the known difference in biotargets. HNO primarily interacts with thiols and ferric complexes [106] while NO readily associates with ferrous complexes and other free radicals [107]. Both HNO and NO can target and post-translationally modify crucial cellular proteins, especially those containing thiols. Direct association of HNO with thiols [73–75] leads to irreversible modification and inhibition of proteins with critical thiols such as aldehyde dehydrogenase and GAPDH [26, 76, 77, 108]. NO does not directly interact with thiols [109], but reaction with reactive oxygen species (ROS) can lead to nitrosating species. The reaction of HNO or NO with O2 forms more deleterious species, but the autoxidation product of HNO is more cytotoxic [110] and is capable of cleaving double strand DNA [110–113].
In addition to interacting with specific chemical targets, HNO donors have been shown to enhance apoptosis and suppress angiogenesis in breast cancer cells [72]. However, the mechanisms for these effects are currently unresolved. To evaluate the mechanism of action involved in cell death, the effects of our NONO-aspirin prodrugs, typically near or below the IC50 values (Table 1), was studied on metabolism, ROS and reactive nitrogen species (RNS) levels, DNA damage and apoptotic markers, angiogenesis and metastasis.
Impact on GAPDH activity
GAPDH is a critical glycolytic enzyme that can play an important role in cancer therapy [114, 115] as solid tumors often utilize glycolytic pathways even during normoxia to meet their energy requirements [116, 117]. Thus, inhibition of GAPDH can diminish ATP availability for tumor cells. GAPDH is also involved in initiation of apoptosis and in DNA repair [118]. As has been shown previously [26, 76, 77], IPA/NO-aspirin inhibited GAPDH activity in a concentration (Figure 3A) and time-dependent-manner (data not shown) in MDA-MB-231 cells. In contrast, IPA/NO-aspirin did not affect GAPDH activity in MCF-10A cells (Figure 3B), indicating that this protein may play a role in the selective cytotoxicity of IPA/NO-aspirin toward carcinogenic cell lines. At up to 100 μM, DEA/NO-aspirin was not inhibitory in either cell line.
Figure 3.

Effect of NONO-prodrugs on inhibition of GAPDH in A) MDA-MB-231 or B) MCF-10A cells. The cells were treated with varied concentrations (0, 25, 50, 75, 100 μM) of IPA/NO-aspirin (blue) or DEA/NO-aspirin (green) for 1 h at 37 °C. GAPDH activity was assessed by measuring fluorescence intensity at 570 nm. Data are plotted as mean ± SD (n = 3).
Assessment of production of oxidative species
Redox active species can modify proteins, DNA and lipids through oxidative, nitrosative and nitrative mechanisms. Redox induced stress is a result of uncontrolled modification of biomolecules once natural cellular defenses, such as the antioxidant system, are overwhelmed [119]. Accumulation of damage can lead to cell death and tissue injury. Redox active species also function as signaling molecules, through modification of individual targets that elicit specific biological responses, such as protection against or response to chemical stress. For example, high levels of NO lead to phosphorylation and stabilization of p53 [120], suggesting a role for NO in cell cycle regulation, DNA repair and apoptosis. Redox active oxygen and nitrogen species can also interact to lead to responses unique to either reactant.
Oxidation of 2′,7′-dichlorofluorescein (DCF) to fluorescein is a convenient indictor of the level of oxidants in cells [121]. In all three breast cancer lines, a significant increase in fluorescence intensity was observed upon exposure of IPA/NO-aspirin (Figure 4; MDA-MB-231 shown as the representative example). Both IPA/NO and DEA/NO also oxidized DCF, but to a much reduced extent. In contrast, aspirin was ineffective compared to control while DEA/NO-aspirin had minimal impact.
Figure 4.

Effect of NONO-aspirin prodrugs, aspirin or parent diazeniumdiolates (100 μM) on oxidation of DCF in MDA-MB-231 cells. DCF loaded cells were treated with IPA/NO-aspirin (blue), DEA/NO-aspirin (green), IPA/NO (magenta), DEA/NO (brown), aspirin (red) or DMSO (0.1%; black) at 37 °C, and the fluorescence at 535 nm was measured in a time-dependent manner. Data are plotted as mean ± SD (n = 4).
These data are consistent with our initial study, which demonstrated that uptake and esterase-mediated cleavage of the prodrugs was significant and that intracellular delivery was more effective at modifying cellular targets than extracellular decomposition [26]. In A549 cells, IPA/NO-aspirin decomposition was apparently complete in approximately 3 h while DEA-NO-aspirin was about three times longer-lived. Here, it is clear that IPA/NO-aspirin has substantial capacity to induce oxidative modifications.
Assessment of DNA damage
Autoxidation of both NO and HNO leads to species that are capable of damaging DNA. Conversion of NO to N2O3 can induce mutations through base deamination [122] while the product of the reaction of HNO with O2 can both oxidize and cleave DNA [111]. To investigate the possibility of RNS-induced DNA damage as a mechanism of cytotoxicity, the effect of the NONO-aspirin prodrugs was studied using the alkaline Comet assay in MDA-MB-231 cells. After 8 h, the damage induced by treatment with 50 μM IPA/NO-aspirin was significant (Figure 5A) and similar to the positive control of H2O2 treatment (Figure 5C, E). Exposure instead to 75 μM DEA/NO-aspirin (Figure 5B) did not differ from the DMSO-treated negative control (Figure 5D, E). These data are consistent with the DCF study that intracellular delivery of HNO can induce substantial oxidative modifications. This may be an important factor in the observed enhanced cytotoxicity by IPA/NO-aspirin.
Figure 5.


Effect of the NONO-aspirin prodrugs on DNA damage in MDA-MB-231 cells as determined using the Comet assay after 8 h at 37 °C: A) 50 μM IPA/NO-aspirin, B) 75 μM DEA/NO-aspirin, C) 0.1% DMSO or D) 100 μM H2O2. Tail length is quantified in E (mean ± SEM, n ≥ 150).
Effect on apoptosis, angiogenesis and metastasis
Upregulation of COX-2 levels in tumor cells, leading to elevated levels of prostaglandin E2 (PGE2) [123] can be an important factor in cancer progression [124–127] by increasing metastasis, cell proliferation [128] and angiogenesis [129–131] and reducing immune response [132] and apoptosis [131]. Both NONO-aspirin prodrugs have previously been shown to decrease the level of PGE2 [26]. Here, we investigated more directly the impact of the prodrugs on apoptosis, angiogenesis and metastasis.
Apoptosis is a biological process that is critical for normal development and tissue homeostasis [133]. Mutations leading to dysfunction in apoptosis can lead to the uncontrolled cell proliferation that is inherent to cancer. Apoptosis is tightly controlled both genetically and by post-translational modifications. NO is an important regulator of apoptosis and shows a concentration-dependent effect on induction of both pro- and anti-apoptotic signaling pathways based on cell type and cellular redox status [33]. For example, NO activates the tumor suppressor p53 [120]. Although HNO has been shown to increase apoptosis [72], the mechanism is not resolved. DNA damage is one of the principle triggers of programmed cell death, leading to elimination of impaired cell. Thus, accumulation of DNA damage as shown in Figure 5 may contribute, but like NO, HNO may also activate apoptotic pathways. Here, we examined the effect of the NO-aspirin prodrugs on the apoptotic markers caspase-3 and cleaved PARP and on p53 stabilization.
Caspases are cysteine-aspartate proteases that can trigger apoptosis through a signaling cascade [134]. Caspase-3 is an effector protease that has active cysteine residues that are susceptible to S-nitrosation. The impact of this modification has been variously reported to lead to reversible inhibition [135, 136] and to activation [137]. IPA/NO-aspirin activated caspase-3 in all three breast cancer lines, with somewhat varied sensitivity (Figure 6). The effect of exposure to DEA/NO-aspirin was more modest in MDA-MB-231 and MCF7 cells.
Figure 6.


Effect of NONO-aspirin prodrugs on caspase-3 activity of A) MDA-MB-231, B) MDA-MB-468 or C) MCF-7 cells. The cells were treated with various concentrations (0, 25, 50, 100 μM) of IPA/NO-aspirin (blue) or DEA/NO-aspirin (green) for 24 h at 37 °C. Caspase-3 activity was determined by recording the fluorescence (λex 485 nm, λem 535 nm) of the product formed by cleavage of the substrate N-Ac-DEVD-N′-MC-R11. Data are plotted as mean ± SD (n = 3).
As an important component of the DNA repair machinery, the PARP family of nuclear enzymes is a major target in the design of anti-cancer agents [138]. PARP is activated by DNA strand breaks and catalyzes the transfer of ADP-ribose to nuclear proteins. When cells are undergoing apoptosis, the DNA repair mechanism is halted by cleavage of PARP, a downstream target of caspase-3 [139]. As a zinc finger protein, PARP is a target for RNS [140]. The inhibitory effect of the HNO donor Angeli’s salt and of DEA/NO on PARP activity has been previously determined in MCF-7 cells that were caspase-3 null [80]. The inhibitory effect was rapid, but required rather high concentrations of donor.
Preliminary analysis in MDA-MB-231 cells indicates that IPA/NO-aspirin increases the level of cleaved PARP in a longer-term fashion while sensitivity to DEA/NO-aspirin was relatively short-lived (Figure 7; n = 1). A similar time sensitivity was observed for phosphorylation of p53 (Figure 8; n = 1). Since a number of chemotherapies and radiation therapy are based on inducing DNA damage in cancer cells, inhibition of PARP by HNO donors may increase the efficacy of these treatments. Furthermore, IPA/NO-aspirin is able to not only induce DNA damage but also to impede DNA repair.
Figure 7.

Effect of NONO-aspirin prodrugs on PARP cleavage in MDA-MB-231 cells. The cells were treated with 50 μM IPA/NO-aspirin (blue) or 75 μM DEA/NO-aspirin (green), respectively at 37 °C, and protein was collected at various time points (1–44 h). Lanes 1–6 (20 μg of protein): IPA/NO-aspirin at 1, 3, 6, 10, 20, 44 h; Lanes 6–12: DEA/NO-aspirin at 1, 6, 10, 20, 30, 44 h; Lane 15: control. Cleaved PARP levels at different time points were quantified by Western blot with respect to control, which was treated as 100%. Protein levels are normalized to the loading control HPRT.
Figure 8.

Effect of NONO-aspirin prodrugs on p53 phosphorylation in MDA-MB-231 cells. The cells were treated with 50 μM IPA/NO-aspirin (blue) or 75 μM DEA/NO-aspirin (green), respectively at 37 °C, and protein was collected at various time points (1–44 h). P-p53 was quantified by Western blot. Lanes 1–6 (20 μg of protein): IPA/NO-aspirin at 1, 3, 6, 10, 20, 44 h; Lanes 6–12: DEA/NO-aspirin at 1, 6, 10, 20, 30, 44 h; Lane 15: control. p-P53 protein at different time points was quantified by Western blot with respect to control, which was treated as 100%. Protein levels are normalized to the loading control HPRT.
Angiogenesis promotes tumor progression by initiating recruitment of blood vessels to supply nutrients and oxygen to growing cancer cells [141]. Angiogenesis also plays an important role in development of metastatic tumors at secondary sites. NO impacts tumor growth in a concentration-dependent manner [33] with low levels promoting cell survival, migration, proliferation and angiogenesis [142, 143] and higher fluxes causing vascular cell growth arrest and cell death [144, 145]. A high concentration of the HNO donor Angeli’s salt inhibited angiogenesis in mouse tumor specimens, decreased serum vascular endothelial growth factor (VEGF) levels and inhibited hypoxia-inducible factor (HIF) 1α in human breast cancer cells [72].
The impact of the NONO-aspirin prodrugs on tube formation in HUVECs after 12 h treatment was examined as a measure of angiogenesis. Both IPA/NO-aspirin and DEA/NO-aspirin reduced angiogenesis as compared to control even at 1 μM (Figure 9). We previously determined that neither IPA/NO-aspirin or DEA/NO-aspirin are appreciably cytotoxic toward HUVECs [26].
Figure 9.

Effect of NONO-aspirin prodrugs on inhibition of angiogenesis in HUVECs. Cells in Matrigel were treated with A) DMSO, B) 1 μM or C) 10 μM IPA/NO-aspirin, D) 1 μM or E) 10 μM DEA/NO-aspirin at 37 °C, and the extent of tube formation was measured after 12 h using a microscope.
Epithelial cadherin (E-cadherin) is calcium-dependent, membrane-associated cellular adhesion glycoprotein. Loss of E-cadherin-mediated cellular adhesion is a prerequisite for invasion and metastasis of tumor cells [146]. NO is an endogenous mediator of endothelial cell migration [147]. NO also has been shown to promote the growth, invasion and metastasis of murine mammary tumors [148]. To our knowledge, the effect of HNO on metastasis has not been studied. IPA/NO-aspirin does not show any significant change in E-cadherin levels, suggesting that reduced metastasis apparent Figure 2 is a result of either activation of a different pathway or an effect at the primary tumor. In contrast, DEA/NO-aspirin did increase E-cadherin expression in a time-dependent manner at both the mRNA and protein levels (Figure 10). This can result in a reversal of tumor cells from an invasive, mesenchymal, to a benign, epithelial phenotype [146].
Figure 10.


Effect of NONO-aspirin prodrugs on E-cadherin expression in MDA-MB-231 cells. The cells were treated with vehicle (black) or 100 μM IPA/NO-aspirin (blue) or DEA/NO-aspirin (green) for 3, 6 or 12 h at 37 °C. Relative A) mRNA or B) protein expression was measured using RT-PCR and Western blot techniques, respectively. E-cadherin mRNA and protein levels were quantified with respect to control (HPRT), which was treated as 1 for mRNA and 100% for protein. In A, data are plotted as mean ± SD (n = 3).
One avenue in development of the next generation of anticancer drugs is to produce a well-tolerated, even nontoxic, agent that has multidimensional modality with existing clinical agents. The data reported here support that NONO-NSAID prodrugs have such advantages. IPA/NO-aspirin in particular is able to damage DNA (Figure 5) including inducing double strand breaks, in a manner similar to radiation therapy and chemotherapies such as doxorubicin. However, in contrast to these treatments, the effects of IPA/NO-aspirin are apparently limited to rapid proliferating cancer cells, with little toxicity observed in normal or primary cells (Figures 1 and 2). The basis for this selectivity has yet to be determined but may be a function of uptake and cleavage preferentially in cancer cells. It is clear that cells are more sensitive to HNO and NO when produced intracellularly by prodrugs such as IPA/NO-aspirin and DEA/NO-aspirin compared to spontaneous donors that decompose systemically and extracellularly (Figure 4).
The DNA damage mediated by HNO donors is driven by oxidative pathways, thus as with radiation therapy, this effect will likely to be limited to oxygenated regions of tumors. HNO can also reduce glycolysis by inhibiting GAPDH activity (Figure 3) through direct binding to a critical thiol. The capacity to inhibit tumor proliferation in both normoxic and hypoxic regions would be a significant advantage in cancer therapy. The direct binding of HNO to thiols can also lead to disruption of PARP-dependent DNA repair pathways (Figure 7) and activation of caspase-3 (Figure 6), suggesting multiple O2-indepdenent mechanisms to induction of apoptosis by IPA/NO-aspirin.
COX-2 is a target for NSAID design to reduce side effects such as gastrointestinal irritation. Although aspirin is not a selective COX-2 inhibitor, the prodrug approach has been shown to not only enhance selective delivery but to reduce NSAID toxicity. COX-2 may also facilitate cancer progression in a variety of ways including activation of the immune response, down regulation of apoptosis and increased proliferation, aggregation, angiogenesis and metastasis. As a heme protein with critical thiols, COX-2 is a potential target for HNO. The observed reduction in PGE2 levels by IPA/NO-aspirin [26] may be in part responsible for the observed antiangiogenic (Figure 9) and antimetastatic effects (Figure 2). In summary, we suggest that IPA/NO-aspirin offers a single compound route to multifaceted targeting of cancer progression and recurrence mechanisms while maintaining a high safety profile.
The NO donor DEA/NO-aspirin lacks the DNA damaging effect of IPA/NO-aspirin and cannot directly lead to modification of thiols. In fact much of the antiproliferative effects of NO are O2-dependent through formation of nitrosative intermediates. However, DEA/NO-aspirin also was selectivity cytotoxic toward cancer cells. The observed decrease in angiogenesis (Figure 9) and inhibition of metastasis (Figures 2 and 10) is expected from NO donors. It is not clear at this time if any of the effects of DEA/NO-aspirin are cGMP-dependent, but intracellular delivery will increase specificity. Since some cancer cells can produce NO via NOS, the impacts of intracellular production of NO by DEA/NO-aspirin require further investigation.
A comparison of HNO and NO intracellular delivery demonstrates stark differences, although both reduce platelet aggregation and blood-born metastasis. We suggest that the HNO donor may lead to cell death while the NO donor reduces proliferation. Further analysis may provide insight into for development of the next generation of anticancer treatments.
Here, we have focused on targets for HNO and NO. The role of aspirin has yet to be elucidated. Furthermore, the effects of substitution with other NSAIDs, perhaps with higher specificity for COX-2, and interactions with other anticancer agents, either in combination or as new prodrugs, have not yet been investigated.
Conclusions
NONO-aspirin derivatives are significantly more effective compared to aspirin or nitrogen oxide donors both in vitro in human breast adenocarcinoma cells as well as in vivo in a nude mouse model. Additionally, the prodrugs were not appreciably cytotoxic toward non-tumorigenic epithelial cells. This suggests a cancer-specific sensitivity that is promising for chemotherapy or chemoprevention of breast cancer. The high cytotoxicity of IPA/NO-aspirin was in part due to increased oxidant levels leading to damage to DNA and to inhibition of GAPDH leading to caspase-3 mediated induction of apoptosis. On the other hand DEA/NO-aspirin is a promising candidate for reduction of metastasis by increasing the level of E-cadherin, which is responsible for cellular adhesion.
Highlights.
Diazeniumdiolate-based aspirin prodrugs reduced growth of breast cancer
The HNO donor also was more cytotoxic than the related NO donor
Cytotoxicity was significantly lower in endothelial cells, indicating cancer-specificity
The antiproliferative and antimetastatic mechanisms were unique for HNO and NO
Acknowledgments
This work is supported in part by a National Institutes of Health grant (R01-GM076247 to KMM) and the Intramural Research Program of the National Institutes of Health, National Cancer Institute and Center for Cancer Research (DAW).
Abbreviations
- A549
human alveolar epithelial carcinoma cells
- COX
cyclooxygenase
- DCF
2′,7′-dichlorofluorescein
- DCF-2DA
4-amino-5-methylamino-2′,7′-dichlorofluorescein diacetate
- DEA/NO
sodium 1-(N,N-diethylamino)diazen-1-ium-1,2-diolate NONOate, diazeniumdiolate
- DEA/NO-aspirin
O2-(acetylsalicyloyloxymethyl)-1-(N,N-diethylamino)-diazen-1-ium-1,2-diolate
- DMSO
dimethyl sulfoxide
- E-cadherin
epithelial cadherin
- ER(−)
estrogen receptor α-negative
- ER(+)
estrogen receptor α-positive
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- HIF
hypoxia-inducible factor
- HNO
nitroxyl
- HPRT
hypoxanthine phosphoribosyltransferase
- HUVECs
human umbilical vein endothelial cells
- iNOS
inducible isoform of NO synthase
- IPA/NO
sodium 1-(N-isopropylamino)diazen-1-ium-1,2-diolate
- IPA/NO-aspirin
O2-(acetylsalicyloyloxymethyl)-1-(N-isopropylamino)-diazen-1-ium-1,2-diolate
- MCF7
human ER(+) breast adenocarcinoma cells
- MCF-10A
non-tumorigenic, human epithelial breast cells
- MDA-MB-231
human ER(−) breast adenocarcinoma cells
- MDA-MB-468
human ER(−) breast adenocarcinoma cells
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NO
nitric oxide
- NSAID
non-steroidal anti-inflammatory drug
- NO-NSAID
NO-donating NSAID derivative
- NONO-NSAID
diazeniumdiolate-NSAID adduct
- NONO-aspirin
diazeniumdiolate-aspirin adduct
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PGE2
prostaglandin E2
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute
- RT-PCR
real-time polymerase chain reaction
- SERM
selective estrogen receptor modulator
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of Interest Disclosure
A patent on use of IPA/NO-aspirin is held by KMM, DAW and DB.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 2.Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287–1294. doi: 10.1056/NEJM199405053301808. [DOI] [PubMed] [Google Scholar]
- 3.The SALT Collaborative Group. Swedish Aspirin Low-Dose Trial (SALT) of 75 mg aspirin as secondary prophylaxis after cerebrovascular ischaemic events. Lancet. 1991;338:1345–1349. [PubMed] [Google Scholar]
- 4.Cryer B, Luk G, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterol. 1999;117:17–25. doi: 10.1016/s0016-5085(99)70545-7. [DOI] [PubMed] [Google Scholar]
- 5.Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med. 1992;327:749–754. doi: 10.1056/NEJM199209103271101. [DOI] [PubMed] [Google Scholar]
- 6.Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterol. 1997;112:1000–1016. doi: 10.1053/gast.1997.v112.pm9041264. [DOI] [PubMed] [Google Scholar]
- 7.Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810–15814. doi: 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- 8.Reitz DB, Li JJ, Norton MB, Reinhard EJ, Collins JT, Anderson GD, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Isakson PC. Selective cyclooxygenase inhibitors: novel 1,2-diarylcyclopentenes are potent and orally active COX-2 inhibitors. J Med Chem. 1994;37:3878–3881. doi: 10.1021/jm00049a005. [DOI] [PubMed] [Google Scholar]
- 9.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everts B, Währborg P, Hedner T. COX-2-specific inhibitors - the emergence of a new class of analgesic and anti-inflammatory drugs. Clin Rheumatol. 2000;19:331–343. doi: 10.1007/s100670070024. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA. 1993;90:11693–11697. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–1642. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 13.Keeble JE, Moore PK. Pharmacology and potential therapeutic applications of nitric oxide-releasing non-steroidal anti-inflammatory and related nitric oxide-donating drugs. Br J Pharmacol. 2002;137:295–310. doi: 10.1038/sj.bjp.0704876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiorucci S, Antonelli E. NO-NSAIDs: from inflammatory mediators to clinical readouts. Inflamm Allergy Drug Targets. 2006;5:121–131. doi: 10.2174/187152806776383161. [DOI] [PubMed] [Google Scholar]
- 15.Wallace JL, Del Soldato P. The therapeutic potential of NO-NSAIDs. Fundam Clin Pharmacol. 2003;17:11–20. doi: 10.1046/j.1472-8206.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- 16.Balligand JL, Feron O, Kelly RA. Role of nitric oxide in myocardial function. In: Ignarro LJ, editor. Nitric Oxide: Biology and Pathobiology. San Diego: Academic Press; 2000. pp. 585–632. [Google Scholar]
- 17.Kubes P, Arfors KE, Granger DN. Platelet-activating factor-induced mucosal dysfunction: role of oxidants and granulocytes. Am J Physiol. 1991;260:G965–G971. doi: 10.1152/ajpgi.1991.260.6.G965. [DOI] [PubMed] [Google Scholar]
- 18.Whittle BJ, Lopez-Belmonte J, Moncada S. Regulation of gastric mucosal integrity by endogenous nitric oxide: interactions with prostanoids and sensory neuropeptides in the rat. Br J Pharmacol. 1990;99:607–611. doi: 10.1111/j.1476-5381.1990.tb12977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacNaughton WK, Cirino G, Wallace JL. Endothelium-derived relaxing factor (nitric oxide) has protective actions in the stomach. Life Sci. 1989;45:1869–1876. doi: 10.1016/0024-3205(89)90540-7. [DOI] [PubMed] [Google Scholar]
- 20.Fiorucci S, Santucci L, Gresele P, Faccino RM, Del Soldato P, Morelli A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: a proof of concept endoscopic study. Gastroenterol. 2003;124:600–607. doi: 10.1053/gast.2003.50096. [DOI] [PubMed] [Google Scholar]
- 21.Wallace JL, McKnight W, Del Soldato P, Baydoun AR, Cirino G. Anti-thrombotic effects of a nitric oxide-releasing, gastric-sparing aspirin derivative. J Clin Invest. 1995;96:2711–2718. doi: 10.1172/JCI118338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velázquez C, Rao PNP, Knaus EE. Novel nonsteroidal antiinflammatory drugs possessing a nitric oxide donor diazen-1-ium-1,2-diolate moiety: design, synthesis, biological evaluation, and nitric oxide release studies. J Med Chem. 2005;48:4061–4067. doi: 10.1021/jm050211k. [DOI] [PubMed] [Google Scholar]
- 23.Velázquez CA, Chen QH, Citro ML, Keefer LK, Knaus EE. Second-generation aspirin and indomethacin prodrugs possessing an O(2)-(acetoxymethyl)-1-(2-carboxypyrrolidin-1-yl)diazenium-1,2-diolate nitric oxide donor moiety: design, synthesis, biological evaluation, and nitric oxide release studies. J Med Chem. 2008;51:1954–1956. doi: 10.1021/jm701450q. [DOI] [PubMed] [Google Scholar]
- 24.Bandarage UK, Chen L, Fang X, Garvey DS, Glavin A, Janero DR, Letts LG, Mercer GJ, Saha JK, Schroeder JD, Shumway MJ, Tam SW. Nitrosothiol esters of diclofenac: synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs. J Med Chem. 2000;43:4005–4016. doi: 10.1021/jm000178w. [DOI] [PubMed] [Google Scholar]
- 25.Jain S, Tran S, El Gendy MA, Kashfi K, Jurasz P, Velázquez-Martínez CA. Nitric oxide release is not required to decrease the ulcerogenic profile of nonsteroidal anti-inflammatory drugs. J Med Chem. 2012;55:688–696. doi: 10.1021/jm200973j. [DOI] [PubMed] [Google Scholar]
- 26.Basudhar D, Bharadwaj G, Cheng RY, Jain S, Shi S, Ridnour LA, Caceres VM, Spadari-Bratfisch RC, Paolocci N, Velázquez-Martínez CA, Wink DA, Miranda KM. Synthesis and comparison of nitroxyl and nitric oxide releasing diazeniumdiolate-based aspirin derivatives. J Med Chem. 2013;56:7804–7820. doi: 10.1021/jm400196q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keefer LK. Nitric oxide-releasing compounds: from basic research to promising drugs. CHEMTECH. 1998;28:0–35. [Google Scholar]
- 28.Saavedra JE, Dunams TM, Flippen-Anderson JL, Keefer LK. Secondary amine/nitric oxide complex ions, R2N[N(O)NO]−. O-Functionalization chemistry. J Org Chem. 1992;57:6134–6138. [Google Scholar]
- 29.Saavedra JE, Shami PJ, Wang LY, Davies KM, Booth MN, Citro ML, Keefer LK. Esterase-sensitive nitric oxide donors of the diazeniumdiolate family: in vitro antileukemic activity. J Med Chem. 2000;43:261–269. doi: 10.1021/jm9903850. [DOI] [PubMed] [Google Scholar]
- 30.Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther. 2003;2:409–417. [PubMed] [Google Scholar]
- 31.Buga GM, Ignarro LJ. Nitric oxide and cancer. In: Ignarro LJ, editor. Nitric Oxide: Biology and Pathobiology. San Diego: Academic Press; 2000. pp. 895–920. [Google Scholar]
- 32.Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol. 2011;67:1211–1224. doi: 10.1007/s00280-011-1654-4. [DOI] [PubMed] [Google Scholar]
- 33.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 34.Lechner M, Lirk P, Rieder J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin Cancer Biol. 2005;15:277–278. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Menon C, Bauer TW, Kelley ST, Raz DJ, Bleier JI, Patel K, Steele K, Prabakaran I, Shifrin A, Buerk DG, Sehgal CM, Fraker DL. Tumoricidal activity of high-dose tumor necrosis factor-alpha is mediated by macrophage-derived nitric oxide burst and permanent blood flow shutdown. Int J Cancer. 2008;123:464–475. doi: 10.1002/ijc.23499. [DOI] [PubMed] [Google Scholar]
- 36.Thomsen LL, Miles DW, Happerfield L, Bobrow LG, Knowles RG, Moncada S. Nitric oxide synthase activity in human breast cancer. Br J Cancer. 1995;72:41–44. doi: 10.1038/bjc.1995.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res. 1995;55:727–730. [PubMed] [Google Scholar]
- 38.Masri FA, Comhair SA, Koeck T, Xu W, Janocha A, Ghosh S, Dweik RA, Golish J, Kinter M, Stuehr DJ, Erzurum SC, Aulak KS. Abnormalities in nitric oxide and its derivatives in lung cancer. Am J Respir Crit Care Med. 2005;172:597–605. doi: 10.1164/rccm.200411-1523OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klotz T, Bloch W, Volberg C, Engelmann U, Addicks K. Selective expression of inducible nitric oxide synthase in human prostate carcinoma. Cancer. 1998;82:1897–1903. [PubMed] [Google Scholar]
- 40.Lagares-Garcia JA, Moore RA, Collier B, Heggere M, Diaz F, Qian F. Nitric oxide synthase as a marker in colorectal carcinoma. Am Surg. 2001;67:709–713. [PubMed] [Google Scholar]
- 41.Hajri A, Metzger E, Vallat F, Coffy S, Flatter E, Evrard S, Marescaux J, Aprahamian M. Role of nitric oxide in pancreatic tumour growth: in vivo and in vitro studies. Br J Cancer. 1998;78:841–849. doi: 10.1038/bjc.1998.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weninger W, Rendl M, Pammer J, Mildner M, Tschugguel W, Schneeberger C, Stürzl M, Tschachler E. Nitric oxide synthases in Kaposi’s sarcoma are expressed predominantly by vessels and tissue macrophages. Lab Invest. 1998;78:949–955. [PubMed] [Google Scholar]
- 43.Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC, Grimm EA. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin Cancer Res. 2000;6:4768–4775. [PubMed] [Google Scholar]
- 44.Ambs S, Merriam WG, Bennett WP, Felley-Bosco E, Ogunfusika MO, Oser SM, Klein S, Shields PG, Billiar TR, Harris CC. Frequent nitric oxide synthase-2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998;58:334–341. [PubMed] [Google Scholar]
- 45.Gallo O, Masini E, Morbidelli L, Franchi A, Fini-Storchi I, Vergari WA, Ziche M. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J Natl Cancer Inst. 1998;90:587–589. doi: 10.1093/jnci/90.8.587. [DOI] [PubMed] [Google Scholar]
- 46.Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, Martin DN, Switzer CH, Hudson RS, Wink DA, Lee DH, Stephens RM, Ambs S. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cook JA, Krishna MC, Pacelli R, DeGraff W, Liebmann J, Mitchell JB, Russo A, Wink DA. Nitric oxide enhancement of melphalan-induced cytotoxicity. Br J Cancer. 1997;76:325–334. doi: 10.1038/bjc.1997.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wink DA, Cook JA, Christodoulou D, Krishna MC, Pacelli R, Kim S, DeGraff W, Gamson J, Vodovotz Y, Russo A, Mitchell JB. Nitric oxide and some nitric oxide donor compounds enhance the cytotoxicity of cisplatin. Nitric Oxide. 1997;1:88–94. doi: 10.1006/niox.1996.0108. [DOI] [PubMed] [Google Scholar]
- 49.Gray LH, Green FO, Hawes CA. Effect of nitric oxide on the radiosensitivity of tumour cells. Nature. 1958;182:952–953. doi: 10.1038/182952a0. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell JB, Wink DA, Degraff W, Gamson J, Keefer LK, Krishna MC. Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res. 1993;53:5845–5848. [PubMed] [Google Scholar]
- 51.Mitchell JB, Cook JA, Krishna MC, DeGraff W, Gamson J, Fisher J, Christodoulou D, Wink DA. Radiation sensitisation by nitric oxide releasing agents. Br J Cancer. 1996;74:S181–S184. [PMC free article] [PubMed] [Google Scholar]
- 52.Bonavida B, Baritaki S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: downregulation of the NF-κB/Snail/YY1/RKIP circuitry. Nitric Oxide. 2011;24:1–7. doi: 10.1016/j.niox.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Glynn SA, Prueitt RL, Ridnour LA, Boersma BJ, Dorsey TM, Wink DA, Goodman JE, Yfantis HG, Lee DH, Ambs S. COX-2 activation is associated with Akt phosphorylation and poor survival in ER-negative, HER2-positive breast cancer. BMC Cancer. 2010;10:626–637. doi: 10.1186/1471-2407-10-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R, Barrow D, Nicholson RI. Cyclooxygenase-2 protein reduces tamoxifen and N-(4-hydroxyphenyl)retinamide inhibitory effects in breast cancer cells. Int J Cancer. 2006;118:290–301. doi: 10.1002/ijc.21355. [DOI] [PubMed] [Google Scholar]
- 55.Harris RE, Beebe-Donk J, Namboodiri KK. Inverse association of non-steroidal anti-inflammatory drugs and malignant melanoma among women. Oncol Rep. 2001;8:655–657. doi: 10.3892/or.8.3.655. [DOI] [PubMed] [Google Scholar]
- 56.Rüegg C, Zaric J, Stupp R. Non steroidal anti-inflammatory drugs and COX-2 inhibitors as anti-cancer therapeutics: hypes, hopes and reality. Ann Med. 2003;35:476–487. doi: 10.1080/07853890310017053. [DOI] [PubMed] [Google Scholar]
- 57.Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade. Oncol Rep. 2005;13:559–583. [PubMed] [Google Scholar]
- 58.Schreinemachers DM, Everson RB. Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994;5:138–146. doi: 10.1097/00001648-199403000-00003. [DOI] [PubMed] [Google Scholar]
- 59.Rostom A, Dubé C, Lewin G, Tsertsvadze A, Barrowman N, Code C, Sampson M, Moher D U.S. Preventive Services Task Force. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146:376–389. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 60.Gallicchio L, Visvanathan K, Burke A, Hoffman SC, Helzlsouer KJ. Nonsteroidal anti-inflammatory drugs and the risk of developing breast cancer in a population-based prospective cohort study in Washington County, MD. Int J Cancer. 2007;121:211–215. doi: 10.1002/ijc.22656. [DOI] [PubMed] [Google Scholar]
- 61.Chang ET, Cronin-Fenton DP, Friis S, Hjalgrim H, Sørensen HT, Pedersen L. Aspirin and other nonsteroidal anti-inflammatory drugs in relation to Hodgkin lymphoma risk in northern Denmark. Cancer Epidemiol Biomarkers Prev. 2010;19:59–64. doi: 10.1158/1055-9965.EPI-09-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Famenini S, Young LC. Aspirin use and melanoma risk: a review of the literature. J Am Acad Dermatol. 2014;70:187–191. doi: 10.1016/j.jaad.2013.09.045. [DOI] [PubMed] [Google Scholar]
- 63.Robak P, Smolewski P, Robak T. The role of non-steroidal anti-inflammatory drugs in the risk of development and treatment of hematologic malignancies. Leuk Lymphoma. 2008;49:1452–1462. doi: 10.1080/10428190802108854. [DOI] [PubMed] [Google Scholar]
- 64.Rigas B, Kashfi K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol Med. 2004;10:324–330. doi: 10.1016/j.molmed.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 65.Rigas B. Novel agents for cancer prevention based on nitric oxide. Biochem Soc Trans. 2007;35:1364–1368. doi: 10.1042/BST0351364. [DOI] [PubMed] [Google Scholar]
- 66.Miranda KM. The chemistry of nitroxyl (HNO) and implications in biology. Coord Chem Rev. 2005;249:433–455. [Google Scholar]
- 67.Paolocci N, Katori T, Champion HC, StJohn ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: independence from β-adrenergic signaling. Proc Natl Acad Sci USA. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feelisch M. Nitroxyl gets to the heart of the matter. Proc Natl Acad Sci USA. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 70.Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- 71.DeMaster EG, Redfern B, Nagasawa HT. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 72.Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, Jackson MI, Fukuto JM, Brooks MN. Nitroxyl inhibits breast tumor growth and angiogenesis. Int J Cancer. 2008;122:1905–1910. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 73.Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 74.Bartberger MD, Fukuto JM, Houk KN. On the acidity and reactivity of HNO in aqueous solution and biological systems. Proc Natl Acad Sci USA. 2001;98:2194–2198. doi: 10.1073/pnas.041481598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doyle MP, Mahapatro SN, Broene RD, Guy JK. Oxidation and reduction of hemoproteins by trioxodinitrate(II): the role of nitrosyl hydride and nitrite. J Am Chem Soc. 1988;110:593–599. [Google Scholar]
- 76.Lopez BE, Rodriguez CE, Pribadi M, Cook NM, Shinyashik M, Fukuto JM. Inhibition of yeast glycolysis by nitroxyl (HNO): mechanism of HNO toxicity and implications to HNO biology. Arch Biochem Biophys. 2005;442:140–148. doi: 10.1016/j.abb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 77.Lopez BE, Wink DA, Fukuto JM. The inhibition of glyceraldehyde-3-phosphate dehydrogenase by nitroxyl (HNO) Arch Biochem Biophys. 2007;465:430–436. doi: 10.1016/j.abb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 78.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 79.Stoyanovsky DA, Schor NF, Nylander KD, Salama G. Effects of pH on the cytotoxicity of sodium trioxodinitrate (Angeli’s salt) J Med Chem. 2004;47:210–217. doi: 10.1021/jm030192j. [DOI] [PubMed] [Google Scholar]
- 80.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of poly(ADP-ribose) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic Biol Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 81.Keefer LK. Nitric oxide (NO)- and nitroxyl (HNO)-generating diazeniumdiolates (NONOates): emerging commercial opportunities. Curr Top Med Chem. 2005;5:625–636. doi: 10.2174/1568026054679380. [DOI] [PubMed] [Google Scholar]
- 82.Dutton AS, Fukuto JM, Houk KN. Mechanisms of HNO and NO production from Angeli’s salt: density functional and CBS-QB3 theory predictions. J Am Chem Soc. 2004;126:3795–3800. doi: 10.1021/ja0391614. [DOI] [PubMed] [Google Scholar]
- 83.Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Cologna SM, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. Comparison of the NO and HNO donating properties of diazeniumdiolates: primary amine adducts release HNO in vivo. J Med Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 84.Dutton AS, Miranda KM, Wink DA, Fukuto JM, Houk KN. Mechanism of pH-dependent decomposition of monoalkylamine diazeniumdiolates to form HNO and NO, deduced from the model compound methylamine diazeniumdiolate, density functional theory, and CBS-QB3 calculations. Inorg Chem. 2006;45:2448–2456. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salmon DJ, Torres de Holding CL, Thomas L, Peterson KV, Goodman GP, Saavedra JE, Srinivasan A, Davies KM, Keefer LK, Miranda KM. HNO and NO release from a primary amine-based diazeniumdiolate as a function of pH. Inorg Chem. 2011;50:3262–3270. doi: 10.1021/ic101736e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Drago RS, Karstetter BR. The reaction of nitrogen(II) oxide with various primary and secondary amines. J Am Chem Soc. 1961;83:1819–1822. [Google Scholar]
- 87.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide - vasorelaxant effects. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 88.American Cancer Society. Cancer Facts and Figures 2015. Atlanta: G. A. C. S; 2015. [Google Scholar]
- 89.Chen GG, Zeng Q, Tse GM. Estrogen and its receptors in cancer. Med Res Rev. 2008;28:954–974. doi: 10.1002/med.20131. [DOI] [PubMed] [Google Scholar]
- 90.Kennedy BJ. Hormone therapy for advanced breast cancer. Cancer. 1965;18:1551–1557. doi: 10.1002/1097-0142(196512)18:12<1551::aid-cncr2820181206>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 91.Anderson WF, Chatterjee N, Ershler WB, Brawley OW. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Cancer Res Treat. 2002;76:27–36. doi: 10.1023/a:1020299707510. [DOI] [PubMed] [Google Scholar]
- 92.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 93.Thürlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Rabaglio M, Smith I, Wardley A, Price KNAG. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 94.Cohen I. Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol Oncol. 2004;94:256–266. doi: 10.1016/j.ygyno.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 95.Yang G, Nowsheen S, Aziz K, Georgakilas AG. Toxicity and adverse effects of tamoxifen and other anti-estrogen drugs. Pharmacol Ther. 2013;139:392–404. doi: 10.1016/j.pharmthera.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 96.Howe LR, Chang SH, Tolle KC, Dillon R, Young LJ, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, Hudis C, Brown AM, Hla T, Subbaramaiah K, Dannenberg AJ. HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res. 2005;65:10113–10119. doi: 10.1158/0008-5472.CAN-05-1524. [DOI] [PubMed] [Google Scholar]
- 97.Nath N, Vassell R, Chattopadhyay M, Kogan M, Kashfi K. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: effects on COX-2 expression and Wnt/beta-catenin/TCF-4 signaling. Biochem Pharmacol. 2009;78:1298–1304. doi: 10.1016/j.bcp.2009.06.104. [DOI] [PubMed] [Google Scholar]
- 98.Huguenin S, Vacherot F, Kheuang L, Fleury-Feith J, Jaurand MC, Bolla M, Riffaud JP, Chopin DK. Antiproliferative effect of nitrosulindac (NCX 1102), a new nitric oxide-donating non-steroidal anti-inflammatory drug, on human bladder carcinoma cell lines. Mol Cancer Ther. 2004;3:291–298. [PubMed] [Google Scholar]
- 99.Huguenin S, Fleury-Feith J, Kheuang L, Jaurand MC, Bolla M, Riffaud JP, Chopin DK, Vacherot F. Nitrosulindac (NCX 1102): a new nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibits proliferation and induces apoptosis in human prostatic epithelial cell lines. Prostate. 2004;61:132–141. doi: 10.1002/pros.20081. [DOI] [PubMed] [Google Scholar]
- 100.Pathi SS, Jutooru I, Chadalapaka G, Sreevalsan S, Anand S, Thatcher GR, Safe S. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol Cancer Res. 2011;9:195–202. doi: 10.1158/1541-7786.MCR-10-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li C, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. Blood. 2007;110:709–718. [Google Scholar]
- 102.McMurtry V, Saavedra JE, Nieves-Alicea R, Simeone AM, Keefer LK, Tari AM. JS-K, a nitric oxide-releasing prodrug, induces breast cancer cell death while sparing normal mammary epithelial cells. Int J Oncol. 2011;38:963–971. doi: 10.3892/ijo.2011.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG, Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. Mechanism of aerobic decomposition of Angeli’s salt (sodium trioxodinitrate) at physiological pH. J Am Chem Soc. 2005;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- 104.Hughes MN, Wimbledon PE. The chemisry of trioxodinitrates. Part I. Decompostion of sodium trioxodinitrate (Angeli’s salt) in aqueous solution. J Chem Soc Dalton Trans. 1976:703–707. [Google Scholar]
- 105.Espey MG, Xavier S, Thomas DD, Miranda KM, Wink DA. Direct real-time evaluation of nitration with green fluorescent protein in solution and within human cells reveals the impact of nitrogen dioxide vs. peroxynitrite mechanisms. Proc Natl Acad Sci USA. 2002;99:3481–3486. doi: 10.1073/pnas.062604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc Natl Acad Sci USA. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 108.Shen B, English AM. Mass spectrometric analysis of nitroxyl-mediated protein modification: comparison of products formed with free and protein-based cysteines. Biochemistry. 2005;44:14030–14044. doi: 10.1021/bi0507478. [DOI] [PubMed] [Google Scholar]
- 109.Pryor WA, Church DF, Govindan CK, Crank G. Oxidation of thiols by nitric oxide and nitrogen dioxide - synthetic utility and toxicological implications. J Org Chem. 1982;47:156–159. [Google Scholar]
- 110.Wink DA, Feelisch M, Fukuto J, Christodoulou D, Jourd’heuil D, Grisham MB, Vodovotz Y, Cook JA, Krishna M, DeGraff WG, Kim S, Gamson J, Mitchell JB. The cytotoxicity of nitroxyl: possible implications for the pathophysiological role of NO. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 111.Ohshima H, Gilibert I, Bianchini F. Induction of DNA strand breakage and base oxidation by nitroxyl anion through hydroxyl radical production. Free Radic Biol Med. 1999;26:1305–1313. doi: 10.1016/s0891-5849(98)00327-x. [DOI] [PubMed] [Google Scholar]
- 112.Chazotte-Aubert L, Oikawa S, Gilibert I, Bianchini F, Kawanishi S, Ohshima H. Cytotoxicity and site-specific DNA damage induced by nitroxyl anion (NO−) in the presence of hydrogen peroxide - implications for various pathophysiological conditions. J Biol Chem. 1999;274:20909–20915. doi: 10.1074/jbc.274.30.20909. [DOI] [PubMed] [Google Scholar]
- 113.Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Further evidence for distinct reactive intermediates from nitroxyl and peroxynitrite: effects of buffer composition on the chemistry of Angeli’s salt and synthetic peroxynitrite. Arch Biochem Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 114.Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem. 2005;95:45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- 115.Xu R, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Deating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 116.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 117.Gatenby RA, Gillies RJ. Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol. 2007;39:1358–1366. doi: 10.1016/j.biocel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 118.Colell A, Green DR, Ricci JE. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009;16:1573–1581. doi: 10.1038/cdd.2009.137. [DOI] [PubMed] [Google Scholar]
- 119.Cadenas E, Sies H. Oxidative stress: excited oxygen species and enzyme activity. Adv Enzyme Regul. 1985;23:217–237. doi: 10.1016/0065-2571(85)90049-4. [DOI] [PubMed] [Google Scholar]
- 120.Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, Quezadol MM, Zurer I, Rotter V, Wink DA, Appella E, Harris CC. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci USA. 2003;100:143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Myhre O, Andersen JM, Aarnes H, Fonnum F. Evaluation of the probes 2′,7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem Pharmacol. 2003;65:1575–1582. doi: 10.1016/s0006-2952(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 122.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS, Keefer LK. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 123.Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 2000;30:3–21. [PubMed] [Google Scholar]
- 124.Howe LR, Subbaramaiah K, Brown AM, Dannenberg AJ. Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer. 2001;8:97–114. doi: 10.1677/erc.0.0080097. [DOI] [PubMed] [Google Scholar]
- 125.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17:1640–1647. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 126.Fulton AM, Zhang SZ, Chong YC. Role of the prostaglandin E2 receptor in mammary tumor metastasis. Cancer Res. 1991;51:2047–2050. [PubMed] [Google Scholar]
- 127.Wang D, DuBois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2009;29:781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Trifan OC, Hla TJ. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. Cell Mol Biol. 2003;7:207–222. doi: 10.1111/j.1582-4934.2003.tb00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leahy KM, Koki AT, Masferrer JL. Role of cyclooxygenases in angiogenesis. Curr Med Chem. 2000;7:1163–1170. doi: 10.2174/0929867003374336. [DOI] [PubMed] [Google Scholar]
- 130.Rüegg C, Dormond O, Mariotti A. Endothelial cell integrins and COX-2: mediators and therapeutic targets of tumor angiogenesis. Biochim Biophys Acta. 2004;1654:51–67. doi: 10.1016/j.bbcan.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 131.Gately S. The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 2000;19:19–27. doi: 10.1023/a:1026575610124. [DOI] [PubMed] [Google Scholar]
- 132.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 133.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 134.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 135.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31134. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 137.Brockhaus F, Brüne B. U937 apoptotic cell death by nitric oxide: Bcl-2 downregulation and caspase activation. Exp Cell Res. 1998;238:33–41. doi: 10.1006/excr.1997.3778. [DOI] [PubMed] [Google Scholar]
- 138.Griffin RJ, Curtin NJ, Newell DR, Golding BT, Durkacz BW, Calvert AH. The role of inhibitors of poly(ADP-ribose) polymerase as resistance-modifying agents in cancer therapy. Biochimie. 1995;77:408–422. doi: 10.1016/0300-9084(96)88154-5. [DOI] [PubMed] [Google Scholar]
- 139.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 140.Messmer UK, Reimer DM, Reed JC, Brüne B. Nitric oxide induced poly(ADP-ribose) polymerase cleavage in RAW 264.7 macrophage apoptosis is blocked by Bcl-2. FEBS Lett. 1996;384:162–166. doi: 10.1016/0014-5793(96)00311-0. [DOI] [PubMed] [Google Scholar]
- 141.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 142.Fukumura D, Jain R. Role of nitric oxide in angiogenesis and microcirculation in tumors. Cancer Metastasis Rev. 1998;17:77–89. doi: 10.1023/a:1005908805527. [DOI] [PubMed] [Google Scholar]
- 143.Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in tumor angiogenesis. Cancer Treat Res. 2004;117:155–167. doi: 10.1007/978-1-4419-8871-3_11. [DOI] [PubMed] [Google Scholar]
- 144.Pipili-Synetos E, Sakkoula E, Haralabopoulos G, Andriopoulou P, Peristeris P, Maragoudakis ME. Evidence that nitric oxide is an endogenous antiangiogenic mediator. Br J Pharmacol. 1994;111:894–902. doi: 10.1111/j.1476-5381.1994.tb14822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pipili-Synetos E, Papageorgiou A, Sakkoula E, Sotiropoulou G, Fotsis T, Karakiulakis G, Maragoudakis M. Inhibition of angiogenesis, tumour growth and metastasis by the NO-releasing vasodilators, isosorbide mononitrate and dinitrate. Br J Pharmacol. 1995;116:1829–1834. doi: 10.1111/j.1476-5381.1995.tb16670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 147.Murohara T, Witzenbichler B, Spyridopoulos I, Asahara T, Ding B, Sullivan A, Losordo DW, Isner JM. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1156–1161. doi: 10.1161/01.atv.19.5.1156. [DOI] [PubMed] [Google Scholar]
- 148.Jadeski LC, Hum KO, Chakraborty C, Lala PK. Nitric oxide promotes murine mammary tumour growth and metastasis by stimulating tumour cell migration, invasiveness and angiogenesis. Int J Cancer. 2000;86:30–39. doi: 10.1002/(sici)1097-0215(20000401)86:1<30::aid-ijc5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]