

Significance
Although metabolic interactions have long been implicated in the assembly of microbial communities, their general prevalence has remained largely unknown. In this study, we systematically survey, by using a metabolic modeling approach, the extent of resource competition and metabolic cross-feeding in over 800 microbial communities from diverse habitats. We show that interspecies metabolic exchanges are widespread in natural communities, and that such exchanges can provide group advantage under nutrient-poor conditions. Our results highlight metabolic dependencies as a major driver of species co-occurrence. The presented methodology and mechanistic insights have broad implications for understanding compositional variation in natural communities as well as for facilitating the design of synthetic microbial communities.
Keywords: community metabolism, syntrophy, cooperation, metabolic modeling, naturalization theory
Abstract
Microbial communities populate most environments on earth and play a critical role in ecology and human health. Their composition is thought to be largely shaped by interspecies competition for the available resources, but cooperative interactions, such as metabolite exchanges, have also been implicated in community assembly. The prevalence of metabolic interactions in microbial communities, however, has remained largely unknown. Here, we systematically survey, by using a genome-scale metabolic modeling approach, the extent of resource competition and metabolic exchanges in over 800 communities. We find that, despite marked resource competition at the level of whole assemblies, microbial communities harbor metabolically interdependent groups that recur across diverse habitats. By enumerating flux-balanced metabolic exchanges in these co-occurring subcommunities we also predict the likely exchanged metabolites, such as amino acids and sugars, that can promote group survival under nutritionally challenging conditions. Our results highlight metabolic dependencies as a major driver of species co-occurrence and hint at cooperative groups as recurring modules of microbial community architecture.
Microbial communities are ubiquitous in nature and exert a large influence on our environment and health (1–5). These communities exhibit a great compositional variety, ranging from hot-spring assemblies with low species diversity (6) to the human gut microbiota harboring hundreds of species (7, 8). Competition for metabolic resources can affect community composition through competitive exclusion or by facilitating niche differentiation (9–11). Cooperative and syntrophic interactions, such as beneficial metabolic exchanges, are also likely to play an important role because they can substantially alter the nutritional quality of the habitat (8, 9, 11–15). For example, cross-feeding of metabolic by-products such as ethanol and acetate is central to the diversity of cellulose-degrading communities (16). However, such metabolic exchanges are difficult to discover in natural communities, because the metabolites in the environment cannot be easily attributed to a particular donor species or to the abiotic sources. Moreover, species can often use and secrete a large number of metabolites (17, 18), further hampering the elucidation of metabolic exchanges. Here, we tackle these challenges by introducing a modeling approach applicable to large microbial communities. Currently available methods for simulating metabolic exchanges (8, 19–22) are not directly relevant to communities occurring in nature. Whereas some of these methods use only topological information, ignoring mass balance and growth constraints, the others require prior knowledge of metabolic objective functions of the member species (i.e., evolutionarily selected beneficial characteristics such as high growth rate or optimal ATP production)—information that is often not available. In contrast, our modeling approach, termed “species metabolic interaction analysis,” or SMETANA, can be readily applied with as little information as species identity and their genome sequences. Starting with a community metabolic model assembled from the member-species-level models, SMETANA maps all possible interspecies metabolic exchanges. The methodology thus provides an unbiased estimate of the metabolic interaction potential of a community as well as identifies likely exchanged metabolites. We used this approach to interrogate over 800 microbial communities and co-occurring subcommunities therein. To capture interacting species modules beyond pairs, we also considered subcommunities with simultaneous co-occurrence of up to four species. Our results highlight metabolic dependencies as a key biotic force shaping the composition of diverse microbial communities in nature.
Results
Sample Communities and Co-occurring Subcommunities.
We used a previously published compilation of 16S ribosomal RNA sequences, spanning habitats as diverse as soil, water, and the human gut, to obtain the species composition for 1,297 communities (261 species in total, see Methods, Fig. S1, and Table S1) (23). To spot functional dependencies between species, we next analyzed co-occurrence patterns in these sample communities (Fig. 1A). To account for the possibilities of higher-order interactions involving more than two species, we broadened the concept of binary co-occurrence (23–25) to simultaneous occurrence of up to four species (Methods). This identified 7,221 significantly co-occurring subcommunities [Fisher’s exact test, false discovery rate (FDR) 0.01]; 95% of these consisted of triplet or quadruplet subcommunities (Table S1). These subcommunities exhibit a variety of inter- as well as intraphylum relationships (Fig. 1B). Although Proteobacteria, Firmicutes, and Actinobacteria represent a large fraction of these communities, we observed significantly higher interphyla relationships than expected by chance (P = 2.6e−12). In terms of species participation, 15% of species are unique to triplets or quadruplets (Fig. 1C), highlighting the recurring patterns of multispecies interactions that cannot be inferred from simple binary associations.
Fig. 1.
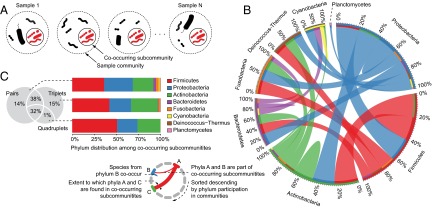
Higher-order species co-occurrence in microbial communities. (A) We consider community composition at two different levels. Sample communities are composed of all species identified by 16S ribosomal RNA in sampling sites. Co-occurring subcommunities are species groups found together more often than expected by chance (Methods), and are thus likely to be functionally dependent. (B) Inter- and intraphyla interactions in co-occurring subcommunities including 381 pairs, 3,322 triplets, and 3,518 quadruplets. (C) Species overlap and distribution of phyla among the co-occurring subcommunities of different size.
Resource Competition Is Predominant in Sample Communities.
We next reconstructed genome-scale metabolic models for the 261 mapped species using the ModelSEED pipeline (26) and curated them to reduce the possibilities of predicting spurious metabolic exchanges (Methods). These species-level models were then combined in multispecies metabolic reconstructions representing all sample communities and co-occurring subcommunities (Methods and Table S2). To assess the degree of metabolic competition in these communities, we devised a metric termed metabolic resource overlap (MRO), defined as the maximum possible overlap between the minimal nutritional requirements of all member species (Fig. 2A and Methods).
Fig. 2.

Degree of resource competition in microbial communities. (A) The concept of MRO—an intrinsic community property providing an upper limit on the degree of resource competition. The algorithm used for the MRO calculation is described in Methods. (B) Biological relevance of the MRO metric. Communities consisting of phylogenetically closely related member species show high resource overlap as expected. Red line indicates the best fit as determined by least squares linear regression analysis. (C) Resource competition is predominant in microbial communities seen as a whole. MRO values for the sample communities of different sizes and random controls are shown. P values were computed using the Wilcoxon rank sum test.
MRO is an intrinsic property of any community, independent of the habitat and robust in view of multiple minimal nutritional requirements for any (or all) member species, and providing an upper bound on the overall degree of resource competition. We observed a significant negative correlation between MRO and phylogenetic relatedness of member species (Fig. 2B, P < 10−15, r = −0.45), supporting the expectation of closely related species to be similar in their nutritional requirements, and so attesting to the biological relevance of the reconstructed metabolic models.
Sample communities of all sizes featured significantly higher resource competition than random assemblies (Fig. 2C, P < 0.05), suggesting a central role for habitat filtering in community assembly. In contrast, resource competition (as reflected in MRO) and phylogenetic diversity in co-occurring subcommunities were found to be only weakly distinguishable from random controls (Fig. S2), prompting us to explore whether species co-occurrence could be explained by their ability to metabolically complement each other.
Co-occurring Subcommunities Feature High Metabolic Interaction Potential.
To quantify the propensity of a given community to exchange metabolites, we devised a metric termed metabolic interaction potential (MIP). MIP is defined as the maximum number of essential nutritional components that a community can provide for itself through interspecies metabolic exchanges (Fig. 3A and Methods). The higher the MIP, the larger the potential of a community to benefit from the complementary biosynthetic capabilities of its member species. Like MRO, MIP is also an intrinsic property of any community, solely determined by the biosynthetic and metabolite transport capabilities of its member species. We note that species pairs within the same taxonomic rank exhibit low MIP and high MRO values (Fig. S3), characteristic of similar metabolic capabilities, and thus suggesting robustness of our results with regard to errors in species mapping. As the collective biosynthetic ability of a community would increase (or remain unchanged) with increasing membership, so would the MIP. Accordingly, we observed a positive relationship between MIP and community size (Fig. 3B). This result was unchanged when including 1,200 additional genome-sequenced species (R2 = 0.98, comparing average MIPs of communities of different size, calculated with and without additional species), suggesting that our analysis is representative of the currently known metabolic diversity.
Fig. 3.

MIP of microbial communities. (A) Illustration of the concept of MIP. A community can use the biosynthetic capabilities of its members to decrease the collective dependence on nutritional availability from the environment. (B) MIP as a function of community size. For each community size, results of simulations based on 1,000 randomly assembled communities are shown. (C) Sample communities display lower than expected interaction potential in line with their high degree of resource competition.
In the case of sample communities, the MIP was significantly lower than expected based on random assemblies (Fig. 3C, P < 0.05). In clear contrast, we find that the interaction potentials of the co-occurring subcommunities are significantly higher than those of the random controls (Fig. 4A and Fig. S4A, P < 10−4, < 10−15, and < 10−15 for pairs, triplets, and quadruplets, respectively), underlining a central role for metabolic exchanges in determining co-occurrence. Because habitat filtering is likely to result in a lower MIP (Fig. 3C), we further verified the enrichment of MIP in co-occurring subcommunities against a more restricted background of habitat-filtered controls (Fig. S4C). In this background, each control community was assembled such that all members together belonged to at least one sample, mimicking habitat-filtered natural communities.
Fig. 4.

Co-occurring subcommunities feature high metabolic interaction potential. (A) MIPs of triplet and quadruplet subcommunities (red density plots) against the background of random assemblies (gray density plots, 10,000 groups). Shown MIP values are normalized by the number of member species. (B) Co-occurring subcommunities (red density plots) show stronger metabolic coupling than non-co-occurring groups (gray density plots, 10,000 groups). (C) Distinction of co-occurring subcommunities in various cooperation (MIP and SMETANA score) and competition (resource overlap and phylogenetic distance) metrics. Error bars mark the 5th and 95th percentile of ratios between these metrics for co-occurring subcommunities and the corresponding values for 1,000 random assemblies. (D) Metabolite classes likely to be exchanged in co-occurring subcommunities as predicted by SMETANA. Numbers mark the scale of log-fold enrichment over non-co-occurring groups. P < 10−7 (amino acids), < 10−5 (carbohydrates), < 10−2 (nucleosides), and 0.039 (organic acids). (E) Removal of mutualistic metabolite exchanges from simulated co-occurring communities diminishes the contrast to random assemblies. Error bars mark the 5th and 95th percentile of ratios between the number of edges in co-occurring communities and 1,000 random assemblies of the same size.
We next ensured that the higher interaction potential of co-occurring subcommunities is a phenomenon independent of uneven species distribution in these groups or across different habitat types. We excluded species frequently participating in the co-occurring subcommunities, either alone or in combination, and observed the same degree of MIP enrichment, confirming that our results are not influenced by a few highly interactive species (Table S3 and Fig. S5 A–D, P < 10−15). Likewise, the discriminating power of MIP was found to be robust toward differences in the gap-filled reactions (reactions with no direct genetic or biochemical evidence) across species models (Fig. S5E). Finally, the enrichment of MIP in co-occurring subcommunities could be detected even in the subset of nutritionally rich habitats where incentive for metabolic cross-feeding is expected to be lower (Fig. S4D and Methods). High MIP thus seems to be a general feature of cross-habitat co-occurrence.
Co-occurring Subcommunities Are Strongly Coupled and Enriched in Mutualistic Interactions.
Metabolic networks of most species contain multiple alternative pathways for producing biomass precursors and secreted by-products. Moreover, multiple community members can secrete a given metabolite. Consequently, numerous scenarios of interspecies metabolic exchanges can equivalently support the growth of member species in any given community. To quantify this cross-feeding plasticity, we applied SMETANA, a mixed-integer linear programming method, to identify metabolic exchanges essential for the survival of the community in a minimal medium. Although the growth of community members was imposed as a constraint, the space of all possible metabolic exchanges was systematically enumerated by solving a series of mixed-integer linear programming problems (Methods). We note that SMETANA is free from arbitrary assumptions of growth optimality, either at the species or the community level. We verified the ability of SMETANA to correctly identify interspecies metabolite exchanges by reproducing experimentally mapped interactions in a well-studied three-species bacterial community (27) (Fig. S6A) and in a recently reported yeast–algal community (28) (Fig. S6B).
The simulation results for each community were summarized as SMETANA score, which estimates the strength of metabolic coupling in the community through enumerating possible metabolite exchanges (Methods). Notably, we observed twofold higher coupling in the co-occurring subcommunities compared with random controls, indicating a high degree of dependency on exchanged metabolites (Fig. 4 B and C and Fig. S4B). To gain mechanistic insight into the exchanged metabolites, we next analyzed which metabolic exchanges are characteristic to the co-occurring subcommunities. We found that the most frequently exchanged metabolites were amino acids and sugars (Fig. 4D), which are essential nutrients for many microbes and have been previously implicated in cross-feeding interactions (16, 29). Obtaining a higher-resolution view of the exchanged metabolites in any given community would require higher accuracy in metabolic models (e.g., in terms of biomass composition) than can be currently achieved owing to limited data. We note that the models of species forming co-occurring subcommunities as well as random controls included transport reactions for these compounds and their exchanges were significantly overrepresented in the subcommunities (P < 10−7 and < 10−5 for amino acids and carbohydrates, respectively). We also observe that the transport reactions associated with the predicted metabolite exchanges are enriched in those with genetic or biochemical evidences (i.e., non–gap-filled reactions, P = 0.04, Fisher’s exact test). Furthermore, the co-occurring subcommunities were strongly enriched in mutualistic metabolite exchanges (Fisher’s exact test; P < 10−4, < 10−15, and < 10−15, odds ratio = 1.68, 2.15, and 2.02 for pairs, triplets, and quadruplets, respectively). Indeed, removal of mutualistic links from co-occurring subcommunities makes them indistinguishable from random controls (Fig. 4E). This enrichment of mutualistic exchanges, together with the strong metabolic coupling, brings forward metabolic interdependency as a distinctive feature of co-occurrence.
Discussion
Accounting for multispecies co-occurrence and the use of a modeling methodology that is independent of any optimality assumptions allowed us to comprehensively map the space of metabolic interactions in diverse microbial communities. Because our simulations were performed under the conditions of limited nutritional availability, the predicted metabolic exchanges represent latent interactions that can manifest in an environment-dependent manner. Several examples of metabolic exchanges arising in nutritionally limiting environment have been reported (12, 28), endorsing the biological relevance of our findings. The enrichment of MIP, even when considering nutritionally rich habitats (Fig. S4D), suggests that the group advantage of metabolic synergy is not limited to poor environments and can also manifest due to, for example, temporal variation in nutritional availability. This finding, together with the mutualistic nature of predicted metabolic exchanges, hints at metabolic cooperation as a key driver of co-occurrence. Our results also provide insight into how competitive and cooperative forces simultaneously act to shape the community composition. Whereas resource competition is apparent in all communities due to habitat filtering, mutualistic interactions are prominent in co-occurring subcommunities.
The observed association between co-occurrence and metabolic dependence suggests a novel interpretation of Darwin’s naturalization hypothesis (30). The naturalization hypothesis implies that co-occurring species are likely to be metabolically dissimilar due to the risk of competitive exclusion. In turn, we find that the distinguishing feature of co-occurrence is not the dissimilarity that reduces resource competition, but rather the dissimilarity leading to complementary biosynthetic capabilities (Fig. 4C). Co-occurring groups can thus make efficient use of limited resources through metabolite exchange, providing a survival advantage and enabling coexistence in diverse niches.
Methods
Species Mapping.
The 16S rRNA sequences, clustered into operational taxonomic units (OTUs, 97% similarity threshold), were obtained from Chaffron et al. (23). These OTUs were mapped, using a stringent sequence similarity criterion (>95% sequence identity, >95% query sequence overlap), to species for which genome sequences were publically available in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (31). The 16S rRNA genes for the species with sequenced genomes were retrieved using the KEGG API. In cases where more than one 16S rRNA gene was present in the genome, we used the longest sequence. Whereas each OTU was mapped uniquely to a single genome using the BLAST bit score, a given genome could be mapped to multiple OTUs (Table S1 and Fig. S1).
Co-occurrence Statistics.
Fisher’s exact test was used to evaluate significance of co-occurrence for all possible combinations of two-, three-, and four- species subcommunities. In the cases of the three- and four-species groups, three and four different contingency tables were built, respectively. For example: in the case of a triplet (A, B, C), the contingency tables included counts of all sites where B and C were present but not A, A and C were present but not B, and where A and B were present but not C. P values were adjusted for multiple testing using the Benjamini–Hochberg procedure (32), as implemented in the multtest package from the Bioconductor toolbox (www.bioconductor.org). Inter- and intraphyla interactions were plotted (Fig. 1C) using Circos software (33).
Phylogenetic Relatedness of Communities.
A phylogenetic tree for 847 species was built based on the National Center for Biotechnology Information (NCBI) taxonomy using a set of 40 ubiquitous, single-copy marker genes (34) as described in Minguez et al. (35). In detail, we extracted the taxonomic tree of the 847 species from the NCBI taxonomy, which is known to be accurate for most taxa (36). Next, we generated alignments using AQUA (37) from 40 universal single copy phylogenetic marker genes (34), and combined the alignments with the tree topology of the NCBI taxonomy tree by using PhyML (38). The resulting tree included branch lengths and was manually curated. Genomes that had an erroneous placement in the NCBI taxonomy tree were removed. The final tree includes 35 eukaryotes, 43 archaea, and 769 bacteria.
Overall phylogenetic distance for a community was calculated as the average of the distances between all species pairs. In a few cases where the species was not present in the phylogenetic tree, another species was randomly chosen from the same genus.
Species-Level Model Reconstruction and Curation.
The ModelSEED pipeline (26) was used to reconstruct genome-scale metabolic models for a total of 1,503 bacterial species. In brief, given the constraints on nutritional availability from the environment, these models can be used to simulate growth and metabolite production capabilities of corresponding species (39). These models were modified to improve reaction directionality and nutrient transport information. These curation steps were necessary to reduce the artifacts of the automated model reconstruction process that can lead to inaccurate prediction of metabolic interactions. The directions for all irreversible reactions in our models were compared with those in the manually reconstructed models (16 models in total, SI Methods). In the case of reactions with inconsistent directions in manual reconstructions, the correct directionality was resolved by majority voting. A mixed-integer linear programming (MILP) routine maximizing the number of corrections was used to ensure that the altered reaction directionalities did not result in infeasible models (i.e., models incapable of biomass production). Finally, amino acid transport reactions were added to the models, if missing, to replace dipeptides represented in the ModelSEED potential nutrient space (SI Methods).
Community Metabolic Modeling.
A conceptual representation of multispecies models was adopted from ref. 19 and extended to include more than two species. All flux simulations were performed under aerobic as well as anaerobic conditions without any observable difference in the results. The presented results are from simulations under aerobic conditions (Table S2). All modeling procedures were implemented in C++ and solved using IBM ILOG CPLEX solver.
MIP Calculation.
For a group of N species, MIP was calculated as the difference between the minimal number of components required for the growth of all members in a noninteracting community (M) and an interacting community (I) (Fig. 3A and Eq. 1). The minimal nutritional requirements were calculated in a similar manner to that described in ref. 20. Inorganic compounds (including water and CO2) were assumed to be always present in the external environment (Table S2). In a noninteracting community, the member species were constrained so as to be exclusively dependent on the nutrients available from the abiotic environment. In contrast, species in an interacting community were free to use metabolites secreted by the other members.
| [1] |
Note that MIP values shown in all density plots are normalized by number of community members.
MRO Calculation.
For every member i in a group of N species, the set of minimal nutritional components required for growth, Mi, was estimated under the interacting community assumption (see Methods, MIP Calculation). The nutrient availability in the abiotic environment was limited to the minimal requirements under the noninteracting conditions (see Methods, MIP Calculation). Nutritional requirement sets Mi were used to compute MRO as per Eq. 2:
| [2] |
SMETANA Score.
The SMETANA score for a community was calculated as the sum of all interspecies dependencies under a given nutritional environment. The growth dependency of species A on metabolite m produced by species B was calculated as a product of three separate scores: (i) species coupling score (SCS), (ii) metabolite uptake score (MUS), and (iii) metabolite production score (MPS). The dependency scores were normalized to range between 0 (complete independence) and 1 (essentiality).
-
i)
The SCS was used to measure the dependency of the growth of a given species A on the presence of another species B in a community of N members. A MILP problem was set up to identify the minimal number of member species necessary to support the growth of the target species A. Once such a set of donor species was identified, this set was eliminated as a potential solution by adding an appropriate constraint to the MILP problem, and subsequently the MILP problem was resolved to identify the next donor set. All possible donor sets were identified in this fashion. SCS was then calculated as the fraction of solutions in which species B was present. Note that this procedure is exhaustive and accounts for direct as well as indirect dependencies.
-
ii)
The MUS was used to measure the growth dependency of a given species A on metabolite m donated by the other community members. MUSs were calculated using a MILP-based algorithm similar to that used for SCS calculation (see above), with the minimal donor sets replaced by the sets of minimal metabolite requirements.
-
iii)
A linear programming (LP) problem was used to calculate the MPS—a binary score indicating whether a given species B can produce metabolite m (MPS = 1) or not (MPS = 0) in the community of N members.
All LP and MILP routines used imposed mass balance constraints on the intracellular as well as the exchanged metabolites. See SI Methods for details.
Curation of Metabolic Uptakes/Exchanges.
Metabolites that cannot be used by microorganisms as primary sources of carbon or nitrogen (e.g., vitamins), but were not restricted from such uses in the automatically reconstructed models obtained from the ModelSEED pipeline, were identified through a systematic analysis using flux balance analysis. Constraints were imposed on the maximum uptake rates of these metabolites as long as the species growth was not limited (as they could use the other, commonly used, C or N sources).
Statistical Tests.
All statistical analyses were performed using the software R (www.r-project.org). Comparisons between the distributions of MIP, MRO, and phylogenetic distances were performed using Wilcoxon rank sum test.
Supplementary Material
Acknowledgments
We thank O. Barabas and S. Sheridan for critical discussions and feedback on the manuscript.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421834112/-/DCSupplemental.
References
- 1.Baker BJ, Banfield JF. Microbial communities in acid mine drainage. FEMS Microbiol Ecol. 2003;44(2):139–152. doi: 10.1016/S0168-6496(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 2.Andersen R, Chapman SJ, Artz RRE. Microbial communities in natural and disturbed peatlands: A review. Soil Biol Biochem. 2013;57:979–994. [Google Scholar]
- 3.Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459(7244):193–199. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 4.Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 5.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 6.Inskeep WP, Jay ZJ, Tringe SG, Herrgård MJ, Rusch DB. YNP Metagenome Project Steering Committee and Working Group Members The YNP Metagenome Project: Environmental parameters responsible for microbial distribution in the Yellowstone geothermal ecosystem. Front Microbiol. 2013;4:67. doi: 10.3389/fmicb.2013.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin J, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci USA. 2013;110(31):12804–12809. doi: 10.1073/pnas.1300926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster KR, Bell T. Competition, not cooperation, dominates interactions among culturable microbial species. Curr Biol. 2012;22(19):1845–1850. doi: 10.1016/j.cub.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8(1):15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. HilleRisLambers J, Adler PB, Harpole WS, Levine JM, Mayfield MM (2012) Rethinking community assembly through the lens of coexistence theory. Annu Rev Ecol Evol Syst 43(1):227–248.
- 12.Morris BE, Henneberger R, Huber H, Moissl-Eichinger C. Microbial syntrophy: Interaction for the common good. FEMS Microbiol Rev. 2013;37(3):384–406. doi: 10.1111/1574-6976.12019. [DOI] [PubMed] [Google Scholar]
- 13.Grosskopf T, Soyer OS. Synthetic microbial communities. Curr Opin Microbiol. 2014;18:72–77. doi: 10.1016/j.mib.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jousset A, Eisenhauer N, Materne E, Scheu S. Evolutionary history predicts the stability of cooperation in microbial communities. Nat Commun. 2013;4:2573. doi: 10.1038/ncomms3573. [DOI] [PubMed] [Google Scholar]
- 15.Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev. 2007;71(4):653–670. doi: 10.1128/MMBR.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato S, Haruta S, Cui ZJ, Ishii M, Igarashi Y. Network relationships of bacteria in a stable mixed culture. Microb Ecol. 2008;56(3):403–411. doi: 10.1007/s00248-007-9357-4. [DOI] [PubMed] [Google Scholar]
- 17.Paczia N, et al. Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb Cell Fact. 2012;11:122. doi: 10.1186/1475-2859-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barve A, Wagner A. A latent capacity for evolutionary innovation through exaptation in metabolic systems. Nature. 2013;500(7461):203–206. doi: 10.1038/nature12301. [DOI] [PubMed] [Google Scholar]
- 19.Stolyar S, et al. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol. 2007;3:92. doi: 10.1038/msb4100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klitgord N, Segrè D. Environments that induce synthetic microbial ecosystems. PLOS Comput Biol. 2010;6(11):e1001002. doi: 10.1371/journal.pcbi.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zomorrodi AR, Maranas CD. OptCom: A multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLOS Comput Biol. 2012;8(2):e1002363. doi: 10.1371/journal.pcbi.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuang K, et al. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011;5(2):305–316. doi: 10.1038/ismej.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaffron S, Rehrauer H, Pernthaler J, von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20(7):947–959. doi: 10.1101/gr.104521.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faust K, et al. Microbial co-occurrence relationships in the human microbiome. PLOS Comput Biol. 2012;8(7):e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6(2):343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry CS, et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28(9):977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 27.Miller LD, et al. Establishment and metabolic analysis of a model microbial community for understanding trophic and electron accepting interactions of subsurface anaerobic environments. BMC Microbiol. 2010;10:149. doi: 10.1186/1471-2180-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hom EF, Murray AW. Plant-fungal ecology. Niche engineering demonstrates a latent capacity for fungal-algal mutualism. Science. 2014;345(6192):94–98. doi: 10.1126/science.1253320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stadie J, Gulitz A, Ehrmann MA, Vogel RF. Metabolic activity and symbiotic interactions of lactic acid bacteria and yeasts isolated from water kefir. Food Microbiol. 2013;35(2):92–98. doi: 10.1016/j.fm.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Darwin C. On the Origin of Species. Appleton; New York: 1871. [Google Scholar]
- 31.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- 33.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciccarelli FD, et al. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311(5765):1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 35.Minguez P, et al. Deciphering a global network of functionally associated post-translational modifications. Mol Syst Biol. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mende DR, Sunagawa S, Zeller G, Bork P. Accurate and universal delineation of prokaryotic species. Nat Methods. 2013;10(9):881–884. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 37.Muller J, Creevey CJ, Thompson JD, Arendt D, Bork P. AQUA: Automated quality improvement for multiple sequence alignments. Bioinformatics. 2010;26(2):263–265. doi: 10.1093/bioinformatics/btp651. [DOI] [PubMed] [Google Scholar]
- 38.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 39.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28(3):245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.