

Abstract
Cellular hypertrophy of adipose tissue underlies many of the proposed proinflammatory mechanisms for obesity-related diseases. Adipose hypertrophy results from an accumulation of esterified lipids (triglycerides) into membrane-enclosed intracellular lipid droplets (LDs). The coupling between adipocyte metabolism and LD morphology could be exploited to investigate biochemical regulation of lipid pathways by monitoring the dynamics of LDs. This article describes an image processing method to identify LDs based on several distinctive optical and morphological characteristics of these cellular bodies as they appear under bright-field. The algorithm was developed against images of 3T3-L1 preadipocyte cultures induced to differentiate into adipocytes. We show that the calculated lipid volumes are in excellent agreement with enzymatic assay data on total intracellular triglyceride content. We also demonstrate that the image processing method can efficiently characterize the highly heterogeneous spatial distribution of LDs in a culture by showing that differentiation occurs in distinct clusters separated by regions of nearly undifferentiated cells. Prospectively, the LD detection method described in this work could be applied to time-lapse data collected with simple visible light microscopy equipment to quantitatively investigate LD dynamics.
Introduction
In adults, body fat essentially consists of white adipose tissue (WAT). Distributed across the body in various depots, WAT performs metabolic and signaling functions critical for whole body homeostasis. The core metabolic functions are to store excess nutrients as esterified lipids (i.e., triglycerides [TGs]), and to mobilize these stores during fasting. The bulk of the WAT cellular mass consists of lipid-laden white adipocytes held in a dense network of fibrous extracellular matrix proteins. In vivo, almost the entire adipocyte volume is filled by a large lipid droplet (LD), which expands or shrinks depending on the body's energy balance. Chronic overfeeding can lead to significant expansion of the adipocyte volume, termed hypertrophy, to accommodate the storage of excess nutrients. Adipocyte hypertrophy correlates with accumulation of proinflammatory immune cells in WAT, which in turn underpins tissue insulin resistance and other metabolic alterations associated with obesity-related metabolic diseases.
Biochemically, the LD volume depends on the balance between lipid synthesis and degradation. There is also a biophysical component, as the LDs interact with various components of the cytoskeleton through vesicle transport-associated proteins.1 How the biochemical and biophysical mechanisms interact to govern the LD fate is an active area of research. To this end, methods are needed for monitoring the dynamics of LDs in relation to cellular metabolism, signaling, and other biochemical processes.
Conventional methods used for LD analysis include enzymatic and dye-based assays. Enzymatic assays offer the benefit of absolute quantitation, but require the cells to be lysed and cannot yield information on the size and spatial distribution of LDs. An alternative is to stain the LDs with lipophilic dyes such as Oil Red O. The stained cells can be analyzed using microscopy, with the dye acting as a contrast agent for the LDs.2 One limitation of using lipophilic dyes is that they also bind other lipids that are not associated with intracellular LDs. Specificity can be improved by staining for LD-associated proteins that colocalize to the surface of the droplets.3 However, this approach requires fixing the cells, which can deform and fuse the LDs and thereby distort the analysis.4 Fixing the cells can be avoided by introducing labeled fatty acid analogs,5 which incorporate into native enzymatic pathways. However, these probes also label other parts of the cell. It has been shown that fluorescent labeling of individual LDs occurs with very different kinetics even when the sizes are identical, which may reflect the degradation kinetics of the probe rather than native lipid metabolism and thus confound the analysis.
In principle, one could avoid the use of dyes or probes by exploiting optical or spectroscopic properties of the LDs. An attractive option is coherent anti-Stokes Raman scattering (CARS), which can be used to image lipids by tuning into the vibrational contrast of C-H bonds that are enriched in aliphatic molecules.6 An especially powerful feature is the ability to characterize the chemical composition of individual LDs, for example, the degree of unsaturation.6,7 On the other hand, CARS requires sophisticated equipment that is not routinely available to many laboratories. A simpler alternative is light microscopy, but visually identifying the LDs is not trivial. Under bright-field (BF), LDs generally appear as the brightest objects. As long as the brightness difference between the LDs and other objects is consistent, this characteristic can be sufficient to identify LDs using a simple threshold filter. This filter can be readily implemented for automated analysis, as recently demonstrated by Or-Tzadikario et al. on images of cultured adipocytes treated with adipogenic or lipogenic factors.8 However, relying solely on a fixed brightness threshold can confound the analysis due to uneven contrast arising from various factors unrelated to LD morphology, such as variations in cell density, location within the well, and shadows caused by cellular debris. Furthermore, LDs of different sizes may appear brighter than others.
In the present study, we extend the algorithm for BF image analysis by incorporating additional features for LD identification with the goal of minimizing both false positives and negatives, while also improving the flexibility to analyze a wide range of microscopy images acquired under different contrast and brightness settings. The algorithm recognizes LDs as objects that are (a) relatively light, (b) circular, and (c) surrounded by a relatively dark boundary, (d) which is also circular. In addition to the manual analysis performed by a trained human expert, we quantitatively compared the lipid volumes calculated from image analysis against enzymatic assay results and found excellent agreement.
Materials and Methods
Materials
3T3-L1 cells were purchased from ATCC (Manassas, VA). Tissue culture reagents, including Dulbecco's modified Eagle's medium (DMEM), calf serum (CS), fetal bovine serum (FBS), human insulin, and penicillin/streptomycin, were purchased from Invitrogen (Carlsbad, CA). Unless otherwise noted, all other chemicals were purchased from Sigma (St. Louis, MO).
Cell culture
Low passage 3T3-L1 preadipocytes were seeded into 48-well plates at a concentration of 2×104 cells per cm2 and cultured in a humidified incubator at 37°C and 10% CO2. The cultures were expanded in a growth medium consisting of DMEM supplemented with 10% v/v CS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2.5 μg/mL amphotericin. The growth medium was changed every 2–3 days until confluence was reached. Two days postconfluence (designated as day 0), the cells were induced to differentiate using an adipogenic cocktail (1 μg/mL insulin, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, and 2 nM triiodothyronine) added to a basal medium (DMEM with 10% FBS and penicillin/streptomycin/amphotericin). After 48 h, the first induction medium was replaced with a second induction medium consisting of the basal adipocyte medium supplemented with only insulin. After another 48 h, the second medium was replaced with the adipocyte basal medium. On days 4, 8, 12, and 16, images were recorded for six randomly selected wells, which were then sacrificed for enzymatic assays of total TG content.
Enzymatic assay
Triglyceride measurements were performed on cell lysates as described previously.9 Briefly, cells were rinsed with warm 1× phosphate-buffered saline after aspirating the medium, and lysed in situ using a 0.1% sodium dodecyl sulfate buffer, followed by sonication. Free glycerol and TG were measured using an enzymatic assay kit from Sigma (catalog number TR0100).
Microscopy
Images were acquired using a Nikon Eclipse TE300 inverted microscope (Melville, NY), an Orca-HR digital CCD camera, and Simple PCI imaging software (HC Image/Hamamatsu, Sewickley, PA). The total area recorded per well was 0.44 mm2, which represents ∼58% of the available culture area. At 200× total magnification, the image pixel to physical distance calibration was ∼3 pixels per μm. On each day, the total number of images recorded was 300 per well, representing an approximately square block of 20×15 contiguous images inscribed in the center of each well. The images were captured using a semiautomated process, where one out of every 25 images was manually focused. Once an image was focused and captured, a 5×5 block of images centered on the manually focused image was captured using programmed microscope stage motion (Prior Proscan, Rockland, MA). This process was repeated 12 times for each well, following a pattern of 4 rows and 3 columns. The amount of time needed to image each well was ∼7 min. Images were acquired under BF, phase contrast (PC), and differential interference contrast (DIC).
Image processing overview
The goal of image processing was to label each pixel in the raw grayscale image as either part of an LD or not such that the processed image displays the location and size of the LDs as white pixels against a black background. The algorithm looks for objects that have the following characteristics of LDs as they appear under BF: a circular shape with a dark boundary and light interior. The algorithm consists of six steps (Fig. 1). First, the original grayscale image is converted into black and white (B/W) using a high threshold to isolate the light interiors of the LDs. Objects that are lighter than the background pixels are labeled as white. Second, the original grayscale image is converted to B/W using a low threshold to identify the dark boundaries. Only regions that are surrounded by a dark boundary are labeled as white. Third, these two images are intersected to yield an image that is white only where objects were both light and surrounded by a dark boundary. Fourth, noncircular objects are eliminated from this image. This step identifies circles based on the area-to-perimeter ratio, as a circle has the highest such ratio of any two-dimensional shape. A second criterion is that a circular object should have boundary pixels that are equidistant from the object's centroid. Fifth, the circle-finding step is applied to the low threshold image to add back potential false negatives. Each white-labeled object is then evaluated based on its solidity and lightness. Sixth, the resulting image is combined with the intersected image (from step 3) to yield the final processed image. All steps were implemented in MATLAB (MathWorks, Natick, MA) using a combination of custom code and built-in functions. The source code is available upon request to the author. The typical runtime for a set of 300 images at a 1024×1280 pixel resolution was 1500 s.
FIG. 1.

Overview of the image processing algorithm. Boxes represent the image after each processing step, starting with the original grayscale image and ending with the final processed image.
Step 1: B/W conversion using high threshold
Each pixel in the original grayscale image has a numerical value between zero (black) and one (white). To generate the high threshold image, all pixels with values greater (lighter) than the threshold are set to white, and pixels less (darker) than the threshold are set to black. Setting the threshold to a fixed value8 was not desirable because the overall brightness and contrast could vary between images. Therefore, the threshold was recomputed automatically for each image based on the pixel value distribution of the image using a built-in function, which uses Otsu's method10 to choose a threshold value that minimizes the intraclass variance of the black and white pixels. However, using this value led to B/W images that still included a substantial amount of background noise (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tec). To better discriminate between LDs and other objects that appear lighter than the background, we compared the pixel value histogram of a representative image containing LDs to an image from a culture that was not induced to differentiate and did not contain LDs. Based on the difference between these two histograms (Supplementary Fig. S2), we set the high threshold 15% higher than the computed value.
Step 2: B/W conversion using low threshold
The original grayscale image was processed using an automatically computed low threshold to generate an image that identifies the dark rings surrounding the LDs. The low threshold value was determined based on a comparison of representative histograms from grayscale images of differentiated and nondifferentiated cultures. The low threshold was set 10% lower than the reference value calculated by Otsu's method. To identify white-labeled objects that are not associated with the dark rings and likely represent the background, the area of every white object in the low threshold image was computed. Using a multiple of the largest LD size as a conservative cutoff, all objects larger than the cutoff were recorded as background. The low threshold image was then inverted and flood filled using the recorded object locations as seeds. The flood-filled image was then reinverted to obtain an image (referred as the low threshold image from here on) where the white pixels represent the dark rings and enclosed areas, that is, LDs, as well as residual noise.
Step 3: Intersecting the B/W images
In the high threshold image, white pixels represent objects that are lighter than the average background pixel in the grayscale image, which include the LDs. In the low threshold image, white pixels represent the LDs and objects that are darker than the average pixel. By intersecting the high and low threshold images, a new image is generated where the white pixels now correspond to pixels that were labeled white in both images. The result is to eliminate most of the noise, that is, background pixels falsely labeled as part of an LD, as these pixels cannot be simultaneously darker and lighter than the low and high thresholds, respectively, and do not overlap in the two images.
Step 4: Identifying circular objects in the intersected image
Much of the remaining noise represents objects that are not circular and are unlikely to be an LD. Therefore, a circle-finding algorithm is applied to the intersected image to eliminate these objects. The circularity of each object is evaluated in two ways. First, the algorithm calculates the area (A)-to-perimeter (P) ratio Z, which is maximal for a circle.
 |
Scaled by 4π, this ratio ranges from zero for a line to one for a circle. However, the Z ratio, which is a measure of roundness, cannot distinguish smooth oval-shaped objects (which probably are not LDs) from circular objects with irregular edges (which very well could be LDs). In a perfect circle, all boundary points are exactly one radius away from the center. Therefore, another score is computed that determines how evenly distant an object's perimeter pixels are from the centroid. After calculating the nominal radius (rn) of an object based on its area and the formula for a circle, the actual radii (ra,i) of boundary pixels are calculated from their coordinates and compared with the nominal radius. The differences between actual and nominal radii are squared, normalized to the nominal radius, and averaged to compute a shape score S:
 |
To keep the computational time reasonable (<10 s), the number of boundary pixels analyzed for each image (N) was fixed at 32. The final circularity score was computed as a ratio of Z to S, such that a more circular object has a larger score. Based on the scores computed for representative LDs, the cutoff was set at 15, that is, objects with circularity scores less than 15 were eliminated.
Step 5: Identifying solid circular objects in the low threshold image
For large LDs (>6 μm in diameter), the presence of a dark ring around the droplet is a more reliable characteristic than the presence of a light interior. It is possible that intersecting the low and high threshold images will erroneously remove large LDs or underestimate the size of these LDs by only retaining the interior regions. To solve this problem, the low threshold image from step 2 is reprocessed to identify large circular objects that should be restored in the final processed image. First, small objects were eliminated. Next, circularity scores are calculated for all remaining objects to identify likely LDs. As was the case for the earlier steps, the parameter values to determine smallness and circularity were chosen based on comparison with manual analysis determined from visual inspection of the original BF images. Finally, any remaining false positives (objects that are erroneously labeled as LDs) were identified based on whether an object is both solid and relatively light. Solidity is defined as the fraction of an object that is contained in its convex hull, and lightness is defined as the mean pixel value of the object normalized to the threshold computed by Otsu's method. The resulting image is referred to as dark ring image from here on.
Step 6: Union of intersected and dark ring images
The final processing step is to form the union of images resulting from steps 3 and 5. This final step restores several large LDs as well as boundaries eliminated during step 3.
Calculating LD size, volume, and distribution
The following statistics were calculated: number of LDs, fraction of image occupied by the LD, average LD size, total LD volume, and a histogram of LD size distribution. All calculations were performed using the final processed image. The LD fraction was calculated by dividing the number of white pixels by the total number of pixels. Average LD size was calculated by dividing the number of white pixels by the number of distinct objects in the image. Total LD volume (in cubic pixels) was calculated by treating each object as a perfect sphere with a nominal radius estimated based on the object's area and the assumption that the area represents a circle defining the equator of the sphere. Size distributions based on the LD volume were computed for each image as well as sets of images representing different time points in the differentiation experiment.
Results
Comparison with manual analysis by a trained human expert
A key desired feature for the image analysis algorithm is to process images that can be readily obtained using standard microscopy equipment without requiring the addition of fixatives, dyes, and/or chemical probes. To this end, we developed the image analysis algorithm against micrographs of cultured 3T3-L1 adipocytes obtained under BF. Starting with a grayscale image (Fig. 2A), the algorithm generates a high threshold image (Fig. 2B) and a low threshold image (Fig. 2C). The latter is inverted and flood filled, then reinverted to obtain a processed low threshold image (Fig. 2D). Intersecting the processed low and high threshold images eliminates much of the background noise (Fig. 2E). However, some noise remains (boxed insert in Fig. 2E), consisting of irregularly shaped objects that are falsely labeled as LDs. To remove these false positives, a circle-finding step is applied. Figure 2F shows an image containing only the circular objects identified in the intersected image. Next, we identify larger solid objects from the processed low threshold image that should be retained as LDs (Fig. 2G). The last step adds these circular objects to the intersected image (Fig. 2F) to form the final image (Fig. 2H). A comparison between Figure 2A and H shows that the final processed image correctly identified nearly all of the LDs recognizable in the original grayscale image.
FIG. 2.

The main processing steps are illustrated on a representative image. (A) Original grayscale image, (B) high threshold image, (C) low threshold image, (D) processed low threshold image, (E) intersected image, (F) circular objects from the intersected image, (G) dark ring image, and (H) final processed image. Boxed insert shows irregularly shaped objects that are falsely labeled as lipid droplets (LDs) in (E).
Comparison with biochemical data
To obtain a more objective and quantitative assessment of the image processing method's performance, we compared the calculated volume of LDs in a culture well against the total TG content measured using an enzymatic assay. For each well, we recorded a contiguous (20×15) block of images covering ∼58% of the culture area. As shown in Figure 3, the calculated LD volume correlates linearly with the corresponding TG data. Interestingly, the linear correlation was stronger (R2=0.991, p<0.01) when the data were averaged for a given time point (n=6 wells). A well-by-well comparison (Fig. 3A) showed a greater amount of scatter (R2=0.723, p<0.0001), especially for wells corresponding to the later time points (days 12 and 16).
FIG. 3.

Comparison of calculated lipid volumes with enzymatically measured triglyceride (TG) content for (A) individual wells and (B) time point averages. Closed squares, open squares, closed circles, and open circles indicate, respectively, day 4, 8, 12, and 16 data. For the individual well comparison, matching images and enzymatic assay results were obtained on the same well. Error bars represent one standard deviation (n=6 wells).
Culture heterogeneity
To investigate the culture heterogeneity suggested by the sample scatter in Figure 3B, we mapped the spatial distribution of LDs in a well based on the calculated total LD volume for each image. The resulting heat maps (Fig. 4) confirm a highly heterogeneous distribution of LDs within a culture well, for all time points. To characterize the intrawell distribution of LDs, the pixel values in each heat map were scaled with respect to the image containing the largest amount of LDs (set to white). A quadratic scale was used for the purpose of visualization. The scaled heat maps indicated that lipid accumulation (and presumably differentiation) occurs unevenly in clusters.
FIG. 4.

Heat maps showing culture heterogeneity on (A) day 8, (B) day 12, and (C) day 16. Each pixel represents a single image with the intensity ranging from black (no lipid) to white (highest lipid content across all samples and time points). Day 4 is not included because lipid accumulation is limited early in the time course.
Size distribution of LDs
In addition to spatial heterogeneity, image analysis can be used to characterize the size distribution of LDs. In one model of LD biogenesis, TG molecules oil out between the leaflets of the bilayer membrane, initiating the formation of tiny primordial LDs.11–14 Once formed, the primordial LDs can fuse to form larger cytosolic LDs independent of TG synthesis. Based on this model, we would expect to see a large number of LDs at early time points following adipogenic induction, with the number of LDs decreasing thereafter as smaller droplets fuse to form larger ones. This trend is indeed reflected in the data, with a steady increase in the number of LDs from day 4 to 12, and a decrease on day 16 (Fig. 5). An alternative model is the incorporation of TG molecules synthesized locally on the droplet's surface.5 Both of these models imply that newly differentiated adipocytes will contain smaller LDs compared with mature adipocytes due to the time required to form larger LDs. Comparing the size distributions of LDs over time, we found a steady increase in the fraction of large LDs with time postinduction (Fig. 6). On day 4, large LDs (>11 μm in diameter, bins 10 and 11) contribute less than 20% of the total lipid volume. In contrast, large LDs contribute more than 70% of the lipid volume on day 16. Similarly, the contribution of the smallest droplets (<4 μm in diameter, bin 1) to the total lipid volume steadily decreases from 47% on day 4 to 5% on day 16.
FIG. 5.

Average number of droplets per image for each time point (n=6 wells). Error bars represent one standard deviation.
FIG. 6.

Droplet size distribution for day 4 (filled bars) and day 16 (open bars). Each droplet was binned according to its contribution to the total lipid volume of the corresponding single image.
Comparison of imaging modalities
In addition to BF, PC and DIC microscopy have been commonly used to observe LDs. We investigated whether our processing method could also analyze these two other types of images. For comparisons, we recorded images of cells in the same field of view using all three modalities (Fig. 7). Overall, the DIC results correlated more strongly with BF than the PC results. For the total lipid volume, the coefficient of determination (R2) for linear regression of DIC on BF was 0.82, whereas the R2 value for linear regression of PC on BF was only 0.55 (Fig. 8). The trend was similar for a number of LDs with R2 values of 0.78 and 0.31 for DIC and PC, respectively (Supplementary Fig. S3). Although values for the total lipid volume were consistent across imaging modalities, the droplet number was consistently underreported for PC and DIC images. In contrast, we did not find any statistically significant differences (based on the two-sample Kolmogorov–Smirnov test) in the distribution of LD sizes calculated from BF, DIC, and PC images (Supplementary Fig. S4). This finding suggests that the missed LDs are likely to be randomly distributed in terms of size.
FIG. 7.
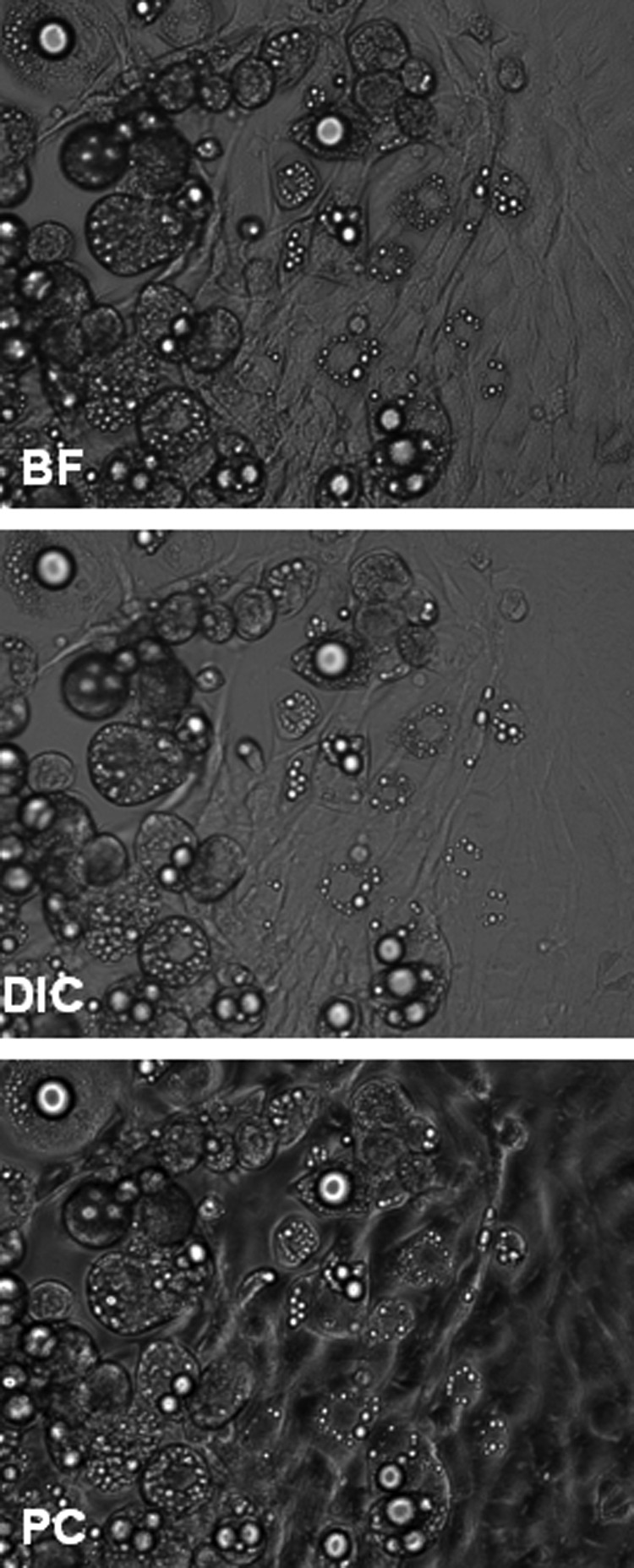
A representative image captured with the bright-field (BF), differential interference contrast (DIC), or phase-contrast (PC) modality.
FIG. 8.

Comparison of the total lipid volume estimated from (A) PC and (B) DIC images compared with BF images. Each data point represents a 5×5 block of images captured at different time points (days 4, 8, 12, and 16).
Discussion
In this article, we present a method for automated image analysis that can be used with BF microscopy to identify LDs in cultured adipocytes. Importantly, the analysis does not require special equipment or labeling. We demonstrate the method by accurately resolving LDs of varying sizes in cells at various stages of adipogenic differentiation. An obvious benefit of automated analysis is efficiency, as manual analysis is time-consuming, especially because large numbers of images need to be analyzed to obtain statistically valid results. Another benefit is consistency. In certain cases, the designation of an object as an LD can be subjective, for example, when only a part of the LD is visible due to spatial overlap with other LDs. Another potential source of error is the determination of LD boundaries, which is necessary to compute the size. Due to practical issues, such as focusing and microscope resolution, manual determination of an LD periphery may yield variable results and thus significantly affect volume calculations. While focusing and resolution influence the image quality, and hence affect the outcomes of both manual and automated analyses, the advantage of using automated processing is that ambiguous objects and LD boundaries will be treated consistently from image to image.
Previous work on automated analysis of label-free images8 identified LDs by applying a brightness threshold as a filter. While this approach is straightforward to implement, we found that the results can be sensitive to slight variations in the brightness and contrast of the acquired image, especially when the image contains a heterogeneous distribution of nonuniformly differentiated cells with a range of LD sizes. To address this issue, our algorithm utilizes two variable thresholds and exploits other characteristic features of LDs. In addition to the lightness of an LD's interior, our method also considers the darkness of the boundary and the circularity of both the LD and its boundary. By accounting for these multiple features, our method can accurately process a variety of complex images with different brightness and contrast.
One particular challenge in analyzing LDs is the treatment of very small objects near the resolution limit of the microscopy instrument. The BF images of this study were captured using a plan fluorite objective with a resolution limit of ∼0.6 μm based on the Rayleigh criterion. In practice, even using manual inspection, we found that LDs with diameters less than 1 μm were difficult to resolve, especially when juxtaposed with other similar sized objects. However, these small droplets accounted for a very small fraction (<1%) of the total LD volume in a given image. Therefore, we tuned the processing parameters (cutoff values for the low and high thresholds and circularity criteria) to focus on larger droplets that quantitatively determine the total lipid volume.
Overall, the automated image analysis results correlated very well with enzymatic assay results (Fig. 3B), but we found that the correlation was weaker for individual culture wells (Fig. 3A). The scatter is likely due to the uneven differentiation and heterogeneous LD distribution within and between wells. As shown by the heat maps (Fig. 4), images with high amounts of lipids tend to cluster, presumably reflecting areas where differentiation has progressed to a greater extent. Thus, sampling depends on both the location of capture and number of images, and it is possible that the culture area covered by the captured images (∼58%) is not always representative of the entire culture surface area. In contrast, the enzymatic assay reflects the total cell contents in a well.
Heterogeneity in the sizes and spatial distribution of LDs in cultured adipocytes has long been recognized.15 A comparison of LD morphologies and key markers for adipogenesis in individual cells and population-averaged trends16 suggested that the heterogeneity could reflect the presence of several subpopulations with distinct phenotypes. In addition, using a single-cell approach, Le and Cheng found that the phenotypic variability in a clonal population of differentiating 3T3-L1 cells was not due to differences in the induction of key adipogenic genes, such as PPARγ and C/EBPα, which were expressed in all cells.17 Rather, the variability was due to cell-to-cell differences in the kinetics of a signaling cascade that determines insulin sensitivity and glucose uptake.
Still, this does not directly explain why adipocytes with microscopically visible LDs tend to cluster. One possible explanation is that the aforementioned kinetic variability between individual cells leads to different rates of LD formation early in the differentiation process, with the differences subsequently amplified through autocrine and paracrine signaling factors that are secreted by the differentiating adipocytes. Enhanced differentiation of preadipocytes due to the production of adipogenic factors by adipocytes has been reported in several in vitro studies involving conditioned media,18 transwell inserts,19,20 and microfluidic devices.21 However, the chemical identities of these adipocyte-derived factors and their effects of LD formation remain to be fully elucidated. In this regard, image analysis techniques that can characterize the time evolution of LDs in conjunction with live-cell microscopy should be useful in investigating the dynamics of LD formation in the context of local signaling between newly differentiated adipocytes and their neighboring cells.
Clearly, the simplicity of the microscopy technique employed in the present study precludes finer analysis on the chemical composition of the LDs. Fine resolution chemical analysis can be performed with more advanced techniques that incorporate spectroscopy, notably CARS.22 However, these techniques require sophisticated equipment not readily available in most laboratories. For certain biological specimens, other light microscopy modalities such as PC and DIC have been shown to provide better resolution and contrast. Surprisingly, this was not the case for LDs in cultured adipocytes. Indeed, DIC images of LDs in adipocytes reported by several other groups23,24 appear quite similar to the BF images we recorded in the present study. Consequently, we were able to use our image analysis algorithm to analyze LDs in DIC images without any parameter adjustments (Fig. 8B). On the whole, the algorithm performed relatively poorly on PC images, generally underestimating the number of LDs in an image. For selected PC images, however, the results were nearly identical to the corresponding BF images (Supplementary Fig. S5), indicating that the main limitation in extending the algorithm to PC images lies in the consistency of image acquisition. In addition to the parameters used to process the images (e.g., high and low threshold values), we also tailored the image acquisition procedure for BF analysis. Specifically, we employed a semiautomated procedure that manually focuses only a subset of the images before they are captured. While this procedure is efficient and enables higher throughput compared to full manual focusing, not all of the captured images are sharply in focus. When the microscope was operated in PC mode, we found that the LDs in the captured images exhibited more obvious variations in morphologies due to the variable focus. In contrast to a BF image that could still be reliably analyzed even if the image was slightly out of focus, a PC image needed to be in sharp focus for the processing algorithm to reliably detect the LDs in the image.
Taken together, our results suggest that the image processing method described in this article can be used to accurately analyze the size and spatial distribution of cellular LDs. As the processing method is capable of detecting LDs in unstained BF images, it should be straightforward to extend the analysis to time-lapse microscopy, enabling studies on the spatiotemporal dynamics of LDs. Prospectively, insights gained from such studies could help better understand the physiological roles of LDs. Studies on LD morphology have revealed, for example, that smaller LDs expose a proportionately greater fraction of their lipid volume to degradation by lipase,25 and that the LDs transition from a clustered to dispersed state in response to hormonal stimulation of lipolysis.26 Growth and remodeling of LDs in hypertrophic WAT are also important in the pathophysiology of obesity and related diseases. In this regard, processing methods that can robustly detect and analyze LDs in microscopy images could facilitate the development of efficient screens for potential therapeutics.
Supplementary Material
Acknowledgment
This work was partially supported by grants from the NIH (1R56DK081768 and 1R56DK088251 to K.L.).
Disclosure Statement
No competing financial interests exist.
References
- 1.Bostrom P., Andersson L., Rutberg M., Perman J., Lidberg U., Johansson B.R., et al. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat Cell Biol 9, 1286, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Mehlem A., Hagberg C.E., Muhl L., Eriksson U., and Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc 8, 1149, 2013 [DOI] [PubMed] [Google Scholar]
- 3.McDonough P.M., Agustin R.M., Ingermanson R.S., Loy P.A., Buehrer B.M., Nicoll J.B., et al. Quantification of lipid droplets and associated proteins in cellular models of obesity via high-content/high-throughput microscopy and automated image analysis. Assay Drug Dev Technol 7, 440, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukumoto S., and Fujimoto T. Deformation of lipid droplets in fixed samples. Histochem Cell Biol 118, 423, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Kuerschner L., Moessinger C., and Thiele C. Imaging of lipid biosynthesis: how a neutral lipid enters lipid droplets. Traffic 9, 338, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Nan X., Cheng J.X., and Xie X.S. Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy. J Lipid Res 44, 2202, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Rinia H.A., Burger K.N., Bonn M., and Muller M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophys J 95, 4908, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Or-Tzadikario S., Sopher R., and Gefen A. Quantitative monitoring of lipid accumulation over time in cultured adipocytes as function of culture conditions: toward controlled adipose tissue engineering. Tissue Eng Part C Methods 16, 1167, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Si Y., Yoon J., and Lee K. Flux profile and modularity analysis of time-dependent metabolic changes of de novo adipocyte formation. Am J Physiol 292, E1637, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Otsu N. Threshold selection method from gray-level hstograms. IEEE Trans Syst Man Cybern 9, 62, 1979 [Google Scholar]
- 11.Beller M., Thiel K., Thul P.J., and Jackle H. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett 584, 2176, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Brasaemle D.L., Dolios G., Shapiro L., and Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem 279, 46835, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Ducharme N.A., and Bickel P.E. Lipid droplets in lipogenesis and lipolysis. Endocrinology 149, 942, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Pol A., Gross S.P., and Parton R.G. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol 204, 635, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green H., and Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell 3, 127, 1974 [DOI] [PubMed] [Google Scholar]
- 16.Loo L.H., Lin H.J., Singh D.K., Lyons K.M., Altschuler S.J., and Wu L.F. Heterogeneity in the physiological states and pharmacological responses of differentiating 3T3-L1 preadipocytes. J Cell Biol 187, 375, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le T.T., and Cheng J.X. Single-cell profiling reveals the origin of phenotypic variability in adipogenesis. PLoS One 4, e5189, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maumus M., Sengenes C., Decaunes P., Zakaroff-Girard A., Bourlier V., Lafontan M., et al. Evidence of in situ proliferation of adult adipose tissue-derived progenitor cells: influence of fat mass microenvironment and growth. J Clin Endocrinol Metab 93, 4098, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Stacey D.H., Hanson S.E., Lahvis G., Gutowski K.A., and Masters K.S. In vitro adipogenic differentiation of preadipocytes varies with differentiation stimulus, culture dimensionality, and scaffold composition. Tissue Eng Part A 15, 3389, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Shillabeer G., Forden J.M., and Lau D.C. Induction of preadipocyte differentiation by mature fat cells in the rat. J Clin Invest 84, 381, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai N., Sims J.K., Jeon N.L., and Lee K. Adipocyte induction of preadipocyte differentiation in a gradient chamber. Tissue Eng Part C Methods 18, 958, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isomaki A., Sillat T., Ainola M., Liljestrom M., Konttinen Y.T., and Hukkanen M. Label-free imaging of adipogenesis by coherent anti-stokes Raman scattering microscopy. Methods Mol Biol 1142, 189, 2014 [DOI] [PubMed] [Google Scholar]
- 23.Nagayama M., Uchida T., and Gohara K. Temporal and spatial variations of lipid droplets during adipocyte division and differentiation. J Lipid Res 48, 9, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Wolins N.E., Quaynor B.K., Skinner J.R., Schoenfish M.J., Tzekov A., and Bickel P.E. S3–12, Adipophilin, and TIP47 package lipid in adipocytes. J Biol Chem 280, 19146, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Walther T.C., and Farese R.V., Jr. The life of lipid droplets. Biochim Biophys Acta 1791, 459, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcinkiewicz A., Gauthier D., Garcia A., and Brasaemle D.L. The phosphorylation of serine 492 of perilipin a directs lipid droplet fragmentation and dispersion. J Biol Chem 281, 11901, 2006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.