

ABSTRACT
Previously, we demonstrated that the efficiency of hepatitis C virus (HCV) E2-p7 processing regulates p7-dependent NS2 localization to putative virus assembly sites near lipid droplets (LD). In this study, we have employed subcellular fractionations and membrane flotation assays to demonstrate that NS2 associates with detergent-resistant membranes (DRM) in a p7-dependent manner. However, p7 likely plays an indirect role in this process, since only the background level of p7 was detectable in the DRM fractions. Our data also suggest that the p7-NS2 precursor is not involved in NS2 recruitment to the DRM, despite its apparent targeting to this location. Deletion of NS2 specifically inhibited E2 localization to the DRM, indicating that NS2 regulates this process. Treatment of cells with methyl-β-cyclodextrin (MβCD) significantly reduced the DRM association of Core, NS2, and E2 and reduced infectious HCV production. Since disruption of the DRM localization of NS2 and E2, either due to p7 and NS2 defects, respectively, or by MβCD treatment, inhibited infectious HCV production, these proteins' associations with the DRM likely play an important role during HCV assembly. Interestingly, we detected the HCV replication-dependent accumulation of ApoE in the DRM fractions. Taking into consideration the facts that ApoE was shown to be a major determinant for infectious HCV particle production at the postenvelopment step and that the HCV Core protein strongly associates with the DRM, recruitment of E2 and ApoE to the DRM may allow the efficient coordination of Core particle envelopment and postenvelopment events at the DRM to generate infectious HCV production.
IMPORTANCE The biochemical nature of HCV assembly sites is currently unknown. In this study, we investigated the correlation between NS2 and E2 localization to the detergent-resistant membranes (DRM) and HCV particle assembly. We determined that although NS2's DRM localization is dependent on p7, p7 was not targeted to these membranes. We then showed that NS2 regulates E2 localization to the DRM, consistent with its role in recruiting E2 to the virus assembly sites. We also showed that short-term treatment with the cholesterol-extracting agent methyl-β-cyclodextrin (MβCD) not only disrupted the DRM localization of Core, NS2, and E2 but also specifically inhibited intracellular virus assembly without affecting HCV RNA replication. Thus, our data support the role of the DRM as a platform for particle assembly process.
INTRODUCTION
Hepatitis C virus (HCV) is a small, enveloped, positive-strand RNA virus belonging to the Flaviviridae family (1, 2). Globally, nearly 200 million people are infected with this virus, which causes severe morbidity and mortality due to its persistent replication in the liver (3, 4). HCV encodes a single open reading frame that is processed by host and viral proteases into three structural proteins, including Core and two envelope proteins (E1 and E2), and seven nonstructural proteins, including p7, NS2 and NS3-5B (NS3, NS4A, NS4B, NS5A, and NS5B) (5). The NS3-NS5B proteins are sufficient to form active replicase complexes involved in viral RNA replication (6). Interestingly, in addition to viral structural proteins, most of the nonstructural proteins were shown to affect HCV particle assembly (7–16). However, detailed mechanisms of HCV assembly are still unclear.
It was shown that the HCV Core protein associates with lipid droplets (LD) following its maturation cleavage by the signal peptide peptidase (17). Core association with the LD is critical for HCV particle assembly, since disrupting this also inhibited infectious HCV production (18, 19). NS5A protein was also shown to be targeted to the LD and interacts with Core and through this interaction promotes virus assembly, at least in part, by recruiting nonstructural proteins in replication complexes to LD-associated endoplasmic reticulum (ER) membranes, which are presumably virus assembly sites (20–22). Recently, we and others have shown that NS2 interacts with both structural and nonstructural proteins (11–13, 16, 23). These interactions correlate with NS2 colocalization with Core, NS5A, and E2 at the punctate virus assembly sites near LD and are important for infectious virus production (11–13, 16, 23). NS2 interactions with these viral proteins probably regulate envelope protein recruitment to the virus assembly sites, since mutations that inhibited NS2 localization to these sites also inhibited E2 localization to these sites (12, 16).
p7 is an ion channel protein. Its ion channel activity was shown to be critical for the release of infectious HCV by potentially protecting nascent virus particles during the maturation process while in transit through acidic intracellular compartments (24). p7 is also involved in the virus particle assembly process by modulating capsid assembly and envelopment of viral particles (25). Although detailed mechanisms of how p7 affects viral assembly have not been characterized, one of the mechanisms may involve p7's regulation of NS2's subcellular localization (10, 26). As we have shown recently, E2-p7 processing modulates infectious virus production by regulating NS2 localization to virus assembly sites, probably by controlling the release of p7 involved in this process (10). Interestingly, Tedbury and colleagues showed that the in-frame expression of p7 in the context of NS2-5B subgenomic replicon induced the NS2 subcellular localization to the punctate sites near replication complexes and detergent-insoluble fractions (DIF) (26). This subgenomic system precludes virus assembly due to the lack of structural proteins. Thus, these results suggest that p7-mediated NS2 subcellular localization change is a preparticle assembly event that may be a prerequisite for infectious virus production.
In aggregate, these previous findings suggest that p7-dependent NS2 and NS2-dependent E2 localization to the virus assembly sites regulates the HCV assembly process. Thus far, most HCV proteins, except p7 and NS2, were shown to associate with detergent-resistant membranes (DRM), including NS3-5B, whose DRM association appears to be critical for efficient HCV RNA replication (27–30). Currently, the role of DRM in the HCV assembly process is still unclear. However, its potential role in this process is supported by the following findings. First, all three structural proteins, including Core, E1, and E2, were shown to associate with the DRM (29, 30), and second, cholesterol and sphingolipid, two major components of DRM, are enriched in the HCV virion envelope and critical for HCV infectivity (29, 31). In this study, we investigated the DRM localization characteristics of virus assembly factors, including p7, NS2, and E2, in cells in which HCV replicates (HCV-replicating cells) and determined the relationship between their DRM localization and infectious virus production by using various well-characterized HCV mutants.
MATERIALS AND METHODS
Cell culture.
FT3-7 and Huh7.5 cells (Huh-7 cell derivatives [7, 32]) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA) at 37°C in 5% CO2 atmosphere.
Plasmids.
Construction of HJ3-5 and its derivatives, including p7(KRAA), E2(AR), E2(AR)/IRES, HAp7, and HAp7/IRES, were described previously (8, 10, 16). To generate Δp7 mutants, the p7 sequence from HJ3-5/p7/IRES (10) was deleted by using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) with primer pairs encoding the C-terminal sequence of E2 and the N-terminal sequence of encephalomyocarditis virus (EMCV) internal ribosomal entry site (IRES): 5′-CATATCCCAAGCGGAGGCGTGAGTTTAAACAGACCACAACGG-3′ and 5′-CCGTTGTGGTCTGTTTAAACTCACGCCTCCGCTTGGGATATG-3′, respectively. To generate ΔNS2 mutants, the NS2 sequence from HJ3-5/p7/IRES (10) was deleted by using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) with primer pairs encoding the C-terminal sequence of EMCV and the N-terminal sequence of NS3: 5′-GAAAAACAGGATAATACCATGGCGCCCATCACGGCGTACG-3′ and 5′-CGTACGCCGTGATGGGCGCCATGGTATTATCCTGTTTTTC-3′, respectively. The sequences of the regions manipulated within each plasmid were verified by DNA sequencing.
In vitro HCV RNA synthesis and transfection.
HCV RNA was transcribed from plasmid DNA and transfected into FT3-7 cells. Briefly, HCV-encoding plasmid DNA was linearized by using restriction enzyme XbaI (New England BioLabs, Ipswich, MA), then transcribed by using a T7 MEGAscript kit (Life Technologies, Carlsbad, CA), and subjected to DNase treatment for 15 min at 37°C followed by RNA purification by using the RNeasy kit (Qiagen). For electroporation, 10 μg of RNA was mixed with FT3-7 cells (1 × 106 cells in 500 μl) and electroporated by using a Gene Pulser system (Bio-Rad, Hercules, CA) once at 270 V and 950 μF in a 0.4-cm-gap width electroporation cuvette (Bio-Rad, Hercules, CA). The cells were then suspended in prewarmed medium and seeded.
Cell fractionations.
Cell fractionations to detergent-soluble and -insoluble fractions were performed according to the methods described previously (26). Briefly, cells grown on 6-well plates were trypsinized, washed twice with phosphate-buffered saline (PBS), and lysed in 200 μl of lysis buffer (1% Triton X-100, 120 mM KCl, 30 mM NaCl, 5 mM MgCl2, 10% glycerol [vol/vol]) supplemented with protease inhibitor cocktail (GenDEPOT, Barker, TX). After incubation on ice for 15 min, a detergent-soluble fraction (supernatant) was collected by centrifugation at 500 × g for 5 min at 4°C. The detergent-insoluble fraction (pellet) was washed twice in lysis buffer. The proteins in the detergent-soluble and -insoluble fractions were then analyzed by Western blotting.
Confocal microscopy.
Electroporated cells were plated on 8-well chamber slides (BD Biosciences, Bedford, MA) at a density of 1 × 104 cells per well. Two days later, the slides were washed with PBS, fixed with 4% formaldehyde for 20 min at room temperature, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Fixed and permeabilized cells were incubated with Huh-7 cell lysate cleared NS2 antibody (1:1,000 dilution) for 2 h at room temperature, followed by incubation with Alexa Fluor 488-conjugated, goat anti-rabbit antibody (Invitrogen, Carlsbad, CA) (1:1,000). Lipid droplets were stained with HCS LipidTOX deep red neutral lipid stain (1:1,000) (Molecular Probes Inc., Eugene, OR) for 30 min at room temperature. Nuclei were labeled with Hoechst stain (Anaspec Inc., Fremont, CA). The slides were examined with a Zeiss LSM 510 Meta laser-scanning confocal microscope.
Membrane flotation assay.
FT3-7 cells electroporated with HCV RNA were seeded on 60-mm dishes. At 48 h, cells were washed with ice-cold PBS and then scraped in 0.4 ml of TNE buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA) containing a protease inhibitor cocktail mix (GenDEPOT, Barker, TX). The cells were then disrupted by passing through a 24-gauge needle 30 times and incubated on ice or at 37°C in the absence or presence of 1% Triton X-100 for 30 min. The cell lysates were then mixed with 0.4 ml of Optiprep (Sigma, St. Louis, MO) to a final concentration of 40% and placed in an ultracentrifuge tube. This mixture was overlaid first with 1.2 ml of 30% Optiprep in cell suspension medium (CSM) (comprising 0.85% NaCl [wt/vol] in 10 mM HEPES [pH 7.4]) and then with 1.2 ml of 26% of Optiprep in CSM, followed by 0.8 ml of 6% Optiprep in CSM. The Optiprep gradient was centrifuged at 42,000 rpm in a Beckman Coulter Optima L-90K ultracentrifuge with an SW60 rotor for 4 h at 4°C. Following ultracentrifugation, 400-μl fractions were collected from the top of the gradient, and proteins in each fraction were precipitated after adding 100 μl of trichloroacetic acid (TCA) (100%, wt/vol). Subsequently, the precipitates were washed twice with cold acetone and solubilized in 2× sodium dodecyl sulfate (SDS) loading buffer (125 mM Tris-HCl [pH 6.8], 2% SDS, 0.005% bromophenol blue, 20% glycerol, 100 mM dithiothreitol [DTT]) before subjecting them to 12% SDS-polyacrylamide gel electrophoresis (PAGE), followed by Western blot analysis.
Western blot analysis.
Proteins separated on a 12% SDS-polyacrylamide gel were transferred to a polyvinylidene difluoride (PVDF) membrane and incubated in Odyssey blocking buffer (Li-COR Biosciences, Lincoln, NE) for 1 h at room temperature. The membranes were then probed at 4°C overnight with primary antibodies for Core (monoclonal mouse anti-Core antibody) (clone C7-50; Thermo Scientific) (1:2,000 dilution), NS2 (polyclonal rabbit anti-NS2 antibody [8]) (1:15,000 dilution), E2 (polyclonal goat anti-E2 antibody; Virostat, Inc., Portland, ME) (1:2,000 dilution), NS3 (monoclonal mouse anti-NS3 antibody) (clone 9-G2; ViroGen, Watertown, MA) (1:2,000 dilution), Calnexin (polyclonal rabbit anticalnexin antibody; Calbiochem) (1:5,000 dilution), Flotilin-1 (monoclonal mouse anti-flotilin-1; BD Transduction Laboratories) (1:2,000 dilution), SPFH2 (also known as Erlin-2 [endoplasmic reticulum lipid raft protein 2]; polyclonal goat anti-SPFH2 antibody, Santa Cruz Biotechnology Inc.) (1:2,000 dilution), and apolipoprotein E (ApoE) (polyclonal goat anti-ApoE antibody; Chemicon International) (1:3,000 dilution). The membranes were then washed three times in PBS supplemented with 0.1% Tween 20 and incubated for 1 h at room temperature with IRdye 800CW-labeled goat anti-mouse, IRdye 680-labeled goat anti-rabbit or IRdye 680-labeled donkey anti-goat antibodies (Li-COR Biosciences, Lincoln, NE), followed by imaging with an Odyssey infrared imaging system (Li-COR Biosciences, Lincoln, NE).
MβCD treatment.
At 48 h after electroporation of HCV RNA, cells grown on the 6-well plates were washed with PBS and preincubated with serum-free DMEM for 2 h and then incubated with DMEM containing methyl-β-cyclodextrin (MβCD) at 1, 5, or 10 mM concentrations for 30 min at 37°C in a 5% CO2 atmosphere. MβCD-treated cells were either directly subjected to membrane flotation analysis after cell lysis in the presence of 1% Triton X-100 as described above or subjected to virus titration and HCV RNA quantification as described below after incubating cells in complete medium (DMEM supplemented with 10% FBS) for 1 h.
Cholesterol quantification.
Cells were labeled with 2 μM TopFluor cholesterol (Avanti Polar Lipids, Alabaster, AL) for 12 h at 37°C in a 5% CO2 atmosphere. At the end of the labeling period, the cells were incubated with serum-free DMEM for 2 h. The cells were then either left untreated or treated with MβCD (10 mM) and harvested for membrane flotation analysis in the presence of cold Triton X-100 as described above. The fluorescence intensity of TopFluor cholesterol in the collected fractions was measured by the GloMax discover system (Promega, San Luis Obispo, CA) with 500- to 550-nm emission and 475-nm excitation filters.
Extracellular and intracellular virus titration.
For extracellular virus titration, culture supernatants of untreated and MβCD-treated FT3-7 cells were harvested and used to infect Huh7.5 cells on 48-well plates (seeded with 1 × 105 cells/well). After 72 h, cells were fixed with methanol-acetone (1:1) and then immunostained for HCV Core expression by using anti-HCV Core monoclonal antibody. The nuclei were stained by using Hoechst (Anaspec Inc., Fremont, CA). For intracellular virus titration, cells were trypsinized, resuspended in 1 ml medium, and lysed by 4 cycles of freeze-thawing. Clarified cell lysates were used to inoculate Huh7.5 cells. Infectivity was determined by counting the clusters of Core-immunostained cells (focus-forming unit [FFU]).
Quantitative real-time RT-PCR.
Viral RNA was detected by a quantitative TaqMan reverse transcription-PCR (RT-PCR) assay (16). Total RNA was isolated from cell lysates by using an RNeasy kit (Qiagen, Valencia, CA) in accordance with the manufacturer's instructions. Quantitative real-time TaqMan RT-PCR analysis was carried out in a Bio-Rad iQ5 real-time PCR detection system (Hercules, CA) by using primer pairs and a probe targeting a conserved 221-base sequence within the 5′ nontranslated RNA segment of the genome: HCV84FP (FP stands for forward primer), 5′-GCCATGGCGTTAGTATGAGTGT-3′; HCV303RP (RP stands for reverse primer), 5′-CGCCCTATCAGGCAGTACCACAA-3′; and HCV146BHQ (BHQ stands for black hole quencher), 5′-FAM-TCTGCGGAACCGGTGAGTACACC-DBH1-3′ (FAM stands for 6-carboxyfluorescein, and DBH1 stands for dual-labeled probe Black Hole Quencher 1). Reaction mixtures were incubated as follows: 50°C for 2 min, 60°C for 45 min, 95°C for 2 min, followed by 40 cycles, with 1 cycle consisting of 95°C for 20 s and 60°C for 1 min.
WST-1 assay.
Following MβCD treatment, cells were washed with PBS and incubated with WST-1 cell proliferation reagent (Roche, Indianapolis, IN) diluted 1:10 in culture medium and further incubated for 1 h at 37°C in 5% CO2 atmosphere. The absorbance at 440 nm was then measured by using POLARstar Omega microplate reader (BGM Labtech, Cary, NC).
Statistical analysis.
A Mann-Whitney test was performed by using GraphPad Prism version 6 software (GraphPad, La Jolla, CA) to determine whether the level of each protein detected at the DRM fractions is significantly higher than the background level of non-DRM protein Calnexin (see Fig. 2, 3, and 4). Student's t test (unpaired, with Welch's correction) was performed by using GraphPad Prism version 6 software (GraphPad, La Jolla, CA) to determine the significance in differences between paired values of nontreated and MβCD-treated samples (see Fig. 6). A P value of <0.05 was considered statistically significant.
FIG 2.

Detection of NS2 in the detergent-resistant membrane (DRM) fractions in wt HCV-replicating cells. (A and B) Membrane flotation assays were performed by using cell lysates harvested from cells electroporated with wt HCV RNA (A) and replication-defective GND mutant RNA (B) at 2 days postelectroporation. The results obtained in the presence or absence of cold 1% Triton X-100 are shown in the middle and left panels, respectively, and the results found in the presence of 1% Triton X-100 at 37°C are shown in the right panel. The DRM fractions (fraction 1 to 4) obtained in the presence of cold and 37°C Triton X-100 were designated DRM and DRM-37, respectively. The percentage of proteins detected in the membrane or DRM fractions is shown below each blot. ADRP, adipose differentiation-related protein. (C) Percentages of each protein from cells electroporated with wt HCV and GND mutant RNA detected in the DRM fractions from at least three different experiments. Proteins with values above the dashed line showed a significantly increased DRM association than that of non-DRM protein Calnexin (the asterisk indicates a P of <0.05 by the Mann-Whitney test).
FIG 3.

p7 mediates DRM localization of NS2 despite the lack of its own DRM association. (A, C, and D) Western blots following membrane flotation assay of cell lysates prepared in the presence of cold 1% Triton X-100 at day 2 postelectroporation of Δp7 (A), HAp7 (C), and HAp7/IRES (D). HAp7 encodes an in-frame hemagglutinin (HA) epitope tag at the N terminus of p7, facilitating the detection of p7 as described previously (10). HAp7/IRES encodes EMCV IRES between HAp7 and NS2 to eliminate the HAp7-NS2 precursor (10). The asterisk at the bottom of the NS2 blot from the Δp7 mutant indicates an additional band detectable by NS2 antibody (α-NS2) from this mutant, as indicated in the Fig. 1 legend. (B and E) Percentage of each protein from the DRM fractions of cells in which wt HCV (B) or Δp7 or wt HCV, HAp7, or HAp7/IRES (E) can replicate from at least three different experiments. Proteins with values above the dashed line showed a significantly increased DRM association compared to that of Calnexin (the asterisk indicates a P of <0.05 by the Mann-Whitney test).
FIG 4.

E2-p7 processing and NS2 regulate E2's association with the DRM. (A to C) Western blotting following membrane flotation assay of cell lysates prepared in the presence of cold 1% Triton X-100 at day 2 postelectroporation of E2(AR) (A), E2(AR)/IRES (B), and ΔNS2 (C). (D) Percentage of each protein from wt HCV, E2(AR), E2(AR)/IRES, and ΔNS2-replicating cells detected in the DRM fractions from at least three different experiments. Proteins with values above the dashed line showed significantly increased DRM association compared to that of Calnexin (the asterisk indicates a P of <0.05 by the Mann-Whitney test).
FIG 6.
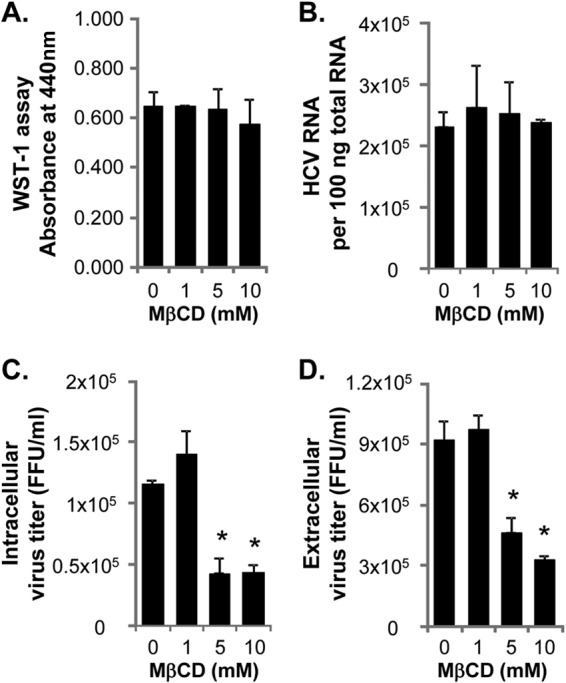
Effect of MβCD treatment on HCV RNA replication and virus production. (A to D) WST-1 assay to measure the cytotoxicity (A), TaqMan RT-PCR assay to measure HCV RNA replication (B), and virus titration assays to measure intracellular (C) and extracellular (D) virus titers were performed at 1 h posttreatment of wt-HCV-replicating cells with 10 mM MβCD for 30 min. Values that are statistically significantly different from the no-treatment values are indicated by an asterisk (P < 0.05 by unpaired Student's t test with Welch's correction).
RESULTS
Correlation between NS2 localization to the punctate virus assembly sites near LD and the DIF in HCV-replicating cells.
The results of our recent study suggested that p7 regulates NS2 localization to punctate sites near lipid droplets (punctate LD [see arrows in Fig. 1C]), which are considered putative virus assembly sites (10, 12, 13, 16, 20), and modulate virus production (10). However, the biochemical characteristics of these virus assembly sites are currently unknown. As mentioned above, a previous report showed that p7 targets NS2 to the detergent-insoluble fraction (DIF) in the context of HCV genotype 1b (gt1b) Con1 and gt2a JFH1 subgenomic replicons that lack viral structural proteins (26). Since NS2 localization to the DIF was also observed in infectious JFH1-replicating cells in the same study, we hypothesized that p7-dependent NS2 localization to the DIF and the virus assembly sites might be related. To test this hypothesis, we performed DIF fractionation experiments with highly infectious HCV (wild-type [wt] HCV, which is a gt1a-2a chimera HJ3-5 [8]) and its p7-defective mutants (Fig. 1).
FIG 1.

E2-p7 processing and p7-dependent NS2 subcellular localization to the detergent-insoluble fraction (DIF) in HCV-replicating cells. (A) Organization of wild-type (wt) HCV and various HCV mutants used in this study. Note that the H77 sequence is shaded within this chimeric HCV encoding gt1a H77 Core to NS2 within the gt2a JFH1 background. (B) Western blots of cold 1% Triton X-100 (1% TX-100) lysates separated into the detergent-soluble (S) and -insoluble (I) fractions. NS2 localization to the punctate foci near lipid droplets (LD) as described before (10) is indicated in the Punctate-LD row as follows: +++, ∼80% of NS2-positive cells with this phenotype; ++, ∼50% of NS2-positive cells with this phenotype; −, background level; N/A, not available. Virus titers were >2 × 10E5 FFU/ml (+++), >2 × 10E4 (++), and undetectable (−). The asterisk at the bottom of NS2 from Δp7 indicates the presence of additional bands detectable by NS2 antibody from this mutant and may represent an aberrant NS2 initiation product by EMCV IRES as described previously (16). (C) Confocal image analysis by using an Zeiss LSM 510 Meta laser-scanning confocal microscope of cells at day 2 postelectroporation with HCV RNA encoding the indicated genomes. Anti-NS2 antibody (green) and LipidTOX deep red neutral lipid stain (red) were used to detect NS2 and lipid droplets. Examples of the punctate-LD phenotype of NS2 are indicated by the white arrows.
The organizations of the HCV constructs used in this study are shown in Fig. 1A. Information regarding the virus production and NS2's punctate-LD localization characteristics of three HCV mutants that affected p7 release from the E2-p7 precursor and/or its ion channel function, including p7(KRAA), E2(AR), and E2(AR)/IRES, were described previously (10, 24) and summarized at the top of Fig. 1B. Briefly, the p7(KRAA) mutant was defective in p7's ion channeling function and in E2-p7 precursor cleavage (10, 24). The E2(AR) mutant also showed an E2-p7 processing defect that blocked p7 release from the E2-p7 precursor. As described in detail previously, these two mutants were defective in both infectious virus production and NS2's punctate-LD localization (Fig. 1B) (10). Restoring the p7 release from the E2(AR) mutant by inserting EMCV IRES between E2(AR) and p7, as in E2(AR)/IRES (Fig. 1A), had led to the restoration of virus production and NS2's punctate-LD localization (Fig. 1B) (10). In this study, we additionally generated a p7 deletion mutant named Δp7 to directly assess the role of p7 on NS2's subcellular localization (Fig. 1A). The Δp7 mutant was defective not only in infectious virus production (data not shown; Fig. 1B, top panel) but also in NS2's punctate-LD localization (Fig. 1C).
Following the fractionation of cell lysates in the presence of cold 1% Triton X-100, we verified that a portion of NS2 from infectious wt HCV was localized to the DIF (Fig. 1B) (10, 26). Further analyses using HCV mutants showed a remarkable correlation between infectious virus production and NS2's localization to the DIF and punctate LD (Fig. 1B). For example, we detected the lack of NS2 localization to the DIF and punctate LD from noninfectious p7(KRAA), E2(AR), and Δp7 mutants (Fig. 1B and C) (10). On the other hand, NS2 was clearly detectable in the DIF from the infectious E2(AR)/IRES mutant, which also showed the punctate-LD phenotype (Fig. 1B) (10).
Under the same conditions, we did not detect significant alteration of DIF fractionation characteristics of HCV Core, Flotilin-1 (a marker of detergent-resistant membranes [DRM]), and Calnexin (a non-DRM ER marker) in the wt and mutants (Fig. 1B). These results indicate that these p7-associated HCV mutants specifically affected NS2's DIF localization characteristics.
NS2 associates with the DRM.
The presence of significant levels of Flotilin-1 in the DIF suggests that this fraction contains proteins associated with the DRM. However, it is unlikely that all the proteins detected in the DIF are associated with the DRM, since detergent insolubility of the proteins could also be caused by protein aggregation or cytoskeletal binding (33). Therefore, to investigate whether NS2 is specifically targeted to the DRM, we performed membrane flotation analysis in the presence of cold detergent (1% Triton X-100) as described previously (34). First, in the absence of the detergent, we detected a majority of NS2 from wt HCV in the top fractions of the membrane flotation gradient (Fig. 2A, left panel). This result indicates that NS2 is associated with the membrane in HCV-replicating cells. Next, we detected ∼17% of NS2 in the DRM fractions following membrane flotation analysis in the presence of cold 1% Triton X-100 (Fig. 2A, middle panel, fractions 1 to 4). This level is significantly higher than the ∼5% background level of non-DRM protein Calnexin and rather close to the ∼18% level of ER-specific DRM protein Erlin-2 (endoplasmic reticulum lipid raft protein 2 [35, 36]) detected in the DRM fractions (Fig. 2A, middle panel, and C). However, only background levels of NS2 were detected at the DRM-37 fractions, which was prepared in the presence of 1% Triton X-100 at 37°C (Fig. 2A, right panel). These results indicate that a significant portion of NS2 is localized to the DRM in infectious HCV-replicating cells. We also detected the DRM localization of other viral proteins, including Core, E2, and NS3, as reported previously (Fig. 2A, middle panel, and C) (9, 14, 21, 22, 37, 38).
Interestingly, we detected an HCV replication-dependent accumulation of Apolipoprotein E (ApoE) in the DRM fractions (compare ApoE levels in the middle panels of Fig. 2A and B derived from wt HCV and replication-defective HCV mutant [GND] RNA electroporated cells, respectively). We also noted a significant increase of ApoE level in the membrane fractions obtained in the absence of any detergent in HCV-replicating cells (compare ApoE levels in the left panels of Fig. 2A and B), which is probably related to this phenotype. Meanwhile HCV replication had no effect on the subcellular localization of other host proteins, including two non-DRM proteins (Calnexin and Caveolin-1), two DRM proteins (Flotilin-1 and Erlin-2), and LD-associated adipose differentiation-related protein (ADRP) (compare Fig. 2A and B) (Fig. 2C).
Indirect role of p7 on NS2's DRM localization.
Consistent with the cell fractionation data shown in Fig. 1B, NS2 from the Δp7 mutant was defective in DRM localization (Fig. 3A and B). These results suggest that p7 plays a critical role in NS2 localization to the DRM. Interestingly, we also detected the lack of E2 localization to the DRM in this p7 deletion mutant. However, it is unlikely that p7 plays a major role in E2's DRM association, since NS2 deletion was sufficient to block E2's DRM association despite the presence of p7 (Fig. 4C). Instead, we speculate that E2's DRM localization defect in the Δp7 mutant was caused by defective NS2. The DRM associations of other viral and host proteins, including Core, NS3, ApoE, Erlin-2, and Calnexin, were not affected in the Δp7 mutant (Fig. 3A and B).
Since p7 and NS2 interacted with each other (16, 39) and also showed partial colocalization (39), we asked whether p7 recruits NS2 to the DRM by associating with the DRM. Due to the lack of a suitable antibody to detect p7, we used HCV encoding the hemagglutinin (HA) epitope-tagged p7 (HAp7-HCV) that we described previously to facilitate its detection (10). First of all, during our preliminary study, only background levels of HAp7 were detected in the DIF prepared from HAp7-HCV (data not shown). Consistent with this result, we detected ∼12% of NS2, but only background levels of HAp7, in the DRM fractions from HAp7-HCV (Fig. 3C and E). These results suggest that p7 does not directly recruit NS2 to the DRM by associating with the DRM. Interestingly, we also detected ∼8% of HAp7-NS2 precursor, which became prominently detectable in HCV encoding the epitope-tagged p7 (10, 39) in DRM fractions (Fig. 3C and E). To determine whether this precursor may have played a role in p7-mediated NS2 recruitment to the DRM, next we performed DRM fractionation experiments by using HAp7/IRES-HCV that encodes EMCV IRES at the junction of HAp7 and NS2 and therefore lacks HAp7-NS2 precursor formation as described previously (10). However, a lack of HAp7-NS2 precursor did not significantly inhibit NS2 localization to the DRM (Fig. 3D and E). This result is consistent with previous findings by Tedbury and colleagues in which they used a subgenomic replicon and showed that NS2 fractionated to the DIF regardless of the presence or absence of p7-NS2 precursor (26). In aggregate, these results suggested that p7 regulates NS2 localization to the DRM by an indirect mechanism that does not involve physical recruitment or p7-NS2 precursor formation. These results also suggest that p7 and NS2 interaction/colocalization likely occur at the non-DRM portion of ER. Then how could p7 have affected NS2 localization to the DRM? One of the potential mechanisms could be the p7-mediated NS2 conformational change. In support of this potential mechanism, p7 mutations altered the NS2 antibody-mediated immunofluorescence detection of NS2 following the differential permeabilization of HCV-replicating cells as well as NS2 pulldown efficiency (10, 16). Alternatively, p7 may have affected NS2 interaction with other proteins that promote NS2's association with DRM. Further study is necessary to determine the exact mechanism of p7-dependent NS2 subcellular localization change.
E2-p7 processing-dependent and NS2-dependent E2 association with the DRM.
Correlating with the lack of significant NS2 in the DIF in the E2-p7 processing-defective E2(AR) mutant (Fig. 1B) (10), we detected background levels of NS2 and E2 in the DRM fractions from this HCV mutant (Fig. 4A and D). On the other hand, reversing the E2-p7 processing defect in the E2(AR) mutant by introducing EMCV IRES between E2 and p7 [as in E2(AR)/IRES] also restored the DRM association levels of both NS2 and E2 similar to those of wt HCV (Fig. 4B and D) (10). The DRM associations of other viral and host proteins, including Core, NS3, ApoE, Erlin-2, and Calnexin, were not affected by either E2(AR) or E2(AR)/IRES mutants. These results imply that E2-p7 processing specifically regulates the DRM association of both E2 and NS2, most likely by controlling the release of p7 involved in this process (10). Interestingly, a previous study showed that E2 was targeted to the DRM whether it was expressed ectopically or in the context of infectious HCV (29). Thus, the defective DRM localization of E2 from E2(AR) could have been caused by the potential defect in the DRM association of E2-p7 precursor. Alternatively, it could have been caused by the defective DRM association of NS2 from this mutant that leads to inhibition of E2's DRM association, since NS2 was shown to regulate E2's subcellular localization (12, 16). To directly test the role of NS2 on the DRM association of E2, we next generated an NS2 deletion mutant (ΔNS2 [Fig. 1A]). As shown in Fig. 4C and D, a lack of NS2 specifically inhibited E2 localization to the DRM without significantly affecting DRM localization of other proteins. These results suggest that NS2 regulates E2's DRM localization.
Cholesterol-extracting agent specifically displaced Core, E2, and NS2 from the DRM and reduced infectious virus assembly.
Since the DRM are enriched in cholesterol, cholesterol depletion agents such as MβCD (methyl-β-cyclodextrin) have been used to disrupt the DRM to study its function (40, 41). However, although MβCD efficiently depleted cholesterol and displaced DRM proteins in the plasma membranes, it hardly affected the DRM association of intracellular DRM proteins such as Erlins and Flotilin-1 when used on intact cells (35, 41–43). We confirmed the latter phenotype upon 10 mM MβCD treatment for 30 min in living cells, as this treatment had no significant impact on the DRM localization of Erlin-2 and Flotilin-1 (Fig. 5A). To ensure that MβCD extracted the cholesterol from the DRM under our experimental conditions, we determined the cholesterol content of DRM fractions following 10 mM MβCD treatment. To facilitate quantification of the cholesterol, we labeled the cells with 2 μM TopFluor cholesterol for 12 h before MβCD treatment for 30 min (see Materials and Methods for details). The result shown in Fig. 5B indicates that MβCD treatment significantly reduced the cholesterol content of the DRM (fraction 3), while having little impact on that of detergent-soluble fractions (fractions 7 to 10). We also detected moderate reduction of cholesterol content from fraction 6, which likely corresponds to the high-density DRM (DRM-H) described previously (44) (Fig. 5B).
FIG 5.
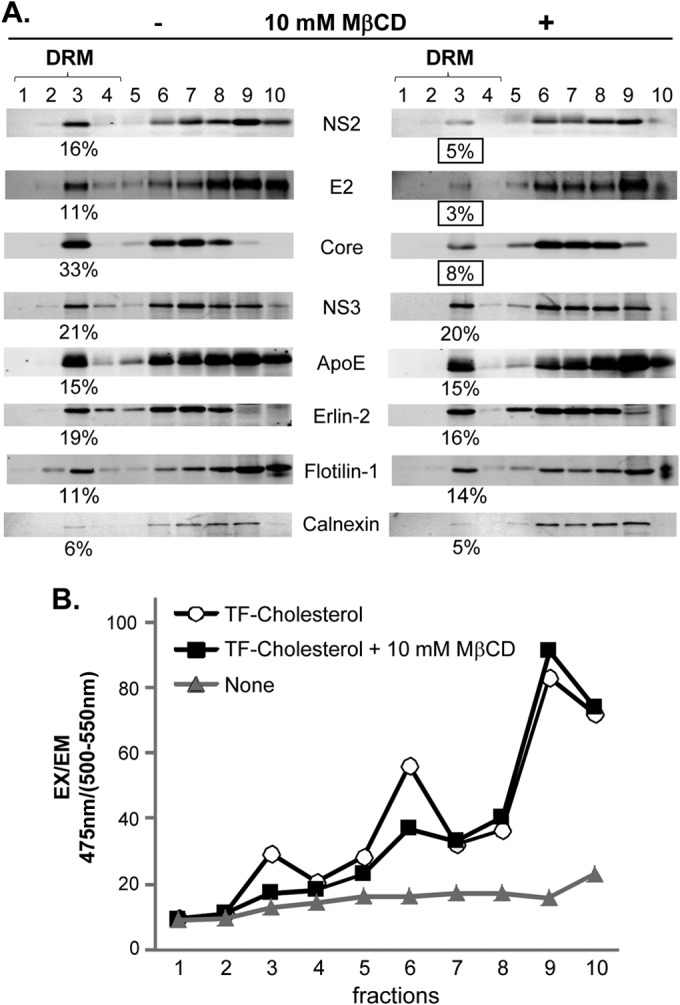
Effect of cholesterol-extracting agent MβCD treatment on viral and cellular protein association with the DRM. (A) Huh-7 cells replicating wt HCV were treated with 10 mM MβCD (+) for 30 min before being lysed in the presence of cold 1% Triton X-100 and subjected to membrane flotation analysis. (B) Cholesterol content of each fraction following membrane flotation analysis in the presence of cold 1% Triton X-100. Cells were labeled with or without 2 μM TopFluor cholesterol (TF-Cholesterol) for 12 h before treatment with 10 mM MβCD for 30 min before subjecting the cell lysates to membrane flotation analyses. The fluorescence was measured with a plate reader at 475-nm excitation (EX) and 500- to 550-nm emission (EM) setting.
A previous study showed that MβCD treatment disrupted the DRM association of ectopically expressed HCV Core protein despite its intracellular localization (30). We confirmed this phenotype with Core expressed from the infectious wt HCV genome following MβCD treatment (Fig. 5A, compare left and right blots). Interestingly, when the same MβCD treatment conditions were used, we detected a substantial reduction in the DRM association of NS2 and E2, but not NS3 and ApoE. These results suggest that the DRM association of Core, NS2, and E2 is especially sensitive to cholesterol content in the DRM compared to that of other proteins. Alternatively, these three viral assembly factors may be recruited to the specific DRM segments, possibly virus assembly sites, which are highly sensitive to MβCD treatment. To understand the significance of the DRM association of Core, NS2, and E2 during HCV replication, we determined the impact of MβCD treatment on HCV replication. We measured the level of viral RNA by using a quantitative RT-PCR assay and intra- and extracellular virus titers by performing fluorescent-focus formation assay 1 h after treatment with 1 to 10 mM MβCD for 30 min. As shown in Fig. 6A and B, MβCD treatment did not affect the cell viability and viral RNA replication under these experimental conditions. However, we observed a significant reduction in both intracellular and extracellular virus titers to a similar extent (Fig. 6C and D). These results suggest that the DRM association of Core, NS2, and E2 is critical for the virus particle assembly step rather than release.
DISCUSSION
Although the detailed mechanism of HCV assembly is unclear, LD and/or LD-associated ER membranes are implicated as playing critical roles in this process. Targeting of viral components involved in virus assembly to these membranes strongly suggest that these membranes correspond to HCV particle assembly sites. First, HCV Core protein associates with the LD upon its maturation cleavage (17). Second, NS5A protein is targeted to LD and interacts with Core (45). Third, the interaction between Core and NS5A on LD seems critical for infectious HCV production (21, 22), probably by recruiting replication complexes (20). Fourth, p7 mediates NS2 localization to these virus assembly sites by an unknown mechanism (10), and NS2 recruits envelope proteins to these sites by scaffolding the interaction between viral envelope proteins and nonstructural proteins (11–13, 16). In this study, we showed a strong correlation between NS2 localizations to the DRM and the putative virus assembly sites near LD (Fig. 1 to 4 and 7) (10, 16). We also showed the defective DRM association of NS2 and E2 in p7- or NS2-deleted HCV mutants that are defective in infectious virus production (Fig. 3 and 4). In addition, we showed that short-term treatment with MβCD specifically disrupted Core, NS2, and E2 association with the DRM and inhibited virus assembly (Fig. 5 and 6). These results suggest that HCV assembly process involves the DRM and that the association of Core, NS2, and E2 with the DRM promotes the virus assembly process (Fig. 7).
FIG 7.
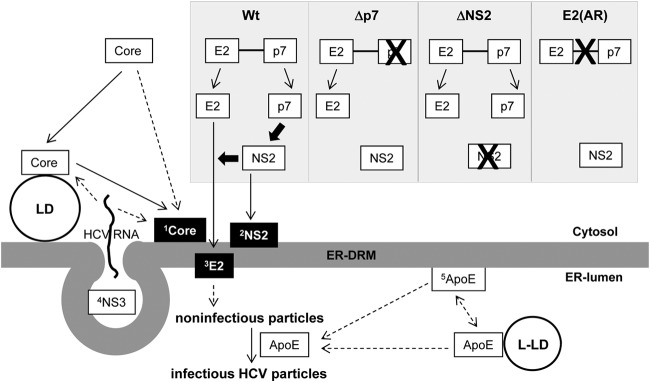
Model for the role of DRM targeting of viral and host proteins during HCV particle assembly. 1Core associates with the DRM (Fig. 1) (30, 34). Core could be targeted to the DRM directly or indirectly following its initial interaction with LD (11, 20, 53, 68). 2NS2 was targeted to the DRM in a p7 (and therefore E2-p7 processing)-dependent manner (Fig. 3A, 4A, and B). 3E2 was targeted to the DRM in an NS2-dependent manner (Fig. 4C). Defects in p7, NS2, or E2-p7 processing block both NS2 and E2 localization to the DRM and virus assembly (Fig. 1, 3, and 4). 4NS3 and other nonstructural proteins (NS4A, NS4B, NS5A, and NS5B) comprising the replication complexes (RC) were targeted to the DRM for HCV RNA replication (27, 28). RC are likely enclosed by membrane invaginations called membranous web (69–71). Positive-strand HCV RNA produced by RC will then interact with Core for encapsidation. Currently, it is unclear whether HCV RNA interacts with Core on the LD, at the DRM or both (50). Encapsidated Core will then interact with envelope proteins (72), and this interaction will likely trigger enveloped particle budding at the ER-DRM into the ER-lumen. 5ApoE incorporation to the enveloped particles (31, 58), potentially through its interaction with envelope proteins (60, 63), is critical for HCV infectivity (62, 64). We speculate that ER-DRM-enriched ApoE facilitates this process. However, further study will be needed to clarify this issue. Solid arrows indicate the pathways supported by experimental evidence from our data and previous literature. Dashed arrows indicate potential or undefined pathways. Thick black arrows indicate the driving forces for NS2 or E2 localization to the DRM. The selective MβCD sensitivity of DRM-associated Core, E2, and NS2 is indicated by a black background box. L-LD, luminal LD.
Our study revealed complex regulations governing E2's DRM localization. This begins with delayed E2-p7 processing that we reported previously (10). Once released from E2-p7 precursor cleavage, p7 will then trigger NS2's DRM localization (Fig. 3). Only NS2 capable of localizing to the DRM (with the help of p7) could promote E2's DRM localization (Fig. 3 and 4). If there was a defect in any of these steps, E2's DRM localization and infectious virus assembly were inhibited (Fig. 1, 3, 4, and 7). These results indicate that tight regulation of E2's DRM localization is critical for the virus assembly process. One of the purposes of this multistep regulation of E2's subcellular localization could be to ensure the late onset of the virus assembly process. As described previously, E2-p7 processing is intrinsically inefficient (10, 46–48). Due to this, the release of p7 and, consequently, p7-dependent NS2 localization to the virus assembly sites/DRM is expected to be delayed as well. In fact, it was shown previously that NS2 localization to the punctate virus assembly sites was minimal at the early phase of HCV replication but gradually increased over time with kinetics that correlate with increased virus production (12, 13). As NS2 regulates E2 subcellular localization, the delay in NS2 localization to the virus assembly sites/DRM should also delay E2 localization to these sites. On the basis of this information, we propose that the late onset of virus particle assembly, which was observed under one-step HCV growth conditions (49), is triggered at least in part by the delayed E2-p7 processing that would also delay the p7-mediated NS2 localization, which would, in turn, regulate the timing of NS2-mediated E2 localization to the virus assembly sites/DRM. On the other hand, a second purpose of this complex regulation of E2 subcellular localization could be to ensure the delivery of E2 near virus RNA replication sites (also localized at the DRM [27]) by utilizing NS2, which interacts with both viral replication proteins, including NS3 and NS5A, and envelope proteins E1 and E2 (11–13, 16). In other words, by viral scaffolding protein NS2 interacting with both envelope and nonstructural proteins, HCV may achieve delayed, but specific, recruitment of envelope proteins near the viral RNA replication sites at the late phase of viral replication cycle to promote effective assembly of actively replicating viral genome.
Currently, it is unclear whether initiation of the HCV assembly process begins with HCV RNA transfer from ER-associated replication complexes to LD-associated Core or Core transfer from LD to LD-associated ER membranes where actively replicating RNA is available (Fig. 7) (50). However, recent studies indicate that efficient virus production is likely associated with Core mobilization from LD to ER membranes. Boson and colleagues showed that the majority of Core from gt2a JFH1 colocalized with LD, while that from its derivatives, which produce much higher titers of virus than JFH1 does, is mainly detected from the ER (51). Their findings further indicate that p7 is sufficient to induce Core localization to the ER and that compatibility between p7 and NS2 regulates this process. Since newly synthesized HCV Core was shown to localize first to the LD even in high-titer HCV-infected cells by live-cell imaging analysis (52, 53), efficient mobilization of Core from LD to ER, rather than direct targeting of Core to ER following its translation, is responsible for Core's ER dominant localization phenotype observed during high-titer HCV replication. Since p7 expression-driven Core mobilization from LD to ER occurred even in the absence of HCV RNA replication (51), these results favor the possibility that Core particle assembly occurs at the LD-associated ER membranes rather than LD. However, more study is needed to prove this possibility. As for the sites of HCV particle envelopment, stronger evidence points to LD-associated ER membranes. Not only do HCV envelope proteins associate with the ER membranes (54) rather than with LD, but in fact, HCV-like particle budding was observed from the LD-associated membranes by electron microscopy (20, 55). Thus, our results of NS2-dependent E2 association with the DRM (Fig. 4C), which correlate with NS2-regulated E2 localization to virus assembly sites (12, 16), may imply that HCV particle envelopment occurs at the LD-associated ER DRM. To support this notion, the HCV virion is enriched with two major lipid components of the DRM, cholesterol and sphingolipid, which were shown to play critical roles in HCV infectivity (29, 31). Also, it is interesting that the localization of Core, NS2, and E2 to the DRM was equally sensitive to short-term treatment with MβCD, which did not significantly disrupt the DRM localization of other viral and host proteins (Fig. 5). These results may indicate that these three viral proteins are targeted to the same DRM compartment that is potentially more sensitive to cholesterol extraction than other DRM compartments in the cells (Fig. 7). If this is the case, this particular DRM compartment could serve as an ideal location for coordinated Core-particle assembly and envelopment. In aggregate, these data support the potential role of the DRM as the platform of HCV assembly and budding. However, more study is necessary to prove this notion.
ApoE is an essential host factor involved in infectious virus production (56, 57). ApoE incorporates onto HCV particles (31, 58) and facilitates viral entry (59–62). Interestingly, HCV E1/E2 complexes were shown to interact with ApoE and colocalize with it only in the ER (63). Since it was shown that the ectodomain of E1, which localizes to the ER lumen, could interact with ApoE (60), the interaction of ApoE with HCV E1/E2 complexes likely occurs at the ER lumen. Therefore, it is reasonable to predict that the interaction of ApoE and E1/E2 complexes in the ER lumen contributes to ApoE incorporation into the virus particles. Interestingly, while ApoE deficiency did not prevent HCV particle envelopment, this condition inhibited infectious virus production, secretion of noninfectious virus particles, and cell-to-cell transmission (64). These results suggest that ApoE contributes to HCV particle assembly at a postenvelopment step (Fig. 7). However, despite the fact that E1/E2 complexes could interact with ApoE even in the absence of other viral proteins (63), this interaction alone may not be sufficient to promote an ApoE-dependent HCV particle assembly event, since previous reports showed that the interaction between NS5A and ApoE is the major determinant of infectious HCV assembly (65, 66). It is interesting that ApoE localizes to the DRM in a HCV RNA replication-dependent manner regardless of E2's DRM localization, since wt level of ApoE was detected in the DRM fractions of HCV mutants that are defective in infectious virus production and E2's DRM localization (Fig. 2 and 4). Considering that NS5A interacts with ApoE and was shown to associate with the DRM whether it was expressed singly in the context of the subgenomic replicon or infectious virus (28, 67), NS5A is likely responsible to recruit ApoE to the DRM. This will then allow the interaction of ApoE and E1/E2 complexes at the DRM, promoting infectious virus assembly. We plan to test this hypothesis in detail in our future studies.
In conclusion, our data indicate that p7-dependent NS2 localization to the DRM correlates with NS2 localization to the putative virus assembly sites near LD and infectious virus production. Our data further indicate that NS2 is responsible for recruiting E2 to the DRM, which correlates with previous reports that suggested NS2-mediated E2 recruitment to the virus assembly sites (12, 16). The similar sensitivities of DRM-localized Core, NS2, and E2 to MβCD treatment suggest the possibility that these viral assembly factors target the same DRM. Since host-derived virus assembly factor ApoE was also detected at the DRM in HCV-replicating cells, we propose that DRM may serve as the HCV particle assembly platform that allows effective coordination of Core particle envelopment and the ApoE-mediated postenvelopment process.
ACKNOWLEDGMENTS
This work is supported by a JSME pilot grant from the University of Texas Medical Branch (UTMB) and grant R01-AI075090 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
We thank Charles Rice and Takaji Wakita for providing Huh7.5 cells and the JFH1 cDNA clone, respectively. We also thank Mardelle Susman for careful reading of the manuscript.
REFERENCES
- 1.Houghton M, Weiner A, Han J, Kuo G, Choo QL. 1991. Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology 14:381–388. doi: 10.1002/hep.1840140227. [DOI] [PubMed] [Google Scholar]
- 2.Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, Netesov S, Nishioka K, Shin IT, Simmonds P, Smith D, Stuyver L, Weiner A. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol 143:2493–2503. [DOI] [PubMed] [Google Scholar]
- 3.Rein DB, Wittenborn JS, Weinbaum CM, Sabin M, Smith BD, Lesesne SB. 2011. Forecasting the morbidity and mortality associated with prevalent cases of pre-cirrhotic chronic hepatitis C in the United States. Dig Liver Dis 43:66–72. doi: 10.1016/j.dld.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 4.McCombs J, Matsuda T, Tonnu-Mihara I, Saab S, Hines P, L'Italien G, Juday T, Yuan Y. 2014. The risk of long-term morbidity and mortality in patients with chronic hepatitis C: results from an analysis of data from a Department of Veterans Affairs Clinical Registry. JAMA Intern Med 174:204–212. doi: 10.1001/jamainternmed.2013.12505. [DOI] [PubMed] [Google Scholar]
- 5.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 6.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 7.Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J Virol 81:629–638. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi M, Ma Y, Yates J, Lemon SM. 2009. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog 5:e1000403. doi: 10.1371/journal.ppat.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol 82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shanmugam S, Yi M. 2013. Efficiency of E2-p7 processing modulates production of infectious hepatitis C virus. J Virol 87:11255–11266. doi: 10.1128/JVI.01807-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 85:1706–1717. doi: 10.1128/JVI.02268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog 6:e1001233. doi: 10.1371/journal.ppat.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popescu CI, Callens N, Trinel D, Roingeard P, Moradpour D, Descamps V, Duverlie G, Penin F, Heliot L, Rouille Y, Dubuisson J. 2011. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog 7:e1001278. doi: 10.1371/journal.ppat.1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gouklani H, Bull RA, Beyer C, Coulibaly F, Gowans EJ, Drummer HE, Netter HJ, White PA, Haqshenas G. 2012. Hepatitis C virus nonstructural protein 5B is involved in virus morphogenesis. J Virol 86:5080–5088. doi: 10.1128/JVI.07089-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinmann E, Pietschmann T. 2010. Hepatitis C virus p7—a viroporin crucial for virus assembly and an emerging target for antiviral therapy. Viruses 2:2078–2095. doi: 10.3390/v2092078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J Virol 85:86–97. doi: 10.1128/JVI.01070-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLauchlan J, Lemberg MK, Hope G, Martoglio B. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J 21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulant S, Targett-Adams P, McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88:2204–2213. doi: 10.1099/vir.0.82898-0. [DOI] [PubMed] [Google Scholar]
- 19.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Ott M. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med 16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 21.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol 82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. 2010. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog 6:e1001087. doi: 10.1371/journal.ppat.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog 9:e1003355. doi: 10.1371/journal.ppat.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tedbury P, Welbourn S, Pause A, King B, Griffin S, Harris M. 2011. The subcellular localization of the hepatitis C virus non-structural protein NS2 is regulated by an ion channel-independent function of the p7 protein. J Gen Virol 92:819–830. doi: 10.1099/vir.0.027441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MMC. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J Virol 77:4160–4168. doi: 10.1128/JVI.77.7.4160-4168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao L, Aizaki H, He JW, Lai MMC. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78:3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MMC, Miyamura T, Wakita T, Suzuki T. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol 82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matto M, Rice CM, Aroeti B, Glenn JS. 2004. Hepatitis C virus core protein associates with detergent-resistant membranes distinct from classical plasma membrane rafts. J Virol 78:12047–12053. doi: 10.1128/JVI.78.21.12047-12053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merz A, Long G, Hiet MS, Brügger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor CM, Coetzee T, Pfeiffer SE. 2002. Detergent-insoluble glycosphingolipid/cholesterol microdomains of the myelin membrane. J Neurochem 81:993–1004. doi: 10.1046/j.1471-4159.2002.00884.x. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto K, Mori Y, Komoda Y, Okamoto T, Okochi M, Takeda M, Suzuki T, Moriishi K, Matsuura Y. 2008. Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J Virol 82:8349–8361. doi: 10.1128/JVI.00306-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Browman DT, Resek ME, Zajchowski LD, Robbins SM. 2006. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci 119:3149–3160. doi: 10.1242/jcs.03060. [DOI] [PubMed] [Google Scholar]
- 36.Browman DT, Hoegg MB, Robbins SM. 2007. The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol 17:394–402. doi: 10.1016/j.tcb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J Virol 83:2163–2177. doi: 10.1128/JVI.01885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vieyres G, Brohm C, Friesland M, Gentzsch J, Wölk B, Roingeard P, Steinmann E, Pietschmann T. 2013. Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J Virol 87:1664–1678. doi: 10.1128/JVI.02782-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang XQ, Paller AS. 2006. Lipid rafts: membrane triage centers. J Invest Dermatol 126:951–953. doi: 10.1038/sj.jid.5700282. [DOI] [PubMed] [Google Scholar]
- 41.Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. 2003. Resistance of cell membranes to different detergents. Proc Natl Acad Sci U S A 100:5795–5800. doi: 10.1073/pnas.0631579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostermeyer AG, Beckrich BT, Ivarson KA, Grove KE, Brown DA. 1999. Glycosphingolipids are not essential for formation of detergent-resistant membrane rafts in melanoma cells. Methyl-beta-cyclodextrin does not affect cell surface transport of a GPI-anchored protein. J Biol Chem 274:34459–34466. [DOI] [PubMed] [Google Scholar]
- 43.Chen X, Resh MD. 2002. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J Biol Chem 277:49631–49637. doi: 10.1074/jbc.M208327200. [DOI] [PubMed] [Google Scholar]
- 44.Nebl T, Pestonjamasp KN, Leszyk JD, Crowley JL, Oh SW, Luna EJ. 2002. Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. J Biol Chem 277:43399–43409. doi: 10.1074/jbc.M205386200. [DOI] [PubMed] [Google Scholar]
- 45.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MMC. 2002. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 292:198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 46.Lin C, Lindenbach BD, Pragai BM, McCourt DW, Rice CM. 1994. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J Virol 68:5063–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carrère-Kremer S, Montpellier C, Lorenzo L, Brulin B, Cocquerel L, Belouzard S, Penin F, Dubuisson J. 2004. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J Biol Chem 279:41384–41392. doi: 10.1074/jbc.M406315200. [DOI] [PubMed] [Google Scholar]
- 48.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keum SJ, Park SM, Park JH, Jung JH, Shin EJ, Jang SK. 2012. The specific infectivity of hepatitis C virus changes through its life cycle. Virology 433:462–470. doi: 10.1016/j.virol.2012.08.046. [DOI] [PubMed] [Google Scholar]
- 50.Bartenschlager R, Penin F, Lohmann V, André P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Boson B, Granio O, Bartenschlager R, Cosset FL. 2011. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog 7:e1002144. doi: 10.1371/journal.ppat.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog 7:e1002302. doi: 10.1371/journal.ppat.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog 8:e1002466. doi: 10.1371/journal.ppat.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cocquerel L, Wychowski C, Minner F, Penin F, Dubuisson J. 2000. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J Virol 74:3623–3633. doi: 10.1128/JVI.74.8.3623-3633.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roingeard P, Hourioux C, Blanchard E, Prensier G. 2008. Hepatitis C virus budding at lipid droplet-associated ER membrane visualized by 3D electron microscopy. Histochem Cell Biol 130:561–566. doi: 10.1007/s00418-008-0447-2. [DOI] [PubMed] [Google Scholar]
- 56.Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol 84:12048–12057. doi: 10.1128/JVI.01063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A 110:9505–9510. doi: 10.1073/pnas.1307527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang J, Wu X, Tang H, Luo G. 2013. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS One 8:e67982. doi: 10.1371/journal.pone.0067982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazumdar B, Banerjee A, Meyer K, Ray R. 2011. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54:1149–1156. doi: 10.1002/hep.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Owen DM, Huang H, Ye J, Gale M. 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol 86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boyer A, Dumans A, Beaumont E, Etienne L, Roingeard P, Meunier JC. 2014. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J Biol Chem 289:18904–18913. doi: 10.1074/jbc.M113.538256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wolk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol 88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol 84:11532–11541. doi: 10.1128/JVI.01021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53. doi: 10.1002/hep.23278. [DOI] [PubMed] [Google Scholar]
- 67.Oakland TE, Haselton KJ, Randall G. 2013. EWSR1 binds the hepatitis C virus cis-acting replication element and is required for efficient viral replication. J Virol 87:6625–6634. doi: 10.1128/JVI.01006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J Biol Chem 282:37158–37169. doi: 10.1074/jbc.M707329200. [DOI] [PubMed] [Google Scholar]
- 69.Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J Biol Chem 278:50301–50308. doi: 10.1074/jbc.M305684200. [DOI] [PubMed] [Google Scholar]
- 70.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakai K, Okamoto T, Kimura-Someya T, Ishii K, Lim CK, Tani H, Matsuo E, Abe T, Mori Y, Suzuki T, Miyamura T, Nunberg JH, Moriishi K, Matsuura Y. 2006. Oligomerization of hepatitis C virus core protein is crucial for interaction with the cytoplasmic domain of E1 envelope protein. J Virol 80:11265–11273. doi: 10.1128/JVI.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]