

ABSTRACT
The Bunyamwera (BUNV) orthobunyavirus NSs protein has proven a challenge to study in the context of viral infection. NSs is encoded in a reading frame that overlaps that of the viral nucleocapsid (N) protein thus limiting options for mutagenesis. In addition, NSs is poorly immunogenic, and antibodies only work in certain techniques while the protein itself is subject to proteasomal degradation. In order to generate a virus that expresses NSs independently of N, an ambisense S RNA segment was designed by mutating the 5′- and 3′-terminal nucleotide sequences. These mutations were previously shown to alter promoter activity so that both replication and transcription were promoted from both the genome and the antigenome RNAs (J. N. Barr et al., J. Virol. 79:12602–12607, 2005). As proof of principle, a recombinant BUNV was created that expressed green fluorescent protein (GFP) in the ambisense orientation. GFP expression was detected throughout at least 10 passages. Recombinant BUNV encoding epitope-tagged versions of NSs in the ambisense orientation expressed NSs via a subgenomic mRNA, and two viruses grew to titers only modestly lower than parental rBUNdelNSs2 virus. The ambisense viruses were temperature sensitive, and NSs was shown to localize to both the nucleus and the cytoplasm during infection. These viruses will be useful in further studies on structure-function relationships of the orthobunyavirus NSs protein.
IMPORTANCE Bunyamwera virus (BUNV) is the type species and model system for both the family Bunyaviridae and the genus Orthobunyavirus, a group that includes many significant human and animal pathogens. Studying the basic molecular biology of these viruses is of great importance to underpin research into vaccines and antivirals. We demonstrate here the plasticity of the BUNV genome by generating recombinant viruses where the normal negative-sense S segment has been converted into an ambisense segment, allowing independent expression of either a foreign gene (green fluorescent protein) or the viral nonstructural NSs protein. These new reagents will allow detailed investigation of NSs, the orthobunyavirus interferon antagonist.
INTRODUCTION
Bunyavirus genomes consist of three RNA segments called large, medium, and small (L, M, and S, respectively) that encode four structural proteins (RNA polymerase or L protein; two glycoproteins, Gn and Gc; and nucleocapsid protein, N), as well as one or two nonstructural proteins (NSm and NSs). Each genome segment has the same basic structure with the coding region flanked by untranslated regions (UTRs) at the 5′ and 3′ ends. Although bunyaviruses are classed as negative-strand viruses, some viruses have genome segments with an ambisense coding strategy. Phleboviruses, for instance, have S genome segments in which the N open reading frame (ORF) is in the negative sense, whereas the NSs ORF is in the positive sense (1–3).
Bunyavirus genomes have been shown to be remarkably flexible. The UTRs of the three segments can be exchanged (4) or can be drastically shortened (5, 6), the ORFs within an ambisense segment can be swapped around (7), the genomes can be lengthened through insertions of epitope tags and additional ORFs (8, 9), and the tripartite genome can even be converted into two-segmented or four-segmented versions (10, 11).
The promoters for replication and transcription of the genome segments are located in the terminal sequences of the UTRs that are largely complementary and form panhandle structures (9, 12, 13). The promoters of both the genomic and antigenomic RNAs of the ambisense Rift Valley phlebovirus S segment direct both replication and transcription. In contrast, the Bunyamwera virus (BUNV) S segment genomic RNA promoter drives both replication to synthesize antigenomic RNA and transcription to generate the N mRNA, whereas the antigenomic S segment promoter drives only replication. In vitro studies have shown that the BUNV termini can be mutated such that both the genome and the antigenome panhandles promote both replication and transcription (14), potentially allowing for the BUNV S segment to have an ambisense coding strategy.
The BUNV NSs protein has proved a difficult target for study. It is small (11 kDa), very hydrophobic, and poorly immunogenic, and it is rapidly degraded during infection (15). Most of the current knowledge about its function is derived from studies using an NSs deletion virus, rBUNdelNSs, which showed that NSs is the viral interferon (IFN) antagonist (16, 17). This antagonism is achieved by a general block of host cell transcription caused by NSs inhibiting RNA polymerase II (RNAPII) activity (18), probably through its interaction with Med8 (19). Transiently expressed tagged NSs was shown to be present in both the cytoplasm and the nucleus by immunofluorescent staining (18, 20). Proteasomal degradation of NSs could be prevented by mutation of its four lysine residues to arginine residues in a recombinant virus called rBUN4KR (15). The increased accumulation of NSs in cells infected with rBUN4KR made the protein easier to detect, while the virus seemed to suffer no adverse affect from these mutations (15).
The biggest challenge for functional studies of BUNV NSs is posed by its expression strategy. The NSs open reading frame (ORF) is fully encompassed by the N protein ORF and is translated from the same mRNA as N protein as the result of alternate AUG initiation codon usage by scanning ribosomes (21). Therefore, only limited mutagenesis of NSs is possible without affecting the sequence of the overlapping N protein. In order to be able to study NSs localization in the context of virus infection and to allow future investigation of interacting partners of NSs, a mutant virus is required in which NSs can be affinity tagged. The studies by Barr et al. (14) suggest that the N and NSs ORFs could be expressed by an ambisense coding arrangement which would enable epitope-tagging or other mutagenesis of NSs. In addition, the degradation-resistant mutant NSs sequence (4KR) could be used to enhance expression levels. Here we report the creation of recombinant BUNV with an ambisense S segment. As proof of principle, we generated a virus expressing green fluorescent protein (GFP) from the opposite orientation to the N protein, followed by the creation of viruses in which the GFP ORF was replaced by epitope-tagged versions of NSs.
MATERIALS AND METHODS
Cells and viruses.
Aedes albopictus C6/36, Vero-E6, BHK-21, BSR-T7/5 (22; kindly provided by K. K. Conzelmann), 2fTGH, A549, and A549-V and A549-NPro (15, 23; the latter two lines both kindly provided by R. Randall) were maintained as described previously (15, 19, 23, 24, 25). Wild-type (wt) BUNV and rBUNdelNSs2 working stocks were prepared as described previously following plaque purification (19). Cells were incubated at 33°C for virus rescue attempts and for producing virus working stocks in order to minimize formation of defective interfering particles. Of the newly rescued viruses described here, AmbiGFP was the only one for which plaques could be picked. The other recombinant ambisense viruses were not plaque purified, but instead supernatants from transfected BSR-T7/5 cells were used as elite stocks. The S segments of the working stocks of all ambisense viruses were sequenced to confirm the mutations in the panhandle, as well as the presence of intact ORFs.
Plasmids.
Plasmids containing full-length cDNAs of BUNV genome segments—pT7ribo-BUNL(+), pT7ribo-BUNM(+), pT7ribo-BUNS(+), and pT7ribo-BUNNdelORF2—have been described previously (26, 27). The ambisense S segments were constructed by the duplication of nucleotides (nt) 889 to 911 (antigenome sense) in pT7ribo-BUNNdelORF2 and inserting in between this duplication a sequence that in the genome sense contained an ORF insertion region (a 5′ NcoI site followed by a linker and a 3′ EagI site), followed by the 3′ UTR sequence of the M segment (Fig. 1B). In addition to these modifications, the termini of the segment were mutated to match the termini of construct AG-CB(25/25)-U/A described by Barr et al. (14) to allow ambisense transcriptional activity. This basic construct [BunSAmbi(M)] was synthesized commercially (GenScript Corp.) and subcloned into the pT7ribo vector, followed by insertion of the GFP ORF to create pT7ribo-AmbiGFP.
FIG 1.

Design of ambisense constructs. (A) Schematic of the wild-type BUNV panhandle in genome and antigenome sense, and the ambisense panhandle as published by Barr et al. (14). The mutated nucleotides in the ambisense panhandles are indicated by lowercase letters. (B) Diagram of the different S segments described in the present study. The mutated termini are indicated with an asterisk (*). The wild-type 3′ UTR (antigenome sense) is arbitrarily divided into regions W, X, Y, and Z (the nucleotide coordinates of these regions are described in the text). Regions X and Y contain sequences that have been reported to be involved in transcription termination of the N mRNA.
The scrambled-codon Flag-tagged NSs sequence (see Results) was also synthesized by GenScript. The GFP ORF in the ambisense construct pT7ribo-AmbiGFP was replaced by the Flag-NSs sequence to yield pT7ribo-AmbiFlNSs. Subsequently, the NSs sequence was mutated by QuikChange PCR to the NSs4KR sequence as described previously (15) to yield pT7ribo-AmbiFl4KR, and finally the Flag tag was replaced by a V5 tag (13) to yield pT7ribo-AmbiV54KR.
Generation of recombinant viruses from cloned cDNA.
Viruses were rescued as described previously (15). In brief, subconfluent BSR-T7/5 cells were transfected with pT7riboBUNL(+), pT7riboBUNM(+), and the appropriate S segment expressing construct (1 μg each per 60-mm dish) using Lipofectamine 2000 (9 μl per 60-mm dish). Supernatant fluids were harvested after 6 days incubation at 33°C. The progress of infection of virus expressing GFP was monitored by UV microscopy using an EVOS FL cell imaging system (Advanced Microscopy Group/Invitrogen). The progress of infection of the other viruses was monitored by observing the cytopathic effects (CPE).
Virus titration by plaque- and focus-forming assays.
The plaque or focus phenotype of viruses and the titers of growth curve and passaging assay samples were determined by plaque or focus-forming assay on BHK-21 cells or other cell types as indicated. Cells were infected with serial 10-fold dilutions of the samples, and after 1 h of incubation at 37 or 33°C, as indicated, an overlay was applied. The assays (see results in Fig. 2A and B) used an overlay of 0.6% agarose and 2% newborn calf serum (NCS) in modified Eagle medium (MEM), and samples were incubated for 8 days (5 days for wt BUNV). The assays depicted in Fig. 2C used an overlay of 0.6% Avicel and 2% NCS in MEM and were incubated for 6 days (3 days for wt BUNV). The cells were fixed in 4% formaldehyde for at least 1 h before staining with Giemsa stain or detection with anti-BUNV N antibodies as described previously (6).
FIG 2.

Plaque- and focus-forming phenotypes of ambisense viruses. Monolayers of BHK-21 cells infected with wt BUNV, AmbiGFP, AmbiFlNSs, AmbiFl4KR, or AmbiV54KR viruses as indicated. (A and B) The cells were incubated under an agarose overlay at 37 or 33°C as indicated, for 5 days for wt BUNV and for 8 days for the ambisense viruses. (A) Giemsa-stained plaques; (B) immunostained foci. (C) The cells were incubated under an Avicel overlay at 33°C for 6 days before immunostaining of foci.
Biological assay for IFN production.
A549 or 2fTGH cells in 35-mm dishes were infected at 2 PFU/cell or 2 focus-forming units (FFU)/cell, as appropriate, and incubated at 33°C for 48 h. After UV inactivation of the virus in the supernatant, the interferon (IFN) content was determined as described previously (15). Briefly, A549-Npro cells were treated with serial 2-fold dilutions of the supernatant for 24 h, and then the cells were infected with encephalomyocarditis virus (EMCV) and incubated for 5 days. The cells were fixed and stained with Giemsa, and the destruction or not of the cell monolayer was recorded. The relative IFN units are expressed as 2N, where N is the number of 2-fold dilutions that protect the reporter cells from EMCV infection.
Passage of virus.
T25 flasks of BHK-21 cells were infected initially with approximately 5,000 FFU of AmbiGFP, AmbiFlNSs, AmbiFl4KR, or AmbiV54KR virus. The supernatant was harvested when clear signs of CPE were observed. Subsequent passages were performed “blindly” (i.e., without titrating the viruses): new flasks were infected with a fixed volume (10 μl) virus per flask and harvested when a CPE was observed. The supernatants were collected and stored at −80°C.
Indirect immunofluorescent staining.
For immunofluorescence studies of NSs protein localization low-confluence BHK-21 cells grown on glass coverslips were infected at 5 FFU per cell, fixed at different times after infection, and analyzed as described previously (7). Proteins were detected using rabbit anti-BUNV N (19), mouse anti-V5 (kindly provided by R. Randall), or mouse anti-Flag (Sigma, catalog no. F3165), goat anti-rabbit antibody conjugated with fluorescein isothiocyanate (FITC; Sigma, catalog no. F9887), and goat anti-mouse antibody conjugated with Cy5 (Chemicon International, catalog no. AP1245).
Viral protein synthesis.
Detection of proteins by Western blotting was performed as described previously (25) using the following antibodies: mouse anti-tubulin (1:5,000; Sigma, catalog no. T5168), rabbit anti-BUNV N (1:5,000) (19), rabbit anti-NSs (1:500) (27), mouse anti-V5 (1:2,000), rabbit anti-Flag (1:2,000; Sigma, catalog no. F7425), anti-rabbit IgG-HRP conjugate (1:5,000; Cell Signaling, catalog no. 7074S), anti-mouse IgG peroxidase conjugate (1:5,000; Cell Signaling, catalog no. 7076). For metabolic labeling, 30 μCi per well of [35S]-EasyTag Express 35S-protein labeling mix in methionine/cysteine-free Dulbecco modified Eagle medium was added, and incubation was continued for 2 h before harvest. After SDS-PAGE, the gels were fixed and dried prior to phosphorimaging.
Northern blotting.
For RNA analysis, infected cells were harvested in TRIzol reagent (Invitrogen, catalog no. 15596), followed by RNA extraction with chloroform and subsequent isopropanol precipitation. Total RNA from a 35-mm well was resuspended in 30 μl of sterile H2O, which would typically give yields in the range of 1 to 2 μg/μl, and 4 μg was used per lane for Northern blot analysis as described previously (15).
For riboprobes to distinguish N mRNA from NSx mRNA, unique N and scrambled-NSs sequences were PCR amplified from plasmid pT7ribo-AmbiFlNSs with one of the primers containing the T7 promoter sequence. After electrophoresis, PCR products of the expected sizes (ca. 450 to 500 nt) were purified from an agarose gel, and digoxigenin (DIG)-labeled riboprobes were produced using DIG labeling mix (Roche) with T7 RNA polymerase (Promega) according to the manufacturer's instructions.
Nucleotide sequencing.
To determine the viral S segment genome sequences, virions were pelleted from 1 ml of supernatant by centrifugation in a benchtop microcentrifuge at 20,000 × g for 1.5 h at 4°C. The pellet was resuspended in 0.5 ml of TRIzol, and the RNA extracted as described above. The RNA was amplified by reverse transcription-PCR (RT-PCR) and purified as described previously (15), and the sequence was determined commercially (Source Biosciences).
The viral S segment termini were analyzed by 3′-RACE (3′-rapid amplification of cDNA ends). Extracted RNA was polyadenylated using a poly(A) tailing kit (Ambion), resulting in 3′-poly(A)-tailed genome sense and antigenome sense RNAs. The sequence of the 5′ end of the antigenome was determined by sequencing of the 3′ end of the genome. cDNA was prepared using oligo(dT)-anchor primer (dT-AP), and both 3′ termini were amplified by using the primer AP plus an appropriate internal primer. After electrophoresis, the amplified fragments were extracted from an agarose gel, and the sequence was determined commercially (Source Biosciences).
RESULTS
Design of ambisense constructs and rescue of AmbiGFP virus.
For clarity, RNA segments are referred to here in the antigenome sense. The ambisense S segment constructs presented in the present study were based on the S segment in the recombinant BUNdelNSs2, where the expression of NSs is abrogated by mutation of the tandem start codons for NSs, as well as a downstream AUG at codon 30 in the NSs ORF (27). Extensive studies of BUNV S segment minigenome constructs with various degrees of complementarity between the termini had previously indicated that a synthetic reporter genome with fully complementary termini was capable of directing ambisense transcription (14). In the ambisense S segments presented here, the termini of the original rBUNdelNSs2 S segment were mutated to match this panhandle in order to achieve transcription from both the genome and the antigenome (Fig. 1A). The 3′ UTR was (arbitrarily) divided into regions W (nt 788 to 849), X (nt 850 to 890), Y (nt 891 to 911), and Z (nt 912 to 936), followed by the 3′ terminus (nt 937 to 961; Fig. 1B). Regions X and Y contain sequences that have been reported to be involved in transcription termination (28, 29), so insertion of the ambisense cassette was designed between the Y and Z regions. However, no virus could be recovered using this construct. Therefore, the 3′ UTR was extended by duplication of the Y region, and the ambisense cassette was inserted between the first and second Y region; this resulted in rescuable virus. The ambisense cassette contained part of the M segment antigenome (ag) 3′ UTR and would allow insertion of different sequences between NcoI and EagI restriction sites. The AUG codons of all the ambisense ORFs had the same “Kozak” context (AcCAUGG; capitals indicate conformation to an optimal Kozak context), which was stronger than the context of the wild-type NSs ORF (uuCAUGa). Figure 1B depicts the original wt BUN S segment and derived ambisense constructs.
As proof of principle to determine whether an ambisense ORF would be expressed, initial reactions aimed to create a virus expressing the enhanced GFP gene. Using the reverse genetics system described previously (30) BSR-T7/5 cells were transfected with pT7ribo-AmbiGFP, pT7riboBUNM(+), and pT7riboBUNL(+) to rescue AmbiGFP virus. GFP expression was observed in transfected cells from 2 days posttransfection, with CPE ensuing. Supernatant containing virus was collected at 4 days posttransfection and used to infect fresh BHK-21 cells. GFP expression was detected in the infected cells and spread through the monolayer over time, followed by CPE, suggesting that infectious AmbiGFP virus had indeed been rescued.
The ability of AmbiGFP virus to infect BHK-21, VeroE6, A549, and A549-NPro cells was tested by plaque- and focus-forming assays. Quadruplicate sets of cells were infected with a dilution series of virus, incubated at either 33 or 37°C, and plaques or foci detected by Giemsa staining or immunostaining with anti-BUNV N antibodies. On A549, A549-Npro, or Vero cells no AmbiGFP virus could be detected at either temperature or with either detection method (data not shown). Detectable plaques or foci were formed only on BHK-21 cells. AmbiGFP formed small plaques on BHK-21 cells at 33°C but not at 37°C, whereas immunostaining revealed small foci at both temperatures (Fig. 2A and B) after 8 days of incubation. By comparison, plaques and foci produced by wt BUNV were much larger, while these were incubated for only 5 days, and there was no difference in plaque size between incubations at 33 and 37°C. AmbiGFP titers obtained on BHK-21 cells were 4 × 105 focus-forming units (FFU) per ml at 33°C and 5 × 104 FFU/ml at 37°C. Given the apparent temperature sensitivity of the ambisense virus, all further experiments, including production of virus stocks, were performed at 33°C unless stated otherwise.
Stability of expression of the ambisense ORF.
To determine whether expression of the ambisense ORF was stably maintained, the working stock (P0) of AmbiGFP virus was passaged 10 times in BHK-21 cells at 33°C, and GFP expression was monitored by UV microscopy. Cells infected with passage 5 (P5) stock displayed various intensities of GFP expression, whereas the cells infected with P9 stock all expressed only low levels of GFP (Fig. 3A).
FIG 3.

Stability of expression from the ambisense ORF. (A) GFP expression in BHK-21 cells infected with P0, P5, or P9 stocks of AmbiGFP. All pictures were taken with the same magnification and light intensity settings. (B) The nucleotide sequence of the S segment of the P10 stock of AmbiGFP. The mutations in the antigenome 5′ terminus that were introduced to create an ambisense panhandle are indicated by lowercase letters in the area marked by a black bar over the sequence. The blue arrow indicates the mutation shortly before the N protein start codon (blue box). The red arrow indicates 3′ terminal nt 9, which is the only nucleotide in the 3′ terminus that was mutated to create the ambisense panhandle. This nucleotide reverted to the wild-type C residue in AmbiGFP P10. Relevant nucleotide positions in the S segment are indicated. (C) Northern blot analysis of total RNA from cells infected with wt BUNV or P0, P6, or P10 stocks of AmbiGFP hybridized with probe S+ that detects antigenome and N mRNA. The positions of the antigenomes (ag) and mRNA are indicated on the left, and size markers are indicated on the right. The asterisk (*) indicates a very faint band that could correspond to the AmbiGFP segment in which the GFP ORF is partially deleted.
RNA was extracted from virions of P0 and P10 stocks and the sequence of the S segment analyzed by RT-PCR and 3′-RACE (3′-RACE on the genome was used to infer the 5′ sequence of the antigenome). The sequence of the S segment of P0 AmbiGFP was identical to that of the input plasmid construct used to rescue the virus, including the mutations in the segment termini and the inserted GFP ORF. The RACE analysis of the P10 stock indicated a uniform population in which the single 3′ mutation to make the segment ambisense had reverted from a U residue (T in the sequence trace) to a C residue as found in the wt S segment (Fig. 3B). The mutations that had been introduced in the 5′ end of the antigenome were all fully intact (Fig. 3B). Interestingly, the P10 stock had acquired a mutation in the triplet preceding the N start codon (UUA-AUG to AUA-AUG), increasing the predicted strength of the Kozak context (31) for N translation initiation (Fig. 3B).
The PCR-amplified cDNA of AmbiGFP P10 S segment appeared as two bands of distinct sizes: a full-length S segment and a truncated one. Nucleotide sequence analysis of the shorter product revealed a 587-nt deletion in the GFP ORF. To confirm the internal deletion in the GFP ORF and investigate the relative abundance of the full-length and truncated forms of the S segment, the RNA from cells infected with AmbiGFP P0, P6, and P10 stocks was analyzed by Northern blotting. The blot was hybridized with probe S+ which detects the BUNV S segment antigenome (961 nt) and N mRNA (∼900 nt) (4). This probe also detected the AmbiGFP antigenome (1,796 nt), as well as the N mRNA (Fig. 3C). Intriguingly, the majority of the P10 antigenomic RNA appeared to be of full length, although there appeared to be a very faint band that might correspond to the size of the antigenome with the GFP deletion (∼1,200 nt; marked with an asterisk in Fig. 3C).
Since only a small proportion of genomes lacked the GFP coding sequence it seemed likely that the reduction in GFP expression was mainly due to reversion to wild type of the antigenome 3′ end. Since all cells infected with the P10 stock appeared to express GFP, albeit to a minimal level, the reverted panhandle apparently has some residual transcriptional activity to produce the ambisense GFP mRNA.
Design of S segments expressing NSs in an ambisense strategy.
After the proof of principle was established and expression from the ambisense ORF appeared relatively stable in the mutant virus AmbiGFP, the GFP ORF was replaced by a tagged NSs ORF. However, since NSs overlaps the N coding region and the original NSs sequence would be largely intact, inserting the NSs sequence in the ambisense orientation would give rise to a long stretch of complementarity within the segment. Anticipating that this might prove inhibitory to virus replication, the codons for the ambisense NSs ORF were scrambled in order to generate a segment with as little complementarity as possible between the original and inserted NSs nucleotide sequences, while the amino acid sequence remained the same. This scrambled-codon NSs nucleotide sequence will be referred to as NSx where necessary to distinguish it from the authentic NSs sequence. The NSx sequence was designed with an N-terminal Flag tag (FlNSs) since N-terminally tagged NSs had previously been shown to be functional in a minireplicon assay (20) to generate construct pT7ribo-AmbiFlNSs (Fig. 1B).
Since NSs expression during infection is limited due to proteasomal degradation (15), pT7ribo-AmbiFlNSs was modified to change the four lysine residues into arginine residues (4KR mutation [15]) to increase the levels of NSs in infected cells. This yielded construct pT7ribo-AmbiFl4KR (Fig. 1B). Finally, the Flag tag in pT7ribo-AmbiFl4KR was changed to a V5 tag (13), creating construct pT7ribo-AmbiV54KR (Fig. 1B). It should be noted that the lysine residue (K) in the V5 tag sequence (GKPIPNPLLGLDST) was replaced by an arginine residue (R) to eliminate any lysine residues from the NSs protein.
Rescue, growth characteristics, and stability of AmbiFlNSs, AmbiFl4KR, and AmbiV54KR viruses.
Using the appropriate modified S segment plasmid constructs, AmbiFlNSs, AmbiFl4KR, and AmbiV54KR viruses were rescued in BSR-T7/5 cells at 33°C. The rescue of the ambisense viruses was not as efficient as that of wt BUNV but was successful on all occasions that the rescue of wt BUNV, performed in parallel, was itself highly efficient, as monitored by visible CPE in the transfected cells within 2 to 3 days. Despite repeated attempts, these viruses could not be plaque purified, so the supernatants from the transfected cells were used as elite stocks from which working stocks were grown up. Like the AmbiGFP virus, the NSs-expressing viruses were initially characterized by plaque and focus formation on BHK-21 cells. Only AmbiFlNSs formed detectable plaques on BHK-21 cells; these were larger than those of AmbiGFP (Fig. 2A). AmbiFlNSs showed temperature sensitivity similar to AmbiGFP, with both plaques and foci notably smaller at 37°C than at 33°C (Fig. 2A and B). Surprisingly, the two recombinant viruses with the tagged and stabilized form of NSs, AmbiFl4KR and AmbiV54KR, did not form detectable plaques on any cell type. The foci formed by these viruses on BHK-21 cells were only detectable at 33°C and were much smaller than those of AmbiFlNSs (Fig. 2C). This was unexpected since the 4KR mutation in a wild-type S segment background did not lead to reduced plaque size (I. van Knippenberg and R. M. Elliott, unpublished results) or titers (15).
The growth kinetics of the ambisense NSs-expressing viruses were compared to those of wt BUNV by a low-multiplicity (0.01 FFU/cell) infection of BHK-21 cells at 33°C (Fig. 4A). After an initial lag in growth of AmbiV54KR, the titers reached by AmbiFlNSs and AmbiV54KR (8.7 × 106 FFU/ml and 8.6 × 106 FFU/ml, respectively) were ∼40 times lower than wt BUNV (3.5 × 108 FFU/ml), indicating that these viruses were impaired in growth. AmbiFl4KR grew much slower than both of the other ambisense viruses and achieved a titer of 1.7 × 104 FFU/ml by 120 h postinfection (p.i.), the end of the experiment.
FIG 4.
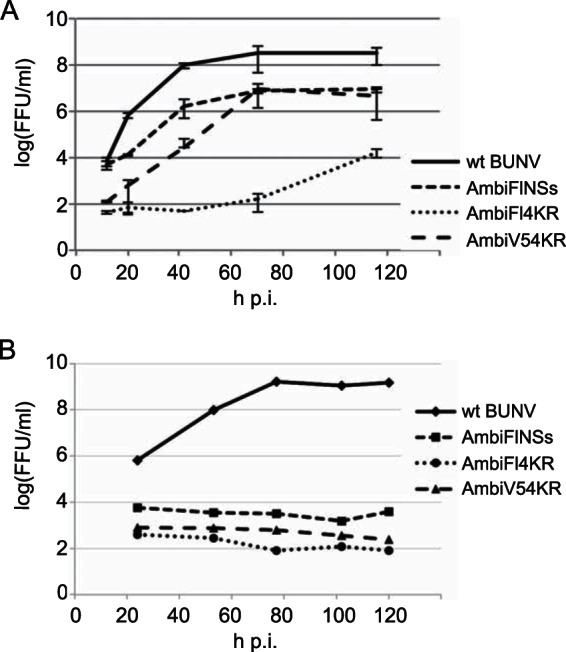
Growth phenotypes of the NSs-expressing ambisense viruses. (A) Growth curves in BHK-21 cells comparing wt BUNV and the three ambisense NSs expressing viruses. Cells were infected with 0.01 FFU per cell and incubated at 33°C. (B) Growth curves in A. albopictus C6/36 cells comparing wt BUNV and the three ambisense NSs-expressing viruses. Cells were infected with 0.01 FFU per cell and incubated at 28°C. Virus released into the supernatant was titrated in BHK-21 cells as described above.
Growth kinetics were also compared in A. albopictus C6/36 cells, but the ambisense viruses (including AmbiGFP [not shown]) did not appear capable of replication in these mosquito cells (Fig. 4B). wt BUNV reached titers of 109 FFU/ml, whereas for the ambisense viruses no increase in titer could be detected. (As previously described, it is difficult to remove the inoculum from C6/36 cells by repeated washing since many cells detach from the dish [25]). It is known that NSs is required for replication in certain mosquito cell lines, but this is not the case for C6/36 cells (25). It is unclear why these ambisense viruses appear incapable of replication in these cells.
The stability of NSs-expressing ambisense viruses was tested by five passages in BHK-21 cells, followed by S-segment nucleotide sequence analysis. Like the P10 AmbiGFP, all P5 viruses showed reversion of the 3′ nucleotide 9 to the wild-type C residue. By analogy to the reduced levels of GFP expression in AmbiGFP P10-infected cells, the P5 NSs-expressing viruses were not expected to express enough NSs to be detectable. Apart from this 3′-terminal mutation, no other mutations were detected in any of the P5 viruses.
Replication, transcription, and packaging of the ambisense S segment.
To identify the different RNA species produced from the S segments of the ambisense viruses RNA was extracted from infected BHK-21 cells (multiplicity of infection [MOI] of 1) and analyzed by Northern blotting. The probes used are depicted schematically in Fig. 5A. The BUN S+ probe detected the wt antigenome of 961 nt, as well as the ambisense antigenomes of 1,402 or 1,425 nt for Flag-tagged and V5-tagged versions, respectively (Fig. 5B, left panel). The BUN S+ probe also detected the N mRNA (∼900 nt) produced by wt BUNV and all three of the ambisense viruses (Fig. 5B, left panel), confirming that the mutations in the panhandle had not impaired transcription from the genomic S segment RNA. The BUN S− probe detected the wt genome, as well as the ambisense genomes, but, as expected, no other subgenomic mRNAs from the ambisense viruses were observed (Fig. 5B, right panel).
FIG 5.

Replication, transcription, and packaging of the ambisense viruses. (A) Diagram of wt BUNV and AmbiFlNSs genomes and expression strategies, indicating the positions of the probes used in Northern blot hybridization (S+, S−, N450, and NSx450). The hatched boxes represent the complement of the coding sense ORFs. The dotted line represents the 3′ end of the putative NSs mRNA whose transcription termination site is unknown. The diagonal line with the dot at the 5′ ends of mRNAs represents the nontemplated capped leader sequence. (B and C) Northern blot analysis. Membranes carrying total RNA from BHK-21 cells infected at an MOI of 1 with wt BUNV, AmbiFlNSs, AmbiFl4KR, or AmbiV54KR as indicated were probed with the S+ and S− probes (B) or N450 and NSx450 probes (C). The positions of the genomes and antigenomes (g/ag) and mRNAs are indicated on the left, and the positions of size markers are indicated on the right. The putative NSs mRNA is indicated by an asterisk (*) in panel C. (D) Northern blot analysis of virion-derived RNA using probes S− and M− to detect genomic S and M segments, respectively.
New probes were designed in an attempt to detect the ambisense NSx sequence and distinguish it from the authentic NSs coding sequence embedded in the N ORF. The similarity between these two sequences was 71%. The probe for NSx, NSx450, encompassed the entire coding sequence for NSx (Fig. 5A). A new probe for the N coding sequence outside the NSs-overlapping region, N450, was also produced, with a similar length to that of the NSx probe, to serve as a control for hybridization conditions (Fig. 5A).
Hybridization of Northern blots containing the same samples as described above with probe N450 (Fig. 5C, left panel) resulted in the same pattern as probing with the S+ probe (Fig. 5B, left panel), confirming that the smaller size of the probe did not result in diminished sensitivity. Probe N450 recognized the wt antigenome and N mRNA, as well as the antigenomes and the N mRNAs from the ambisense viruses. Probe NSx450 did not recognize any wt RNA, as expected, but clearly reacted with the ambisense genomes, indicating that the modest sequence variation was sufficient to distinguish scrambled NSs from wt NSs sequence and that this probe was highly specific for the inserted NSx sequence (Fig. 5C, right panel). For AmbiFl4KR, all RNAs appeared to be less intense than for the other two ambisense viruses, which seemed to correlate with the lower rates of replication observed in the growth curves (Fig. 4B). The NSx450 probe also reacted with a subgenomic RNA from the ambisense viruses of ∼600 nt, representing most likely the (ambisense) NSs mRNA (marked by an asterisk in Fig. 5C). This was surprising, since no specific termination signal for the NSs mRNA was included in the design of the ambisense constructs. The size of the putative subgenomic mRNA suggested that transcription termination occurred ∼150 nt downstream of the stop codon.
Packaging of the ambisense S segments was analyzed by Northern blotting using RNA extracted from virions (Fig. 5D). The signals for the M and S segments appeared to be of comparable intensity for wt BUNV, whereas the AmbiGFP and AmbiFlNSs virions clearly contained relatively less M segment. AmbiFl4KR and AmbiV54KR also appeared to have less M than S, although the difference seemed less marked compared to the other ambisense viruses. Quantification of the bands by densitometry from different blots and different virus preparations showed overall S/M ratios of ∼1.2 for wt BUNV, 3.3 for AmbiGFP and AmbiFlNSs, and 2.7 for AmbiFl4KR and AmbiV54KR. Given that AmbiGFP and AmbiFlNSs appear to be very similar in this respect, it can be concluded that any difference in packaging is most likely the result of the mutations in the panhandle rather than the absence or presence of NSs.
Functional analysis of NSs expressed by ambisense viruses.
The expression of tagged NSs protein was monitored by Western blotting of infected BHK-21 cell lysates (MOI of 1). Similar to wt BUNV-infected cells, expression of N protein was first detected in AmbiFlNSs-infected cells at 12 h p.i. (Fig. 6A) and accumulated to 48 h p.i. The expression of NSs in these cells was also detected at 12 h p.i. but decreased markedly thereafter. The detection of NSs was greatly increased in the presence of epoxomicin, a proteasome inhibitor. In AmbiV54KR-infected cells, the amount of tagged, stabilized NSs protein was greatly increased compared to AmbiFlNSs-infected cells and even slightly increased compared to AmbiFlNSs infection in the presence of epoxomicin (Fig. 6A). AmbiFl4KR protein expression levels deviated from those of the other ambisense viruses. The N protein was barely detectable at 24 h p.i. and remained low at 48 h p.i., and yet there was a very strong signal of NSs by 48 h p.i. The data from AmbiFl4KR-infected cells all point to impaired viral replication with a slower growth rate, smaller amounts of viral RNA, and smaller amounts of N protein produced.
FIG 6.

Functional expression of NSs. (A) Western blot analysis of lysates from BHK-21 cells infected at an MOI of 1 with wt BUNV, AmbiFlNSs (in the absence or presence of epoxomicin), AmbiFl4KR, or AmbiV54KR as indicated. Viral proteins or tubulin were detected with appropriate antibodies as indicated. The blots were exposed for 3 min to detect NSs, 30 s to detect N, and 10 s to detect tubulin. (B) Total cell labeling. BHK-21 cells were infected at an MOI of 0.25 with wt BUNV, AmbiFlNSs, AmbiFl4KR, AmbiV54KR or AmbiGFP as indicated. At 24 or 48 h p.i., the cells were incubated for 2 h with medium containing 35S-labeled cysteine-methionine. The cell monolayers were then lysed for analysis by SDS-PAGE, followed by exposure to a phosphorimager. (C and D) IFN production in A549 (C) and 2fTGH (D) cells infected at an MOI of 2 with wt BUNV, rBUNdelNSs2, AmbiFlNSs, AmbiFl4KR, or AmbiV54KR as indicated. Below the bar graphs are the Western blot analyses performed on lysates of the same cells to detect tubulin and N protein as indicated.
The relative levels of viral proteins expressed, as well as the shutoff of host gene expression, was investigated by metabolic labeling. Cells infected with wt BUNV, AmbiFlNSs, AmbiFl4KR, AmbiV54KR, or AmbiGFP were incubated for 2 h with medium containing 35S-labeled cysteine-methionine at 24 h or 48 h p.i. (Fig. 6B). As expected, no shutoff was observed in AmbiGFP-infected cells, where no NSs was expressed. AmbiFlNSs and AmbiV54KR showed some degree of shutoff, although this was slower than for wt BUNV. By 48 h p.i. AmbiFlNSs appeared to have achieved nearly complete shutoff (compared to wt BUNV), whereas shutoff was still lower in AmbiV54KR-infected cells. AmbiFl4KR displayed only a modest level of shutoff, a finding consistent with its overall slower growth and viral protein expression rates. The relative levels of N and Gc proteins produced in the 2-h labeling period were similar for AmbiFlNSs and AmbiV54KR. Both of these viruses produced more N protein than did wt BUNV in the same period, but relatively less Gc protein.
The functionality of the tagged NSs expressed by ambisense viruses was further tested by a biological IFN assay (32) in A549 cells and 2fTGH cells. Due to the apparent temperature sensitivity of the ambisense viruses, the infections were carried out at 33°C, although it should be noted that A549 cells produce less IFN at this temperature than at 37°C (15). After infection (MOI of 2) and virus inactivation, the IFN content of the supernatant was measured by testing its ability to protect A549-NPro cells from EMCV infection. No IFN was produced in mock-infected A549 cells or in cells infected with wt BUNV, whereas infection with rBUNdelNSs2 resulted in a marked induction of IFN (Fig. 6C). AmbiFlNSs inhibited the IFN response to some extent, although less than was observed with wt virus (Fig. 6C). Western blot analysis of the infected A549 cells showed that the level of N protein expressed by AmbiFlNSs was considerably lower than that in wt BUNV-infected cells (Fig. 6C). This might be an indication that NSs levels also were lower, which could account for the less-complete inhibition of the IFN response (the NSs levels were too low in all samples to detect by Western blotting). The ambisense viruses expressing the 4KR version of NSs behaved like rBUNdelNSs2 virus, which lacks expression of the IFN antagonist and induced high levels of IFN (Fig. 6C). This was unexpected since rBUN4KR, like wt BUNV, had previously been shown to completely inhibit the IFN response (15). Western blotting showed that no N protein could be detected in AmbiFl4KR-infected cells, and hardly any could be detected in AmbiV54KR-infected cells (Fig. 6C), suggesting that these viruses were severely impaired in replication in A549 cells. The lack of IFN antagonism observed for these viruses, therefore, seemed most likely due to lack of viral replication rather than the lack of NSs functionality.
2fTGH cells (another human lung epithelial line) seemed more permissive to replication of the ambisense viruses since larger amounts of N protein were detected (Fig. 6D; a very low level of N protein was detected in AmbiFl4KR-infected cells, although this is not readily visible in this exposure of the blot). These cells appeared to produce less IFN than A549 cells (rBUNdelNSs2 induced roughly half the amount of IFN in 2fTGH cells compared to A549; compare Fig. 6C and D). In 2fTGH cells, all of the NSs expressing ambisense viruses inhibited the IFN response, with AmbiV54KR being the most effective (Fig. 6D). Together, these results indicate that all ambisense viruses expressed fully functional NSs proteins and the epitope tags did not interfere with NSs function.
Intracellular localization of NSs.
The intracellular localization of tagged NSs proteins was studied by indirect immunofluorescent staining in BHK-21 cells. Unfortunately, no consistent signal above background could be obtained for AmbiFlNSs or AmbiFl4KR (not shown). BHK-21 cells infected with wt BUNV (MOI of 3) showed clear cytoplasmic localization of N protein and produced no signal with anti-V5 antibodies (Fig. 7). In cells infected with AmbiV54KR at an MOI of 3, N protein showed the same cytoplasmic localization as in wt BUNV-infected cells. V5-tagged NSs4KR appeared to be localized to both the cytoplasm and the nucleus; in some cells, this localization was predominantly nuclear, and in others, the localization was equally distributed between both the cytoplasm and the nucleus (Fig. 7).
FIG 7.

Localization of NSs. BHK-21 cells were infected (MOI of 3) with wt BUNV or AmbiV54KR and fixed at 24 or 52 h p.i., respectively. The cells were stained using mouse anti-V5 and rabbit anti-BUNV N antibodies, as indicated, followed by anti-mouse-Cy5-conjugated and anti-rabbit-FITC-conjugated antibodies. The DAPI (4′,6′-diamidino-2-phenylindole) signal is omitted from the merge so as not to mask the V5 signal. Representative images are shown.
DISCUSSION
The orthobunyavirus NSs protein is a major virulence factor and functions as the viral IFN antagonist. Although much has been learned about the mechanisms of action of NSs (reviewed in reference 33), detailed mutagenic studies to define functional domains or the roles of key amino acid residues are hampered by the overlapping coding strategy used to produce NSs (21). We have investigated various strategies to address this problem and here report the successful manipulation of the BUNV S segment to express N and NSs (or an alternative gene, such as GFP) using an ambisense arrangement as used by other members of the Bunyaviridae family such as the phleboviruses. The use of an ambisense variant of a negative sense virus has also been demonstrated for the nonsegmented Sendai and rabies viruses (34, 35). Based on mutations in the S segment genome termini to generate a panhandle structure that can promote both replication and transcription not only from the genome sense but also from the antigenome sense (14), a set of S segments was designed to generate viruses expressing GFP or tagged NSs from an ambisense ORF.
The recombinant virus with an ambisense S segment encoding GFP proved to be viable but had a small plaque phenotype and was temperature sensitive. In the absence of any selection pressure, GFP expression was maintained over at least 10 passages, although the level of expression diminished at later passages. At P10, a partial internal deletion of the GFP ORF was detected, but Northern blot analysis showed this had occurred only in a small subpopulation of the virus. In addition, reversion of the 3′ position 9 nt from a U to a C residue was also observed. It seems likely that this mutation was the main cause of the reduced GFP expression levels.
Having shown the viability of AmbiGFP, we next generated viruses expressing a modified NSs gene (designated NSx) to avoid internal complementarity between the authentic N/NSs coding region and the ambisense NSs ORF. Although the NSs and NSx sequences were 71% similar, this was sufficiently divergent to generate a Northern blot hybridization probe that could distinguish between these two sequences. Using this probe, a putative NSx mRNA of ∼600 nt was detected. This would place the transcription termination site at ∼150 nt downstream from the NSx stop codon. The termination signal(s) identified for BUNV have been subject to discussion (28) but appear to conform to the consensus sequence 5′-GCN1-3GC-3′ as found for Simbu group orthobunyaviruses (36). Such a motif is present in the ambisense genome at approximately 100, 130, and 250 nt downstream from the ambisense ORF stop codon.
Previously, we showed that in a wild-type background, the 4KR version of NSs confers resistance to proteasomal degradation, resulting in increased levels of NSs in infected cells while not interfering with its IFN antagonist properties or virus fitness (15). It was anticipated that introducing this version in the ambisense virus would similarly facilitate the detection of NSs, while not having any negative effects on virus fitness. However, the 4KR-expressing ambisense viruses exhibited a smaller plaque phenotype compared to plaques produced by AmbiFlNSs. This would suggest that the K-to-R mutations caused the smaller plaque phenotype, but this was not seen when comparing rBUN4KR with wt BUNV (unpublished observations). It is also unlikely to be the result of the tag since two different tags were used and the AmbiFlNSs forms larger plaques than AmbiFl4KR.
The mutations that were introduced into the termini of the S segment to accomplish ambisense expression appeared to have an impact on the packaging efficiency of the S segment relative to the M segment. The S/M segment ratio of virions was increased >2-fold for all ambisense viruses regardless of expressing NSs or GFP. Possibly, the same mutations also contributed to the altered expression rates of N to Gc protein, since this ratio appeared even more affected than the segment packaging ratio (Fig. 6B).
The NSs proteins expressed by the ambisense viruses were functional both in inducing shutoff of host protein expression and in counteracting the IFN response. This demonstrated that the presence of a N-terminal tag does not inhibit these functions of NSs. Although no IFN antagonism could be detected with the 4KR viruses in A549 cells, it was close to wt levels in 2fTGH cells, indicating that these viruses expressed functional NSs proteins. The lack of IFN antagonism observed in A549 cells presumably reflects the poor infection efficiency these viruses have in this cell line, as demonstrated also by the very low levels of N protein produced. The reasons these viruses replicate so poorly in A549 cells and, in particular, why the 4KR viruses would be restricted in A549 cells more so than AmbiFlNSs remain unclear.
Since NSs inhibits RNAPII function, it would be expected to localize to the nucleus, and this was indeed demonstrated for transiently expressed tagged NSs (18, 20). However, no formal proof existed that NSs is localized to the nucleus in BUNV-infected cells. Here, immunofluorescent staining of BHK-21 cells infected with AmbiV54KR demonstrated that a proportion, at least, of NSs was located in the nuclei of infected cells. NSs was also detected in the cytoplasm, and the ratio of nuclear and cytoplasmic NSs appeared to vary from cell to cell, with no clear relation to the time postinfection.
In summary, we were able to create recombinant viruses expressing the N and NSs proteins as independent genes using an ambisense coding strategy. Despite these viruses being slightly attenuated for growth in BHK-21 cells and temperature sensitive at 37°C compared to 33°C, AmbiFlNSs exhibited IFN antagonism. This virus will be a helpful tool to further explore structure-function relationships of the NSs protein in IFN antagonism. The successful rescue and apparent relative stability of ambisense BUNV recombinants underscores the genome plasticity of this family of viruses.
ACKNOWLEDGMENTS
We thank V. Anspaugh, E. Koudriakova, B. Mazel-Sanchez, and A. McLees for technical assistance.
This study was supported by grants from the Wellcome Trust to R.M.E., who is a Wellcome Trust Senior Investigator.
REFERENCES
- 1.Elliott R, Schmaljohn C. 2013. Bunyaviridae, p 1244–1282. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott/The Williams & Wilkins Co, Philadelphia, PA. [Google Scholar]
- 2.Elliott RM, Blakqori G. 2011. Molecular biology of orthobunyaviruses, p 1–40. In Plyusnin A, Elliott RM (ed), Bunyaviridae: molecular and cellular biology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 3.Elliott RM. 2014. Orthobunyaviruses: recent genetic and structural insights. Nat Rev Microbiol 12:673–685. doi: 10.1038/nrmicro3332. [DOI] [PubMed] [Google Scholar]
- 4.Lowen AC, Boyd A, Fazakerley JK, Elliott RM. 2005. Attenuation of bunyavirus replication by rearrangement of viral coding and noncoding sequences. J Virol 79:6940–6946. doi: 10.1128/JVI.79.11.6940-6946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowen AC, Elliott RM. 2005. Mutational analyses of the nonconserved sequences in the Bunyamwera orthobunyavirus S segment untranslated regions. J Virol 79:12861–12870. doi: 10.1128/JVI.79.20.12861-12870.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazel-Sanchez B, Elliott RM. 2012. Attenuation of Bunyamwera orthobunyavirus replication by targeted mutagenesis of genomic untranslated regions and creation of viable viruses with minimal genome segments. J Virol 86:13672–13678. doi: 10.1128/JVI.02253-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan B, Welch SR, Elliott RM. 2014. The consequences of reconfiguring the ambisense S genome segment of Rift Valley fever virus on viral replication in mammalian and mosquito cells and for genome packaging. PLoS Pathog 10:e1003922. doi: 10.1371/journal.ppat.1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brennan B, Li P, Elliott RM. 2011. Generation and characterization of a recombinant Rift Valley fever virus expressing a V5 epitope-tagged RNA-dependent RNA polymerase. J Gen Virol 92:2906–2913. doi: 10.1099/vir.0.036749-0. [DOI] [PubMed] [Google Scholar]
- 9.Shi X, Elliott RM. 2009. Generation and analysis of recombinant Bunyamwera orthobunyaviruses expressing V5 epitope-tagged L proteins. J Gen Virol 90:297–306. doi: 10.1099/vir.0.007567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan B, Welch SR, McLees A, Elliott RM. 2011. Creation of a recombinant Rift Valley fever virus with a two-segmented genome. J Virol 85:10310–10318. doi: 10.1128/JVI.05252-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wichgers Schreur PJ, Oreshkova N, Moormann RJM, Kortekaas J. 2014. Creation of Rift Valley fever viruses with four-segmented genomes reveals flexibility in bunyavirus genome packaging. J Virol 88:10883–10893. doi: 10.1128/JVI.00961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott RM, Schmaljohn CS, Collett MS. 1991. Bunyaviridae genome structure and gene expression. Curr Top Microbiol Immunol 169:91–141. [DOI] [PubMed] [Google Scholar]
- 13.Southern JA, Young DF, Heaney F, Baumgärtner WK, Randall RE. 1991. Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J Gen Virol 72:1551–1557. doi: 10.1099/0022-1317-72-7-1551. [DOI] [PubMed] [Google Scholar]
- 14.Barr JN, Rodgers JW, Wertz GW. 2005. The Bunyamwera virus mRNA transcription signal resides within both the 3′ and the 5′ terminal regions and allows ambisense transcription from a model RNA segment. J Virol 79:12602–12607. doi: 10.1128/JVI.79.19.12602-12607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Knippenberg I, Fragkoudis R, Elliott RM. 2013. The transient nature of Bunyamwera orthobunyavirus NSs protein expression: effects of increased stability of NSs protein on virus replication. PLoS One 8:e64137. doi: 10.1371/journal.pone.0064137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridgen A, Weber F, Fazakerley JK, Elliott RM. 2001. Bunyamwera bunyavirus nonstructural protein NSs is a nonessential gene product that contributes to viral pathogenesis. Proc Natl Acad Sci U S A 98:664–669. doi: 10.1073/pnas.98.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber F, Bridgen A, Fazakerley JK, Streitenfeld H, Kessler N, Randall RE, Elliott RM. 2002. Bunyamwera bunyavirus nonstructural protein NSs counteracts the induction of alpha/beta interferon. J Virol 76:7949–7955. doi: 10.1128/JVI.76.16.7949-7955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas D, Blakqori G, Wagner V, Banholzer M, Kessler N, Elliott RM, Haller O, Weber F. 2004. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J Biol Chem 279:31471–31477. doi: 10.1074/jbc.M400938200. [DOI] [PubMed] [Google Scholar]
- 19.Léonard VHJ, Kohl A, Hart TJ, Elliott RM. 2006. Interaction of Bunyamwera orthobunyavirus NSs protein with mediator protein MED8: a mechanism for inhibiting the interferon response. J Virol 80:9667–9675. doi: 10.1128/JVI.00822-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber F, Dunn EF, Bridgen A, Elliott RM. 2001. The Bunyamwera virus nonstructural protein NSs inhibits viral RNA synthesis in a minireplicon system. Virology 281:67–74. doi: 10.1006/viro.2000.0774. [DOI] [PubMed] [Google Scholar]
- 21.Fuller F, Bhown AS, Bishop DH. 1983. Bunyavirus nucleoprotein, N, and a nonstructural protein, NSS, are coded by overlapping reading frames in the S RNA. J Gen Virol 64:1705–1714. doi: 10.1099/0022-1317-64-8-1705. [DOI] [PubMed] [Google Scholar]
- 22.Buchholz UJ, Finke S, Conzelmann K-K. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale BG, Knebel A, Botting CH, Galloway CS, Precious BL, Jackson D, Elliott RM, Randall RE. 2009. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 383:6–11. doi: 10.1016/j.virol.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Kohl A, Clayton RF, Weber F, Bridgen A, Randall RE, Elliott RM. 2003. Bunyamwera virus nonstructural protein NSs counteracts interferon regulatory factor 3-mediated induction of early cell death. J Virol 77:7999–8008. doi: 10.1128/JVI.77.14.7999-8008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szemiel AM, Failloux A-B, Elliott RM. 2012. Role of Bunyamwera orthobunyavirus NSs protein in infection of mosquito cells. PLoS Negl Trop Dis 6:e1823. doi: 10.1371/journal.pntd.0001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bridgen A, Elliott RM. 1996. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc Natl Acad Sci U S A 93:15400–15404. doi: 10.1073/pnas.93.26.15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart TJ, Kohl A, Elliott RM. 2009. Role of the NSs protein in the zoonotic capacity of orthobunyaviruses. Zoonoses Public Health 56:285–296. doi: 10.1111/j.1863-2378.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 28.Blakqori G, Lowen AC, Elliott RM. 2012. The small genome segment of Bunyamwera orthobunyavirus harbours a single transcription-termination signal. J Gen Virol 93:1449–1455. doi: 10.1099/vir.0.042390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barr JN, Rodgers JW, Wertz GW. 2006. Identification of the Bunyamwera bunyavirus transcription termination signal. J Gen Virol 87:189–198. doi: 10.1099/vir.0.81355-0. [DOI] [PubMed] [Google Scholar]
- 30.Lowen AC, Noonan C, McLees A, Elliott RM. 2004. Efficient bunyavirus rescue from cloned cDNA. Virology 330:493–500. doi: 10.1016/j.virol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Kozak M. 2002. Pushing the limits of the scanning mechanism for initiation of translation. Gene 299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohamed M, McLees A, Elliott RM. 2009. Viruses in the Anopheles A, Anopheles B, and Tete serogroups in the Orthobunyavirus genus (family Bunyaviridae) do not encode an NSs protein. J Virol 83:7612–7618. doi: 10.1128/JVI.02080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eifan S, Schnettler E, Dietrich I, Kohl A, Blomström A-L. 2013. Nonstructural proteins of arthropod-borne bunyaviruses: roles and functions. Viruses 5:2447–2468. doi: 10.3390/v5102447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finke S, Conzelmann K-K. 1997. Ambisense gene expression from recombinant rabies virus: random packaging of positive- and negative-strand ribonucleoprotein complexes into rabies virions. J Virol 71:7281–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Mercier P, Garcin D, Hausmann S, Kolakofsky D. 2002. Ambisense Sendai viruses are inherently unstable but are useful to study viral RNA synthesis. J Virol 76:5492–5502. doi: 10.1128/JVI.76.11.5492-5502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coupeau D, Claine F, Wiggers L, Martin B, Kirschvink N, Muylkens B. 2013. Characterization of messenger RNA termini in Schmallenberg virus and related simbuviruses. J Gen Virol 94:2399–2405. doi: 10.1099/vir.0.055954-0. [DOI] [PubMed] [Google Scholar]