

ABSTRACT
Hepatitis C virus (HCV) enters its target cell via clathrin-mediated endocytosis. AP-2-associated protein kinase 1 (AAK1) and cyclin G-associated kinase (GAK) are host kinases that regulate clathrin adaptor protein (AP)-mediated trafficking in the endocytic and secretory pathways. We previously reported that AAK1 and GAK regulate HCV assembly by stimulating binding of the μ subunit of AP-2, AP2M1, to HCV core protein. We also discovered that AAK1 and GAK inhibitors, including the approved anticancer drugs sunitinib and erlotinib, could block HCV assembly. Here, we hypothesized that AAK1 and GAK regulate HCV entry independently of their effect on HCV assembly. Indeed, silencing AAK1 and GAK expression inhibited entry of pseudoparticles and cell culture grown-HCV and internalization of Dil-labeled HCV particles with no effect on HCV attachment or RNA replication. AAK1 or GAK depletion impaired epidermal growth factor (EGF)-mediated enhanced HCV entry and endocytosis of EGF receptor (EGFR), an HCV entry cofactor and erlotinib's cancer target. Moreover, either RNA interference-mediated depletion of AP2M1 or NUMB, each a substrate of AAK1 and/or GAK, or overexpression of either an AP2M1 or NUMB phosphorylation site mutant inhibited HCV entry. Last, in addition to affecting assembly, sunitinib and erlotinib inhibited HCV entry at a postbinding step, their combination was synergistic, and their antiviral effect was reversed by either AAK1 or GAK overexpression. Together, these results validate AAK1 and GAK as critical regulators of HCV entry that function in part by activating EGFR, AP2M1, and NUMB and as the molecular targets underlying the antiviral effect of sunitinib and erlotinib (in addition to EGFR), respectively.
IMPORTANCE Understanding the host pathways hijacked by HCV is critical for developing host-centered anti-HCV approaches. Entry represents a potential target for antiviral strategies; however, no FDA-approved HCV entry inhibitors are currently available. We reported that two host kinases, AAK1 and GAK, regulate HCV assembly. Here, we provide evidence that AAK1 and GAK regulate HCV entry independently of their role in HCV assembly and define the mechanisms underlying AAK1- and GAK-mediated HCV entry. By regulating temporally distinct steps in the HCV life cycle, AAK1 and GAK represent “master regulators” of HCV infection and potential targets for antiviral strategies. Indeed, approved anticancer drugs that potently inhibit AAK1 or GAK inhibit HCV entry in addition to assembly. These results contribute to an understanding of the mechanisms of HCV entry and reveal attractive host targets for antiviral strategies as well as approved candidate inhibitors of these targets, with potential implications for other viruses that hijack clathrin-mediated pathways.
INTRODUCTION
Hepatitis C virus (HCV) is a major global health problem, estimated to infect 170 million people worldwide (1, 2). HCV persistence results in severe liver disease, including cirrhosis, liver failure, and hepatocellular carcinoma (reviewed in reference 3). No effective vaccine is currently available, and although the combination of interferon-ribavirin-based regimens with HCV protease or polymerase inhibitors as well as interferon-free regimens significantly improves response rates, HCV resistance and drug-drug interactions are among the ongoing challenges (4–6). A cocktail of drugs, each targeting an independent function, will likely offer the best pharmacological control. Hence, there is an ongoing need to better understand the HCV life cycle in order to identify drugs directed at novel targets. No FDA-approved inhibitors of HCV cell entry are currently available even though viral entry represents a potential target for antiviral strategies.
HCV is an enveloped, positive, single-stranded RNA virus from the Flaviviridae family. Its 9.6-kb genome encodes a single polyprotein, which is proteolytically cleaved into three structural proteins (core and the glycoproteins, E1 and E2) and seven nonstructural (NS) proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (7–9). Specific interactions between viral proteins and cell surface molecules facilitate HCV entry into host cells and define HCV tropism (reviewed in reference 10). The important roles of these interactions were initially defined using recombinant E1 and E2 envelope glycoproteins and HCV pseudoparticles (HCVpp). HCVpp are lentiviral vectors that incorporate the HCV glycoproteins on the viral envelope and measure only viral entry (11–13). The establishment of an infectious HCV cell culture system (HCVcc) (14) has facilitated studies of HCV entry under more authentic conditions of viral replication.
HCV particles circulate in the blood associated with lipoproteins (15–19). Low-density lipoprotein receptor (LDLR) and cell surface glycosaminoglycans, including heparan sulfate, are thought to play a role in the initial attachment of HCV to target cells (20–23). HCV internalization into the cell is mediated by a complex set of receptors, including the tetraspanin CD81 (24–27), scavenger receptor B1 (SR-BI) (28–31), and the tight junction proteins occludin (OCDN) (32–34) and members of the claudin (CLDN) family (11, 35–37). Additional cellular molecules identified as HCV entry factors include the two receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin type A receptor 2 (EPHA2) (38), the cholesterol uptake molecule Niemann-Pick C1-like 1 (NPC1L1) (39), and transferrin receptor 1 (TFR1) (40). CD81-bound HCV particles have been shown to traffic laterally on the plasma membrane to tight junctions, where they form stable CD81-CLDN1 complexes, and these activities are promoted by protein kinase A (PKA), Rho, and EGFR/HRas signaling (38, 41–44). Multiple lines of evidence support the finding that HCVpp and HCVcc enter the cell via clathrin-mediated endocytosis (13, 45–49). HCV colocalizes with clathrin prior to internalization (49), and suppression of components of the clathrin machinery disrupts viral entry (47, 49). Following internalization, HCV is thought to traffic to an endosomal compartment, where fusion of the viral envelope with endosomal membranes facilitates release of the HCV RNA genome into the cytoplasm (13, 45–48, 50, 51). Indeed, HCV entry is dependent on endosomal acidification (46–50). HCV appears to traffic to and fuse specifically with early endosomes, as indicated by the transport of HCV particles to green fluorescent protein (GFP-RAB5A)-positive endosomes (49) and the inhibitory effect of dominant-negative mutants of early, but not late, endosomal markers on HCVpp entry (47). The mechanisms that regulate clathrin-mediated endocytosis of HCV are, however, incompletely characterized.
Clathrin-mediated endocytosis is dependent on the action of oligomeric clathrin and adaptor protein complexes (52), which coordinate the specific recruitment and assembly of clathrin into a polyhedral lattice at the plasma membrane, as well as its coupling to endocytic cargo (53). The heterotetrameric clathrin adaptor protein 2 complex, AP-2, is the major component of clathrin-coated vesicles (CCVs) derived from the plasma membrane destined for fusion with early endosomes (54). AP2M1, the μ2 subunit of AP-2, recognizes tyrosine- or dileucine-based sorting signals, also known as internalization signals, within the cytoplasmic domains of transmembrane receptors (54–58). The two host cell kinases AP-2-associated protein kinase 1 (AAK1) and cyclin G-associated kinase (GAK) are regulators of clathrin-mediated endocytosis. AAK1 and GAK recruit clathrin and AP-2 to the plasma membrane (59) and phosphorylate a T156 residue in AP2M1, thereby stimulating its binding to cargo proteins and enhancing cargo recruitment, vesicle assembly, and efficient internalization (59–65). AAK1 and GAK also regulate clathrin-mediated endocytosis of cellular receptors via alternate sorting adaptors that collaborate with AP-2 (reviewed in reference 66): AAK1 phosphorylates NUMB (67), and GAK activates EPN1 (59). In addition, GAK regulates uncoating of CCVs (59), thereby enabling recycling of clathrin back to the cell surface, while clathrin-stimulated AAK1 activity mediates the rapid recycling of receptors from early/sorting endosomes back to the plasma membrane (68). Notably, GAK is a key regulator of EGFR internalization, known to promote EGF uptake (59) and possibly also to affect EGFR signaling (69). AAK1 has also been implicated in the regulation of EGFR internalization and recycling to the plasma membrane via its effect on and interactions with alternate endocytic adaptors (67, 70–74). Multiple lines of evidence support a critical role for EGFR in HCV entry (38). Binding of HCVcc particles to cells induces EGFR activation (75), EGFR ligands enhance HCV entry (38, 75), and EGFR/HRas signaling promotes CD81-CLDN1 binding (41). Moreover, EGFR ligands were shown to increase colocalization of EGFR with CD81 and their localization to early endosomes, suggesting a potential role for EGFR in a step following CD81-CLDN1 engagement (75). Nevertheless, the role of AAK1 and GAK in regulation of viral entry has not been reported in any viral infection to date.
We have recently reported that AAK1- and GAK-regulated AP2M1 activity is absolutely essential for HCV assembly (76). Since AAK1 and GAK are regulators of clathrin-mediated endocytosis, we hypothesized that they regulate HCV entry in addition to HCV assembly and that these mechanisms can be translated into the development of novel antiviral strategies. Here, we provide evidence that AAK1 and GAK regulate HCV at a postbinding step independently of their role in HCV assembly and that regulation of EGFR endocytosis and phosphorylation of AP2M1 and NUMB are mechanisms involved in AAK1- and GAK-mediated HCV entry. Furthermore, we show that approved anticancer drugs that inhibit AAK1 or GAK significantly inhibit HCV entry in addition to assembly, that their combination is synergistic, and that their antiviral effect is reversed by overexpression of either AAK1 or GAK.
MATERIALS AND METHODS
Plasmids.
Open reading frames encoding AP2M1, AAK1, GAK, EGFR, and NUMB were picked from the Human ORFeome library of cDNA clones (77) (Open Biosystems) and recombined into a pGLuc vector for Gaussia princeps luciferase fragment (Gluc) tagging (78), using gateway technology (Invitrogen). AP2M1 and NUMB mutations were introduced by site-directed mutagenesis using a QuikChange kit (Stratagene). pFL-J6/JFH(p7-Rluc2A) was a gift from Charles M. Rice (48). Plasmids used in the HCVpp entry assays (pNL4-3.Luc.R-E, pcDM8, and pcDM8-E1E2) were a gift from Shoshana Levy.
Cells.
Huh-7.5 cells (Apath LLC) and 293T cells (ATCC) were grown in Dulbecco's modified minimal essential medium supplemented with nonessential amino acids (Gibco), 1% l-glutamine (Gibco), and 1% penicillin-streptomycin (Gibco) and maintained in 5% CO2 at 37°C.
Antibodies and compounds.
Rabbit anti-AAK1, -AP-1, and -AP-4 antibodies were purchased from Abcam. Mouse anti-GAK antibodies were purchased from MBL International Corporation. Rabbit and goat anti-AP2M1 antibodies were purchased from Santa Cruz Biotechnology.
Rabbit anti-phospho-AP2M1 (T156), p21-activated kinase 1 (PAK1), and EGFR antibodies were purchased from Cell Signaling. Rabbit anti-SDC1 antibodies were purchased from One World Lab, and mouse anti-CD81 was from BD Biosciences. Mouse anti-NUMB, anti-beta actin, and anti-alpha tubulin antibodies were from Sigma. Sunitinib malate and heparin were purchased from Sigma, erlotinib was from LC Laboratories, calyculin A was from Cell Signaling, EGF was from Millipore, and IPA-3 was from SelleckChem.
RNA interference (RNAi).
Small interfering RNAs (siRNAs) (50 to 200 nM) were transfected into cells using silMPORTER (Upstate, Millipore) 48 h prior to HCVcc or HCVpp infection. Sequences are as follows: siAAK1#1, GGUGUGCAAGAGAGAAAUCTT (Ambion) (68); siAAK#2, GAGCCGUCUCAAGUUUAAACUUACA (Invitrogen) (68); siGAK, AACGAAGGAACAGCUGAUUCA (Dharmacon) (79); siNUMB#1, CAGCCACUGAACAAGCAGA (Dharmacon) (80); siNUMB#2, GGACCTCATAGTTGACCAG (Dharmacon) (80); siAP2M1#1, AGUUUGAGCUUAUGAGGUA; siPAK1, GCAUCAAUUCCUGAAGAUU (Sigma); and siSDC1, CCAAACAGGAGGAAUUCUA (Dharmacon). siAP1M1 (identification number S17033), siAP1M2 (S19545), siAP4M1 (S17539), and Silencer Select negative control 1 siRNA (4390844) were from Life Technologies. Mission lentiviral transduction particles harboring short hairpin RNAs (shRNAs) targeting distinct sites in the AP2M1 RNA (TRCN0000060239 and TRCN0000060242; Sigma) and a control shRNA were used to transduce Huh-7.5 cells according to the manufacturer's protocol (76).
HCVpp production and entry assays.
HCVpp (H77c strain, genotype 1a) were generated as described previously (12, 13). Briefly, 293T cells were transfected with a 1:1 ratio of plasmids encoding HIV provirus expressing luciferase and HCV E1E2 envelope glycoproteins. Supernatants were harvested at 48 h posttransfection and filtered. Huh-7.5 cells were infected with HCVpp and 8 mg/ml Polybrene (Sigma) for 4 h. Cell lysates were collected at 48 or 72 h after HCVpp infection, and firefly luciferase (Promega) activity was measured using a Tecan luminometer (Tecan).
HCVcc generation.
HCV genotype 2a infectious clone J6/JFH(p7-Rluc2A) RNA was generated and delivered into Huh-7.5 cells, as previously described (14, 81). Virus titers were determined by limiting dilution and immunohistochemical staining using anti-core protein antibodies. The 50% tissue culture infectious dose (TCID50) was calculated, as described previously (14). Results are expressed as TCID50/ml.
HCVcc infection.
A total of 6 × 103 Huh-7.5 cells seeded in 96-well plates were infected in triplicates with HCVcc J6/JFH(p7-Rluc2A) (titer, 4.5 × 105 TCID50/ml) at a multiplicity of infection (MOI) of 0.1 to 1. At 4 h postinfection, cells were washed, and medium was replaced. HCVcc infection was measured by standard luciferase assays at 6, 9, 12, and/or 24 h postinfection, using a Renilla luciferase substrate and a Tecan luminometer (Tecan) according to the manufacturer's protocols.
HCV RNA replication by luciferase assays.
HCV RNA replication was measured at 6 and 72 h postelectroporation, as described previously (76, 81). Electroporated cells plated in quadruplicates in 96-well plates were washed twice with phosphate-buffered saline (PBS) and lysed with 30 μl of Renilla lysis buffer (Promega). Following 15 min of shaking at room temperature, luciferase activity was quantified by standard luciferase assays.
EGF-mediated HCV entry assays.
A total of 5 × 105 Huh-7.5 cells were transfected with 100 nM concentrations of the siRNAs indicated in the figures, as described above. At 24 h posttransfection cells were seeded at 6 × 103 cells/well in 96-well plates and incubated for 24 h. As previously reported (38), cells were then serum starved for 2 h to reduce ligand-receptor binding and the basal activation level of the EGFR pathways, followed by a 1-h treatment with various concentrations of EGF (0.01 to 10 μg/ml) or PBS. Next, cells were infected with HCVpp or HCVcc at an MOI of 0.1 in the presence of EGF. At 4 h postinfection, unbound virus was removed by washing, and fresh growth medium was added. HCVpp or HCVcc infection was measured by luciferase activity following 68 or 20 h of incubation at 37°C, respectively.
Pharmacological inhibition.
To determine the effect of inhibitors on HCV entry, Huh-7.5 cells seeded in 96-well plates were washed once with ice-cold PBS and infected with HCVpp or HCVcc. Following a 1-h incubation on ice to allow attachment of viral particles to cell surface receptors, cells were treated with various concentrations of the inhibitors either individually or in combination or with dimethyl sulfoxide (DMSO) in preheated medium and transferred to 37°C for a 4-h incubation. Residual drugs and unbound virus were then washed off, and cells were incubated in complete medium in the absence of drugs for 72 h (HCVpp) or 24 h (HCVcc) at 37°C and subsequently subjected to viability assays and standard luciferase reporter gene assays. Drug effects on AP2M1 phosphorylation were determined as previously described (76).
Gain-of-function and overexpression experiments.
Gain-of-function assays were performed by transducing AP-2M1-, AAK1-, or GAK-depleted cells with a lentivirus expressing AP-2M1 for 24 h prior to HCVcc infection or by transfecting AAK1-, GAK-, or EGFR-expressing vectors in Huh-7.5 cells for 48 h prior to HCVcc infection and drug treatment. AP2M1 and NUMB overexpression experiments were conducted by overexpressing wild-type (WT) or AP2M1 and NUMB mutants for 48 h prior to infection.
Attachment assays.
At 48 h after siRNA transfection, 6 × 104 Huh-7.5 cells in 96-well plates were incubated with HCVcc at an MOI of 3. Cells transfected with a nontargeting (NT) siRNA remained untreated or were treated with 2 unit/ml of heparinase I (Sigma) in a buffer containing 20 mM Tris-HCl (pH 6.8), 50 mM NaCl, 4 mM CaCl2, and 0.01% bovine serum albumin (BSA) at 37°C for 1 h prior to viral inoculation (82). Plates were spun (1,000 rpm) at 4°C for 1 h and 45 min and incubated on ice for another 15 min. Unbound HCV was then removed by washing with PBS three times. Total RNA was extracted with NucleoSpin RNA XS (Macherey-Nagel) and used to quantify cell-bound HCV RNA by quantitative reverse transcription-PCR (qRT-PCR).
Time-of-addition studies.
Huh-7.5 cells were seeded at 5 × 103 cells/well in 96-well plates. The following day, cells were incubated with HCVcc at an MOI of 0.3 at 4°C for 1 h, washed three times with Dulbecco's modified Eagle's medium (DMEM), and shifted to 37°C. IgG, anti-CD81 antibody (10 μg/ml), DMSO, sunitinib (2 μM), erlotinib (10 μM), or heparin was added at different time points either prior to or after the temperature shift (20, 40, 60, 120, 150, and 180 min). After 4 h the cells were washed and incubated in fresh medium at 37°C. HCVcc infection was measured at 24 h using a Renilla luciferase assay system (Promega).
Highly infectious stock preparation and Dil labeling.
Viral supernatants were concentrated, and HCV viral particles were labeled with Dil (Dil-HCV), as previously described (14, 49). Briefly, filtered viral supernatants were incubated overnight with polyethylene glycol 8000 (PEG) (final concentration, 8% [wt/vol]), followed by centrifugation (8,000 × g for 20 min), and resuspended in 10 ml of supernatant. Virus was centrifuged (8,000 × g for 15 min) and resuspended in a final volume of 1 ml of cell culture DMEM. Virus was labeled by the addition of 5 μl (5 μM final concentration) of the lipophilic dye Dil (Invitrogen) (excitation, 549 nm; emission, 565 nm) to 1 ml of concentrated virus. Virus and dye were incubated protected from light for 1 h with shaking at room temperature. Labeled virus was loaded onto a 10 to 60% (wt/vol) continual sucrose gradient (in PBS) and centrifuged for 16 h at 4°C and 34,000 rpm. Ten 1-ml fractions were collected and subjected to viral titration assays by focus formation assays, viral RNA measurements by qRT-PCR (as described below), and density measurements. Dil-HCV was isolated by ultracentrifugation at 36,000 rpm for 2 h from gradient fraction 3, which harbored the peak of infectivity and had a density of 1.06 g/ml, and resuspended in 1 ml of complete medium for subsequent imaging studies.
Quantitative immunofluorescence (IF) microscopy.
For imaging individual infectious Dil-HCV particles, Huh-7.5 cells were transfected with an AAK1, GAK, or NT siRNA. After 24 h, cells were transduced with CellLight early endosomes-GFP BacMam (Life Technologies) for RAB5 labeling. After a 24-h incubation, cells were seeded on a Nunc Lab-Tek II chamber slide system (Thermo Scientific) (4 × 104 cells per chamber) and allowed to grow for 24 h. The medium was then replaced with ice-cold 3% fetal bovine serum (FBS)-DMEM containing Dil-labeled HCV particles at an MOI of 3, followed by incubation on ice for 1 h with protection from light. Unbound virus was washed off with cold PBS, DMEM–3% FBS medium was added, and cells were transferred to a37°C incubator. Cells were fixed in 4% paraformaldehyde in PBS at 1 h after the temperature shift, washed with PBS, covered with ProLong Gold antifade reagent (Invitrogen), and kept at −20°C until imaged. Wide-field epifluorescence image z-stacks were acquired on a DeltaVision OMX Blaze system (Applied Precision-GE, Inc.) equipped with three electron-multiplying charge-coupled devices (emCCDs; Evolve, Photometrics Inc.) using an Olympus UPlanApo 100× (numerical aperture [NA], 1.40) oil immersion objective. Image acquisition was set at 80 nm per pixel. Excitation was with an InsightSSI illuminator (excitation wavelengths of 405 nm, 488 nm, 568 nm) and standard emission filter sets (435/31 nm, 528/48 nm, and 609/37 nm). Image stacks were acquired with a 200-nm z-step size. Nonblind, iterative constrained deconvolution, image channel alignment, and maximum intensity projections were done using SoftWoRx software (Applied Precision-GE, Inc.). After deconvolution, subresolution viral particles were visualized at or near diffraction-limited resolution and covered an area of approximately 3 by 3 pixels, with a single x, y pixel dimension of 80 nm. Approximately 80 to 120 Dil-HCV particles were analyzed by Volocity Software (PerkinElmer) for their colocalization with RAB5 in each group using Pearson's colocalization coefficient.
To assess the role of AAK1 and GAK in EGFR endocytosis, Huh-7.5 cells were transfected with an siRNA targeting AAK1 or GAK or with an NT siRNA and seeded onto coverslips. At 48 h posttransfection, cells were serum starved for 2 h and then either treated with EGF at 1 μg/ml or with PBS for 1 h or infected with HCVcc in the presence of EGF at 1 μg/ml for 1 h. Cells were washed with PBS and immediately fixed with 4% paraformaldehyde in PBS, washed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked for 1 h in PBS containing 1% BSA. Fixed cells were incubated with primary antibodies against EGFR and EEA1 at room temperature for 1 h. Secondary antibodies (Invitrogen) were incubated for 1 h at room temperature. Coverslips were then washed three times with PBS and mounted with ProLong Gold antifade reagent (Invitrogen). Slides were analyzed using Zeiss LSM 510 confocal microscope. Colocalization was quantified in 10 to 15 randomly chosen cells from each sample using ImageJ (JACoP) colocalization software and Manders' colocalization coefficients (MCCs). Threshold values were determined using auto-threshold (plug-in; ImageJ). Only pixels whose red and green intensity values were both above their respective thresholds were considered to be pixels with colocalized probes. MCCs were then calculated as the fractions of total fluorescence in the region of interest that occurs in these colocalized pixels (with a higher value representing more colocalization). Manders' M2 coefficient values represented as mean percent colocalization are shown.
RNA extraction and qRT-PCR.
Total RNA was isolated from cells using DNA, RNA, and a protein purification kit (Macherey-Nagel). qRT-PCR mixtures were assembled in triplicates using 0.5 μg or 4 μl of RNA and a high-capacity RNA-to-cDNA kit (Applied Biosystems). Amplification and analysis were performed using a StepOnePlus real-time PCR system (Applied Biosystems) (76). S18 RNA was used as a control. To quantify HCV RNA in the sucrose gradient fractions (Dil-HCV entry experiment), RNA from 100 μl was purified from each fraction, and 9 μl of RNA was subjected to two-step qRT-PCR as described above. Levels of RNA were determined using a standard curve of known amounts of HCV RNA.
Viability assays.
Following treatment with inhibitory compounds, siRNA silencing, or overexpression of various proteins, cells were incubated for 2 to 4 h at 37°C in the presence of 10% alamarBlue reagent (Trek Diagnostic Systems). Plates were then scanned, and fluorescence was detected by a FLEXstation II 384 (Molecular Devices, Inc.).
Western blotting.
Cell lysates were subjected to Western analyses using primary antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Band intensity was quantified using NIH ImageJ software.
Data analysis of combination drug treatment.
The MacSynergy II program (kindly provided by M. N. Prichard) was used to analyze data according to the Bliss independence model (83, 84). The combination's effect is determined by subtracting the theoretical additive values from the experimental values (85). A three-dimensional differential surface plot demonstrates synergy as peaks above a theoretical additive plane and antagonism as depressions below it (85). Matrix data sets in four replicates were assessed at the 95% confidence level for each experiment (83–85). Synergy (volume under the curve) and log volume were calculated. As suggested by Prichard et al. (84), such data sets should be interpreted as follows: volumes of synergy or antagonism at values of <25 μM2% are insignificant, those of 25 to 50 μM2% are minor but significant, those of 50 to 100 μM2% are moderate and probably important in vivo, and those of >100 μM2% are strong and likely to be important in vivo.
Statistical analysis.
Fifty percent effective concentrations (EC50s) were measured by fitting data to a three-parameter logistic curve, as described previously (76). P values were calculated using a two-tailed unpaired Student's t test.
RESULTS
AAK1 and GAK are critical host factors for HCV entry.
To test our hypothesis that AAK1 and GAK are involved in the regulation of HCV entry, we measured HCVpp entry and HCVcc infection in Huh-7.5 cells depleted of AAK1 or GAK by small interfering RNAs (siRNAs) (Fig. 1). Cells were transfected with individual siRNAs targeting either AAK1 or GAK or with a nontargeting (NT) sequence (68, 76, 79). Two AAK1 siRNAs and a single GAK siRNA effectively suppressed protein expression, as evidenced by Western blotting (Fig. 1A), with no apparent effect on cellular viability measured by alamarBlue-based assays (Fig. 1B). These AAK1- and GAK-depleted cells were infected with HCVpp, and luciferase activity measurements were determined at 48 h postinfection (11, 12). As shown in Fig. 1C, silencing of AAK1 or GAK expression reduced the entry of HCVpp by ∼1 to 2 logs. The effect of AAK1 siRNAs on HCV entry correlated with the level of AAK1 suppression. To validate these results in the more authentic HCVcc system, AAK- and GAK-depleted cells were infected for 4 h with J6/JFH(p7-Rluc2A), a Renilla luciferase-containing reporter virus that replicates and produces high titers of virus in Huh-7.5 cells (81), and luciferase assays at 6, 9, 12, and 24 h postinfection were performed. Depletion of either AAK1 or GAK reduced HCVcc infection by ∼0.5 to 3 logs at each of the studied time points (Fig. 1D). In contrast, RNAi-mediated silencing of the expression of PAK1 (p21-activated kinase 1), a kinase that regulates macropinocytosis (an alternative, clathrin-independent, endocytosis pathway) (86), had no apparent effect on HCVpp entry and HCVcc infection, as recently reported (87) (Fig. 1A to D). In contrast to its effect on HCV entry, AAK1 and GAK depletion had no effect on HCV RNA replication, as measured by luciferase assays 6 and 72 h following electroporation of Huh-7.5 cells with in vitro-transcribed J6/JFH(p7-Rluc2A) RNA, consistent with our prior observation (76) (Fig. 1E). These results indicate that AAK1 and GAK are host factors required for the regulation of HCV entry and that this role is independent of their role in HCV assembly.
FIG 1.

AAK1 and GAK are essential for HCV entry. Huh-7.5 cells were transfected with siRNAs targeting either AAK1 (1 or 2), GAK, or PAK1 or with a nontargeting (NT) sequence. (A) Cells were lysed at 48 h posttransfection, and the indicated protein levels were measured by quantitative Western analysis. Representative membranes are shown. Numbers represent relative ratios of AAK1, GAK, or PAK1 to actin protein normalized to the NT control level from at least two independent measurements. (B) Cellular viability was measured in these cells by alamarBlue-based assays. Data are relative fluorescence values normalized to the NT control. (C) AAK1-, GAK-, or PAK1-depleted cells and NT controls were infected with HCV pseudoparticles (HCVpp) expressing a luciferase reporter gene for 4 h. Luciferase measurements were taken at 48 h postinfection. RLU, relative light units. (D) The indicated depleted cells and NT controls were infected with cell culture-grown HCV (HCVcc) expressing a luciferase reporter gene for 4 h. Luciferase measurements were taken at the indicated time points postinfection. (E) Huh-7.5 cells depleted of AAK1 or GAK and the NT controls were electroporated with in vitro-transcribed HCV RNA encoding a luciferase reporter. Luciferase measurements were taken at 6 and 72 h postelectroporation as a measurement of HCV RNA replication. Data represent means and standard deviations (error bars) from three experiments in triplicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
AAK1 and GAK regulate HCV entry at a postbinding step.
To further probe the role of AAK1 and GAK in HCV entry, we performed attachment assays (88). Cells depleted of AAK1, GAK, or syndecan 1 (SDC1) were incubated with HCVcc at 4°C for 2 h, followed by washing for removal of unbound virus, RNA extraction, and quantification of cell-bound HCV RNA by qRT-PCR. AAK1 or GAK depletion had no effect on HCV attachment (Fig. 2A). In contrast, depletion of SDC1, a known receptor for HCV attachment, or treatment of NT cells with heparinase I to remove heparin sulfate from the cell surface reduced HCV attachment with no effect on cellular viability, as previously reported (82, 88).
FIG 2.

AAK1 and GAK regulate HCV entry at a postbinding step. (A) Cells transfected with siRNAs targeting AAK1, GAK, and SDC1 or an NT sequence as well as NT cells pretreated with heparinase I were infected with HCVcc at 4°C for 2 h. This was followed by removal of unbound virus, RNA extraction, and quantification of cell-bound HCV RNA by qRT-PCR as a measurement of HCV attachment. Shown are SDC1 protein levels in the indicated cells and HCV RNA levels relative to the NT control level. (B) Purification of highly infectious Dil-HCV particles. Concentrated HCV was labeled with Dil, layered on a 10 to 80% sucrose gradient, and purified by ultracentrifugation. The top of the gradient is fraction 1. Infectious HCV (solid line, left y axis) was quantified by focus formation (FF) assays, and HCV RNA copy numbers (dashed line, right y axis) were determined by RT-PCR. Dil-HCV was isolated from gradient fraction 3, which harbored the peak of infectivity and had a density of 1.06g/ml. (C to F) Quantitative immunofluorescence analysis of Dil-HCV internalization. Huh-7.5 cells transfected with an NT (D), AAK1 (E), or GAK (F) siRNA and expressing the early endosomal marker RAB5-GFP were infected with Dil-HCV for 1 h on ice and shifted to 37°C. Cells were fixed 1 h postinfection. Images were acquired on a DeltaVision OMX Blaze system equipped with three emCCDs. Colocalization of Dil-HCV and RAB5 was quantified by Pearson's colocalization coefficient (C). Each symbol represents an individual Dil-HCV particle; horizontal lines indicate the means ± standard deviations (error bars) (n = 121, 78, and 84 Dil-HCV particles in the NT and AAK1- and GAK-depleted cells, respectively). Data are from two independent experiments. Two representatives of Dil-HCV (red), RAB5 (green), and merged images at a magnification of ×100 are shown for each group (D to F). The small frames at the bottom correspond to boxed areas and areas with arrows. Scale bar, 10 μm. Arrows indicate Dil-HCV-RAB5 colocalization. Boxed areas indicate Dil-HCV particles that are not colocalizing with RAB5.
To further pinpoint the step of HCV entry that is mediated by AAK1 and GAK, we measured single HCV particle entry events in Huh-7.5 cells depleted of AAK1 or GAK. Highly infectious viral stocks were prepared, and virions were labeled with the membrane-permeable lipophilic dye Dil (49). It was previously shown that incorporation of Dil does not reduce HCV infectivity and that purification of a highly infectious fraction greatly increases the specific infectivity and allows tracking of relevant entry events of fluorescent HCV particles (49). To purify a highly infectious fraction, Dil-HCV was separated by sucrose gradient density ultracentrifugation, fractions were collected, and fraction density, infectious HCV, and HCV RNA copy numbers were measured (Fig. 2B). As previously described (14, 15, 49, 89), the majority of HCV RNAs sedimented at a density of ∼1.15 g/ml (fraction 8), while a highly infectious fraction of HCV (fraction 3) was associated with a lower density of 1.06 g/ml. To test our hypothesis that AAK1 and GAK regulate HCV internalization, Huh-7.5 cells were transfected with an siRNA targeting AAK1 or GAK or with an NT siRNA, incubated for 24 h and transduced with baculovirus-GFP fusion of RAB5, an early endosomal marker. At 48 h posttransfection, cells were infected with Dil-HCV isolated from fraction 3 at 4°C for 1 h to allow binding of the virus, shifted to 37°C to allow HCV entry, and fixed at 1 h postinfection. Wide-field epifluorescence z-stacks were acquired and deconvolved on a DeltaVision OMX Blaze system (Applied Precision-GE, Inc.) equipped with three emCCDs (Evolve, Photometrics Inc.). Randomly chosen HCV-Dil particles (80 to 120) were analyzed for their localization to early endosomes in each group by quantitative immunofluorescence (IF) analysis. As described by other investigators, GFP-RAB5 was observed on punctate cytoplasmic structures that vary in size (due to fusion events) (90, 91). AAK1 or GAK depletion significantly reduced the colocalization of Dil-HCV particles to early endosomes from a mean Pearson's colocalization coefficient of 0.6 ± 0.27 in NT cells to 0.32 ± 0.32 and 0.25 ± 0.3 in AAK1- and GAK-depleted cells, respectively (P values of 9.3 × 10−10 and 4.7 × 10−16, respectively) (Fig. 2C to F). The majority of Dil-labeled viral particles had an equivalent size based on fluorescence emissions, suggesting that we were imaging single viral particles (Fig. 2D to F). Importantly, depletion of either AAK1 or GAK did not appear to alter the number, intensity, and intracellular distribution of RAB5-positive endosomes, as determined by an IF analysis, and had no effect on GFP-RAB5 expression, as measured by Western analysis (data not shown). Last, while transduction with baculovirus GFP-RAB5 slightly reduced (<15%) HCVcc infection compared with a Polybrene control in NT cells, a comparable reduction was measured in baculovirus GFP-RAB5-transduced AAK1- or GAK-depleted cells compared to a Polybrene control in these cells (data not shown). Together, these results indicate that AAK1 and GAK regulate HCV entry at a postbinding step.
AAK1 and GAK regulate EGF-mediated enhanced HCV entry and EGFR endocytosis.
Next, we sought to determine the mechanism by which AAK1 and GAK regulate HCV entry. Both AAK1 and GAK have been implicated in the regulation of EGFR internalization and/or its recycling to the plasma membrane (59, 67, 70–74). We therefore hypothesized that AAK1 and GAK regulate EGFR-mediated HCV entry. To test this hypothesis, we first studied the effect of AAK1 and GAK depletion on EGF-mediated enhanced HCV entry. Serum-starved Huh-7.5 cells depleted of AAK1 or GAK by siRNA and NT controls were incubated with various concentrations of EGF or PBS for 1 h. Cells were subsequently infected with HCVpp or HCVcc for 4 h, unbound virus was washed off, and luciferase activity was measured at 72 or 24 h postinfection, respectively. As previously reported (38, 75), increasing concentrations of EGF enhanced HCVpp entry and HCVcc infection in control NT cells in a dose-dependent manner (Fig. 3A and B). Suppression of either AAK1 or GAK markedly reduced this effect on both HCVpp entry and HCVcc infection. Nevertheless, a high concentration of EGF (10 μg/ml) appeared to either partially or fully compensate for the effect of AAK1 or GAK depletion, respectively, on fold increase in HCVcc infection but not HCVpp entry (Fig. 3A and B).
FIG 3.

AAK1 and GAK regulate EGFR-mediated HCV entry. (A and B) Huh-7.5 cells depleted of AAK1 or GAK by siRNAs and NT controls were serum starved and treated with PBS or EGF at the indicated concentrations for 1 h. Cells were subsequently infected with HCVpp (A) or HCVcc (B) expressing a luciferase reporter gene for 4 h, unbound virus was washed off, and luciferase signal was measured at 72 or 24 h postinfection, respectively. Data are relative luciferase values normalized to the corresponding PBS-treated controls (AAK1-depleted, GAK-depleted, or NT) representing fold increase in HCV entry. Data represent means and standard deviations (error bars) from at least three experiments in triplicates. (C to I) Huh-7.5 cells transfected with NT (C and F), AAK1 (D and G), or GAK (E and H) siRNA and treated with either PBS (C to E) or EGF (1 μg/ml) (F to H) were subjected to quantitative confocal IF analysis of EGFR-EEA1 colocalization. Percent colocalization of the indicated signals was measured by a quantitative colocalization analysis of panels C to H using Manders' coefficients (I). Values indicate mean M2 values represented as percent colocalization (the fraction of red intensity that coincides with green intensity) ± standard deviation (error bars; n = 10 to 15). ***, P < 0.001. Representative images at a ×60 magnification of EGFR (red) and the early endosomal marker EEA1 (green) and merged images are shown. Enlargements of insets are shown at the bottom. Scale bar, 10 μm. EE, EEA1.
We then used quantitative confocal IF analysis to test our hypothesis that the defect in EGF-mediated enhanced HCV entry, resulting from AAK1 or GAK silencing, correlates with alterations in the localization of EGFR to early endosomes. Cells depleted of AAK1 or GAK and NT controls were serum starved and treated with EGF or PBS for 1 h or infected for 1 h with HCVcc in the presence of EGF or a PBS control. Cells were then fixed and stained for EGFR and the early endosomal marker EEA1. A total of 10 to 15 randomly chosen cells from each category were analyzed for the degree of localization of EGFR to early endosomes using ImageJ (JACoP) software and Manders' colocalization coefficients (MCCs) (92). In the absence of EGF, EGFR minimally (<1%) colocalized with EEA1 in NT cells and in cells depleted of AAK1 or GAK (Fig. 3C to E, I). EGF treatment dramatically increased this colocalization in NT cells, with 25.5% ± 7% of early endosomes staining positive for EGFR (Fig. 3F and I). Depletion of either AAK1 or GAK, however, significantly reduced EGFR localization to early endosomes in the face of EGF treatment, with 9.85% ± 4% or 15% ± 4% of early endosomes being positive for EGFR, respectively (P value of 0.0003 or 0.00079) (Fig. 3G to I,). Similar results were demonstrated in cells infected with HCVcc (data not shown). AAK1 or GAK depletion had no effect on the expression level of EGFR in the presence or absence of EGF, as measured by Western analysis (data not shown).
Together, these results indicate that regulation of clathrin-mediated endocytosis of EGFR represents one of the mechanisms by which AAK1 and GAK mediate HCV entry.
Efficient HCV entry is dependent on AP2M1 and its phosphorylation.
AAK1 and GAK mediate their role in clathrin-mediated endocytosis in part by phosphorylating AP2M1 (59–65). To determine whether AP2M1 and its phosphorylation play a role in HCV entry, we generated stable Huh-7.5 clones harboring short hairpin RNA (shRNA) lentiviral constructs targeting distinct regions in the AP2M1 gene or an NT sequence. Effective suppression of AP2M1 levels was achieved (Fig. 4A and B) without apparent cytostatic or cytotoxic effects. These clones were infected with either HCVpp or HCVcc and subjected to luciferase assays at various time points postinfection. As shown in Fig. 4C and D, silencing AP2M1 expression markedly reduced HCVpp entry and HCVcc infection in these clones compared to levels in NT controls. The magnitude of the defect in HCV entry correlated with the degree of AP2M1 depletion. The effect of silencing endogenous AP2M1 on HCV entry was rescued by ectopic expression of shRNA-resistant wild-type (WT) AP2M1 harboring a wobble mutation within the site targeted by the shRNA, largely excluding off-target effects (Fig. 4E). In addition, transient depletion of AP2M1 by siRNA reduced HCVpp entry (Fig. 4F to H). In contrast, depletion of AP1M1, AP1M2, and AP4M1, the μ subunits of the adaptor protein complexes AP-1A, AP-1B, and AP-4, respectively, which sort in the secretory but not endocytic pathway (93), had no effect on HCVpp entry (Fig. 4F to H). Both our stable and transient RNAi approaches thus demonstrate that AP2M1 is an essential adaptor for efficient HCV entry.
FIG 4.

Efficient HCV entry is dependent on AP2M1 and its phosphorylation. (A) Stable Huh-7.5 clones harboring shRNA lentiviral constructs targeting the AP2M1 gene and an NT sequence were established. RNA levels were measured by qRT-PCR. Data are relative AP2M1/S18 RNA ratios normalized to NT control levels. (B) AP2M1 protein levels were measured by quantitative Western analysis. A representative membrane and combined data from three independent measurements are shown. The y axis represents the relative AP2M1/actin protein ratio normalized to the NT control value. (C) These clones were infected with HCVpp, and the luciferase signal was measured at 72 h postinfection. Data are relative luciferase values normalized to the NT control. (D) The clones were infected with HCVcc, and luciferase signal was measured at the indicated time points postinfection. RLU, relative light units. (E) HCVcc infection relative to the NT control level and levels of AP2M1 protein by a Western blot analysis in cells concurrently transduced with lentiviruses expressing an shRNA targeting AP2M1 and shRNA-resistant WT AP2M1 cDNA (AP2M1-WT). (F) Huh-7.5 cells were transfected with siRNAs targeting either AP2M1, AP1M1, AP1M2, or AP4M1 or with an NT sequence. Adaptor protein (AP) levels were measured by Western analysis. Representative membranes are shown. Numbers represent relative AP/actin protein ratios normalized to the NT control. (G) Cellular viability was measured in these cells by alamarBlue-based assays. Data are relative fluorescence values normalized to the NT control. (H) Adaptor protein-depleted cells and NT controls were infected with HCVpp. Luciferase measurements were taken at 72 h postinfection. (I to L) Huh-7.5 cells were transfected with plasmids encoding an empty plasmid or the WT or T156A AP2M1 mutant and infected with HCVpp or HCVcc. Levels of AP2M1 protein expression were determined by a Western blot analysis (I). Cellular viability was measured at 48 h posttransfection by alamarBlue-based assays. Data are relative fluorescence values normalized to the empty plasmid control (J). HCVpp entry (K) and HCVcc infection (L) in these cells were measured by luciferase assays at 72 and 24 h postinfection, respectively. Data are relative luciferase values normalized to the empty plasmid control signal measured at the respective time point (72 h postinfection for HCVpp and 24 h for HCVcc). (M) HCVpp entry relative to that of the NT control at 72 h postinfection in cells depleted of AAK1 or GAK and transduced with lentiviruses expressing WT AP2M1 cDNA. Means and standard deviations (error bars) of results from three independent experiments are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
A threonine residue in position 156 within AP2M1 undergoes phosphorylation by AAK1 and GAK (60–62), which then stimulates AP2M1 binding to tyrosine signals within cargo proteins. To determine whether AP2M1 regulation by AAK1 and GAK plays a role in HCV entry, we studied the effect of overexpression of WT AP2M1 and AP2M1 harboring a T156A mutation on HCVpp entry and HCVcc infection. Huh-7.5 cells were transfected with an empty control plasmid or plasmids encoding the WT or AP2M1 T156A mutant and subsequently infected with either HCVpp or HCVcc, and luciferase assays were performed at 72 and 24 h, respectively. Overexpression of these plasmids had no effect on cellular viability (Fig. 4I and J). HCVpp entry and HCVcc infection were greater following overexpression of WT AP2M1 than that of an empty vector (Fig. 4K and L). Moreover, overexpression of the T156A AP2M1 mutant significantly reduced both HCVpp entry and HCVcc infection compared with the WT AP2M1 level (Fig. 4K and L), consistent with a dominant negative effect of this mutant on HCV entry. Last, the effect of silencing endogenous AAK1 or GAK on HCVpp entry (Fig. 4M) and HCVcc infection (data not shown) was rescued by ectopic expression of WT AP2M1. These data not only support our RNAi data in respect to the functional relevance of AP2M1 for HCV entry but also indicate a role for AAK1 and GAK in the regulation of AP2M1-mediated HCV entry.
We have previously reported that AP2M1 depletion by RNAi and overexpression of the T156A AP2M1 mutant dramatically inhibited HCV assembly (76). These conditions had absolutely no effect on HCV RNA replication, as measured by luciferase assays 72 h following electroporation of in vitro transcribed J6/JFH(p7-Rluc2A) RNA (76). Together, our RNAi, overexpression, and dominant interfering approaches thus indicate a role for AP2M1 and its phosphorylation in HCV entry that is independent of its role in HCV assembly (76).
Efficient HCV entry is dependent on NUMB and its phosphorylation.
NUMB is the only other endocytic clathrin adaptor known to be a target for AAK1 phosphorylation besides AP-2 to date (67). AAK1 activates NUMB by phosphorylating its threonine residue in position 102 (67). We therefore sought to determine the role of NUMB in HCV entry. We first measured HCVpp entry and HCVcc infection in Huh-7.5 cells depleted of NUMB by siRNAs. Two NUMB siRNAs suppressed protein expression, as evidenced by Western blotting (Fig. 5A), with no apparent effect on cellular viability, as measured by alamarBlue-based assays (Fig. 5B). These NUMB-depleted cells were infected with either HCVpp or HCVcc and subjected to luciferase assays at 72 or 9 h postinfection, respectively. Silencing NUMB expression reduced HCVpp entry and HCVcc infection in these clones compared to levels in NT controls (Fig. 5C and D). The magnitude of the defect in HCV entry correlated with the degree of NUMB depletion. Next, we tested our hypothesis that activation of NUMB by AAK1 plays a role in HCV entry. To do so, we studied the effect of overexpression of the WT or a T102A NUMB mutant that lacks the AAK1 phosphorylation site (67) on HCVpp entry and HCVcc infection. Huh-7.5 cells were transfected with plasmids encoding either an empty plasmid, the WT NUMB, or the NUMB T102A mutant and subsequently infected with either HCVpp or HCVcc, followed by luciferase assays at 72 and 24 h, respectively. Overexpression of the WT NUMB plasmid had no effect on cellular viability (Fig. 5E and F) but significantly increased HCVpp entry and HCVcc infection compared with the empty vector, thereby supporting our siRNA data (Fig. 5G and H). Moreover, overexpression of the T102A NUMB mutant significantly reduced HCVpp entry and HCVcc infection compared with the level with WT NUMB (Fig. 5G and H). Nevertheless, this NUMB mutant demonstrated a dominant negative effect on HCVpp entry but not HCVcc infection. While the exact reason for this difference remains to be elucidated, differences between the HCVcc and HCVpp systems, both in the binding and postbinding steps of HCV entry, have been previously reported (25, 94, 95). Functional differences, such as dependency on cellular receptors, have been attributed to differences in oligomerization and/or conformation of the glycoproteins and on sequence variations between the two relevant genotypes. It is thus possible that in the context of HCVcc infection a redundant regulatory mechanism is activated, subjecting NUMB to regulation by another host kinase, such as CaM-KI (96), in addition to AAK1.
FIG 5.

Efficient HCV entry is dependent on NUMB and its phosphorylation. (A) Huh-7.5 cells were transfected with an siRNA targeting NUMB or an NT control. NUMB protein levels were measured by a quantitative Western analysis. A representative membrane and combined data from two independent measurements are shown. Numbers represent relative NUMB/actin protein ratios normalized to the level of the NT control. (B) Cellular viability was measured in the indicated cells at 48 h posttransfection by alamarBlue-based assays. Data are relative fluorescence values normalized to the value of the NT control. (C and D) These cells were infected with either HCVpp or HCVcc, and luciferase activity was measured at 72 and 9 h postinfection, respectively. Data are relative luciferase values normalized to the NT control signal measured at the respective time point. (E) Huh-7.5 cells were transfected with an empty plasmid or a plasmid encoding the WT or T102A NUMB mutant and infected with HCVpp or HCVcc 48 h posttransfection. (F) Cellular viability was measured at 48 h posttransfection by alamarBlue-based assays. Data are relative fluorescence values normalized to the level of the WT NUMB control. (G and H) HCVpp entry and HCVcc infection in these cells were measured by luciferase assays at 72 and 24 h postinfection, respectively. Data are relative luciferase values normalized to the WT NUMB control signal measured at the respective time point. Means and standard deviations (error bars) of results from three independent experiments are shown. RLU, relative light units. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Together, these results indicate that, like AP2M1, NUMB is a functional adaptor for HCV entry, and they suggest that NUMB phosphorylation by AAK1 may be critical for the regulation of HCV entry.
Pharmacological inhibitors of AAK1 and GAK inhibit HCV entry at a postbinding step, and their combination is synergistic.
Using analysis of heat maps and affinity assays of kinase inhibitors, we have previously identified compounds, such as sunitinib and erlotinib, which bind AAK1 and GAK, respectively, with high affinities (nM range) (76, 97, 98). To determine the effect of these compounds on HCV entry, we studied the effect of a 4-h treatment with various concentrations of sunitinib, erlotinib, or DMSO on HCVpp entry and HCVcc infection (MOI of 0.1) using luciferase reporter-linked assays. Sunitinib inhibited HCVpp entry and HCVcc infection in a dose-dependent manner (Fig. 6A and B), with half-maximal effective concentrations (EC50s) of 0.87 ± 0.178 μM (P = 0.0081) and 0.67 ± 0.18 μM (P = 0.048), respectively. Erlotinib had a similar effect on HCVpp entry and HCVcc infection, with EC50s of 1.22 ± 0.32 μM (P = 0.0189) and 3.93 ± 0.75 μM (P = 0.0139), respectively (Fig. 6A and B), consistent with a prior report wherein erlotinib's effect on HCV entry was attributed to EGFR inhibition (38). In contrast, IPA-3, a selective allosteric inhibitor of the macropinocytosis regulator PAK1 (99), had no significant effect on HCVcc infection, in line with our siRNA data and a recent report (87) (Fig. 6B). These drugs had no apparent cellular toxicity at the concentration range used (Fig. 6A and B). Increasing the MOI from 0.1 to 0.5 and 1 had no apparent effect on the dose response of sunitinib's or erlotinib's effect on HCVcc at 24 h postinfection (Fig. 6C and D).
FIG 6.

Pharmacological inhibitors of AAK1 and GAK inhibit HCV entry at a postbinding step. (A and B) Huh-7.5 cells were infected with HCVpp or HCVcc for 1 h on ice, followed by a temperature shift to 37°C, a 4-h treatment with various concentrations of sunitinib, erlotinib, IPA-3, or DMSO, and medium replacement. Data represent dose-response curves of the effect of the indicated inhibitors on HCVpp entry and HCVcc infection (MOI of 0.1) by luciferase assays at 72 and 24 h postinfection, respectively. The y axes at left (black lines) represent relative luciferase values normalized to the DMSO-treated controls. These compounds had no effect on cellular viability at the concentration range used, as measured by alamarBlue-based assays. The y axes at right (gray lines) represent relative fluorescence values normalized to DMSO-treated controls. Data represent means and standard deviations (error bars) from at least three experiments in triplicates. (C and D) Dose-response curves of the effects of sunitinib and erlotinib on HCVcc infection by luciferase assays at 24 h postinfection at the indicated MOIs. (E to G) Time-of-addition experiments, depicted graphically in panel E, were performed by incubation of cells with HCVcc (MOI of 0.2) for 1 h at 4°C and transfer to 37°C. Serum, IgG, anti-CD81 antibody, DMSO, erlotinib, sunitinib, or heparin was added either during the binding step or at various time points after the temperature switch. Data are relative luciferase values normalized to the value of the DMSO control after inhibition of binding and postbinding steps (F) and the time course of HCVcc infection following inhibition with the indicated compounds (G). (H) Supernatants collected from Huh-7.5 cells following HCVcc infection and either a single 4-h or 72-h daily treatment were used to inoculate naive cells. Data represent dose-response curves of the drugs' effects on infectious virus production measured by luciferase assay. (I) Huh-7.5 cells were infected with HCVcc, treated with sunitinib and erlotinib at the indicated concentrations or with DMSO in the presence of calyculin A (an AP2M1 dephosphorylation inhibitor [117]) and subsequently lysed. The effect of the inhibitors on AP2M1 phosphorylation was measured by Western analysis in these cell lysates. A representative membrane blotted with anti-phospho-AP2M1 (p-AP2M1), anti-AP2M1, and anti-actin antibodies and quantitative analysis from three experiments are shown. The y axis represents the relative p-AP2M1/total AP2M1 protein ratio normalized to the value of the DMSO controls. (J) Combinations of sunitinib and erlotinib at the indicated concentrations were used to treat HCVcc-infected cells. Data represent the differential surface analysis at the 95% confidence level (CI) (MacSynergy II). Peaks above the theoretical additive plane indicate synergy, whereas depressions below it indicate antagonism. The level of synergy or antagonism is as indicated by the color code on the figure. Results of a representative experiment (out of three) are shown.
We then sought to define the entry steps targeted by sunitinib and erlotinib. To discriminate between virus binding and postbinding events, HCVcc binding to Huh-7.5 cells was performed for 1 h at 4°C in the presence or absence of the drugs, DMSO, serum, IgG, or anti-CD81 antibody before the temperature was shifted to 37°C to initiate infection (Fig. 6E). Similarly to anti-CD81 antibodies, sunitinib and erlotinib had comparable effects on HCVcc infection when added prior to or following binding of the virus to the target cell, indicating that they inhibit a postbinding step (Fig. 6F). In addition, we performed kinetic entry assays (38, 50, 75) (Fig. 6E and G). Either sunitinib, erlotinib, heparin (an attachment inhibitor), DMSO, or IgG was added at different time points prior to or following viral binding. Residual inhibitors were removed at 4 h postbinding, and luciferase assays were performed at 24 h (Fig. 6G). Sunitinib was still effective when given at 120 min postbinding. As previously described, erlotinib and anti-CD81 antibodies inhibited HCV entry when added as late as 40 to 60 min postbinding (38) (Fig. 6G). Together, these data indicate that these compounds inhibit HCV entry at a postbinding step.
As we previously reported, these drugs had no effect on HCV RNA replication (76) (data not shown). Moreover, the inhibitory effect of sunitinib and erlotinib on infectious virus production measured by luciferase assays in naive cells inoculated with cell culture supernatants was greater following daily treatment with the drugs for 72 h than with a single 4-h treatment (Fig. 6H), in line with their inhibitory effect on both entry and assembly. The EC50 values for the effects of the drugs on infectivity were 0.85 ± 0.27 and 0.4 ± 0.1 μM for sunitinib at 4 and 72 h, respectively, and 2.2 ± 0.8 and 0.37 ± 0.13 μM for erlotinib's effect, respectively (P values < 0.05). In addition, the antiviral effect of these drugs correlated with a decrease or loss of AP2M1 phosphorylation (Fig. 6I).
These results provide pharmacological validation of the requirement for AAK1 and GAK in the regulation of HCV entry and suggest that they mediate a postbinding step. Moreover, sunitinib and erlotinib represent candidate compounds that target two steps of the HCV life cycle: entry and assembly.
Since both AAK1 and GAK regulate HCV entry, we hypothesized that the combination of an AAK1 inhibitor with a GAK inhibitor would increase the antiviral effect over that achieved with each drug alone. Luciferase reporter-linked HCVcc entry assays were used to study the antiviral effects of combinations of these drugs. Data were analyzed using the MacSynergy program (84, 85) based on the Bliss independence model for synergy, as we have previously described (100). Six-by-five matrix data sets in four replicates were assessed at the 95% confidence level for each of the three experiments performed. The sunitinib-erlotinib combination had antiviral effects that were significantly more potent than the theoretical additive effects, which supported the idea that this combination was synergistic (Fig. 6J). No evidence of significant antiviral antagonism was seen with any of the tested doses. The calculated synergy and log volume were 40.11 μM2% and 10.04 μM2%, respectively, with a calculated antagonism of −0.71 μM2% and −0.18 μM2%. According to the criteria suggested by Prichard et al. (84) (see Materials and Methods), this level of synergy is considered minor but significant. Importantly, because there was no cellular toxicity with either drug alone at the studied concentrations and no increase in cytotoxicity when they were used in combination, the measured synergy was indeed specific and did not reflect synergistic toxicity.
AAK1 and GAK are molecular targets underlying the sunitinib and erlotinib antiviral effect.
While our data indicate that AAK1 and GAK are potential targets for antiviral treatment, it is important to validate that AAK1 and GAK are indeed molecular targets underlying the antiviral effect of sunitinib and erlotinib, respectively. This is particularly relevant because of the known inhibitory effect of erlotinib on EGFR-mediated HCV entry (38). We therefore performed gain-of-function studies. Huh-7.5 cells plated in six-well plates were transfected with plasmids encoding AAK1, GAK, or EGFR or an empty plasmid. At 24 h posttransfection these cells were trypsinized and plated in either 6- or 96-well plates. Effective overexpression was demonstrated by Western blotting in cell lysates (in six-well plates) at 48 h posttransfection (Fig. 7A), and there was no effect on cellular viability by alamarBlue-based assays (data not shown). In parallel, cells plated in 96-well plates were infected with HCVcc for 1 h on ice, treated with sunitinib, erlotinib, or DMSO for 4 h, and incubated in fresh medium for 20 h prior to luciferase assays. Overexpression of AAK1, sunitinib's target, either completely or partially reversed the antiviral effect of sunitinib, as predicted (Fig. 7B). Similarly, overexpression of GAK, erlotinib's target, either completely or partially reversed the antiviral effect of erlotinib (Fig. 7B). Interestingly, overexpression of GAK partially reversed the antiviral effect of sunitinib, and overexpression of AAK1 completely reversed the antiviral effect of erlotinib (Fig. 7B). Last, overexpression of EGFR partially reversed the antiviral effect of both sunitinib and erlotinib (Fig. 7B). These results demonstrate some functional overlap between the kinase activities of AAK1 and GAK in HCV entry. Importantly, they establish the link between the antiviral effect of sunitinib and erlotinib and their molecular targets and indicate that erlotinib inhibits two cellular factors critical for HCV entry: EGFR and its regulator GAK.
FIG 7.
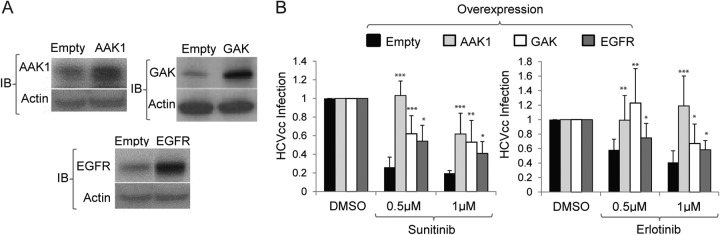
AAK1 and GAK are molecular targets underlying sunitinib and erlotinib antiviral effects. Huh-7.5 cells plated in six-well plates were transfected with plasmids encoding AAK1, GAK, or EGFR or with an empty plasmid. At 24 h posttransfection cells were trypsinized and plated in 6- and 96-well plates. (A) Western analysis was performed in cell lysates (obtained from the six-well plate) at 48 h posttransfection to assess the level of overexpression. Representative membranes immunoblotted (IB) with antibodies targeting the indicated proteins and actin are shown. (B) At 48 h posttransfection cells plated in 96-well plates were infected with HCVcc for 1 h on ice, followed by a temperature shift to 37°C and 4-h treatment with sunitinib (left) or erlotinib (right) at 0.5 μM and 1 μM or with a DMSO control. Medium was then replaced for removal of residual drugs, and unbound virus and luciferase signals were measured following a 20-h incubation. Data are relative luciferase values normalized to the values of the DMSO controls overexpressing the respective protein. Means and standard deviations (error bars) of results from three independent experiments are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
We combined molecular virology, RNAi, dominant interfering, and pharmacological approaches to test the hypothesis that regulators of clathrin-mediated endocytosis, AAK1 and GAK, and their phosphorylation targets, the clathrin adaptors AP-2 and NUMB, are required for HCV entry. Our findings support a role of these host factors and reveal key mechanisms by which they regulate HCV entry. Moreover, we identified inhibitors that disrupt these mechanisms and inhibit HCV entry.
Suppression of AAK1 or GAK expression by siRNAs dramatically disrupted HCVpp (genotype 1a) entry and HCVcc (genotype 2a) infection, indicating that these host kinases are critical for HCV entry and that this requirement is conserved at least across two genotypes. By performing attachment assays, kinetic assays, and tracking single Dil-HCV particles, we demonstrated that, in line with their role in cellular membrane trafficking (59–65, 67, 68), AAK1 and GAK mediate a postbinding step of HCV entry, likely internalization. While auxilin (DNAJC6) has been previously reported to mediate entry of vesicular stomatitis virus (VSV), the role of AAK1 and GAK (auxilin 2) in viral entry has not been reported in any viral infection to date. We previously reported that AAK1 and GAK regulate core protein-AP2M1 binding and HCV assembly, with no effect on HCV RNA replication (76). Our results therefore indicate that AAK1 and GAK regulate two independent, temporally distinct steps in the HCV life cycle: entry and assembly. AAK1 and GAK thus appear to represent “master regulators” of HCV infection and potential targets for antiviral treatment (Fig. 8). We predict that the requirement for AAK1 and GAK is broadly conserved among viruses that hijack clathrin-mediated pathways for either entry or late steps of their life cycle.
FIG 8.

Proposed model. AAK1 and GAK regulate two distinct steps in the HCV life cycle: entry and assembly. For HCV entry, AAK1 and GAK are critical for the regulation of HCV internalization. AAK1 and GAK phosphorylate the clathrin adaptor AP-2, and AAK1 phosphorylates the alternate adaptor NUMB. AP-2 and/or NUMB then collaborates with other endocytic factors, possibly also subjected to AAK1 and/or GAK regulation, to enhance endocytosis and/or recycling of EGFR and potentially an additional HCV coreceptor(s). In HCV assembly, AAK1 and GAK mediate HCV assembly independently of their role in HCV entry by regulating AP-2 binding to the HCV core protein on lipid droplets (LD) (76). Sunitinib inhibits AAK1, and erlotinib inhibits both EGFR and its regulator GAK, thereby disrupting both HCV entry and assembly. Numbers represent Kd values of binding of the drugs to the respective targets. Solid arrows represent target phosphorylations that we show are critical for HCV entry. Dashed lines represent protein-protein interactions. ER, endoplasmic reticulum.
Although alternate clathrin adaptors can sustain endocytic uptake of certain receptors in the absence of AP-2, optimal coat assembly and trafficking evidently require AP-2 (66, 101–104). Indeed, almost all clathrin-coated pits at the cell surface contain AP-2, and the surface density of clathrin coats in AP-2-deficient cells is <10% of normal (104). One would therefore predict that AP2M1 is essential for any virus that hijacks the clathrin-mediated endocytosis pathway for its entry. This, however, does not appear to be the case: clathrin-mediated endocytosis of VSV (105) is dependent on AP-2, whereas that of influenza A virus is AP-2 independent yet dependent on EPN1 (106, 107). Using several approaches, we show that AP2M1 is a functional adaptor for HCV entry, consistent with results of two independent siRNA screens (49, 108). Moreover, we reveal a novel role for the alternate adaptor, NUMB, in HCV entry. Combined with earlier reports (47, 49, 108), these data indicate that, like endocytosis of various receptors such as P-selectin and LRP1 (109), HCV entry is dependent on both AP-2 and alternate adaptors. These findings likely reflect this apparent redundancy in adaptor usage as well as the requirement for multiple receptors in HCV entry. It will be important to identify the specific cellular HCV coreceptor(s) that is internalized in an AP-2- and/or alternate adaptor-dependent manner. Investigating whether the differences in clathrin adaptor dependence between viruses reflects usage of distinct receptors or sorting to distinct early endosomal compartments is also of interest.
AP2M1 and NUMB are the only known targets for AAK1 and/or GAK phosphorylation in the endocytic pathway to date. Their phosphorylation by AAK1 and/or GAK induces their activation and plays an essential role in the regulation of clathrin-mediated endocytosis of cell surface receptors (59–65). Our data indicate that these mechanisms mediate HCV entry. First, depletion of AP2M1 or NUMB reduced HCV entry. Second, consistent with the previously reported dominant negative effects of AP2M1 and NUMB (67, 76, 110), overexpression of either an AP2M1 or NUMB phosphorylation site mutant reduced HCV entry, albeit the NUMB mutant exhibited such a phenotype in HCVpp but not in HCVcc infection. Third, AP-2 overexpression rescued the effect of AAK1 and GAK depletion on HCV entry. Fourth, the effect of the pharmacological inhibitors of AAK1 and GAK, sunitinib and erlotinib, respectively, on HCV entry was associated with reduced AP2M1 phosphorylation. These findings not only support the functional relevance of AP2M1 and NUMB for HCV entry but also indicate a key role for their phosphorylation by AAK1 and/or GAK in HCV entry. Moreover, we have recently reported that AP2M1 is recruited by a tyrosine motif within the HCV core protein to assembly sites on lipid droplets and that this interaction is absolutely critical for HCV assembly (76). Combined, these results thus indicate that HCV hijacks AP2M1 to mediate two distinct steps in its life cycle, entry and assembly, both of which are subjected to regulation by AAK1 and GAK (Fig. 8).
Using an RNAi approach, we show that GAK depletion reduces both EGF-mediated enhanced HCV infection and localization of EGFR to early endosomes. These results indicate that GAK regulates EGFR-mediated HCV entry and are in line with the established role of GAK as a regulator of EGFR internalization (59). GAK's role in EGFR endocytosis appears to be mediated by its target, AP-2, and the alternate endocytic adaptor, EPN1 (59, 111) (Fig. 8). Since, like AP-2, EPN1 is also essential for HCV entry (49, 108), its regulation may represent an additional mechanism by which GAK mediates HCV entry. Interestingly, stable depletion of GAK by shRNA was shown to alter EGFR downstream signaling, suggesting that GAK may function in receptor signaling (69). It will therefore be interesting to determine whether, in addition to receptor trafficking, GAK regulates EGFR/HRas signaling, which is reported to be critical for HCV entry (41). siRNAs targeting AAK1 reduced both EGF-mediated enhanced HCV infection and localization of EGFR to early endosomes, suggesting that, like GAK, AAK1 is also involved in regulating EGFR-mediated HCV entry. Further support to these findings comes from our gain-of-function assays, wherein EGFR overexpression partially but significantly reversed the antiviral effect of the AAK1 inhibitor, sunitinib. The exact mechanism by which AAK1 regulates EGFR-mediated HCV entry is an area of further investigation. We speculate that AAK1 mediates this process by interacting with and/or phosphorylating endocytic adaptors that act downstream of EGFR to mediate its internalization and/or recycling to the plasma membrane, such as NUMB and EPS15 (EGFR pathway substrate 15) (70–72). Indeed, NUMB deletion mutants completely block EGFR internalization (71). Moreover, EPS15, a binding partner of AAK1 (73), interacts with AP-2 (112), EPN1 (113), and NUMB (72) to internalize EGFR and regulate its levels at the cell surface (74, 114, 115), and intriguingly EPS15 was previously shown to be critical for HCV entry (47). Together, these data suggest that AAK1 and GAK mediate HCV entry by regulating several endocytic proteins, which may collaborate to promote EGFR internalization (Fig. 8). Pharmacological inhibition of AAK1 and GAK or their depletion by siRNAs did not reduce the level of expression of either EGFR or the HCV receptors: CD81, occludin, claudin, and SR-B1 (data not shown). Nevertheless, it is likely that, in addition to EGFR regulation, AAK1 and GAK may mediate HCV entry by regulating internalization or recycling of some of these or other relevant cellular receptors. Interestingly, internalization and/or recycling of the HCV entry factors TFR1 and LDLR has been shown to be regulated by AAK1 and/or GAK (40, 59, 62, 67).
Prompted by the finding that AAK1 and GAK regulate core protein-AP2M1 binding and are essential for HCV assembly, we previously discovered that sunitinib and erlotinib, approved anticancer drugs that potently inhibit AAK1 and GAK, respectively, disrupt core protein-AP2M1 binding and HCV assembly (76). Here, we show that these compounds also inhibit HCV entry and that this effect is independent of their effect on assembly. Erlotinib was previously identified as an inhibitor of HCV entry (38). Its effect on HCV entry, however, was attributed solely to its effect on EGFR (38). Although GAK is not erlotinib's known cancer target, it is inhibited by this drug with a comparable potency to EGFR (Kd [dissociation constant] of 3.4 nM versus 1 nM) (97, 98). Importantly, as discussed above, GAK is a key regulator of EGFR internalization (59, 111). Indeed, we show that overexpression of either GAK or EGFR can reverse the inhibitory effect of erlotinib on HCV entry. Our results thus indicate that erlotinib inhibits two cellular factors critical for HCV entry, EGFR and its regulator GAK (Fig. 8), and validate GAK, in addition to EGFR, as a molecular target underlying erlotinib's antiviral effect. Further support of these findings and of the role of GAK in HCV entry comes from our recent data demonstrating that novel highly selective GAK inhibitors, with chemical structures that are unrelated to erlotinib and with no anti-EGFR activity, inhibit HCV entry (unpublished data). Our data also indicate that AAK1 is the molecular target underlying sunitinib's anti-HCV activity. Together, these results establish the link between the antiviral effect of the drugs and their underlying molecular targets.
While the kinase functions of AAK1 and GAK appear to have partially overlapping roles in clathrin-mediated endocytosis, other functions of these proteins, such as CCV uncoating mediated by a GAK auxilin-like motif, are thought to be distinct (62). We show that pharmacological inhibition of the kinase activity of either AAK1 or GAK by sunitinib and erlotinib impairs AP2M1 phosphorylation and HCV entry. These results suggest that, unlike noninfected cells (62), in the context of HCV infection, the endogenous level of one of the enzymes, AAK1 or GAK, is not sufficient to compensate for the loss of the kinase function of the other and that the observed defect in endocytosis is in part dependent on AP2M1. Moreover, the combination of sunitinib and erlotinib had a synergistic effect on HCV entry. Synergy between antimicrobial drugs often results from a sequential blocking mechanism, implying a block of two steps in a common pathway that is critical for viral replication (100, 116). The observed synergy thus suggests that, in the context of HCV entry, some of the AAK1- and GAK-phosphorylated targets may be distinct. Inhibition of other drug targets by erlotinib (e.g., EGFR) and/or sunitinib could also contribute to the observed synergy. Last, overexpression of either AAK1 or GAK partially or completely reversed the antiviral effects of both sunitinib and erlotinib, reflecting some functional overlap between the kinase activities of both enzymes in HCV entry. Together, these data suggest that both overlapping and distinct mechanisms of AAK1 and GAK likely play a role in mediating HCV entry.
Based on this study, we propose a model (Fig. 8) wherein AAK1 and GAK regulate HCV entry at multiple points by phosphorylating both AP2M1 and NUMB and possibly activating additional endocytic proteins that collaborate to mediate EGFR-dependent entry. In addition, by regulating two temporally distinct steps of the HCV life cycle, entry and assembly, AAK1 and GAK represent master regulators of HCV infection and attractive targets for antiviral strategies. Last, sunitinib and erlotinib inhibit the kinase function of AAK1 and GAK, thereby inhibiting these two distinct steps in the HCV life cycle. Taken together, these results provide insight into mechanisms of HCV entry and the design of novel antiviral strategies, with potential implications for other viruses that enter via clathrin-mediated endocytosis.
ACKNOWLEDGMENTS
This work was supported by grants from Stanford Bio-X, the Stanford SPARK program, award number 1U19AI10966201 (CETR) from the National Institute of Allergy and Infectious Diseases, grant 2013100 from the Doris Duke Charitable Foundation, and grant RSG-14-11 0-0 1-MPC from the American Cancer Society to S.E. It was also supported by award number 1S10OD01227601 (SIG) from the National Center for Research Resources to J.M. G.N. is supported by the Child Health Research Institute, Lucile Packard Foundation for Children's Health, as well as the Stanford CSTA (grant number UL1 TR000093).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
We thank Charles M. Rice for kindly providing the pFL-J6/JFH(p7-Rluc2A) plasmid, Apath LLC (St. Louis, MO) for providing Huh-7.5 cells, Shoshana Levy for providing the plasmids used in the HCVpp entry assays (pNL4-3.Luc.R-E, pcDM8, and pcDM8-E1E2), and Mark N. Prichard for providing the MacSynergy II program. We also thank the Stanford Cell Sciences Imaging Facility for acquiring the images of the Dil-HCV internalization assays and helping with data analysis.
REFERENCES
- 1.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int 29(Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med 132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 3.Lemon SM, Walker C, Alter MJ, Yi M. 2007. Hepatitis C viruses, p 1253–1304. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Soriano V, Vispo E, Poveda E, Labarga P, Martin Carbonero L, Fernandez Montero J, Barreiro P. 2011. Directly acting antivirals against hepatitis C virus. J Antimicrob Chemother 66:1673–1686. doi: 10.1093/jac/dkr215. [DOI] [PubMed] [Google Scholar]
- 5.Pawlotsky J-M. 2013. Treatment of chronic hepatitis C: current and future. Curr Top Microbiol Immunol 369:321–342. doi: 10.1007/978-3-642-27340-7_13. [DOI] [PubMed] [Google Scholar]
- 6.Lange CM, Zeuzem S. 2013. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol 58:583–592. doi: 10.1016/j.jhep.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Lohmann V. 2000. Replication of hepatitis C virus. J Gen Virol 81:1631–1648. [DOI] [PubMed] [Google Scholar]
- 8.Branch AD. 2000. Hepatitis C virus RNA codes for proteins and replicates: does it also trigger the interferon response? Semin Liver Dis 20:57–68. doi: 10.1055/s-2000-9502. [DOI] [PubMed] [Google Scholar]
- 9.Reed KE, Rice CM. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol 242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 10.Lindenbach B, Rice C. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drummer HE, Maerz A, Poumbourios P. 2003. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett 546:385–390. doi: 10.1016/S0014-5793(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 12.Bartosch B, Dubuisson J, Cosset F-L. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 15.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomssen R, Bonk S, Propfe C, Heermann KH, Köchel HG, Uy A. 1992. Association of hepatitis C virus in human sera with β-lipoprotein. Med Microbiol Immunol 181:293–300. doi: 10.1007/BF00198849. [DOI] [PubMed] [Google Scholar]
- 17.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. 2002. characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol 76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz O, Delers F, Maynard M, Demignot S, Zoulim F, Chambaz J, Trépo C, Lotteau V, André P. 2006. Preferential association of hepatitis C virus with apolipoprotein B48-containing lipoproteins. J Gen Virol 87:2983–2991. doi: 10.1099/vir.0.82033-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol 80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barth H, Schnober EK, Zhang F, Linhardt RJ, Depla E, Boson B, Cosset F-L, Patel AH, Blum HE, Baumert TF. 2006. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol 80:10579–10590. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morikawa K, Zhao Z, Date T, Miyamoto M, Murayama A, Akazawa D, Tanabe J, Sone S, Wakita T. 2007. The roles of CD81 and glycosaminoglycans in the adsorption and uptake of infectious HCV particles. J Med Virol 79:714–723. doi: 10.1002/jmv.20842. [DOI] [PubMed] [Google Scholar]
- 22.Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, Roitelman J, Barbaras R, Graber P, Ghersa P, Smolarsky M, Funaro A, Malavasi F, Larrey D, Coste J, Fabre J-M, Sa-Cunha A, Maurel P. 2007. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol 46:411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Monazahian M, Böhme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. 1999. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol 57:223–229. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 25.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol 81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Randall G, Higginbottom A, Monk P, Rice CM, McKeating JA. 2004. CD81 is required for hepatitis C Virus glycoprotein-mediated viral infection. J Virol 78:1448–1455. doi: 10.1128/JVI.78.3.1448-1455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKeating JA, Zhang LQ, Logvinoff C, Flint M, Zhang J, Yu J, Butera D, Ho DD, Dustin LB, Rice CM, Balfe P. 2004. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J Virol 78:8496–8505. doi: 10.1128/JVI.78.16.8496-8505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreux M, Dao Thi VL, Fresquet J, Guarin M, Julia Z, Verney G, Durantel D, Zoulim F, Lavillette D, Cosset F-L, Bartosch B. 2009. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog 5:e1000310. doi: 10.1371/journal.ppat.1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scarselli E, Ansuini H, Cerino R, Roccasecca R, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeisel MB, Koutsoudakis G, Schnober EK, Haberstroh A, Blum HE, Cosset F-L, Wakita T, Jaeck D, Doffoel M, Royer C, Soulier E, Schvoerer E, Schuster C, Stoll-Keller F, Bartenschlager R, Pietschmann T, Barth H, Baumert TF. 2007. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 46:1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 31.Grove J, Huby T, Stamataki Z, Vanwolleghem T, Meuleman P, Farquhar M, Schwarz A, Moreau M, Owen JS, Leroux-Roels G, Balfe P, McKeating JA. 2007. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J Virol 81:3162–3169. doi: 10.1128/JVI.02356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benedicto I, Molina-Jiménez F, Bartosch B, Cosset F-L, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernández A, Aldabe R, López-Cabrera M, Majano PL. 2009. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol 83:8012–8020. doi: 10.1128/JVI.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 36.Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset F-L, Dragic T. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J Virol 82:3555–3560. doi: 10.1128/JVI.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471. doi: 10.1128/JVI.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lupberger J, Zeisel M, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee C, Turek M, Gorke S, Royer C, Fischer B, Zahid M, Lavillette D, Fresquet J, Cosset F-L, Rothenberg SM, Pietschmann T, Patel A, Pessaux P, Doffol M, Raffelsberger W, Poch O, McKeating J, Brino L, Baumert T. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sainz B, Barretto N, Martin D, Hiraga N, Imamura M, Hussain S, Marsh K, Yu X, Chayama K, Alrefai W, Uprichard S. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A 110:10777–10782. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zona L, Lupberger J, Sidahmed-Adrar N, Thumann C, Harris Helen J, Barnes A, Florentin J, Tawar Rajiv G, Xiao F, Turek M, Durand Sarah C, Duong François HT, Heim Markus H, Cosset F-L, Hirsch I, Samuel D, Brino L, Zeisel Mirjam B, Le Naour F, McKeating Jane A, Baumert Thomas F. 2013. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Farquhar MJ, Hu K, Harris HJ, Davis C, Brimacombe CL, Fletcher SJ, Baumert TF, Rappoport JZ, Balfe P, McKeating JA. 2012. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J Virol 86:4305–4316. doi: 10.1128/JVI.06996-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farquhar MJ, Harris HJ, Diskar M, Jones S, Mee CJ, Nielsen SU, Brimacombe CL, Molina S, Toms GL, Maurel P, Howl J, Herberg FW, van IJzendoorn SCD, Balfe P, McKeating JA. 2008. Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J Virol 82:8797–8811. doi: 10.1128/JVI.00592-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. 2008. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J Virol 82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Codran A, Royer C, Jaeck D, Bastien-Valle M, Baumert TF, Kieny MP, Pereira CA, Martin J-P. 2006. Entry of hepatitis C virus pseudotypes into primary human hepatocytes by clathrin-dependent endocytosis. J Gen Virol 87:2583–2593. doi: 10.1099/vir.0.81710-0. [DOI] [PubMed] [Google Scholar]
- 46.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meertens L, Bertaux C, Dragic T. 2006. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80:11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coller K, Berger K, Heaton N, Cooper J, Yoon R, Randall G. 2009. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog 5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moradpour D, Penin Fo Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 52.Conner S, Schmid S. 2003. Regulated portals of entry into the cell. Nature 422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 53.Brodsky F. 2012. Diversity of clathrin function: new tricks for an old protein. Annu Rev Cell Dev Biol 28:309–336. doi: 10.1146/annurev-cellbio-101011-155716. [DOI] [PubMed] [Google Scholar]
- 54.Owen D, Collins B, Evans P. 2004. Adaptors for clathrin coats: structure and function. Annu Rev Cell Dev Biol 20:153–191. doi: 10.1146/annurev.cellbio.20.010403.104543. [DOI] [PubMed] [Google Scholar]
- 55.Ohno H. 2006. Clathrin-associated adaptor protein complexes. J Cell Sci 119:3719–3721. doi: 10.1242/jcs.03085. [DOI] [PubMed] [Google Scholar]
- 56.Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- 57.Pearse BM. 1988. Receptors compete for adaptors found in plasma membrane coated pits. EMBO J 7:3331–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boll W, Ohno H, Songyang Z, Rapoport I, Cantley LC, Bonifacino JS, Kirchhausen T. 1996. Sequence requirements for the recognition of tyrosine-based endocytic signals by clathrin AP-2 complexes. EMBO J 15:5789–5795. [PMC free article] [PubMed] [Google Scholar]
- 59.Lee D-w, Zhao X, Zhang F, Eisenberg E, Greene LE. 2005. Depletion of GAK/auxilin 2 inhibits receptor-mediated endocytosis and recruitment of both clathrin and clathrin adaptors. J Cell Sci 118:4311–4321. doi: 10.1242/jcs.02548. [DOI] [PubMed] [Google Scholar]
- 60.Ricotta D, Conner SD, Schmid SL, von Figura K, Honing S. 2002. Phosphorylation of the AP2 μ subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol 156:791–795. doi: 10.1083/jcb.200111068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korolchuk VI, Banting G. 2002. CK2 and GAK/auxilin2 are major protein kinases in clathrin-coated vesicles. Traffic 3:428–439. doi: 10.1034/j.1600-0854.2002.30606.x. [DOI] [PubMed] [Google Scholar]
- 62.Zhang CX, Engqvist-Goldstein ÅE, Carreno S, Owen DJ, Smythe E, Drubin DG. 2005. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic 6:1103–1113. doi: 10.1111/j.1600-0854.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 63.Umeda A, Meyerholz A, Ungewickell E. 2000. Identification of the universal cofactor (auxilin 2) in clathrin coat dissociation. Eur J Cell Biol 79:336–342. doi: 10.1078/S0171-9335(04)70037-0. [DOI] [PubMed] [Google Scholar]
- 64.Conner S, Schröter T, Schmid S. 2003. AAK1-mediated micro2 phosphorylation is stimulated by assembled clathrin. Traffic 4:885–890. doi: 10.1046/j.1398-9219.2003.0142.x. [DOI] [PubMed] [Google Scholar]
- 65.Zhao X, Greener T, Al-Hasani H, Cushman SW, Eisenberg E, Greene LE. 2001. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J Cell Sci 114:353–365. [DOI] [PubMed] [Google Scholar]
- 66.Traub LM. 2003. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol 163:203–208. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sorensen E, Conner S. 2008. AAK1 regulates Numb function at an early step in clathrin-mediated endocytosis. Traffic 9:1791–1800. doi: 10.1111/j.1600-0854.2008.00790.x. [DOI] [PubMed] [Google Scholar]
- 68.Henderson DM, Conner SD. 2007. A Novel AAK1 splice variant functions at multiple steps of the endocytic pathway. Mol Biol Cell 18:2698–2706. doi: 10.1091/mbc.E06-09-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Gjoerup O, Roberts TM. 2004. The serine/threonine kinase cyclin G-associated kinase regulates epidermal growth factor receptor signaling. Proc Natl Acad Sci U S A 101:10296–10301. doi: 10.1073/pnas.0403175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith CA, Dho SE, Donaldson J, Tepass U, McGlade CJ. 2004. The cell fate determinant Numb interacts with EHD/Rme-1 family proteins and has a role in endocytic recycling. Mol Biol Cell 15:3698–3708. doi: 10.1091/mbc.E04-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santolini E, Puri C, Salcini AE, Gagliani MC, Pelicci PG, Tacchetti C, Di Fiore PP. 2000. Numb is an endocytic protein. J Cell Biol 151:1345–1352. doi: 10.1083/jcb.151.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salcini AE, Confalonieri S, Doria M, Santolini E, Tassi E, Minenkova O, Cesareni G, Pelicci PG, Di Fiore PP. 1997. Binding specificity and in vivo targets of the EH domain, a novel protein-protein interaction module. Genes Dev 11:2239–2249. doi: 10.1101/gad.11.17.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta-Rossi N, Ortica S, Meas-Yedid V, Heuss S, Moretti J, Olivo-Marin J-C, Israël A. 2011. The adaptor-associated kinase 1, AAK1, is a positive regulator of the Notch pathway. J Biol Chem 286:18720–18730. doi: 10.1074/jbc.M110.190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chi S, Cao H, Wang Y, McNiven MA. 2011. Recycling of the epidermal growth factor receptor is mediated by a novel form of the clathrin adaptor protein Eps15. J Biol Chem 286:35196–35208. doi: 10.1074/jbc.M111.247577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Diao J, Pantua H, Ngu H, Komuves L, Diehl L, Schaefer G, Kapadia SB. 2012. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol 86:10935–10949. doi: 10.1128/JVI.00750-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. 2012. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog 8:e1002845. doi: 10.1371/journal.ppat.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rual J-F, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A, Li N, Rosenberg J, Lamesch P, Vidalain P-O, Clingingsmith T, Hartley J, Esposito D, Cheo D, Moore T, Simmons B, Sequerra R, Bosak S, Doucette-Stamm L, Le Peuch C, Vandenhaute J, Cusick M, Albala J, Hill D, Vidal M. 2004. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res 14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cassonnet P, Rolloy C, Neveu G, Vidalain PO, Chnatier T, Pellet J, Jones L, Muller M, Demeret C, Gaud G, Vuillier F, Lotteau V, Tangy F, Favre M, Jacob Y. 2011. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat Methods 8:990–992. doi: 10.1038/nmeth.1773. [DOI] [PubMed] [Google Scholar]
- 79.Kametaka S, Moriyama K, Burgos PV, Eisenberg E, Greene LE, Mattera R, Bonifacino JS. 2007. Canonical interaction of cyclin G associated kinase with adaptor protein 1 regulates lysosomal enzyme sorting. Mol Biol Cell 18:2991–3001. doi: 10.1091/mbc.E06-12-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, Zurrida S, Maisonneuve P, Viale G, Di Fiore PP. 2004. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray CL, Jones CT, Tassello J, Rice CM. 2007. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J Virol 81:10220–10231. doi: 10.1128/JVI.00793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol 86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prichard MN, Shipman C. 1996. Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther 1:9–20. [PubMed] [Google Scholar]
- 84.Prichard MK, Aseltine KR, Shipman C Jr. 1993. MacSynergy II, version 1.0 User's manual. University of Michigan, Ann Arbor, MI. [Google Scholar]
- 85.Prichard MN, Shipman C. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. doi: 10.1016/0166-3542(90)90001-N. [DOI] [PubMed] [Google Scholar]
- 86.Liberali P, Kakkonen E, Turacchio G, Valente C, Spaar A, Perinetti G, Böckmann RA, Corda D, Colanzi A, Marjomaki V, Luini A. 2008. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J 27:970–981. doi: 10.1038/emboj.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsuda M, Suzuki R, Kataoka C, Watashi K, Aizaki H, Kato N, Matsuura Y, Suzuki T, Wakita T. 2014. Alternative endocytosis pathway for productive entry of hepatitis C virus. J Gen Virol 95:2658–2667. doi: 10.1099/vir.0.068528-0. [DOI] [PubMed] [Google Scholar]
- 88.Shi Q, Jiang J, Luo G. 2013. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol 87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hijikata M, Shimizu YK, Kato H, Iwamoto A, Shih JW, Alter HJ, Purcell RH, Yoshikura H. 1993. Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J Virol 67:1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M. 1999. Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol 1:376–382. doi: 10.1038/14075. [DOI] [PubMed] [Google Scholar]
- 91.Helmuth JA, Burckhardt CJ, Greber UF, Sbalzarini IF. 2009. Shape reconstruction of subcellular structures from live cell fluorescence microscopy images. J Struct Biol 167:1–10. doi: 10.1016/j.jsb.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 92.Dunn KW, Kamocka MM, McDonald JH. 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park Sang Y, Guo X. 2014. Adaptor protein complexes and intracellular transport. Biosci Rep 34:e00123. doi: 10.1042/BSR20140069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Helle F, Vieyres G, Elkrief L, Popescu C-I, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol 84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sainz B Jr, Barretto N, Uprichard SL. 2009. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One 4:e6561. doi: 10.1371/journal.pone.0006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tokumitsu H, Hatano N, Inuzuka H, Sueyoshi Y, Yokokura S, Ichimura T, Nozaki N, Kobayashi R. 2005. Phosphorylation of Numb family proteins: possible involvement of Ca2+/calmodulin-dependent protein kinases. J Biol Chem 280:35108–35118. doi: 10.1074/jbc.M503912200. [DOI] [PubMed] [Google Scholar]
- 97.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. 2008. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 98.SuperNova Life Science. 2008. Human kinome heat map. SuperNoval Life Science, Auburn, WA: http://www.supernovalifescience.com/HM/HM%2041.pdf. [Google Scholar]
- 99.Viaud J, Peterson JR. 2009. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol Cancer Ther 8:2559–2565. doi: 10.1158/1535-7163.MCT-09-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Einav S, Sobol Hadas D, Gehrig E, Glenn JS. 2010. The hepatitis C virus (HCV) NS4B RNA binding inhibitor clemizole is highly synergistic with HCV protease inhibitors. J Infect Dis 202:65–74. doi: 10.1086/653080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT. 2001. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 102.Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. 2001. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J Biol Chem 276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- 103.Mishra SK, Keyel PA, Hawryluk MJ, Agostinelli NR, Watkins SC, Traub LM. 2002. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J 21:4915–4926. doi: 10.1093/emboj/cdf487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Motley A, Bright NA, Seaman MNJ, Robinson MS. 2003. Clathrin-mediated endocytosis in AP-2–depleted cells. J Cell Biol 162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lakadamyali M, Rust MJ, Zhuang X. 2006. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen C, Zhuang X. 2008. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci U S A 105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Trotard M, Lepère-Douard C, Régeard M, Piquet-Pellorce C, Lavillette D, Cosset FL, Gripon P, Le Seyec J. 2009. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J 23:3780–3789. doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 109.Traub LM. 2009. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol 10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- 110.Motley AM, Berg N, Taylor MJ, Sahlender DA, Hirst J, Owen DJ, Robinson MS. 2006. Functional Analysis of AP-2 α and μ2 subunits. Mol Biol Cell 17:5298–5308. doi: 10.1091/mbc.E06-05-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kazazic M, Bertelsen V, Pedersen KW, Vuong TT, Grandal MV, Rødland MS, Traub LM, Stang E, Madshus IH. 2009. Epsin 1 is involved in recruitment of ubiquitinated EGF receptors into clathrin-coated pits. Traffic 10:235–245. doi: 10.1111/j.1600-0854.2008.00858.x. [DOI] [PubMed] [Google Scholar]
- 112.Benmerah A, Bègue B, Dautry-Varsat A, Cerf-Bensussan N. 1996. The ear-of-adaptin interacts with the COOH-terminal domain of the Eps15 protein. J Biol Chem 271:12111–12116. doi: 10.1074/jbc.271.20.12111. [DOI] [PubMed] [Google Scholar]
- 113.Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, De Camilli P. 1998. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 394:793–797. doi: 10.1038/29555. [DOI] [PubMed] [Google Scholar]
- 114.Carbone R, Fré S, Iannolo G, Belleudi F, Mancini P, Pelicci PG, Torrisi MR, Di Fiore PP. 1997. eps15 and eps15R are essential components of the endocytic pathway. Cancer Res 57:5498–5504. [PubMed] [Google Scholar]
- 115.van Bergen en Henegouwen PM. 2009. Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell Commun Signal 7:24. doi: 10.1186/1478-811X-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Black ML. 1963. Sequential blockage as a theoretical basis for drug synergism. J Med Chem 6:145–153. doi: 10.1021/jm00338a014. [DOI] [PubMed] [Google Scholar]
- 117.Ricotta D, Hansen J, Preiss C, Teichert D, Höning S. 2008. Characterization of a protein phosphatase 2A holoenzyme that dephosphorylates the clathrin adaptors AP-1 and AP-2. J Biol Chem 283:5510–5517. doi: 10.1074/jbc.M707166200. [DOI] [PubMed] [Google Scholar]