

Abstract
Objective
Lipid peroxides and their reactive aldehyde derivatives (LPPs) have been linked to obesity-related pathologies, but whether they have a causal role has remained unclear. Glutathione peroxidase 4 (GPx4) is a selenoenzyme that selectively neutralizes lipid hydroperoxides, and human gpx4 gene variants have been associated with obesity and cardiovascular disease in epidemiological studies. This study tested the hypothesis that LPPs underlie cardio-metabolic derangements in obesity using a high fat, high sucrose (HFHS) diet in gpx4 haploinsufficient mice (GPx4+/−) and in samples of human myocardium.
Methods
Wild-type (WT) and GPx4+/− mice were fed either a standard chow (CNTL) or HFHS diet for 24 weeks, with metabolic and cardiovascular parameters measured throughout. Biochemical and immuno-histological analysis was performed in heart and liver at termination of study, and mitochondrial function was analyzed in heart. Biochemical analysis was also performed on samples of human atrial myocardium from a cohort of 103 patients undergoing elective heart surgery.
Results
Following HFHS diet, WT mice displayed moderate increases in 4-hydroxynonenal (HNE)-adducts and carbonyl stress, and a 1.5-fold increase in GPx4 enzyme in both liver and heart, while gpx4 haploinsufficient (GPx4+/−) mice had marked carbonyl stress in these organs accompanied by exacerbated glucose intolerance, dyslipidemia, and liver steatosis. Although normotensive, cardiac hypertrophy was evident with obesity, and cardiac fibrosis more pronounced in obese GPx4+/− mice. Mitochondrial dysfunction manifesting as decreased fat oxidation capacity and increased reactive oxygen species was also present in obese GPx4+/− but not WT hearts, along with up-regulation of pro-inflammatory and pro-fibrotic genes. Patients with diabetes and hyperglycemia exhibited significantly less GPx4 enzyme and greater HNE-adducts in their hearts, compared with age-matched non-diabetic patients.
Conclusion
These findings suggest LPPs are key factors underlying cardio-metabolic derangements that occur with obesity and that GPx4 serves a critical role as an adaptive countermeasure.
Keywords: Glutathione peroxidase 4, Lipid peroxidation, Obesity, Mitochondria, Inflammation, Human heart
Abbreviations: PUFA, polyunsaturated fatty acids; LPPs, lipid peroxidation end products; GPx4, glutathione peroxidase 4; HFHS, high fat, high sucrose; WT, wild type; CNTL, control; BMI, body mass index; 4-HNE, 4-hydroxynonenal; ROS, reactive oxygen species; RNS, reactive nitrogen species; HDL, high-density lipoprotein; TG, triglycerides; EF, ejection fraction; FS, fractional shortening; Nrf2, nuclear factor (erythroid-derived 2)-like 2; IL-1β, interleukin-1 beta; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; iNOS, inducible nitric oxide synthase; RAGE, receptor for advanced glycation end products; Coll1a1, collagen, type I, alpha; Coll4a1, collagen, type IV, alpha 1; β-MHC, β myosin heavy chain; TGF-β1, transforming growth factor beta 1; TGF-β2, transforming growth factor beta 2
Graphical abstract
Highlights
-
•
Role of GPx4 and lipid-derived aldehydes (LPPs) in obesity was studied.
-
•
Cardio-metabolic pathologies were exacerbated in GPx4+/− mice with obesity.
-
•
Cardiac fibrosis and mitochondrial abnormalities more severe in obese GPx4+/−.
-
•
Myocardium from diabetic patients exhibit less GPx4 and more LPPs than non-diabetic.
-
•
An adaptive role for GPx4 to protect against LPPs in obesity is proposed.
1. Introduction
The prevalence of obesity is rapidly spreading throughout the world with over 28% of adults having a body mass index (BMI) of ≥25 kg/m2 [1,2]. In addition to the increased risk for type 2 diabetes, a growing number of studies have reported a high prevalence of cardiomyopathy in obese patients (∼40% by some estimates) [3], with ‘preclinical’ myocardial damage [4] and diastolic dysfunction already evident in young adult (≤35 yrs) obese/diabetic patients [5] and even children [6]. Importantly, these cardiac derangements appear in the absence of detectable coronary disease or hypertension. Together, these findings strongly indicate a need for more careful examination of potential mechanisms that underlie cardiomyopathy in obese individuals.
Polyunsaturated fatty acids (PUFAs) are continuously oxidized in vivo through both enzymatic and non-enzymatic reactions [7,8], forming lipid peroxidation end products (LPPs) such as isoprostanes, isofurans, thromboxanes, and α,β-unsaturated aldehydes [9], all of which have biological effects [10–13]. These aldehydes cause post-translational modifications of proteins through Michael addition with lysines and histidines, and through covalent modification of sulfhydryl groups (e.g., cysteines) to form carbonyl adducts [14,15]. Due to this high degree of reactivity, many of these aldehydes have been proposed to be etiologic agents in disease [7,14,16–21]. 4-hydroxynonenal (HNE) is a α,β-unsaturated aldehyde derived from n-6 PUFA peroxidation, and HNE levels increase proportionally with ROS/RNS levels [22]. One recent report showed that HNE-modified albumin is significantly increased in the serum of type 2 diabetic patients [23].
Mitochondrial membranes are abundant with unsaturated fatty acids, which are prone to peroxidation [24–26]. The mitochondrion is also the primary source of cellular ROS, making it a major source of LPPs [27]. As a countermeasure, mitochondria contain an elaborate system of antioxidant and LPP-detoxifying enzymes. One of these is glutathione peroxidase 4 (GPx4), which resides in the mitochondrial inner membrane (in addition to nucleus and cytosol) to specifically scavenge lipid hydroperoxides [28–30]. GPx4 is one of a few antioxidant enzymes known to neutralize both simple and complex lipid hydroperoxides (e.g., Cholesterol hydroperoxide) [31,32]. It is the only member of the GPx super-family that is indispensable during development, with embryonic lethality of GPx4 null mice occurring at stage E7.5 [33–35]. In recent genetic and epidemiological studies, variants of gpx4 that result in diminished content and/or catalytic activity have been associated with obesity [36], cardiovascular disease [37,38] and inflammation [39,40]. However, no experimental studies to date have explored GPx4 in the context of obesity or its related pathologies, and this may be a significant oversight given the well-known association between mitochondrial-derived oxidative stress and metabolic disease [41,42]. In this study, we tested the hypothesis that LPPs are a causal factor underlying cardio-metabolic derangements in obesity by investigating the effect of a long-term high fat, high sucrose (HFHS) diet in a mouse model of gpx4 haploinsufficiency (GPx4+/−) and in samples of human atrial myocardium obtained from non-diabetic and diabetic patients undergoing elective heart surgery.
2. Results
2.1. GPx4 deficiency in obesity leads to enhanced lipid peroxidation and carbonyl stress in liver, exacerbating insulin resistance and steatosis
To test our hypothesis that LPPs underlie obesity-related pathologies we used a GPx4-deficient (GPx4+/−) mouse model. GPx4+/− mice are phenotypically indistinguishable from WT in the absence of an exogenous stressor but more susceptible to damage from radiation and environmental toxicants [43,44]. The rationale for using this mouse model was that it mimics the effect of previously identified gpx4 gene variants on GPx4 enzyme levels and activity in human cells [36,38,45]. Following HFHS diet no significant differences in adiposity or weight gain were observed between WT and GPx4+/− mice, although obese GPx4+/− mice had substantial dyslipidemia and fasting hyperglycemia with HFHS diet (Table 1). Whole body energy expenditure, determined by VO2 and VCO2 using indirect calorimetry, was not different between WT and GPx4+/− mice, either with CNTL or HFHS diet (data not shown). Glucose intolerance was exacerbated in the GPx4+/− mice compared with WT (Figure 1A), although serum insulin levels were increased with HFHS diet to equal extent regardless of genotype (Figure 1B). GPx4 enzyme levels in liver of WT mice increased significantly as a result of HFHS diet, although GPx4+/− mice contain significantly less enzyme than WT (Figure 1C and Supplemental Figure 1A). In parallel with the increase in GPx4, levels of HNE-adducts in liver increased in WT with HFHS diet but were significantly more pronounced in GPx4+/− mice compared with WT. To determine whether the increased lipid peroxidation in the GPx4+/− mice corresponded to a more severe liver pathology following HFHS diet, liver triglycerides and collagen were measured by oil red O and picrosirius red staining, respectively. GPx4+/− mice displayed greater liver steatosis (Figure 1D) and fibrosis (Figure 1E) than WT mice following HFHS diet.
Table 1.
Body composition and metabolic parameters.
| Variables | WT CNTL |
WT HFHS |
GPx4+/-CNTL |
GPx4+/− HFHS |
p-value |
|---|---|---|---|---|---|
| Terminal body weight (g) | 34.5 ± 1.3 | 48.0 ± 2.2∗ | 35.8 ± 4.2 | 48.5 ± 1.2∗§ | <0.0001 |
| Fat mass (g) | 8.4 ± 1.0 | 17.7 ± 0.9∗ | 10.0 ± 2.9¤ | 19.4 ± 0.8∗§ | 0.0001 |
| Lean mass (g) | 21.6 ± 0.4 | 26.0 ± 1.2∗ | 22.3 ± 1.4¤ | 24.9 ± 0.4∗ | 0.004 |
| Body fat (%) | 24.0 ± 2.1 | 36.9 ± 0.5∗ | 31.3 ± 3.0∗ | 40.0 ± 0.8∗§ | <0.0001 |
| Fasting glucose (mg/dL) | 141.3 ± 4.1 | 168.6 ± 14.2∗ | 148.0 ± 3.7 | 204.8 ± 11.3∗¤§ | 0.007 |
| Cholesterol (mg/dL) | 51.0 ± 8.6 | 72.8 ± 6.5 | 34.0 ± 8.7 | 103.5 ± 17.8∗§ | 0.01 |
| Triglycerides (mg/dL) | 46.8 ± 6.3 | 44.0 ± 10.6 | 55.5 ± 1.5 | 99.5 ± 11.3∗¤§ | 0.003 |
| HDL cholesterol (mg/dL) | 74.9 ± 8.0 | 96.7 ± 7.2 | 53.1 ± 9.4 | 118.4 ± 14.4∗§ | 0.009 |
All values expressed as mean ± S.E.M., n = 4–6 per group. * P<0.05 vs. WT-CNTL vs. ¤ P<0.05 vs. WT-HFHS. § P<0.05 vs. GPX4+/- CNTL.
Figure 1.
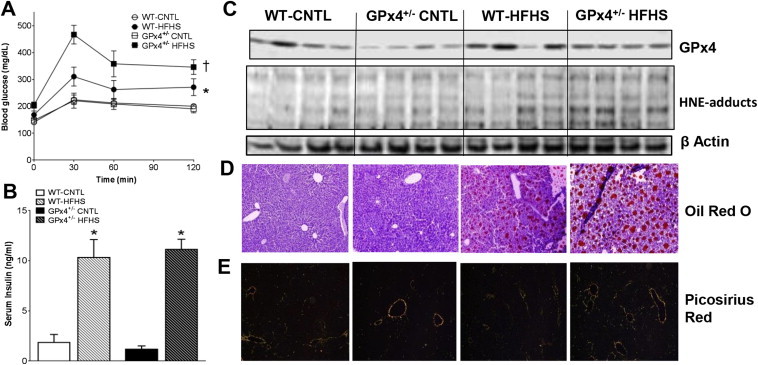
Glycemic control and liver biochemistry/pathology. Shown in A are oral glucose tolerance tests, along with fed-state serum insulin levels B after 24 weeks on the diet. In C are representative immunoblots of GPx4 and HNE-adducts in liver from 4 individual mice in each group, along with the corresponding β-actin loading control. Representative images of liver triglycerides stained with oil red O D, and collagen stained with picrosirius red under polarized light E are shown for each group. Images are representative of 16 image fields captured per mouse, n = 2–3 mice per treatment group. Data shown as mean ± S.E.M., n = 7–8 mice per group. *P < 0.05 vs. CNTL within genotype, †P < 0.05 vs. WT-HFHS.
2.2. Cardiac structural remodeling and fibrosis are exacerbated by GPx4 deficiency in obesity
Previous studies have shown that HFHS diet-induced obesity causes significant cardiac hypertrophy and left ventricular (LV) diastolic dysfunction in rodent models [46–48]. Here, HFHS diet caused an increase in cardiac mass to a similar extent between WT and GPx4+/− mice (Figure 2A,E). However, compared with WT, GPx4+/− mice display significantly greater cardiomyocyte diameter (Figure 2E,F) and fibrosis (Figure 2B,C) following HFHS diet. Obese mice also have increased levels of serum brain natriuretic peptide (BNP, Figure 2G), indicative of ventricular wall stress. This evidence of cardiomyopathy exists in the absence of any changes in LV systolic function (Figure 2H), blood pressure (Figure 2I) or heart rate (Figure 2J) in the mice following HFHS diet, indicating that any cardiomyopathy which might be present in these mice is not due to increased afterload.
Figure 2.
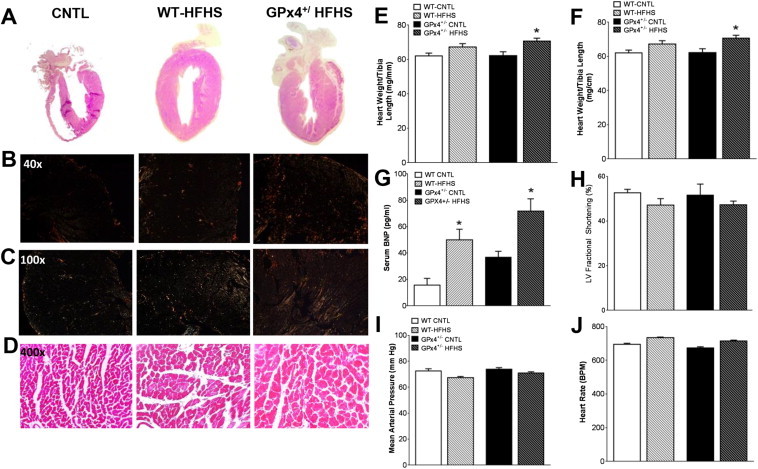
Cardiac structural and functional parameters. Panels shown are representative images of whole hearts A, cardiac collagen stained with picosirius red under polarized light B,C, and Masson's Trichrome stained cardiac tissue D, from mice within each study group. Shown in E are heart weight/tibia length ratio, cardiomyocyte diameter F, and serum BNP levels G. Cardiac contractility H, mean arterial pressure I, and heart rate J are shown for each group Images are representative of 16 image fields captured per mouse, n = 2–3 mice per treatment group. Data in E-J are shown as mean ± S.E.M., n = 6–8 mice per group. *P < 0.05 vs. WT-CNTL, †P < 0.05 vs. WT-HFHS, §P < 0.05 vs. GPx4+/--CNTL.
Recently, in two independent experimental models of cardiac hypertrophy it was observed that GPx4 and Thioredoxin Reductase (TxnRd) were the only antioxidant seleno-enzymes to increase in the heart, suggesting that they are critical for cardiac compensatory response to hypertrophic stimuli [49,50]. A significant increase in GPx4 mRNA (Figure 3A) and enzyme content (Figure 3B and Supplemental Figure 1B) in myocardium of WT mice with HFHS diet was observed. Conversely, GPx4+/− mice had lower levels of mRNA that did not correspond to any significant change in GPx4 enzyme levels following HFHS diet. Levels of HNE-adducts (Figure 3C) and protein carbonyls (Figure 3C) do increase moderately in WT hearts with HFHS diet, although GPx4 deficiency leads to a striking increase in this protein carbonylation with obesity. To determine if GPx4 deficiency and augmented carbonyl stress in obesity triggered a compensatory redox adaptive response in the heart, expression of a number of antioxidant genes were examined. GPx4+/− mice were found to have increased expression of several antioxidant genes following HFHS diet (Figure 3D).
Figure 3.
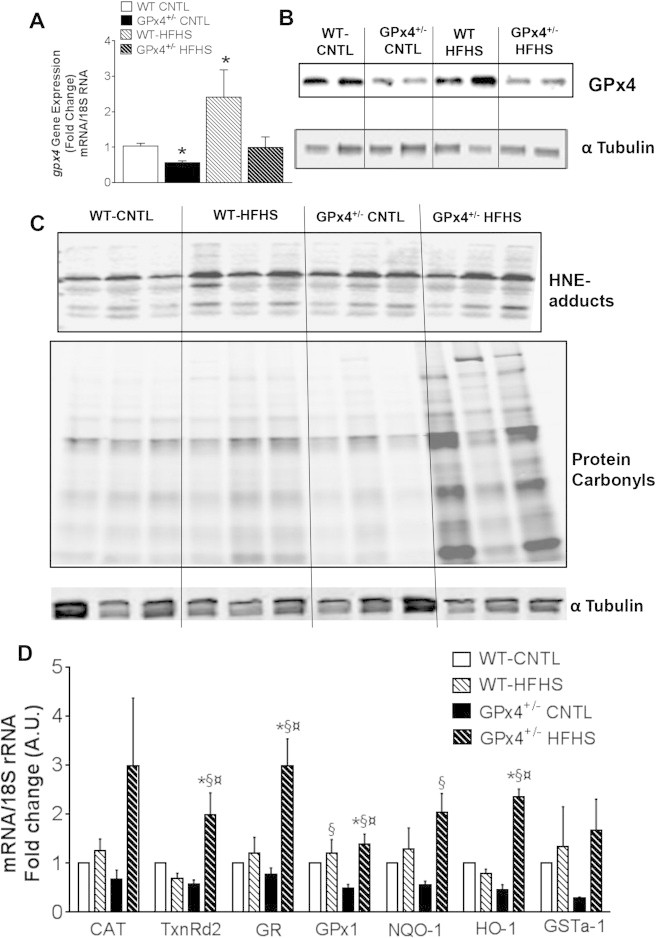
Cardiac GPx4, protein carbonylation and redox signaling. Shown here is GPx4 mRNA expression A and protein content B in whole hearts from mice used in this study. Representative immunoblot of HNE-adducts and hydrazide-labeled protein carbonyls C are shown from 3 individual mice in each group, Expression of redox and phase II detoxifying genes are shown in D. Data shown in A & D are means ± S.E.M, n = 5–7 per group. *P < 0.05 vs. WT-CNTL, ¤ P < 0.05 vs. WT-HFHS, §P < 0.05 vs. GPx4+/--CNTL. CAT-Catalase; TrxII- Thioredoxin reductase II; GR- Glutathione Reductase; GPX1- Glutathione Peroxidase1; NQO1- NAD(P)H:quinine oxidoreductase; HO-1- Heme oxygenase-1; GSTA1-glutathione s-transferase-1.
2.3. GPx4 deficiency causes mitochondrial abnormalities and up-regulation of cardiac inflammation and fibrosis signaling pathways with obesity
Mitochondrial dysfunction, as characterized by decreased ATP and/or respiration combined with increased ROS, has been linked to several obesity-related pathologies, including insulin resistance and cardiomyopathy, and recent studies have implicated a role for mitochondrial protein carbonylation in mediating this dysfunction [51–53]. Parameters of mitochondrial function in permeabilized LV myofibers were examined to assess the relationship between GPx4 deficiency, carbonyl stress, and mitochondria in the obese myocardium. GPx4 enzyme content was measured in the mitochondrial fraction isolated from whole hearts. GPx4 increased in cardiac mitochondria of WT mice with HFHS diet, while total levels of GPx4 were lower and remained that way following HFHS diet in the GPx4+/− mice (Figure 4A and Supplemental Figure 2A).
Figure 4.
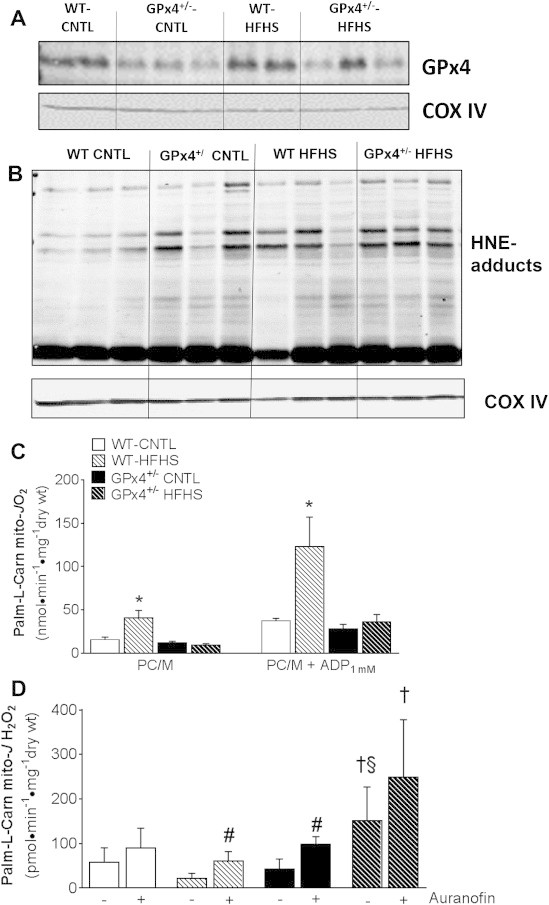
Cardiac mitochondrial GPx4 and functional parameters. A representative immunoblot of GPx4 protein A and HNE-adducts B along with corresponding COX IV loading control are shown of isolated cardiac mitochondria obtained from mice used in this study. In C are maximal ADP-stimulated rates of mitochondrial respiration (JO2) supported by palmitoyl-l-carnitine in permeabilized cardiac myofibers from these mice. Shown in D are rates of mitochondrial H2O2 emission (mito-JH2O2) in phosphorylating state supported by palmitoyl-l-carnitine + 100 μM ADP, in the absence (−) and presence (+) of the TxnRd2 inhibitor Auranofin. Data shown in C & D are means ± S.E.M, N = 4–6 per group. *P < 0.05 vs. all other groups for each respiratory state, †P < 0.05 vs. WT-HFHS, §P < 0.05 vs. GPx4+/--CNTL, #P < 0.05 for Auranofin effect within group.
It was expected that mitochondrial GPx4 deficiency would potentially lead to a greater degree of mitochondrial membrane lipid peroxidation and reactive aldehyde formation. Here, the amount of mitochondrial HNE-adducts were greater in GPx4+/− hearts than WT, and the HFHS diet increased the relative amount of these adducts in both WT and GPx4+/− mice (Figure 4B and Supplemental Figure 2B). These increased HNE-adducts may have functional consequences since recent studies have documented that mitochondrial HNE-adducts correspond to decreased oxidative phosphorylation in diabetic heart mitochondria [74,75]. Moreover, another report highlights the importance of mitochondrial fatty acid β-oxidation as a metabolic ‘sink’ for HNE [72]. In WT hearts, both basal and maximal ADP-stimulated palmitoyl-l-carnitine supported respiration was markedly increased following HFHS diet, while these rates were unchanged with HFHS diet in the GPx4+/− mice (Figure 4B).
In a previous study we reported that the selenoenzyme thioredoxin reductase-2 (TxnRd2) increased in rat heart with HFHS diet [54], corresponding to a decreased rate of mitochondrial H2O2 emission in the obese rat heart. Other groups have also reported decreased mitochondrial ROS with obesity/diabetes in mouse [55] and rat models [56]. Given our previous findings, combined with the fact that TxnRd2 expression was increased in GPx4+/− hearts with HFHS diet (Figure 3E), we sought to ascertain whether a similar adaptation in redox buffering was occurring in cardiac mitochondria of these mice. Here, using paired H2O2 emission experiments in the absence and presence of the TxnRd2 inhibitor auranofin, a dramatic (∼4-fold) increase in total H2O2 emission was observed in permeabilized myofibers prepared from obese GPx4+/− mice fed HFHS diet, while a decrease in this ROS emission was observed in the obese WT mice following HFHS diet (Figure 4C). Auranofin exposure significantly increased the rate of mitochondrial ROS in the obese WT mice but not obese GPx4+/− mice.
Chronic, low-grade inflammation in the obese heart is known to cause profibrotic and hypertrophic signaling. Although it has been difficult to fully ascertain the exact nature of the relationship between inflammation and cardiac remodeling, some investigators have proposed that activation of the receptor for advanced glycation end products (RAGE) may play a role. RAGE belongs to a family of pattern recognition receptors for AGE and has been studied extensively in the context of diabetes-related pathologies [57,58]. Increased expression of RAGE is present in the hearts of obese GPx4+/− mice (Figure 5A), and this is consistent with the increased carbonyl stress present in these hearts since reactive aldehydes such as HNE are known to potentiate AGE formation [59]. Activation of RAGE has been shown to induce chronic activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), which, in turn, activates further inflammatory pathways. Furthermore, there is extensive crosstalk between the RAGE and Transforming Growth Factor-Beta (TGF-β) family of proteins [60–62]. Notably, the hearts of obese GPx4+/− mice also displayed greater pro-inflammatory cytokine expression and genes involved in fibrosis and hypertrophy compared with WT following HFHS diet, while only a modest increase in a few select genes was observed with HFHS diet in WT hearts (Figure 5A & B).
Figure 5.
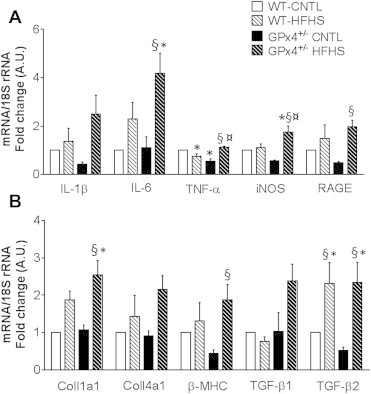
Cardiac inflammatory and pro-fibrotic/hypertrophic gene expression. Expression of pro-inflammatory A, pro-fibrotic and hypertrophy genes B are shown in hearts from all four groups of mice in this study. Β-MHC- β-Myosin Heavy Chain; Coll1a1-Collagen 1a1; Coll4a1-Collagen 4a1; TGFβ1 and 2 -Transforming Growth Factorβ 1 and 2; IL-β-Interleukin-1 Beta; IL-6 -Interleukin −6; TNF-α-Tumor Necrosis Factor-alpha; iNOS-Inducible nitric oxide synthase; RAGE- Receptor for Advanced Glycation End Products. All target genes normalized to 18S ribosome RNA. Data shown are means ± S.E.M, N = 4–6 per group. *P < 0.05 vs. WT-CNTL, ¤ P < 0.05 vs. WT-HFHS, §P < 0.05 vs. GPx4+/--CNTL.
2.4. Cardiac GPx4 deficiency is associated with increased HNE-adducts and relative risk (RR) for diabetes in humans
Next, we sought to determine whether there is an association between GPx4 and lipid-derived carbonyl stress in human heart and, also, to examine whether there is a connection between these parameters and human metabolic disease. In a small pilot study of ∼20 patients, we previously reported greater levels of HNE-protein adducts in the hearts of hyperglycemic type 2 diabetic patients, and these increased HNE-adducts were accompanied by depletion of cardiac glutathione and increased mitochondrial ROS production [63]. Here, using samples of atrial myocardium obtained from patients (n = 103) undergoing elective cardiac surgery (Table 2), we observed that diabetic patients had significantly less cardiac GPx4 than non-diabetic patients (Figure 6A), and this corresponded with greater levels of HNE-adducts in the diabetic hearts (Figure 6B). A multivariable regression analysis was performed on quartiles of cardiac GPx4 and tertiles of HNE-adducts, and cardiac GPx4 was found to be negatively associated while HNE-adducts positively associated with RR for diabetes in these patients (Table 3). To determine if hyperglycemia was associated with cardiac GPx4 or HNE-adducts in these patients, a multiple linear regression model was applied, adjusting for sex. No significant association between cardiac GPx4 or HNE-adduct levels and HbA1c was observed in these patients (Table 3 and Supplemental Figure 1).
Table 2.
Patient clinical and demographic characteristics.
| Variables | Diabetes n (%) |
No diabetes n (%) |
p-value |
|---|---|---|---|
| Age | |||
| Mean ± SD | 64 ± 9.28 | 61 ± 11 | 0.49 |
| Median (IQR) | 65 (16) | 60 (17) | |
| Sex | |||
| Female | 7 (14) | 1 (2) | |
| Male | 42 (85) | 53 (97) | 0.026 |
| Race | |||
| White | 41 (83) | 47 (86) | 0.78 |
| Black | 8 (16) | 7 (13) | |
| BMI | |||
| Mean ± SD | 31 ± 6.6 | 30 ± 5.3 | 0.46 |
| Median (IQR) | 30 (7.4) | 29 (4.5) | |
| Preoperative medications | |||
| β-blockers | 34 (69) | 41 (75) | 0.87 |
| ACEi/ARBs | 15 (31) | 22 (41) | 0.79 |
| Diuretics | 21 (43) | 20 (37) | 0.69 |
| CCBs | 10 (20) | 13 (24) | 0.89 |
| Diabetic medications | 43 (87) | 2 (4) | <0.0001 |
| Insulin | 32 (65) | 2 (4) | |
| Metformin | 15 (31) | 0 | |
| Sulfonylureas | 4 (8) | 0 | |
| Glitazones | 1 (2) | 0 | |
| GLP-1 agonist | 2 (4) | 0 | |
| DDP-4 inhibitor | 3 (6) | 0 | |
| Statins | 41 (83) | 46 (84) | 0.94 |
| Nitrates | 19 (39] | 32 (59) | 0.65 |
| Heart failure | 8 (16) | 5 (9) | 0.31 |
| LVEF (%) | 51.8 ± 3.9 | 53.6 ± 6.4 | 0.87 |
Sample size N = 103 for GPx4, N = 61 for HNE-adduct.
Figure 6.
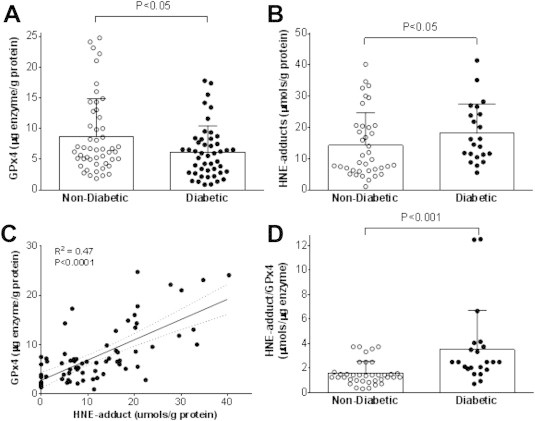
GPx4 content and HNE-adducts in human myocardium. Total GPx4 enzyme content A and HNE-adducts B are shown in atrial myocardium obtained from non-diabetic and diabetic patients undergoing elective heart surgery. Shown in C is association between HNE-adducts and GPx4 enzyme in these heart samples, and the ratio of HNE-adduct to GPx4 enzyme are depicted in D. Each symbol corresponds to one individual patient (N = 103, see Table 2 for demographics and clinical variables). P-value computed using Friedman's nonparametric test for central tendency, adjusting for sex.
Table 3.
Multivariable analysis of GPx4 and HNE-adducts in human heart.
| GPx4 content (ug/ml) | |||
| Diabetes n (%) |
No diabetes n (%) |
RR (95% CI)a | |
| Mean ± SD | 6.2 ± 4.2 | 8.7 ± 5.9 | P = 0.034b |
| Median (IQR) | 6.0 (5.2) | 6.7 (6.6) | |
| Q1 (≤3.9) | 17 (35) | 9 (17) | 1.0 Referent |
| Q2 (>3.9–6.3) | 10 (20) | 15 (28) | 0.65 (0.37–1.1) |
| Q3 (>6.3–9.4) | 13 (27) | 13 (24) | 0.74 (0.47–1.2) |
| Q4 (>9.4) | 9 (18) | 17 (31) | 0.58 (0.31–1.1) |
| Ptrend = 0.063c | |||
| HNE content (mM) | |||
| Mean ± SD | 18 ± 8.3 | 14 ± 9.8 | P = 0.028b |
| Median (IQR) | 16 (11) | 11 (12) | |
| T1 (≤9.0) | 3 (10) | 21 (47) | 1.0 Referent |
| T2 (>9.0–17) | 14 (47) | 11 (24) | 4.2 (1.4–13) |
| T3 (>17) | 13 (43) | 13 (29) | 3.9 (1.2–12) |
| Ptrend = 0.050c | |||
Relative Risk (RR) and 95% Confidence Interval (CI), adjusted for sex.
P-value computed using Friedman's Nonparametric test for central tendency, adjusting for sex.
P-value computed using the likelihood ratio trend test, adjusting for sex.
Similar to what was observed with the WT-HFHS mice, cardiac GPx4 content was positively correlated with cardiac HNE-adducts in these patients (Figure 6C), suggesting that GPx4 is an adaptive response of the tissue to persistent oxidative stress. Interestingly, the ratio of HNE-adducts/GPx4 was much higher in diabetic than non-diabetic hearts (Figure 6D), indicating that the GPx4 adaptive response has been over-whelmed or compromised in these patients.
3. Discussion
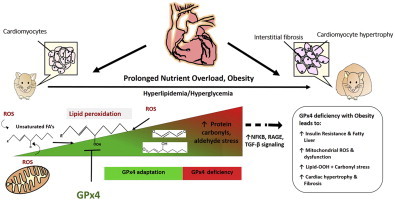
Only an increased understanding of the mechanisms underlying obesity-related pathologies will enable physicians to better manage diseases associated with this emerging epidemic. Lipid peroxides and their reactive aldehyde derivatives have been postulated to play a causal role in obesity-related cardio-metabolic diseases, and emerging epidemiologic data has reported a link between polymorphisms and mutations in gpx4 and human diseases, including obesity. Together, these previous findings suggest that GPx4 may be an adaptive component that serves a protective role against the persistent oxidative stress in obesity. The present study tested this hypothesis in a translational model of HFHS diet-induced obese GPx4+/− (i.e. GPx4 deficient) mice, combined with cardiac tissue samples obtained from patients undergoing elective cardiac surgery. The findings from this study demonstrate that GPx4 deficiency with diet-induced obesity leads to significant increases in lipid peroxide-derived aldehydes, corresponding to more severe cardio-metabolic derangements including glucose intolerance, dyslipidemia, liver steatosis, and cardiac hypertrophy and fibrosis. Abnormalities in mitochondrial function in hearts from the obese GPx4+/− mice implicate a role for the enzyme in protecting that critical organelle from oxidative damage in obesity. Furthermore, the adverse cardiac remodeling observed in the obese GPx4+/− mice was accompanied by up-regulation of genes involved in cardiac inflammation and remodeling, illustrating the importance of carbonyl stress as a causal factor in these pathways. Lastly, the relationship between decreased cardiac GPx4 and increased HNE-adducts and human metabolic disease was confirmed by studying cardiac tissue samples obtained from non-diabetic and diabetic patients. Collectively, these observations allow for a proposed model of GPx4 as a protective adaptation in obesity (as shown in the Graphical Abstract). Clearly, when lipid peroxidation increases as a result of persistent oxidative stress in obesity/nutrient overload, GPx4 is upregulated to compensate (Figures 1 and 3). However, over time and possibly due to underlying genetic causes, GPx4 becomes overwhelmed in an obese individual and HNE-adducts increase (along with numerous other LPPs that were not measured in this study). The consequence of this is increased inflammation, mitochondrial dysfunction (and likely potentiation of mitochondrial ROS), and the broad cardio-metabolic pathologies that are known to follow (e.g., diabetes, cardiomyopathy, liver disease).
These findings stand in contrast to preceding studies of diet-induced obesity in antioxidant-deficient models. One recent study of high fat-fed GPx1-deficient mice (whole body GPx1−/−) found that systemic oxidative stress increased with obesity as expected, but these mice were actually protected from insulin resistance and liver steatosis [98]. In a study using high fat diet in SOD2+/− mice (also exhibiting greater oxidative stress), insulin sensitivity was again unchanged with obesity, although a modest effect on pancreatic β-cell release of insulin was observed [99]. Another intriguing study used high fat diet in a glutathione-deficient (Gclm−/−) mouse model. The authors found that despite a huge increase in systemic oxidative stress with the diet, these mice were also protected from increased adiposity, insulin resistance and steatosis, possibly due to an increase in basal energy expenditure [100]. Given these recent findings, the fact that GPx4 haploinsufficiency alone can induce such a damaging effect on cardio-metabolic parameters with HFHS diet implies that lipid peroxides and their LPP derivatives are a distinctive form of oxidative stress that must play a very prominent etiological role in obesity-related cardio-metabolic diseases.
Deleterious effects of lipid-derived aldehydes in biological systems have been well known for many years. For example, nucleophilic attack of cysteine residues by aldehydes has negative ramifications on metabolic pathways in the cell [14,64]. A recent mass spectrometric analysis of cysteine oxidation status in hearts from mice fed a diabetogenic diet for 8 months showed that ∼40% of total oxidized cysteines are mitochondrial in origin [65]. Proteomic analysis of mitochondria from diabetic rat hearts reveals an increased formation of carbonyl adducts (e.g. HNE, MDA) with complexes of the electron transport chain and respiratory complexes, and these adducts correspond to a reduction in enzyme activity [18]. Our data suggest that GPx4 protects against cardiac mitochondrial dysfunction with obesity as the obese GPx4+/− mice had very high levels of carbonyl stress in their hearts (Figure 3), particularly HNE-adducts in the cardiac mitochondria (Figure 4B), accompanied by mitochondrial dysfunction, as compared with obese WT mice. Several studies have shown that GPx4 preserves ATP production and attenuates cytochrome c release in the mitochondria under oxidative stress [66–68]. Cardiac-specific overexpression of mitochondrial GPx4 is protective in models of cardiac ischemia/reperfusion injury and in a streptozotocin (STZ)-induced diabetes model [69,70]. GPx4 exists as both short (i.e., mitochondrial-localized) and long (i.e., cytosolic-localized) isoforms, yet only deletion of the former is lethal in mice [67]. Thioredoxin Reductase is the only other essential selenoenzyme identified to date, and this enzyme also has both mitochondrial (TxnRd2) and cytosolic (TxnRd1) isoforms [71]. In the present study, increased mitochondrial H2O2 emission was observed in hearts of lean GPx4+/− mice following the inhibition of TxnRd2 with auranofin (Figure 4C), although auranofin did not significantly affect the rate in the obese GPx4+/− mice (which was already much greater than all other groups). This suggests that mitochondrial lipid peroxide-scavenging with GPx4 and the thioredoxin redox couple are both important adaptive responses of cardiac mitochondria in obesity-related cardiac hypertrophy and that TxnRd2 cannot appropriately compensate for GPx4 deficiency within cardiac mitochondria if animals are obese.
A recent study showed that fatty acid β-oxidation is a novel HNE clearance pathway in heart [72]. Considering the diminished rates of maximal palmitoyl carnitine-supported respiration observed in the obese GPx4+/− hearts, it is plausible that the elevated carbonyl stress observed in the hearts of these mice is a result of both increased mitochondrial ROS production (which initiates more lipid peroxidation) as well as a disruption of aldehyde detoxification pathways. The decreased GPx4 and correspondingly increased HNE-adducts seen here in myocardium of diabetic patients support our previous observations of decreased palmitoyl-l-carnitine supported respiration and increased mitochondrial ROS in diabetic human hearts [42,73]. A link between mitochondrial HNE-adduct formation and decreased oxidative phosphorylation in diabetic heart mitochondria has recently been made [74,75], and our findings suggest that GPx4 deficiency may underlie these oxidative modifications. High variability in dietary fat may also be a contributor to the greater levels of cardiac HNE-adducts in the diabetic patients, as it has been reported that diets consisting of high fat mixed with carbohydrate, or high fat alone, can lead to drastically different outcomes in HNE accumulation in oxidative tissues [101]. Other studies directed at examining the role that alternative aldehyde-producing (e.g., monoamine oxidase) and detoxifying enzymes (e.g., aldehyde dehydrogenases) may have in pathologies of obese/diabetic hearts may also be useful [64]. In addition, an improved understanding of the regulation of selenocysteine status may serve as a major clue to uncovering the mechanisms that govern the progression to maladaptive remodeling in hearts of patients with metabolic disease. Epidemiological studies of Keshan's disease, a cardiomyopathy endemic to regions of China, confirm that the heart is highly susceptible to injury resulting from disturbances to selenocysteine enzymes. Cardiomyopathy in Keshan's disease manifests as acute cardiac fibrosis and mitochondrial dysfunction [76], a phenotype that is strikingly similar to the GPx4+/− mice in the present study.
Aside from measurements of liver steatosis and serum lipids, our study did not explore the potential contribution of altered lipid metabolism that might exist in these mice. Lipotoxicity is a consistent feature of cardio-metabolic disease in both humans and animal models of obesity [77–82] and therefore may contribute to the severe liver and cardiac pathology observed in the obese GPx4+/− mice. The obese GPx4+/− mice do not possess significantly greater adiposity than obese WT, as determined by % body fat. However, we cannot exclude that there may have been differences in adipose distribution between groups. It is also known that GPx4 directly modulates the lipoxygenase and cyclooxygenase II enzyme systems, and, when viewed in this context, the increased cardiac inflammation present in obese GPx4+/− mice supports a potential role for prostaglandins and other oxidized lipids in mediating the severe metabolic pathologies in this model. Thus, we cannot exclude the contribution of these mediators in the obese GPx4+/− mice. Other studies have documented that proteolysis and autophagy become compromised with carbonyl stress, and the diminished activity of these cellular processes may also explain the more severe phenotype observed in the obese GPx4+/− hearts [83,84]. None of these alternative pathways were explored in the present study.
It is anticipated that these findings will serve to lay a foundation for future mechanistic studies directed at understanding how LPPs contribute to obesity-related pathologies and the adaptive role of GPx4. Considering that the most severe effects observed in the obese GPx4+/− mice were evident in oxidative tissues (i.e., liver, heart, muscle), it will be very important in future studies to identify the cell- and tissue-specific role of GPx4 in mediating lipid peroxidation and carbonyl stress with obesity. Furthermore, our findings have clinical implications as they suggest that patients exhibiting decreased GPx4 expression and activity might be predisposed to obesity-related cardio-metabolic diseases. Indeed, clinical studies and human cell models have shown that GPx4 expression and activity vary widely in response to environmental and nutritional cues [38,45,85–88]. Finally, although our understanding of the role of lipid-derived aldehydes and carbonyl stress in the etiology of obesity-related diseases is in its infancy, it is important to recognize that compounds that scavenge HNE and other lipid-derived aldehydes specifically, such as histidine-containing dipeptides, may be very useful in this regard [89,90,102] and have experienced some success in pre-clinical testing and even clinical trials [91–93]. Thus, further research into novel therapeutics targeting carbonyl stress will be important for mitigating the numerous cardio-metabolic diseases that accompany this rising epidemic of obesity.
4. Methods
4.1. Mouse model and study design
Animal care and experimental procedures were performed with approval from the Institutional Animal Care and Use Committee of East Carolina University and were in compliance with the National Institutes of Health's Guide for Care and Use of Laboratory Animals. C57BL6/J female mice (Jackson Laboratory) were crossed to male GPx4+/− mice and pups were genotyped by polymerase chain reaction (PCR) using primers described in [43]. At 8–12 weeks, WT and GPx4+/− male and female age-matched littermates (n = 6) were randomly assigned to groups and individually housed. Mice were fed either control (CNTL-TD110367) or high fat high sucrose (HFHS-TD110365) diet from Harlan-Teklad Laboratories (Madison, WI) ad libidum for 25 weeks. The composition of this diet was a special formulation consisting of mixed saturated and n-6 PUFA (44.6% kcal/g fat), with high sucrose (34% kcal/g) content [54]. Mice were housed at controlled temperature and a 12hr light/dark cycle was maintained. At the end of the diet period, all live animal metabolic and echocardiographic measurements were made 1–2 weeks prior to tissue collection to allow for a ‘wash-out period’ to minimize artifact attributable to additional stress that may have been introduced by these procedures.
4.2. Patient enrollment and myocardial tissue collection
The Institutional Review Board of the Brody School of Medicine at East Carolina University approved all aspects of this study. Adult patients undergoing primary, non-emergent elective coronary artery bypass graft (CABG) or CABG/valve surgery between January 2011 and December 2014 were enrolled for this study. Demographic and clinical variables for these patients, including preoperative cardiovascular and metabolic drugs, are listed in Table 2 according to diabetes status. Those having prior cardiac surgery, history of arrhythmia, or severely enlarged atria (>4.0 cm) were excluded from the study. Atrial appendage biopsies were obtained from each patient as previously described [94], directly prior to institution of cardiopulmonary bypass. Myocardium was dissected from the endocardial side of the biopsy, rinsed briefly and frozen in liquid N2. All samples were rapidly processed this way to minimize protein and mRNA degradation across patients.
4.3. Metabolic parameters
Body weight of the mice was recorded on a weekly basis throughout HFHS diet. Body composition was analyzed by nuclear-MRI (EchoMRI 700 Echo Medical Systems, Houston, TX). An oral glucose tolerance test (dose-0.5 g/kg) was performed following a ∼4 h fast in week 22 of the HFHS diet period. Glucose levels were measured using a standard glucometer (OneTouch UltraMini, LifeScan, Milpitas, CA) in blood collected from a tail nick. Serum cholesterol, high-density lipoprotein (HDL), and triglycerides (TG) were analyzed using UniCel DxC 600 (Beckman Coulter, Indianapolis, IN).
4.4. Assessment of cardiovascular function
At end of the diet intervention, blood pressure was determined by non-invasive tail-cuff plethysmography (SC1000, Hattaras Instruments, Cary, NC). Mice were acclimated to the procedure for 3 days, and the average of blood pressure readings made over 3 consecutive days was recorded. A single recording was an average of 10 measurements after 5 acclimatization cycles, with duration no longer than 10 min, at approximately the same time every day. Also at this time, high-resolution echocardiography was performed on fully conscious mice using a 30 MHz transducer (Vevo 2100, VisualSonics, Toronto, ON) to assess cardiac function and structural parameters in vivo. Parasternal long- and short-axis views were captured in M-mode and recordings analyzed using system software.
4.5. Liver and cardiac histology
Mice were anesthetized with isoflurane. Hearts (n = 2/group) were stopped with KCl, fixed in Zinc Fixative then embedded in paraffin. Next, they were cut cross-sectionally, stained in Hematoxylin and eosin or picosirus red (collagen), then viewed under polarized light. Livers from these animals were fixed, embedded and stained in a similar fashion, with the exception that oil red O was used for liver triglycerides. Images of each heart were captured at 20X. The cardiomyocyte diameter of 8–10 myocytes from 5 captured images per animal was analyzed using Image J (http://imagej.nih.gov/ij/) by an individual blinded to the treatment group.
4.6. Preparation of permeabilized cardiac myofibers and mitochondrial function measurements
All procedures for these measurements have been described in detail by our group previously [42,54,73,95]. The mice were anesthetized with isoflurane and rapidly euthanized by pneumothorax. Blood was collected for serum analysis via cardiac puncture. Whole hearts were dissected, weighed, and a small portion of the left ventricle (LV) was cut for preparation of permeabilized myofibers (∼30 mg) and placed in Buffer X containing (in mM): 7.23 K2EGTA, 2.77 CaK2EGTA, 20 Imidazole, 20 Taurine, 5.7 ATP, 14.3 Phosphocreatine, 6.56 MgCl2-6H2O and 50 MES (pH 7.1, 295 mOsm). Of the remaining LV/septum, one portion was used for standard mitochondrial isolation, and another was snap frozen in liquid N2 and stored at −80 °C for further biochemical analysis. Following permeabilization in 50 μg/ml saponin for 30 min, fibers were transferred to Buffer Z containing (in mM): 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO2, 3 MgCl2-6H2O, and 5 mg/ml BSA (pH 7.4, 295 mOsm) and remained in Buffer Z with 20uM Blebbstatin on a rotator at 4 °C until analysis (<1.5 h). Fibers were then transferred to an Oroboros O2K (Oroboros Instruments, Innsbruck, Austria) where respiration was recorded in Buffer Z + 20uM Blebbistatin, 20 mM Creatine at 30 °C. Respiration (JO2) measurements were performed following addition of 125uM palmitoyl-carnitine + 1 mM malate followed by 4 mM ADP. H2O2 emission was measured using a spectrofluorometer (Photon Technology Instruments, Birmingham, NJ) in Buffer Z + 20uM Blebbistatin, 10 μM Amplex Red, 1 U/ml horseradish peroxidase, 25 U/ml superoxide dismutase. For H2O2 measurements, respiratory substrates used were 125 palmitoyl-carnitine, 2 mM Malate +100 μM ADP, 5 mM glucose and 1 U/ml hexokinase (i.e., phosphorylating state) followed by sequential addition of 5 mM glutamate then 5 mM succinate. Once mitochondrial experiments completed, fibers were collected, rinsed in ddH2O to remove excess salts, and lyophilized (Labconco, Kansas City, MO). Fibers were then weighted on a microbalance (Mettler Toledo, Denver, CO) and mitochondrial JO2 (oxygen consumption rate) and JH2O2 (peroxide emission rate) were normalized to dry weight.
4.7. Real-time qPCR of gene expression
Cardiac samples (∼10 mg) were homogenized in glass grinders (Kimble Chase, Vineland, NJ) then subjected to Proteinase K digestion. All materials used in RNA extraction were provided in RNeasy® Fibrous Tissue Mini Kit (Qiagen Inc, Valencia, CA cat# 74704). Reverse Transcription was performed using the iScript ™ cDNA Kit (Biorad Laboratories, Hercules, CA cat# 170–8891). The SsoAdvanced ™ SYBER ® Green reaction cocktail was used in the amplification and detection of DNA in RT-PCR. All protocols were performed according to product specifications unless otherwise stated. Cycle threshold (C(t)) values were converted to relative gene expression levels using the 2-ΔΔC(t) method and normalized to the level of 18S ribosomal RNA. Data are reported as fold-change in gene expression (arbitrary units) ± S.E.M. relative to WT-CNTL mice (n = 4–5 per group). The sequences and references for the primers used in qRT-PCR are listed in Supplemental Table 1.
4.8. Mitochondria isolation
The technique applied has been modified from methods previously described [96,97]. Briefly, ∼100 mg of cardiac tissue was excised and added to a petri dish with ice. The tissue was minced for 4–5 min, until it could pass through a pipette, and subjected to a brief (2 min) trypsin incubation. The mixture was transferred to a 50 mL conical tube and allowed to settle. The supernatant was removed and discarded. The remaining mixture was resuspended in 3 mL of Mitochondria Isolation Medium (MIM) (300 mM Sucrose, 10 mM Na-HEPES, 0.2 mM EDTA) with BSA, then transferred to a pre-chilled Dounce homogenizer and slowly homogenized for a total of ∼10–12 counts. The homogenate was then subjected to a series of differential centrifugation steps. The fractions of isolated mitochondria obtained (mixed subsarcolemmal and intermyofibrillar) were resuspended in a small volume of MIM + BSA and frozen at −80° for biochemical analysis.
4.9. Immunoblot and enzyme-linked immunosorbent assay (ELISA)
Cardiac tissue was homogenized in TEET Buffer (10 mM Tris, 1 mM EDTA, 1 mM EGTA + 0.5% Triton X-100), then loaded on 4–20% pre-cast polyacrylamide SDS gel (Biorad, Hercules, CA) under reducing conditions. Protein was transferred to PVDF membranes (Millipore, Bellerica, CA) and incubated with primary antibodies for GPx4 and α-tubulin (Abcam, Cambridge, UK), COX IV and β-actin (Cell Signaling Technology, Danvers MA), and HNE-adduct (Percipio Biosciences). Membranes were incubated using infrared fluorophore-conjugated secondary antibodies (LiCor Biosciences, Lincoln, NE), scanned using Odyssey Clx Infrared Imaging System (Li-Cor) and analyzed by densitometry using Image J (NIH). The absolute quantities of GPx4 and HNE-modified protein adducts in human cardiac tissue was determined by a quantitative ELISA approach developed in our lab, as described previously [54]. Fixed concentrations of HNE-adducts were made by incubating 4-hydroxynonenal (Cayman Chemical, Ann Arbor, MI) with BSA. A standard curve of both recombinant GPx4 (AbFrontier, Seoul, Korea) and HNE-modified BSA were then incubated on immunolon-coated 96-well assay plate (Fisher Scientific) along with diluted cardiac protein. Samples were incubated overnight at 4 °C, and subsequently washed with PBS+0.05% Tween-20 and blocked for 2 h with BSA or NB4025 (NOF America, White Plains, NY) at 37 °C. Samples were then incubated with primary antibodies for GPx4 and HNE adduct for 2 h at 37 °C. Samples were washed with PBS+0.05% Tween-20 and incubated with HRP-conjugated secondary antibody for 2 h at 27 °C. Following this incubation, samples were washed as before and incubated with Amplex Red (10 μM). Concentrations of GPx4 and HNE-adducts in the samples were determined by fitting to the standard curve within each plate and normalized to total protein concentration.
4.10. Hydrazide labeling of carbonyl-modified proteins
Hydrazide Cy5.5 dye (GE Healthcare Bio-Sciences, Pittsburgh, PA) was diluted in dimethyl sulfoxide (DMSO) at a stock concentration 1 mg/ml. Tissue was homogenized under an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) in 10 mM sodium phosphate buffer, containing 0.1% Triton X-100, and de-gassed using dry vacuum pump (Welch No. 25228–01, Monroe, LA). Dye was added to half of the homogenate (1:10). The samples were left under anaerobic conditions for 2 h and incubated overnight on orbital shaker at 4 °C. The other aliquot of tissue homogenate was used to quantify protein content. Portions of the labeled cardiac tissue homogenate were subjected to SDS-PAGE on 10% SDS polyacrylamide gel. The gels were scanned using Odyssey Clx Infrared Imaging System (Li-Cor) and Cy5.5-labeled proteins were analyzed by densitometry using Image J (NIH).
4.11. Statistical analysis
All animal data are presented as mean ± SEM. Statistical analysis on mouse model variables were performed with GraphPad Prism (GraphPad Prism, La Jolla, Ca.). One way ANOVA was performed on continuous variables followed by Newman–Keuls post test, with α < 0.05 considered statistically significant. In the human data analysis, categorical variables are presented as frequency and percentage and continuous variables were presented as mean ± standard deviation, mediation, median and interquartile range. Fisher exact and Chi (χ2) procedures were used to compute statistical significance of group comparisons for categorical variables. Deuchler-Wilcoxon was used for continuous variables. Biochemical variables were divided into quartiles (GPx4) or tertiles (HNE) and analyzed using a robust Poisson regression model (with relative risk as the measure of association). Missing values for all clinical and biochemical variable were imputed using the iterative expectation-maximization (EM) algorithm as described in a recent study [94]. Variables that were statistically significant in the univariable analysis were included into the multivariable analysis. P-value was computed using Friedman's Nonparametric test for central tendency while adjusting for age. Ptrend was computed using likelihood ratio trend test, adjusting for age and sex. Statistical significance was defined as P < 0.05. SAS Version 9.3 was used for all analyses of human biochemical and clinical variables.
Funding
This research was supported in part by National Institutes of Health grant 1R01HL122863 to E.J.A
Conflict of interest
The authors have no conflict of interest to declare.
Acknowledgments
This authors would like to thank Amy Bullock for assistance with animal care and breeding and Joany Oswald Zary for assistance with histology.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Ng M., Fleming T., Robinson M., Thomson B., Graetz N., Margono C. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the global burden of disease study 2013. Lancet. 2014;384(9945):766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danaei G., Finucane M.M., Lu Y., Singh G.M., Cowan M.J., Paciorek C.J. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 3.Kiencke S., Handschin R., von Dahlen R., Muser J., Brunner-Larocca H.P., Schumann J. Pre-clinical diabetic cardiomyopathy: prevalence, screening, and outcome. European Journal of Heart Failure. 2010;12(9):951–957. doi: 10.1093/eurjhf/hfq110. [DOI] [PubMed] [Google Scholar]
- 4.Selvin E., Lazo M., Chen Y., Shen L., Rubin J., McEvoy J.W. Diabetes mellitus, prediabetes, and incidence of subclinical myocardial damage. Circulation. 2014;130(16):1374–1382. doi: 10.1161/CIRCULATIONAHA.114.010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan J.N., Wilmot E.G., Leggate M., Singh A., Yates T., Nimmo M. Subclinical diastolic dysfunction in young adults with type 2 diabetes mellitus: a multiparametric contrast-enhanced cardiovascular magnetic resonance pilot study assessing potential mechanisms. European Heart Journal-Cardiovascular Imaging. 2014 doi: 10.1093/ehjci/jeu121. jeu121. [DOI] [PubMed] [Google Scholar]
- 6.Porcar-Almela M., Codoner-Franch P., Tuzon M., Navarro-Solera M., Carrasco-Luna J., Ferrando J. Left ventricular diastolic function and cardiometabolic factors in obese normotensive children. Nutrition, Metabolism & Cardiovascular Diseases. 2015;25(1):108–115. doi: 10.1016/j.numecd.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Catala A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chemistry and Physics of Lipids. 2009;157(1):1–11. doi: 10.1016/j.chemphyslip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Guichardant M., Chantegrel B., Deshayes C., Doutheau A., Moliere P., Lagarde M. Specific markers of lipid peroxidation issued from n-3 and n-6 fatty acids. Biochemical Society Transactions. 2004;32(Pt 1):139–140. doi: 10.1042/bst0320139. doi: 10.1042/ [DOI] [PubMed] [Google Scholar]
- 9.Guichardant M., Bacot S., Moliere P., Lagarde M. Hydroxy-alkenals from the peroxidation of n-3 and n-6 fatty acids and urinary metabolites. Prostaglandins Leukotrienes and Essential Fatty Acids. 2006;75(3):179–182. doi: 10.1016/j.plefa.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Refsgaard H.H., Tsai L., Stadtman E.R. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proceedings of National Academy of Sciences USA. 2000;97(2):611–616. doi: 10.1073/pnas.97.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.West J.D., Marnett L.J. Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chemical Research in Toxicology. 2005;18(11):1642–1653. doi: 10.1021/tx050211n. [DOI] [PubMed] [Google Scholar]
- 12.Uchida K. 4-hydroxy-2-nonenal: a product and mediator of oxidative stress. Progress in Lipid Research. 2003;42(4):318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 13.Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radical Biology and Medicine. 2000;28(12):1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 14.LoPachin R.M., Gavin T., Petersen D.R., Barber D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chemical Research in Toxicology. 2009;22(9):1499–1508. doi: 10.1021/tx900147g. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Januszewski A.S., Alderson N.L., Metz T.O., Thorpe S.R., Baynes J.W. Role of lipids in chemical modification of proteins and development of complications in diabetes. Biochemical Society Transactions. 2003;31(Pt 6):1413–1416. doi: 10.1042/bst0311413. doi: 10.1042/ [DOI] [PubMed] [Google Scholar]
- 16.Blair I.A. DNA adducts with lipid peroxidation products. Journal of Biological Chemistry. 2008;283(23):15545–15549. doi: 10.1074/jbc.R700051200. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siems W., Grune T. Intracellular metabolism of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24(4–5):167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 18.Chavez J.D., Wu J., Bisson W., Maier C.S. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. Journal of Proteomics. 2011 doi: 10.1016/j.jprot.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minko I.G., Kozekov I.D., Harris T.M., Rizzo C.J., Lloyd R.S., Stone M.P. Chemistry and biology of DNA containing 1, N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chemical Research in Toxicology. 2009;22(5):759–778. doi: 10.1021/tx9000489. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier C.S., Chavez J., Wang J., Wu J. Protein adducts of aldehydic lipid peroxidation products identification and characterization of protein adducts using an aldehyde/keto-reactive probe in combination with mass spectrometry. Methods in Enzymology. 2010;473:305–330. doi: 10.1016/S0076-6879(10)73016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair J., Barbin A., Velic I., Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutation Research. 1999;424(1–2):59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 22.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology and Medicine. 1991;11(1):81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 23.Shimozu Y., Hirano K., Shibata T., Shibata N., Uchida K. 4-hydroperoxy-2-nonenal is not just an intermediate but a reactive molecule that covalently modifies proteins to generate unique intramolecular oxidation products. Journal of Biological Chemistry. 2011;286(33):29313–29324. doi: 10.1074/jbc.M111.255737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paradies G., Petrosillo G., Pistolese M., Di Venosa N., Serena D., Ruggiero F.M. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radical Biology and Medicine. 1999;27(1–2):42–50. doi: 10.1016/s0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 25.Fry M., Green D.E. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. Journal of Biological Chemistry. 1981;256(4):1874–1880. [PubMed] [Google Scholar]
- 26.Chicco A.J., Sparagna G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. American Journal of Physiology – Cell Physiology. 2007;292(1):C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 27.Anderson E.J., Katunga L.A., Willis M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clinical and Experimental Pharmacology and Physiology. 2012;39(2):179–193. doi: 10.1111/j.1440-1681.2011.05641.x. 10.1111/j.1440-1681.2011.05641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai H., Hirao F., Sakamoto T., Sekine K., Mizukura Y., Saito M. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochemical and Biophysical Research Communications. 2003;305(2):278–286. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 29.Brigelius-Flohe R., Maiorino M. Glutathione peroxidases. Biochimica et Biophysica Acta. 2012 doi: 10.1016/j.bbagen.2012.11.020. 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 30.Brigelius-Flohe R. Tissue-specific functions of individual glutathione peroxidases. Free Radical Biology and Medicine. 1999;27(9–10):951–965. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- 31.Maiorino M., Thomas J.P., Girotti A.W., Ursini F. Reactivity of phospholipid hydroperoxide glutathione peroxidase with membrane and lipoprotein lipid hydroperoxides. Free radical research communications. 1991;1(12–13 Pt):131–135. doi: 10.3109/10715769109145777. [DOI] [PubMed] [Google Scholar]
- 32.Maiorino M., Gregolin C., Ursini F. Phospholipid hydroperoxide glutathione peroxidase. Methods in Enzymology. 1990;186:448–457. doi: 10.1016/0076-6879(90)86139-m. [DOI] [PubMed] [Google Scholar]
- 33.Scimeca M.S., Lisk D.J., Prolla T., Lei X.G. Effects of gpx4 haploid insufficiency on GPx4 activity, selenium concentration, and paraquat-induced protein oxidation in murine tissues. Experimental Biology and Medicine(Maywood) 2005;230(10):709–714. doi: 10.1177/153537020523001003. [DOI] [PubMed] [Google Scholar]
- 34.Morgan E.E., Faulx M.D., McElfresh T.A., Kung T.A., Zawaneh M.S., Stanley W.C. Validation of echocardiographic methods for assessing left ventricular dysfunction in rats with myocardial infarction. American Journal of Physiology - Heart and Circulatory Physiology. 2004;287(5):H2049–H2053. doi: 10.1152/ajpheart.00393.2004. doi: 287/5/H2049 [pii] 10.1152/ajpheart.00393.2004. [DOI] [PubMed] [Google Scholar]
- 35.Ran Q., Van Remmen H., Gu M., Qi W., Roberts L.J., 2nd, Prolla T. Embryonic fibroblasts from Gpx4+/- mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radical Biology and Medicine. 2003;35(9):1101–1109. doi: 10.1016/s0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- 36.Ruperez A.I., Olza J., Gil-Campos M., Leis R., Mesa M.D., Tojo R. Association of genetic polymorphisms for glutathione peroxidase genes with obesity in spanish children. Journal of Nutrigenetics and Nutrigenomics. 2014;7(3):130–142. doi: 10.1159/000368833. doi: 000368833. [DOI] [PubMed] [Google Scholar]
- 37.Polonikov A.V., Vialykh E.K., Churnosov M.I., Illig T., Freidin M.B., Vasil'eva O.V. The C718T polymorphism in the 3'-untranslated region of glutathione peroxidase-4 gene is a predictor of cerebral stroke in patients with essential hypertension. Hypertension Research. 2012;35(5):507–512. doi: 10.1038/hr.2011.213. doi: 10.1038/hr.2011.213. [DOI] [PubMed] [Google Scholar]
- 38.Crosley L.K., Bashir S., Nicol F., Arthur J.R., Hesketh J.E., Sneddon A.A. The single-nucleotide polymorphism (GPX4c718t) in the glutathione peroxidase 4 gene influences endothelial cell function: interaction with selenium and fatty acids. Molecular Nutrition & Food Research. 2013;57(12):2185–2194. doi: 10.1002/mnfr.201300216. doi: 10.1002/mnfr.201300216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du X.H., Dai X.X., Xia Song R., Zou X.Z., Yan Sun W., Mo X.Y. SNP and mRNA expression for glutathione peroxidase 4 in kashin-beck disease. British Journal of Nutrition. 2012;107(2):164–169. doi: 10.1017/S0007114511002704. [DOI] [PubMed] [Google Scholar]
- 40.Villette S., Kyle J.A., Brown K.M., Pickard K., Milne J.S., Nicol F. A novel single nucleotide polymorphism in the 3' untranslated region of human glutathione peroxidase 4 influences lipoxygenase metabolism. Blood Cells, Molecules & Diseases. 2002;29(2):174–178. doi: 10.1006/bcmd.2002.0556. doi: S1079979602905565. [DOI] [PubMed] [Google Scholar]
- 41.Houstis N., Rosen E.D., Lander E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi: 10.1038/nature04634. doi: nature04634. [DOI] [PubMed] [Google Scholar]
- 42.Anderson E.J., Lustig M.E., Boyle K.E., Woodlief T.L., Kane D.A., Lin C.T. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yant L.J., Ran Q., Rao L., Van Remmen H., Shibatani T., Belter J.G. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biology and Medicine. 2003;34(4):496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 44.Ran Q., Liang H., Ikeno Y., Qi W., Prolla T.A., Roberts L.J., 2nd Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2007;62(9):932–942. doi: 10.1093/gerona/62.9.932. doi: 62/9/932. [DOI] [PubMed] [Google Scholar]
- 45.Meplan C., Crosley L.K., Nicol F., Horgan G.W., Mathers J.C., Arthur J.R. Functional effects of a common single-nucleotide polymorphism (GPX4c718t) in the glutathione peroxidase 4 gene: interaction with sex. American Journal of Clinical Nutrition. 2008;87(4):1019–1027. doi: 10.1093/ajcn/87.4.1019. [DOI] [PubMed] [Google Scholar]
- 46.Qin F., Siwik D.A., Luptak I., Hou X., Wang L., Higuchi A. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation. 2012;125(14):1757–1764. doi: 10.1161/CIRCULATIONAHA.111.067801. S1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgmaier M., Sen S., Philip F., Wilson C.R., Miller C.C., 3rd, Young M.E. Metabolic adaptation follows contractile dysfunction in the heart of obese zucker rats fed a high-fat “western” diet. Obesity (Silver Spring) 2010;18(10):1895–1901. doi: 10.1038/oby.2009.500. doi: 10.1038/oby.2009.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson C.R., Tran M.K., Salazar K.L., Young M.E., Taegtmeyer H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of wistar rats. Biochemical Journal. 2007;406(3):457–467. doi: 10.1042/BJ20070392. doi: BJ20070392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann F.W., Hashimoto A.S., Lee B.C., Rose A.H., Shohet R.V., Hoffmann P.R. Specific antioxidant selenoproteins are induced in the heart during hypertrophy. Archives of Biochemistry and Biophysics. 2011;512(1):38–44. doi: 10.1016/j.abb.2011.05.007. 10.1016/j.abb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berndt C., Lillig C.H., Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. American Journal of Physiology – Heart and Circulatory Physiology. 2007;292(3):H1227–H1236. doi: 10.1152/ajpheart.01162.2006. doi: 01162.2006. [pii] 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 51.Curtis J.M., Hahn W.S., Stone M.D., Inda J.J., Droullard D.J., Kuzmicic J.P. Protein carbonylation and adipocyte mitochondrial function. Journal of Biological Chemistry. 2012;287(39):32967–32980. doi: 10.1074/jbc.M112.400663. doi: 10.1074/jbc.M112.400663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frohnert B.I., Bernlohr D.A. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Advances in Nutrition. 2013;4(2):157–163. doi: 10.3945/an.112.003319. doi: 10.3945/an.112.003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill B.G., Dranka B.P., Zou L., Chatham J.C., Darley-Usmar V.M. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochemical Journal. 2009;424(1):99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher-Wellman K.H., Mattox T.A., Thayne K., Katunga L.A., La Favor J.D., Neufer P.D. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. Journal of Physiology. 2013;591(Pt 14):3471–3486. doi: 10.1113/jphysiol.2013.254193. 10.1113/jphysiol.2013.254193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rindler P.M., Plafker S.M., Szweda L.I., Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. Journal of Biological Chemistry. 2013;288(3):1979–1990. doi: 10.1074/jbc.M112.412890. doi: 10.1074/jbc.M112.412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhatt N.M., Aon M.A., Tocchetti C.G., Shen X., Dey S., Ramirez-Correa G. Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. American Journal of Physiology – Heart and Circulatory Physiology. 2015;308(4):H291–H302. doi: 10.1152/ajpheart.00378.2014. doi: 10.1152/ajpheart.00378.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie J., Mendez J.D., Mendez-Valenzuela V., Aguilar-Hernandez M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE) Cell Signaling. 2013;25(11):2185–2197. doi: 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 58.Fritz G. RAGE: a single receptor fits multiple ligands. Trends in Biochemical Sciences. 2011;36(12):625–632. doi: 10.1016/j.tibs.2011.08.008. 10.1016/j.tibs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 59.Thorpe S.R., Baynes J.W. Maillard reaction products in tissue proteins: new products and new perspectives. Amino Acids. 2003;25(3–4):275–281. doi: 10.1007/s00726-003-0017-9. [DOI] [PubMed] [Google Scholar]
- 60.Biernacka A., Dobaczewski M., Frangogiannis N.G. TGF-beta signaling in fibrosis. Growth Factors. 2011;29(5):196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobaczewski M., Gonzalez-Quesada C., Frangogiannis N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. Journal of Molecular and Cellular Cardiology. 2010;48(3):504–511. doi: 10.1016/j.yjmcc.2009.07.015. 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frangogiannis N.G. Matricellular proteins in cardiac adaptation and disease. Physiological Reviews. 2012;92(2):635–688. doi: 10.1152/physrev.00008.2011. 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson E.J., Kypson A.P., Rodriguez E., Anderson C.A., Lehr E.J., Neufer P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. Journal of the American College of Cardiology. 2009;54(20):1891–1898. doi: 10.1016/j.jacc.2009.07.031. doi: S0735-1097(09)02758-2 [pii] 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LoPachin R.M., Gavin T. Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chemical Research in Toxicology. 2014;27(7):1081–1091. doi: 10.1021/tx5001046. doi: 10.1021/tx5001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Behring J.B., Kumar V., Whelan S.A., Chauhan P., Siwik D.A., Costello C.E. Does reversible cysteine oxidation link the western diet to cardiac dysfunction? FASEB Journal. 2014;28(5):1975–1987. doi: 10.1096/fj.13-233445. doi: 10.1096/fj.13-233445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang H., Ran Q., Jang Y.C., Holstein D., Lechleiter J., McDonald-Marsh T. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radical Biology and Medicine. 2009;47(3):312–320. doi: 10.1016/j.freeradbiomed.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang H., Yoo S.E., Na R., Walter C.A., Richardson A., Ran Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. Journal of Biological Chemistry. 2009;284(45):30836–30844. doi: 10.1074/jbc.M109.032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cole-Ezea P., Swan D., Shanley D., Hesketh J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free Radical Biology and Medicine. 2012;53(3):488–497. doi: 10.1016/j.freeradbiomed.2012.05.029. 10.1016/j.freeradbiomed.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 69.Baseler W.A., Dabkowski E.R., Jagannathan R., Thapa D., Nichols C.E., Shepherd D.L. Reversal of mitochondrial proteomic loss in type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2013;304(7):R553–R565. doi: 10.1152/ajpregu.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dabkowski E.R., Williamson C.L., Hollander J.M. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radical Biology and Medicine. 2008;45(6):855–865. doi: 10.1016/j.freeradbiomed.2008.06.021. 10.1016/j.freeradbiomed.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 71.Conrad M. Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochimica et Biophysica Acta. 2009;1790(11):1575–1585. doi: 10.1016/j.bbagen.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Li Q., Sadhukhan S., Berthiaume J.M., Ibarra R.A., Tang H., Deng S. 4-hydroxy-2(E)-nonenal (HNE) catabolism and formation of HNE adducts are modulated by beta oxidation of fatty acids in the isolated rat heart. Free Radical Biology and Medicine. 2013;58:35–44. doi: 10.1016/j.freeradbiomed.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anderson E.J., Rodriguez E., Anderson C.A., Thayne K., Chitwood W.R., Kypson A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. American Journal of Physiology – Heart and Circulatory Physiology. 2011;300(1):H118–H124. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lashin O.M., Szweda P.A., Szweda L.I., Romani A.M. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radical Biology and Medicine. 2006;40(5):886–896. doi: 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 75.Sverdlov A.L., Elezaby A., Behring J.B., Bachschmid M.M., Luptak I., Tu V.H. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. Journal of Molecular and Cellular Cardiology. 2015;78:165–173. doi: 10.1016/j.yjmcc.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loscalzo J. Keshan disease, selenium deficiency, and the selenoproteome. New England Journal of Medicine. 2014;370(18):1756–1760. doi: 10.1056/NEJMcibr1402199. [DOI] [PubMed] [Google Scholar]
- 77.Stanley W.C., Dabkowski E.R., Ribeiro R.F., Jr., O'Connell K.A. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circulation Research. 2012;110(5):764–776. doi: 10.1161/CIRCRESAHA.111.253104. 10.1161/CIRCRESAHA.111.253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghosh S., Rodrigues B. Cardiac cell death in early diabetes and its modulation by dietary fatty acids. Biochimica et Biophysica Acta. 2006;1761(10):1148–1162. doi: 10.1016/j.bbalip.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 79.Borisov A.B., Ushakov A.V., Zagorulko A.K., Novikov N.Y., Selivanova K.F., Edwards C.A. Intracardiac lipid accumulation, lipoatrophy of muscle cells and expansion of myocardial infarction in type 2 diabetic patients. Micron. 2008;39(7):944–951. doi: 10.1016/j.micron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Sharma S., Adrogue J.V., Golfman L., Uray I., Lemm J., Youker K. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB Journal. 2004;18(14):1692–1700. doi: 10.1096/fj.04-2263com. doi: 18/14/1692 [pii] 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 81.Stanley W.C., Recchia F.A. Lipotoxicity and the development of heart failure: moving from mouse to man. Cell Metabolism. 2010;12(6):555–556. doi: 10.1016/j.cmet.2010.11.016. S1550-4131(10)00408-0 [pii] 10.1016/j.cmet.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Opie L.H., Knuuti J. The adrenergic-fatty acid load in heart failure. Journal of the American College of Cardiology. 2009;54(18):1637–1646. doi: 10.1016/j.jacc.2009.07.024. 10.1016/j.jacc.2009.07.024; 10.1016/j.jacc.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 83.Hill B.G., Haberzettl P., Ahmed Y., Srivastava S., Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochemical Journal. 2008;410(3):525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 84.Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., Uchida K. 4-hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. identification of proteasomes as target molecules. Journal of Biological Chemistry. 1999;274(34):23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 85.Imai H. Biological significance of lipid hydroperoxide and its reducing enzyme, phospholipid hydroperoxide glutathione peroxidase, in mammalian cells. Yakugaku Zasshi. 2004;124(12):937–957. doi: 10.1248/yakushi.124.937. [DOI] [PubMed] [Google Scholar]
- 86.Nakamura T., Imai H., Tsunashima N., Nakagawa Y. Molecular cloning and functional expression of nucleolar phospholipid hydroperoxide glutathione peroxidase in mammalian cells. Biochemical and Biophysical Research Communications. 2003;311(1):139–148. doi: 10.1016/j.bbrc.2003.09.183. [DOI] [PubMed] [Google Scholar]
- 87.Sneddon A.A., Wu H.C., Farquharson A., Grant I., Arthur J.R., Rotondo D. Regulation of selenoprotein GPx4 expression and activity in human endothelial cells by fatty acids, cytokines and antioxidants. Atherosclerosis. 2003;171(1):57–65. doi: 10.1016/j.atherosclerosis.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 88.Frustaci A., Francone M., Petrosillo N., Chimenti C. High prevalence of myocarditis in patients with hypertensive heart disease and cardiac deterioration. European Journal of Heart Failure. 2013;15(3):284–291. doi: 10.1093/eurjhf/hfs169. [DOI] [PubMed] [Google Scholar]
- 89.Aldini G., Orioli M., Rossoni G., Savi F., Braidotti P., Vistoli G. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in zucker obese rats. Journal of Cellular and Molecular Medicine. 2011;15(6):1339–1354. doi: 10.1111/j.1582-4934.2010.01101.x. 10.1111/j.1582-4934.2010.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Menini S., Iacobini C., Ricci C., Scipioni A., Blasetti Fantauzzi C., Giaccari A. D-carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a western diet through reduction of carbonyl stress and inflammation. British Journal of Pharmacology. 2012;166(4):1344–1356. doi: 10.1111/j.1476-5381.2012.01834.x. 10.1111/j.1476-5381.2012.01834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kass D.A., Shapiro E.P., Kawaguchi M., Capriotti A.R., Scuteri A., deGroof R.C. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104(13):1464–1470. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 92.Little W.C., Zile M.R., Kitzman D.W., Hundley W.G., O'Brien T.X., Degroof R.C. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. Journal of Cardiac Failure. 2005;11(3):191–195. doi: 10.1016/j.cardfail.2004.09.010. doi: S1071916404009017. [DOI] [PubMed] [Google Scholar]
- 93.Tsujita K., Shimomura H., Kaikita K., Kawano H., Hokamaki J., Nagayoshi Y. Long-term efficacy of edaravone in patients with acute myocardial infarction. Circulation Journal. 2006;70(7):832–837. doi: 10.1253/circj.70.832. doi: JST.JSTAGE/circj/70.832. [DOI] [PubMed] [Google Scholar]
- 94.Anderson E.J., Efird J.T., Davies S.W., O'Neal W.T., Darden T.M., BS Thayne K.A., Katunga L.A. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. Journal of the American Heart Association (JAHA) 2014;3(1):e000713. doi: 10.1161/JAHA.113.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anderson E.J., Neufer P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. American Journal of Physiology. Cell physiology. 2006;290(3):C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 96.Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature Protocols. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. doi: nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 97.Gostimskaya I., Galkin A. Preparation of highly coupled rat heart mitochondria. Journal of visualized experiments. 2010;(43) doi: 10.3791/2202. pii: 2202. doi(43):10.3791/2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Merry T.L., Tran M., Stathopoulos M., Wiede F., Fam B.C., Dodd G.T. High-fat-fed obese glutathione peroxidase 1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxid Redox Signal. 2014 May 10;20(14):2114–2129. doi: 10.1089/ars.2013.5428. [DOI] [PubMed] [Google Scholar]
- 99.Kang L., Dai C., Lustig M.E., Bonner J.S., Mayes W.H., Mokshagundam S. Heterozygous SOD2 deletion impairs glucose-stimulated insulin secretion, but not insulin action, in high-fat-fed mice. Diabetes. 2014 Nov;63(11):3699–3710. doi: 10.2337/db13-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kendig E.L., Chen Y., Krishan M., Johansson E., Schneider S.N., Genter M.B. Lipid metabolism and body composition in Gclm(-/-) mice. Toxicology and Applied Pharmacology. 2011 Dec 15;257(3):338–348. doi: 10.1016/j.taap.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li Q., Tomcik K., Zhang S., Puchowicz M.A., Zhang G.F. Dietary regulation of catabolic disposal of 4-hydroxynonenal analogs in rat liver. Free Radical Biology and Medicine. 2012 Mar 15;52(6):1043–1053. doi: 10.1016/j.freeradbiomed.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aldini G., Carini M., Yeum K.J., Vistoli G. Novel molecular approaches for improving enzymatic and nonenzymatic detoxification of 4-hydroxynonenal: toward the discovery of a novel class of bioactive compounds. Free Radical Biology and Medicine. 2014 Apr;69:145–156. doi: 10.1016/j.freeradbiomed.2014.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.