

Abstract
Objective:
To determine the genetic cause of neurodegeneration in a family with myeloneuropathy.
Methods:
We studied 5 siblings in a family with a mild, dominantly inherited neuropathy by clinical examination and electrophysiology. One patient had a sural nerve biopsy. After ruling out common genetic causes of axonal Charcot-Marie-Tooth disease, we sequenced 3 tRNA synthetase genes associated with neuropathy.
Results:
All affected family members had a mild axonal neuropathy, and 3 of 4 had lower extremity hyperreflexia, evidence of a superimposed myelopathy. A nerve biopsy showed evidence of chronic axonal loss. All affected family members had a heterozygous missense mutation c.304G>C (p.Gly102Arg) in the alanyl-tRNA synthetase (AARS) gene; this allele was not identified in unaffected individuals or control samples. The equivalent change in the yeast ortholog failed to complement a strain of yeast lacking AARS function, suggesting that the mutation is damaging.
Conclusion:
A novel mutation in AARS causes a mild myeloneuropathy, a novel phenotype for patients with mutations in one of the tRNA synthetase genes.
Charcot-Marie-Tooth disease (CMT) and hereditary motor and sensory neuropathy (HMSN) are alternative names for inherited neuropathies that are not part of more complex syndromes. With an estimated prevalence of 1 in 2,500 persons, CMT/HMSN is one of the commonest neurogenetic diseases and is subdivided according to clinical, electrophysiologic, histologic, and genetic features.1 CMT2/HMSN-II is a dominantly inherited axonal neuropathy, with nerve conduction velocities greater than 38 m/s.
Mutations in more than 20 different genes cause autosomal dominant CMT2. With the exception of some MPZ mutations, these mutations are thought to produce a neuropathy through their direct effects in neurons. Mutations in genes that encode aminoacyl-tRNA synthetases (ARS) cause some forms of CMT2. These enzymes charge tRNAs with their cognate amino acids, establishing the genetic code. Mutations in the glycyl-tRNA synthetase (GARS) gene were the first to be described, found in patients with dominant purely motor (hereditary motor neuropathy 5A) or dominant motor predominant (CMT2D) neuropathy.2 Mutations in 5 additional ARS loci have been found in various forms of CMT disease—tyrosyl-tRNA synthetase (YARS) in dominant intermediate CMT type C (DI-CMTC)3; alanyl-tRNA synthetase (AARS) in dominant CMT2N4–7; lysyl-tRNA synthetase (KARS) in recessive, intermediate CMT (CMTRIB)8; histidyl-tRNA synthetase (HARS) in a patient with sporadic, presumed dominant neuropathy9; and methionyl-tRNA synthetase (MARS) in late-onset, dominant CMT2 with incomplete penetrance.10 A growing number of rare recessive developmental disorders have also been linked to tRNA synthetase genes, including the mitochondrial alanyl tRNA synthetase gene, AARS2.11–25 Several of these recessive disorders involve the CNS and non-nervous organ systems, but the dominant tRNA synthetase mutations predominantly affect the peripheral nervous system (PNS).21 An example of this pronounced distinction is seen with the glycyl-tRNA synthetase gene, GARS, where dominant mutations cause distal axonopathies2 and compound heterozygous mutations cause systemic mitochondrial disease.24
METHODS
Standard protocol approvals, registrations, and patient consents.
Institutional review board approval was obtained from the University of Pennsylvania for these studies. Written informed consent was obtained from each patient who participated.
Clinical data and sample collection.
The family was seen by one of the authors (S.S.S.) in an outpatient clinic, where clinical neurophysiology was also performed to generate a Charcot-Marie-Tooth neuropathy score (CMTNS).26 A sural nerve biopsy was performed as part of the proband's prior diagnostic workup from another institution. We obtained the epoxy blocks and recut and imaged semi-thin and thin sections with transmitted light and electron microscopes.
Mutation analysis.
A collection of 94 families with dominant forms of peripheral neuropathies (17 CMT1, 37 CMT2, 9 CMTDI, 31 unspecified type) were screened for mutations in GARS, AARS, and YARS by Sanger sequencing. Genomic DNA was extracted from peripheral blood using standard procedures. All exons and exon-intron boundaries were amplified using primers designed with Primer3 v0.4.0 (primer sequences and PCR conditions available upon request). PCR products were purified with ExoSAP-IT (USB, Cleveland, OH) and bidirectionally sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA). Fragments were electrophoretically separated on an ABI3730xl DNA Analyzer (Applied Biosystems) and analyzed with SeqMan II (DNASTAR Inc., Madison, WI). The numerical amino acid position for the human mutation is based on GenBank accession number BAA06808.1, and the equivalent yeast amino acid positions were determined by a ClustalW comparison to the sequence for GenBank accession number EDV10897.1. Our description of the mutation follows HGVS nomenclature guidelines (http://www.hgvs.org/mutnomen).
Yeast complementation assay.
Yeast complementation assays were performed as previously described.4 Briefly, mutation-containing oligonucleotides were designed and used with the QuickChange II XL Site-Directed Mutagenesis Kit (per the manufacturer's instructions; Stratagene, Santa Clara, CA) using forward (5′-TTTTTTGAAATGCTGCGTAACTGGTCGTTTG-3′) and reverse (5′-CAAACGACCAGTTACGCAGCATTTCAAAAAA-3′) primers to model the p.Gly102Arg AARS mutation in the yeast ortholog ALA1, with a c.316G>C nucleotide change, in a pDONR221 Gateway entry clone (Invitrogen, Carlsbad, CA). Plasmids were isolated from individual clones and sequenced to confirm mutagenesis and exclude polymerase errors. The p.Gly102Arg ALA1/pDONR221 entry clone was recombined into a Gateway-compatible LEU2-bearing pRS315 destination vector. Resulting clones were purified and digested with BsrGI (New England Biolabs, Ipswich, MA) to confirm recombination. The Δala1 haploid yeast strain (harboring a pRS316 maintenance vector to express wild-type ALA1 and URA3) was transformed with wild-type or mutant ALA1 in a LEU2-bearing pRS315 vector and selected on medium lacking uracil and leucine (Teknova, Hollister, CA). For each transformation, 2 colonies were selected for further analysis. Each colony was grown to saturation in selection media for 48 hours. Next, 10 μL of undiluted and diluted (1:10 and 1:100) samples from each culture were spotted on plates containing 0.1% 5-FOA complete medium or SD -leu -ura growth medium (Teknova) and incubated at 30°C for 72 hours.
RESULTS
Clinical assessment reveals evidence of peripheral neuropathy and myelopathy.
The family pedigree is shown in figure 1A. There are 5 affected members of the family: the proband (II-5), her 3 brothers (II-2, II-3, and II-4), and their mother (who is deceased but had severe neuropathic pain). When first seen in our clinic at 22 years old, the proband reported frequent ankle sprains during her adolescence, including a fractured left ankle at age 21 that ultimately led to a triple arthrodesis at age 25. At age 21 years, she developed burning pain (worse in arms than legs) and painful electric shocks (worse in legs than arms) in her extremities. Over the next several years she tried amitriptyline, nortriptyline, gabapentin, carbamazepine, and tramadol before subsequently getting some relief with long-acting opioids.
Figure 1. Pedigree and genotype of family.
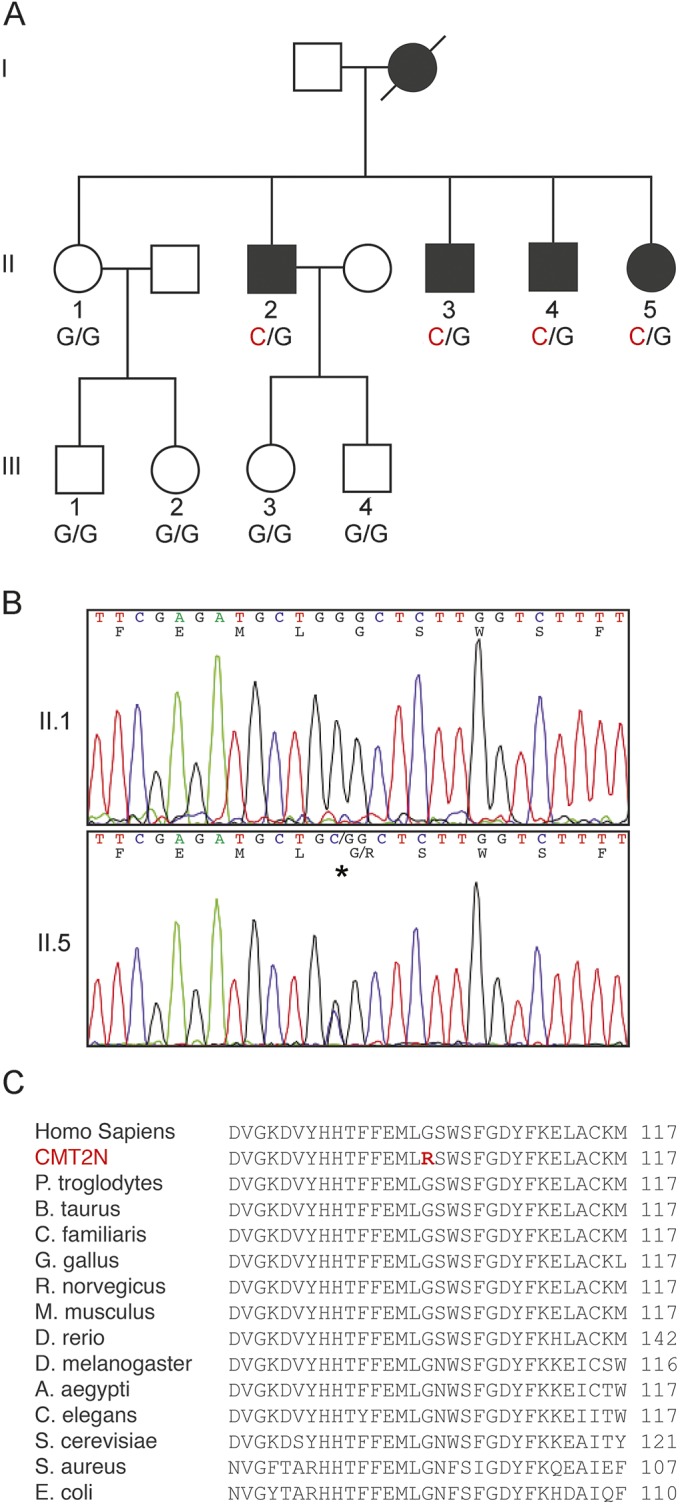
(A) Four of 5 siblings in the second generation (II) have a c.304G>C substitution and have clinical and electrophysiologic evidence of neuropathy. The genotypes of individuals whose DNA was collected are indicated below their respective symbol. (B) Normal sequence of the AARS gene is shown from the proband's sister (II-1), who does not have myeloneuropathy. Sequence of the proband's AARS gene demonstrates a heterozygous c.304G>C substitution, which is predicted to result in p.Gly102Arg. (C) The sequences of the activation domains of the AARS proteins from a range of divergent species, compared with ClustalW multiple sequence alignment tool. The mutated region of the protein, and in particular the residue that is mutated in this family, is highly conserved (the substitution is shown in red).
Examination at age 22 years revealed slight difficulty standing on the heels, slight weakness in extensor hallucis longus (4+/5), a stocking-type pattern of decreased cold and pinprick, and reduced vibratory sensation at the toes. Reflexes were 3+ at the knees with crossed adduction and absent at the ankles. Plantar responses were muted. The examination raised the possibility of a superimposed myelopathy. MRIs of the brain and cervical spine were unremarkable, and a B12 level was normal. While the patient's motor findings have remained largely unchanged from 1999 to 2013, her sensory deficits progressed: pinprick sensation is now decreased to above her knees and vibration sensation is absent in the toes. Plantar responses have remained flexor. Her CMTNS at age 34 years is 14.
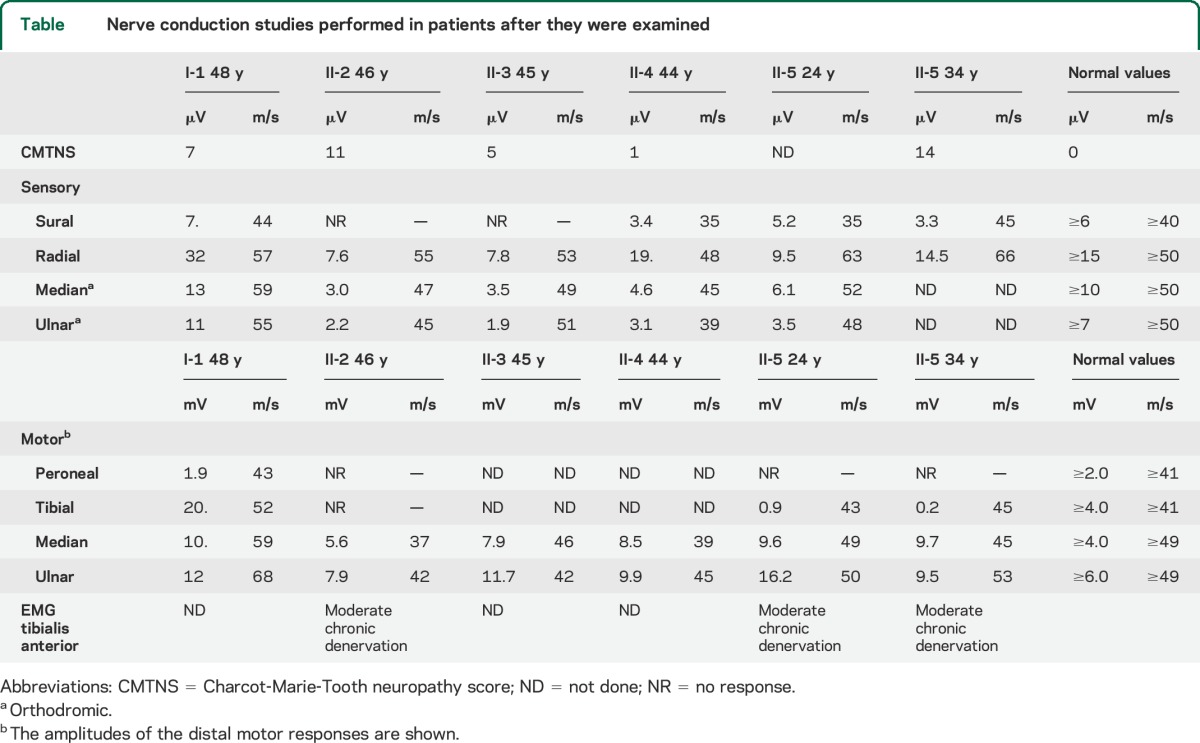
At age 24 years, the tibial motor responses had reduced amplitudes, and the peroneal response was absent. All of the measured sensory amplitudes were reduced. Repeat conduction studies at age 34 years showed little progression—the sensory amplitudes were stable, and 2 of 3 motor amplitudes were smaller (table). EMG of the tibialis anterior showed moderate, chronic denervation at both 24 and 34 years. The above findings support the diagnosis of a mild to moderate axonal neuropathy and a possible superimposed myelopathy.
Table.
Nerve conduction studies performed in patients after they were examined
Evaluation at age 21 years included a sural nerve and quadriceps muscle biopsy. The muscle biopsy was reported as showing neurogenic changes. We recut semi-thin and thin sections of the sural nerve biopsy (figure 2). These revealed a mildly reduced density of normally myelinated axons; no myelin debris was observed, indicating that the axonal loss was indolent. There are a few clusters of regenerated axons. Electron microscopy revealed Schwann cell processes that were not related to unmyelinated axons, indicating prior loss of myelinated or unmyelinated axons.
Figure 2. Sural nerve biopsy.

Digital electron micrographs from the sural nerve biopsy of the proband (patient II-5) at age 21 years. (A) This micrograph shows a cluster of regenerated axons (arrows) and a myelinated axon with myelin that is proportionally thicker than expected (asterisk). (B) This micrograph shows Schwann cell processes that are not associated with axons (arrowheads), and some that partially surround collagen fibers (double arrowheads). Scale bars: 2 µm.
The oldest brother (II-2) reported difficulty with his gait and balance since age 25 years and was aware of numbness in the distal calves. He previously had ankle surgery. At age 45 years, he was strong except for difficulty standing on his heels; bilateral tibialis anterior and abductor pollicus brevis were mildly weak (4+/5). Vibration sensation (scored throughout this work with a Rydell-Seiffer tuning fork) was not felt from the toes, and was reduced at the ankles (3), but not at the knees (5). Pinprick was decreased to the distal calves. Reflexes were 2+ at the ankles, 3+ at the knees (with crossed adduction), and 3+ at the biceps. Plantar responses were extensor. The radial, median, and ulnar sensory responses had reduced amplitudes; the tibial and peroneal motor responses and the sural sensory response were absent (table). His CMTNS was 11. Thus, in addition to a mild axonal neuropathy, his clinical examination indicated that he had a myelopathy. A B12 level and a cervical MRI were normal.
The second brother (II-3) did not report any neurologic symptoms, but examination at age 46 years revealed mild weakness (4+/5) in bilateral tibialis anterior and absent vibration at the toes (0), reduced at the ankles (<5), and normal at the knees (>5). Reflexes were 2+ at the ankles, 3+ at the knees (with crossed adduction), and 3+ at the biceps. Plantar responses were extensor on the right and muted on the left. His radial, median, and ulnar sensory amplitudes were reduced, and sural sensory response was absent. His CMTNS was 5. Thus, in addition to a mild axonal neuropathy, his clinical examination indicated that he had a myelopathy.
The third brother (II-4) did not report any motor or sensory deficits. At age 44 years, his examination had normal results, except for diminished vibration in his toes (less than 5). Reflexes were trace at the ankles and 1+ at the knees and biceps. Plantar responses were muted. His sural, median, and ulnar sensory responses, however, had reduced amplitudes. His CMTNS was 1. Thus, in spite of a lack of symptoms, his electrophysiology demonstrated that he has a mild axonal neuropathy.
The proband's sister (II-1) was evaluated at age 48 years. She reported numbness in her toes for several years. Her strength was normal, and reflexes were present and symmetric. Pinprick sensation was reduced to the knees, and vibration was reduced at the knees (4), ankles (3), and toes (2). Plantar responses were flexor. Her nerve conductions were normal except for borderline amplitudes of the peroneal motor response (table). Her CMTNS was 7. Her B12 and glucose tolerance tests were normal. Although she had sensory symptoms that resulted in her CMTNS of 7, her sensory amplitudes were normal, and we did not consider her to be affected like her other siblings; we made this assessment before the genetic testing was done.
Sequencing of tRNA synthetase genes reveals a novel mutation in AARS.
Genetic testing for known CMT loci was conducted on the proband. GJB1, MPZ, NEFL, GDAP1, and MFN2 mutations were not found upon sequencing in 2007. At this point, the family was added to a group of samples for sequencing of GARS, YARS, and AARS. No mutations were identified in GARS or YARS, but a novel sequence variant c.304G>C was found in AARS (figure 1B). This variant is present in all living, affected members of the family and absent in unaffected family members. The variant is not present in 186 Caucasian control chromosomes, dbSNP, or any sequences included in the NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/), 1000 genomes (http://www.1000genomes.org/), or Genome Variant Database for Human Disease. Thus, c.304G>C AARS is a rare variant that segregates with electrophysiologic evidence of peripheral neuropathy and has not been observed in the general population.
This nucleotide variant results in a missense substitution of the glycine residue at position 102 with an arginine (p.Gly102Arg). This mutation is in the highly conserved activation domain of the AARS protein, where ATP is attached to alanine to make an alanyl adenylate (Ala-AMP) intermediate, completing the first step in tRNA charging. This residue is highly conserved and is present in species as evolutionarily distant as Saccharomyces cerevisiae and Escherichia coli (figure 1C). In silico predictions indicated that the mutation was probably damaging (PolyPhen2 score = 1.00027; SIFT score = 0)28 and disease-causing (MutationTaster score = 0.999) (http://www.mutationtaster.org/).
Mutant alanyl-tRNA synthetase is unable to complement yeast lacking ALA1.
To determine whether the p.Gly102Arg amino acid substitution alters the function of the AARS protein, we used a yeast complementation assay (figure 3), which has been used to identify loss-of-function properties of CMT-associated AARS mutations.4 To assess the functional consequences of p.Gly102Arg AARS, we modeled this mutation in the yeast ortholog ALA1 (residue Gly102 in AARS is equivalent to residue Gly106 in ALA1, and hereafter referred to by the human substitution) and tested for the ability to support yeast cell growth compared to wild-type and p.Arg329His ALA1.4 A previously validated haploid yeast strain (with the endogenous ALA1 gene deleted and a vector that expresses wild-type ALA1 and URA3 to maintain viability) was transformed with a separate vector harboring a LEU2 selection marker and either no insert, wild-type ALA1, p.Gly102Arg ALA1, or the previously reported CMT-associated p.Arg329His ALA1 allele. Yeast were then selected on media containing 5-fluoroorotic acid (5-FOA), which is toxic to yeast expressing URA3 and thus selects for cells that have spontaneously lost the maintenance vector. Only yeast cells expressing a functional ALA1 allele from the LEU2-bearing vector will survive on 5-FOA. The wild-type ALA1 expression vector sustained yeast viability, while the empty vector was unable to complement the knockout allele, consistent with ALA1 being an essential gene (figure 3). Yeast expressing p.Gly102Arg or p.Arg329His ALA1 were unable to survive on 5-FOA media (figure 3). These results suggest that p.Gly102Arg AARS represents a loss-of-function allele.
Figure 3. Yeast assay: p.Gly102Arg ALA1 fails to support yeast cell growth.

Haploid Δala1 yeast strains were transformed with a vector containing no insert (pRS315 Empty) or an insert to express wild-type, p.Gly102Arg or pArg329His ALA1. Cultures resulting from each transformation condition are spotted undiluted and diluted (1:10 and 1:100) on plates containing 0.1% 5-fluoroorotic acid (5-FOA) complete medium or SD -leu -ura growth medium. Experiments were performed using 2 independently generated ALA1 expression constructs (labeled p.Gly102Arg A and B) for each allele.
DISCUSSION
We report a novel mutation in AARS that is associated with an indolently progressive, mild myeloneuropathy—a phenotype not previously associated with AARS mutations. The p.Gly102Arg mutation affects a conserved residue in an important domain, and segregates with disease in a pedigree with 9 living individuals. This phenotype is different from the p.Arg329His AARS mutation that was first identified in 2 French families5 and subsequently in an Australian family.4 All 3 families had a dominantly inherited axonal neuropathy affecting motor and sensory axons. A different mutation, p.Asn71Tyr, was found in a large Chinese family that was said to have late-onset, mild CMT2, but the relevant clinical and electrophysiologic findings demonstrating sensory axon involvement, and thus justifying the diagnosis of CMT2, were not reported.7 Thus, the p.Asn71Tyr mutation could be a distal hereditary motor neuropathy, which was the reported phenotype of the p.Asp893Asn mutation in another Chinese family.29 Like the p.Gly102Arg mutation, the p.Asn71Tyr and the p.Arg329His mutations affect highly conserved amino acids.
The electrophysiologic data demonstrated that 4 of 5 siblings had a mild axonal neuropathy, and we made the clinical diagnosis of a myelopathy in 3 of them. Interestingly, upgoing toes or hyperactive reflexes have been noted in 2 kindreds of patients with GARS mutations.30,31 In these cases, like the family we report, one supposes that both descending axons from the cortex and brainstem, as well as the PNS axons themselves, are affected in a length-dependent manner. Involvement of both PNS and CNS axons is not surprising: an axonal neuropathy is a common feature of many forms of hereditary spastic paraplegias, spinocerebellar ataxias, and more complex syndromes. Rather, it is surprising that a myelopathy is not more commonly noted in CMT2 patients, as most of the genes that are implicated in this disorder are expressed in the neurons of the CNS and PNS, and for most disorders, there is no known reason why CNS neurons would be unaffected. AARS is expressed in all cells and performs a central role in protein translation. Others have hypothesized that as the genetic underpinnings of CMT2 and hereditary spastic paraplegias and are linked to genes in similar pathways, the pathophysiologic and phenotypic distinctions may blur to become a spectrum of axonal diseases.32,33 Whereas the clinical and electrophysiologic findings enable us to deduce that the longest sensory and motor axons are most affected in typical axonal neuropathies (including in this family), exactly which axons are responsible for the clinical phenotype of spasticity is more difficult to infer. In Friedreich ataxia, amyotrophic lateral sclerosis, and hereditary spastic paraplegias,34 the loss of myelinated axons in the lateral corticospinal tract (which contains myelinated axons of various diameters35) is thought to account for the spasticity, so we postulate that this holds for the family we report. It should be noted, however, that this clinical teaching is at odds with the observation that surgical disruption of the corticospinal track at the pyramidal decussation does not cause spasticity in other primates.36
Results from our yeast complementation assay demonstrated loss-of-function characteristics of p.Gly102Arg AARS, which also suggests that the mutation causes disease. Importantly, while tRNA synthetases harboring disease-causing mutations have successfully complemented yeast lacking the equivalent tRNA synthetase, no benign missense variants have been shown to cause a loss of function in this assay. Therefore, loss-of-function properties in yeast are a reliable predictor of those tRNA synthetase mutations that are associated with peripheral neuropathy. The tRNA synthetase mutations that fail to complement their yeast orthologs (yeast genes in parentheses) GARS (GRS1), YARS (TYSI), AARS (ALA1), HARS (HTS1), KARS (KRS1), and MARS (MES1) also demonstrate loss of function in cell-free tRNA charging assays (for those mutations that have been examined using both techniques), which suggests that this assay is a corollary for tRNA charging function. Now 3 pathogenic AARS mutations that have been examined using this assay demonstrated loss-of-function properties (p.Arg329His, p.Asn71Tyr, and now p.Gly102Arg), while the fourth (p.Asp893Asn) has not been examined.
There is little insight into how mutations in alanyl-tRNA synthetase cause neuropathy or a host of other conditions.37 We favor a shared pathogenic mechanism between AARS mutation and other tRNA synthetase mutations that cause peripheral neuropathy. The most insight into the mechanism of tRNA synthetase–linked neuropathy comes from the study of GARS-linked dominantly inherited CMT2D.2 Two Gars mutations cause peripheral neuropathy in mice,38,39 and overexpression of wild-type human GARS failed to mitigate severity of the disease in these models, suggesting a toxic gain-of-function mechanism.40 Despite this, the mechanism of toxicity has not been identified and loss-of-function assays in yeast continue to be among the most specific tests for disease pathogenesis.
Identification of new disease-associated mutations with strong genetic evidence for disease causation and careful evaluation of clinical phenotypes is important to further understand the mechanism of tRNA synthetase-associated CMT disease. Our findings indicate that additional functional characterization of this variant is warranted and may help uncover the relative vulnerabilities of peripheral and CNS axons to tRNA synthetase mutations.
ACKNOWLEDGMENT
The authors thank Drs. Jian Li and Xi-tian Xu for assistance with the electron microscopy. The authors also thank the proband and her family for participation in the study.
GLOSSARY
- 5-FOA
5-fluoroorotic acid
- ARS
aminoacyl-tRNA synthetases
- CMT
Charcot-Marie-Tooth disease
- CMTNS
Charcot-Marie-Tooth neuropathy score
- HMSN
hereditary motor and sensory neuropathy
- PNS
peripheral nervous system
AUTHOR CONTRIBUTIONS
Dr. Motley: study concept and design, analysis and interpretation of data, preparation and revision of the manuscript. L.B. Griffin: acquisition of data, analysis and interpretation of data, preparation and critical revision of the manuscript. I. Mademan: acquisition of data, analysis and interpretation of the data. Dr. Baets: study concept and design, acquisition of data, analysis and interpretation of the data. E. De Vriendt: acquisition of data, analysis and interpretation of the data. Dr. De Jonghe: study concept and design, data acquisition, analysis and interpretation of the data. Dr. Antonellis: study supervision, analysis and interpretation of data, critical revision of the manuscript. Dr. Jordanova: study supervision, study concept and design, analysis and interpretation of the data, critical revision of the manuscript. Dr. Scherer: study concept and design, analysis and interpretation of the data, preparation and revision of the manuscript, study supervision.
STUDY FUNDING
Supported by the Judy Seltzer Levenson Memorial Fund for CMT Research and the NIH (U54 NS065712); the Research Fund of the University of Antwerp (TOP BOF 29069 to A.J.); the Fund for Scientific Research-Flanders (FWO, to A.J.); the Association Belge contre les Maladies Neuromusculaires (ABMM; to A.J., J.B.); and the Association Française contre les Myopathies (AFM, to A.J.). A.A. was supported by a grant from the Muscular Dystrophy Association (294479). L.B.G. was supported by NIH grant F30 NS092238, NIH Cellular and Molecular Biology Training Grant T32 GM007315, and NIH Medical Scientist Training Program Training Grant T32 GM07863. I.M. is supported by a PhD fellowship of the agency for Innovation by Science and Technology of Flanders (IWT).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Shy ME, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic, and pathologic features. In: Dyck PJ, Thomas PK, eds. Peripheral Neuropathy, 4th ed Philadelphia: Saunders; 2005:1623–1658. [Google Scholar]
- 2.Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 2003;72:1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jordanova A, Irobi J, Thomas FP, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 2006;38:197–202. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin HM, Sakaguchi R, Giblin W, et al. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat 2012;33:244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latour P, Thauvin-Robinet C, Baudelet-Mery C, et al. A major determinant for binding and aminoacylation of tRNAAla in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 2010;86:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Z, Hashiguchi A, Hu J, et al. Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 2012;78:1644–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin KP, Soong BW, Yang CC, et al. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 2011;6:e29393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLaughlin HM, Sakaguchi R, Liu CP, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 2010;87:560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vester A, Velez-Ruiz G, McLaughlin HM, et al. A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat 2013;34:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez M, McLaughlin H, Houlden H, et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 2013;84:1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayat V, Thiffault I, Jaiswal M, et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol 2012;10:e1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet 2011;88:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007;81:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet 2012;21:4521–4529. [DOI] [PubMed] [Google Scholar]
- 15.Gotz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 2011;88:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 2011;108:6543–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet 2010;87:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos-Cortez RL, Lee K, Azeem Z, et al. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet 2013;93:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–539. [DOI] [PubMed] [Google Scholar]
- 20.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain 2012;135:1387–1394. [DOI] [PubMed] [Google Scholar]
- 21.Taft RJ, Vanderver A, Leventer RJ, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet 2013;92:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Ling J, Barcia G, et al. Mutations in QARS, encoding elutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet 2014;94:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dallabona C, Diodato D, Kevelam SH, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014;82:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMillan HJ, Schwartzentruber J, Smith A, et al. Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med Genet 2014;15:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolf NI, Toro C, Kister I, et al. DARS-associated leukoencephalopathy can mimic a steroid-responsive neuroinflammatory disorder. Neurology 2014;84:226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shy ME, Blake J, Krajewski K, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64:1209–1214. [DOI] [PubMed] [Google Scholar]
- 27.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 2001;11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Hashiguchi A, Hu J, et al. Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 2012;78:1644–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivakumar K, Kyriakides T, Puls I, et al. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain 2005;128:2304–2314. [DOI] [PubMed] [Google Scholar]
- 31.Klein CJ, Middha S, Duan X, et al. Application of whole exome sequencing in undiagnosed inherited polyneuropathies. J Neurol Neurosurg Psychiatry 2014;85:1265–1272. [DOI] [PubMed] [Google Scholar]
- 32.Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol 2013;246:14–25. [DOI] [PubMed] [Google Scholar]
- 33.Fridman V, Murphy SM. The spectrum of axonopathies: from CMT2 to HSP. Neurology 2014;83:580–581. [DOI] [PubMed] [Google Scholar]
- 34.Oppenheimer DR. Diseases of the basal ganglia, cerebellum and motor neurons. In: Adams JH, Corselllis JAN, Duchen LW, eds. Greenfield's Neuropathology, 4th ed New York: John Wiley & Sons; 1984:699–747. [Google Scholar]
- 35.Brodal A. Neurological Anatomy, 3rd ed New York: Oxford University Press; 1981. [Google Scholar]
- 36.Beck CH, Chambers WW. Speed, accuracy, and strength of forelimb movement after unilateral pyramidotomy in rhesus monkeys. J Comp Physiol Psychol 1970;70:1–22. [DOI] [PubMed] [Google Scholar]
- 37.Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med 2013;5:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 2006;51:715–726. [DOI] [PubMed] [Google Scholar]
- 39.Achilli F, Bros-Facer V, Williams HP, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech 2009;2:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motley WW, Seburn KL, Nawaz MH, et al. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. Plos Genet 2011;7:e1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]