

Significance
A deleterious mutation that is recessive is hidden in individuals containing only one copy (i.e., heterozygotes); however, individuals containing two copies (i.e., homozygotes) suffer negative effects. This class of mutation is responsible for a number of human genetic disorders, including cystic fibrosis and Tay-Sachs, in addition to causing the widespread phenomenon of inbreeding depression. Evidence suggests that recessive deleterious mutations may be abundant in nature, likely due to their ability to persist for long timescales at moderate frequencies. It is thus possible that when an adaptive mutation occurs, it lands on a chromosome containing a recessive deleterious mutation. We propose a model for this and find that recessive deleterious mutations can significantly slow adaptation and alter signatures of adaptation.
Keywords: recessive, hitchhiking, inbreeding depression, adaptation, selective sweep
Abstract
Recessive deleterious mutations are common, causing many genetic disorders in humans and producing inbreeding depression in the majority of sexually reproducing diploids. The abundance of recessive deleterious mutations in natural populations suggests they are likely to be present on a chromosome when a new adaptive mutation occurs, yet the dynamics of recessive deleterious hitchhikers and their impact on adaptation remains poorly understood. Here we model how a recessive deleterious mutation impacts the fate of a genetically linked dominant beneficial mutation. The frequency trajectory of the adaptive mutation in this case is dramatically altered and results in what we have termed a “staggered sweep.” It is named for its three-phased trajectory: (i) Initially, the two linked mutations have a selective advantage while rare and will increase in frequency together, then (ii), at higher frequencies, the recessive hitchhiker is exposed to selection and can cause a balanced state via heterozygote advantage (the staggered phase), and (iii) finally, if recombination unlinks the two mutations, then the beneficial mutation can complete the sweep to fixation. Using both analytics and simulations, we show that strongly deleterious recessive mutations can substantially decrease the probability of fixation for nearby beneficial mutations, thus creating zones in the genome where adaptation is suppressed. These mutations can also significantly prolong the number of generations a beneficial mutation takes to sweep to fixation, and cause the genomic signature of selection to resemble that of soft or partial sweeps. We show that recessive deleterious variation could impact adaptation in humans and Drosophila.
In diploids, the fitness effect of having a single copy of a mutation depends not only on the mutation’s selective effect, s, but also on its heterozygous effect, h. A fully recessive mutation () is hidden in the heterozygote (), and a fully dominant mutation () is completely exposed (). Although it is generally agreed that beneficial mutations that reach fixation tend to be dominant (i.e., ) (1, 2), both empirical data and theoretical models (3–6) suggest that many strongly and even moderately deleterious mutations are likely to be recessive (i.e., ). For example, studies of de novo mutations from both mutation accumulation and mutagenesis experiments in Drosophila melanogaster, Saccharomyces cerevisiae, and Caenorhabditis elegans have repeatedly found that strongly deleterious mutations tend to be completely recessive () and more weakly deleterious mutations tend to be partially recessive () (7–13). Furthermore, studies of natural populations have found that inbreeding depression is pervasive across sexually reproducing diploids and is mainly caused by recessive deleterious variation (reviewed in ref. 14). For example, in natural populations of Drosophila, approximately of chromosomes carry a recessive lethal, and chromosomes that do not carry a recessive lethal suffer from at least depression in homozygous fitness (15–24). These data suggest that many, if not most, deleterious mutations are likely to be fully or partially recessive, and such mutations can have a moderate to strong fitness effect in the homozygote.
It is thus possible that when a new adaptive mutation occurs, it will land on a chromosomal background containing one or more recessive deleterious mutations. It is well established that in finite populations, the fate of a new adaptive mutation should be affected by its genetically linked neighbors. For example, the rate of fixation of beneficial mutations at a single site will be lower if there are additional sites subject to positive or negative selection along the chromosome (25–30). This “Hill−Robertson” or “linkage” interference can be alleviated by recombination, allowing adaptive mutations to combine onto the same background or escape deleterious neighbors (31–35). In addition to affecting the rate of fixation of beneficial mutations, a deleterious mutation genetically linked to a beneficial mutation can “hitchhike” to high frequency or even fixation (36, 37). These theoretical predictions are beginning to be borne out in empirical data (38–47).
Despite the large body of work on hitchhiking and observations of recessive deleterious mutations in real organisms, there is a gap in our understanding of how recessive deleterious mutations affect adaptation. Models of hitchhiking have primarily focused on mutations with codominant effects, thus necessarily emphasizing the hitchhiking of weak deleterious mutations with stronger advantageous ones. Although recessive strongly deleterious mutations are expected to have a lower probability of hitchhiking to fixation than weaker codominant ones (48), recessive deleterious mutations can still have a profound impact on the dynamics of adaptation.
Here we develop a model for the dynamics of a dominant beneficial mutation that is initially genetically linked to a recessive deleterious mutation of larger effect (), with varying rates of recombination. Provided the recessive deleterious mutation is sufficiently hidden in the heterozygote (), hitchhiking occurs even when the beneficial mutation has a smaller fitness effect than its deleterious hitchhiker. We show that the frequency trajectory of a beneficial mutation in this case is dramatically altered, causing what we have termed a “staggered sweep,” whereby the linked mutations are balanced for a period before recombination unlinks them. This balancing selection is a type of associative overdominance. However, instead of the classic case of two recessive deleterious mutations in repulsion or a neutral mutation linked to a single overdominant one (49, 50), our case results from a recessive deleterious mutation linked to a dominant beneficial mutation of weaker selective effect.
Using our model, we show that recessive deleterious mutations can substantially lower the fixation probability of nearby beneficial mutations, thus creating zones around recessive deleterious mutations where adaptation is suppressed. Although it has been observed before in models of balancing selection that alleles balanced at low frequencies in finite populations can drift out of a population with higher probability than a neutral mutation (51–53), we have derived, for the first time (to our knowledge), an analytic solution for the distribution of extinction times. In addition to affecting the fixation probability of a beneficial mutation, we demonstrate that a recessive deleterious hitchhiker can lengthen the duration of a selective sweep and alter the genomic sweep signature, compared with a classic hard sweep. We show that these effects are strongest in small populations, at low recombination rates, and when the recessive deleterious hitchhiker is much stronger than the beneficial mutation in homozygotes. We end by estimating how common this effect could be in D. melanogaster and humans, showing that it may play a potentially important role in adaptation for both natural and laboratory populations.
Results
We first develop a heuristic understanding of staggered sweeps as an interaction between three effects: balancing selection, recombination, and genetic drift. We then derive analytic predictions for the dynamics of staggered sweeps and compare our results with forward simulations. Throughout our analysis, we emphasize scaling and parameter dependence over the details of constant factors. This is intentional. Scaling properties hold generally across different models studied, whereas the constant factors typically do not. For example, the fixation probability of a beneficial mutation is frequently quoted as ; however, the constant 2 depends on the details of the stochastic model (it is inversely proportional to the variance in offspring number). The scaling with the selective effect, s, however, is general. For this reason, we do not distinguish between constant factors and instead emphasize the scaling (e.g., fixation probability of ∼s).
Model.
Consider a population of N diploid individuals in which two sites are genetically linked along a chromosome such that an ancestral haplotype with no mutations is denoted . The first site can harbor a beneficial mutation with selective effect and heterozygous effect , the second site can harbor a deleterious mutation with effects and , and there is a recombination rate between them (base pair distance recombination rate per base pair per generation). When a new adaptive mutation lands on a chromosome harboring an existing recessive deleterious mutation, it generates a haplotype, and in the absence of recombination (for the moment) a diploid will have heterozygote advantage. This is apparent from the fitnesses of the diploids,
| [1] |
| [2] |
| [3] |
where we assume selection coefficients to be small (, ) and the selection coefficient of the deleterious mutation to be much larger than that of the beneficial mutation (), and, for the final approximation, we further assume the beneficial mutation to be completely dominant and the deleterious mutation to be completely recessive (, ). We emphasize that our model and analytic predictions can be extended to cases of partial dominance (i.e., , ), as long as the effect of the beneficial mutation in the heterozygote is stronger than that of the deleterious mutation () but its effect in the homozygote is weaker () (SI Text, sections 1 and 2).
The new beneficial mutation will thus be subject to balancing selection on the haplotype and held at an equilibrium frequency. However, this balanced state is temporary (Fig. 1). If recombination generates a haplotype, unlinking the beneficial mutation from the recessive deleterious hitchhiker, then the beneficial mutation can complete the sweep to fixation, sometimes with a substantial delay (Fig. 1A). We call this a staggered sweep. Alternatively, the haplotype may drift to extinction before the beneficial mutation is able to escape via recombination (Fig. 1 B and C, red trajectories). Extinction is particularly probable if the number of copies of the haplotype at equilibrium is small and therefore drift is strong. As these simulations suggest, the probability of fixation and the sweep duration for a new beneficial mutation with a recessive deleterious hitchhiker can be significantly altered depending on the interaction of balancing selection, recombination, and drift, which we now quantify.
Fig. 1.

Frequency trajectories of a beneficial mutation genetically linked to a recessive deleterious hitchhiker. Plotted trajectories are from 50 simulations that reached an equilibrium frequency of , where blue indicates simulations that fixed the beneficial mutation and red indicates simulations in which it goes extinct (red tick marks below frequency zero mark the generation of extinction). (A) For , selection dominates, and the haplotype staggers stably at the equilibrium frequency waiting for a recombination event that allows the beneficial mutation to escape on a haplotype to fixation. In this regime, loss of the beneficial mutation is very rare, and the staggered sweep can last for a substantial time. (B) For , selection and drift are both important, and the haplotype staggers at the equilibrium frequency but with strong frequency fluctuations due to drift that can drive the haplotype to extinction (but not fixation) before an escape event occurs. (C) For , drift dominates, and the haplotype never stably staggers; thus frequency changes are dominated by drift. Note that it cannot drift to fixation because of the recessive deleterious mutation (Fig. 2). Unless a recombination event occurs very early, the beneficial mutation will fluctuate to extinction on the haplotype.
Balancing selection.
At low frequencies, the haplotype will primarily occur in heterozygotes where the recessive deleterious allele is hidden. As a result, the haplotype will be subject to positive selection. In contrast, at high frequencies, the haplotype will primarily occur in homozygotes where the recessive strongly deleterious allele is exposed, resulting in selection that drives the haplotype down in frequency. These opposing forces cause balancing selection. The change in frequency per generation due to selection, , is thus frequency dependent,
| [4] |
(valid for ; SI Text, section 1). The first term reflects selection on heterozygotes with fitness ≈ sb that occur with probability ≈ p, and the second term reflects selection on homozygotes with fitness ∼−sd that occur with probability ∼p2 (note that the term will be ignored from this point forward as it is very small for ). The stable fixed point or equilibrium frequency can be found by setting ,
| [5] |
(derivations of and for arbitrary selection and dominance coefficients can be found in SI Text, section 1). In the absence of drift and recombination, a haplotype that reaches equilibrium frequency will remain there indefinitely.
Escape via recombination.
The beneficial mutation can reach fixation if two conditions are met. First, there must be a recombination event in a heterozygote individual that creates a BO haplotype. Second, this newly created BO haplotype must survive the effects of drift (to “establish” in the population) and proceed toward fixation. We call this “escape” of the beneficial mutation. In each generation, the probability of escape is proportional to the number of copies of the haplotype [∼N p(t)], multiplied by the probability of a recombination event creating a haplotype (∼rl), and the probability of establishment of this new haplotype (∼sb). Thus, the total probability of an escape event by time t is
| [6] |
(which is valid for a probability of escape ). Provided is known, we can calculate the probability that the beneficial mutation escapes its deleterious background. We note that if the haplotype is held stably at equilibrium frequency, then . However, as discussed in the Drift section below, this is not always the case.
Drift.
Despite the balancing selection driving the haplotype toward equilibrium frequency, sometimes the haplotype can go extinct before a recombination event occurs. This extinction event, which we call “loss,” is driven by random fluctuations in frequency due to genetic drift. The variance in frequency per generation due to drift, , is
| [7] |
To compare the relative effects of drift and selection, one can consider two characteristic timescales. The first, , is the time for selection alone to change the frequency of the balanced haplotype by . The second, , is the time for drift alone to change the frequency of the balanced haplotype by . The ratio of these two timescales tells one which of the two processes is faster, and hence which dominates the dynamics,
| [8] |
Because we are interested in changes in frequency that lead to extinction, the natural choice for is approximately p (SI Text, section 2). With this simplification, , which we can now use in Fig. 2 to understand the transition points between frequency ranges that are drift dominated or selection dominated. Selection will be more important whenever (solid curve) is larger than (dashed lines), such that drift dominates for frequencies near , and (light shading) and selection dominates in the alternate intervals (dark shading). Positive selection is strongest (causing the largest positive changes in frequency) at a frequency approximately midway to equilibrium, such that at frequency the strength of selection is . As the relative strength of drift to selection becomes larger, the size of the drift intervals widens, even to the point where drift dominates for all frequencies below equilibrium (Fig. 2 A vs. B).
Fig. 2.

A model of drift and balancing selection (without recombination). (A) Schematic of regime, where the maximum positive effect of balancing selection () is stronger than the effect of drift (). In this regime, the haplotype can balance at an equilibrium frequency and then either escape to fixation via recombination (as in Fig. 1 A and B, blue trajectories) or drift to extinction (as in Fig. 1B, red trajectories). The rate of drifting to extinction from equilibrium depends primarily on . (B) Schematic of regime, where the maximum positive effect of balancing selection () is weaker than the effect of drift (), and thus frequency dynamics of a balanced haplotype are always dominated by drift near (or below) equilibrium frequency. In this regime, the haplotype neither establishes nor stably balances, and thus primarily drifts to extinction before an escape event occurs (as in Fig. 1C).
If drift is sufficiently strong, the haplotype can fluctuate to extinction. The probability that this occurs depends on the maximum strength of positive selection () relative to the strength of drift () (SI Text, section 2). We call this ratio α,
| [9] |
The dynamics of the haplotype are qualitatively different for and . In the regime, there is a region below where selection dominates (Fig. 2A), and thus the haplotype can establish in the population and stagger at the equilibrium frequency with relatively small fluctuations due to drift. In the regime, there is no region below where selection dominates over drift (Fig. 2B), in which case there is no true establishment or balanced phase for the haplotype and it can easily go extinct. There is a crossover where in which selection and drift are of similar magnitudes. Below, we derive analytic expressions to predict the beneficial mutation’s probability of fixation and total sweep duration in the two distinct regimes ( and ) under the forces of selection, drift, and recombination. We confirm our results with simulations.
Predictions for the Regime of Strong Selection and Weak Drift ().
Probability of fixation.
In order for the beneficial mutation to reach fixation from equilibrium, it must avoid fluctuating to extinction before a recombination event can unlink it from its deleterious hitchhiker. The probability of fixation is therefore determined by which event occurs first: loss or escape.
Loss of the haplotype occurs only if it can fluctuate over the relatively high selective barrier (). The process is similar to chemical reactions, where there is an “activation energy” and the reaction rate depends exponentially on the ratio of the barrier height to the strength of noise (e.g., Arrhenius’ equation for chemical reaction rates). Thus, it can be shown (SI Text, section 2.1) that the time to loss, independent of escape via recombination, is an exponentially distributed random variable with rate and mean
| [10] |
which we have verified using simulations (SI Text, section 4.1).
Escape of the beneficial mutation from equilibrium to fixation can also be modeled as an exponential process. In this regime of strong selection and weak drift, the haplotype is held closely to equilibrium; thus the probability of escape from Eq. 6 simplifies to . In this case, escape, independent of loss via drift, occurs at rate with mean ,
| [11] |
which we have verified using simulations (SI Text, section 4.2).
The overall probability of fixation for a new beneficial mutation that lands on a recessive deleterious background is then the product of the probability that a new haplotype establishes and reaches equilibrium (∼sb) and the probability that the first escape event occurs before the first loss event [],
| [12] |
which we have verified using simulations [Fig. 3 D–F (α ≫ 1) and SI Text, section 4.4]. The probability of fixation of the adaptive mutation will be reduced relative to an identical adaptive mutation with no recessive deleterious hitchhiker whenever , which is when loss is faster than escape.
Fig. 3.

Analytics predict simulation results for the fraction of staggered sweeps that reach fixation. The y axis is the fraction of simulations in which the beneficial mutation reaches fixation relative to a one-locus control with no hitchhiker, and the x axis is base pair distance between the linked sites. The population size (N) used in each panel is indicated by the figure row headings, and the beneficial mutation effect size (sb) used in each panel is indicated by the figure column headings, such that A–C fall into the α < 1 regime, and D–F fall into the α > 1 regime. Points represent results of 1,000/sb simulations (where bars indicate 95% binomial proportion confidence interval), solid lines indicate our analytic predictions (A−C use Eq. S31 due to , and D−F use Eq. 12 due to ). Red dashed lines are analytic predictions for the distance below which the probability of fixation becomes suppressed (A−C use Eq. S37 and D−F use Eq. S33). All simulations used . We have translated recombination rate between the sites into base pair distance using a human recombination rate per base pair per generation .
Sweep time.
If the beneficial mutation is not driven to extinction, the duration of a beneficial mutation’s sweep to fixation can be substantially extended if it’s genetically linked to a recessive deleterious hitchhiker (Fig. 1A). To understand when this occurs, consider that the total time of a successful sweep will, on average, be prolonged by the time it takes for an escape event to occur,
| [13] |
which we verified with simulations [Fig. 4 C–F (α ≫ 1) and SI Text, section 4.5]. The first term corresponds to the sweep time for a single adaptive mutation with no hitchhiker, and the second term is the extension in sweep time due to the staggered phase (for more discussion of the leveling off of sweep times at low recombination rates, as in Fig. 4 C and D, see SI Text, section 4.5).
Fig. 4.

Analytics predict simulation results for the mean sweep time of staggered sweeps. The y axis is the fold increase in the beneficial mutation’s sweep time relative to a one-locus control with no hitchhiker, and the x axis is the base pair distance between the linked sites. The population size (N) used in each panel is indicated by the figure row headings, and the beneficial mutation effect size (sb) used in each panel is indicated by the figure column headings, such that A and B have α ≤ 1, and C–F have α > 1. Points represent results of 500 simulations in which fixation of the beneficial mutation occurred (required to calculate its sweep time), where bars indicate ±SE and solid lines indicate our analytic predictions (A and B have and thus no increase in sweep time; C−F have and thus use Eq. 13). Red dashed lines are analytic predictions for the distance at which the mean sweep time becomes extended (Eq. S35 and SI Text, section 3.2). For more discussion of the leveling off of sweep times at low recombination rates in C and D, see SI Text, section 4.5. All simulations used . We have translated recombination rate between the sites into base pair distance using a human recombination rate per base pair per generation .
Zones of altered adaptation around a recessive deleterious allele.
We can understand the effect of recessive deleterious variation on the genome in terms of a base pair distance around every recessive deleterious mutation within which the dynamics of adaptation are altered (SI Text, section 3). A recessive deleterious hitchhiker will drastically suppress the probability of fixation of a linked beneficial mutation whenever , translating to a distance, , around the deleterious mutation within which new beneficial mutations of effect size (or smaller) have a reduced chance of fixation (, red dashed lines in Fig. 3 D−F). This distance can be substantial for values of α that approach unity (Fig. 3D). Similarly, using Eq. 13, we can derive a distance, , around a recessive deleterious mutation within which new beneficial mutations of effect size have an extended sweep time (, red dashed lines in Fig. 4 C−F). Again, this distance is largest for smaller values of α (Fig. 4 C and D); however, the sweep duration is longest in large α regimes at small recombination rates (Fig. 4 E and F). Note that the overall impact of recessive deleterious mutations on adaptation will depend on the density and strength of these deleterious mutations and whether their zones of altered adaptation might overlap (further explored in Discussion).
Predictions for the Regime of Weak Selection and Strong Drift ().
Probability of fixation.
In this regime, selection for the beneficial mutation is not strong enough to hold the haplotype close to equilibrium frequency (as in Fig. 1C). Instead, the dynamics of the staggered sweep are dominated by a combination of drift (due to ) and selection against the recessive deleterious mutation. In this case, the beneficial mutation on the haplotype can easily drift to extinction, but it is very unlikely to drift to fixation. It can be shown (SI Text, section 2.3) that the upper limit to which the haplotype will typically drift is copies, at which point selection against homozygotes becomes stronger than drift and pushes the haplotype back down to lower frequencies. Thus, the distribution of extinction times of the haplotype will be approximately neutral (power-law distributed as ), but it cannot be much longer than generations (verified using simulations, SI Text, section 4.3).
We are interested in the probability that escape occurs before loss of the haplotype. Consider an interval in which there is a fraction of haplotypes that go extinct (which scales as from neutrality), and a fraction of haplotypes that escape via recombination (which scales as from the total number of haplotypes that have existed by time t). The product of these two gives a constant probability of escape (). Recalling that the haplotype will typically drift for only generations, we can use Eq. 6 to calculate the probability of fixation for the beneficial mutation to be (SI Text, section 2.3). We have verified with simulations that this predicts the probability of fixation of the beneficial mutation [Fig. 3 A–C (α ≪ 1) and SI Text, section 4.4].
Sweep time.
Although the probability of fixation for the beneficial mutation can be significantly decreased in the regime, the total sweep time of the beneficial mutation is not expected to substantially change. This is because if it is to escape at all, it must do so in the first generations, which is generally a small fraction of the classic sweep time . This prediction of no alteration in sweep time in the regime was confirmed with simulations (Fig. 4 A and B and SI Text, section 4.5).
Zones of altered adaptation around a recessive deleterious allele.
We can again understand the effect of recessive deleterious variation on the genome in terms of a base pair distance around every recessive deleterious mutation within which the dynamics of adaptation are altered. Because the sweep time is not significantly altered in this regime of weak selection and no drift, there is no relevant distance within which sweep time is extended. However, a recessive deleterious hitchhiker will drastically suppress the probability of fixation of a linked beneficial mutation whenever (SI Text, sections 3.2 and 4.4). The distance in this case () can be substantial for realistic N, , and r. For example, all panels in the regime in Fig. 3 (Fig. 3 A−C) show that a beneficial mutation will have a reduction in its fixation probability even when it is ∼1 Mb away from the recessive deleterious hitchhiker.
Simulations.
To test our predictions, we conducted two-locus Wright−Fisher forward simulations (Figs. 3 and 4 and SI Text, sections 2 and 4). Simulations start by seeding the haplotype at 1 copy and the haplotype at copies, and recording the frequency of the four possible haplotypes over time. Simulations were performed over a wide range of selection coefficients (, ), recombination rates (), and population sizes (), corresponding to ranges of . All simulations used and heterozygous effects of and such that the equilibrium frequency (analytics were changed appropriately to allow for ). We also included a one-locus control of an adaptive mutation with no deleterious hitchhiker for comparison. Note that figures are plotted using (corresponding to 1 cM/Mbp).
Figs. 3 and 4 show that our analytic expressions (black lines) predict simulation results (data points) and thus accurately capture the parameter dependence. Furthermore, the simulations confirm that an adaptive mutation that lands within a distance of the recessive deleterious mutation (red dashed line in Fig. 3) has a reduced probability of fixation, and an adaptive mutation that lands within a distance of a recessive deleterious mutation (red dashed line in Fig. 4) has an increased sweep time.
The effect of the recessive deleterious mutation on the probability of fixation of linked beneficial mutations can extend for very large genomic distances, especially for weakly adaptive sites (Fig. 3 and SI Text, sections 3.1, 3.2, and 4.4). For instance, when and , any adaptive mutation with will satisfy and thus have a fixation probability that is substantially reduced within base pairs of the recessive deleterious mutation (Fig. 3C). However, if the effect size of the adaptive mutation is increased to , the probability of fixation recovers to about the same level as an adaptive mutation with no deleterious hitchhiker (Fig. 3F). In general, any recessive deleterious mutation will suppress fixation of nearby adaptive mutations if is satisfied.
The sweep time of adaptive mutations that reach fixation, on the other hand, is impacted for small α values that still satisfy (Fig. 4 and SI Text, sections 3.1, 3.2, and 4.5). In this regime, the adaptive mutation can reach a stable equilibrium frequency (due to ), where it has a slow rate of loss () but the rate of new recombinants being generated in the population is not so large that the staggered sweep is resolved quickly (). For example, when a new adaptive mutation of effect lands base pairs away from a recessive deleterious mutation of effect in a population of , the mean sweep time is ∼15 times as long compared with a new adaptive mutation with no hitchhiker (Fig. 4E). In this scenario, where , an extension in sweep time will occur even if the adaptive mutation lands within a distance base pairs of the recessive deleterious mutation. As α increases, this distance decreases such that the duration of staggered sweeps are most extended for intermediate values of α (Fig. 4E).
Discussion
Using a two-locus model, we have shown that recessive deleterious mutations can (i) decrease the rate of fixation and (ii) increase the sweep time of linked adaptive mutations with similar or weaker fitness effects. Here we discuss to what extent these two effects are likely to occur in real populations and how they can alter signatures of selection.
Impact of Recessive Deleterious Mutations in Real Populations.
Every recessive deleterious mutation has a zone around it within which adaptive events are suppressed (SI Text, section 3), and thus their overall effect on adaptation will critically depend on the density and strength of these deleterious mutations. Although these mutations are likely to appear in functional regions of the genome, their actual densities are not yet well understood. Thus, focusing on coding genes, we use what data can be found for the densities of deleterious mutations for both D. melanogaster and humans to derive order-of-magnitude estimates for the impact of recessive deleterious variation on adaptation in real populations (Table 1 and SI Text, section 3.5).
Table 1.
Estimates of the the proportion of the genome in which the probability of fixation of the beneficial mutation is decreased for Drosophila melanogaster and humans
| Organism | Number of coding genes | Recessive deleterious effect (), % | Zone of reduced adaptation [genes in zone] | Beneficial effect () impacted, % | Density of recessive deleterious | Proportion of adaptive mutations impacted, % |
| Drosophila (wild, N = 106) | ∼12,000 | 100 | 100 kb [20 genes] | ≤0.10 | 1/(genome) | ∼0.1 |
| 1 | 10 kb [3 genes] | ≤0.01 | 1/(30 genes) | ∼10 | ||
| Drosophila (laboratory, N = 103) | ∼12,000 | 100 | 3 Mb [400 genes] | ≤3.00 | 1/(genome) | ∼3 |
| 1 | 300 kb [40 genes] | ≤0.30 | 1/(30 genes) | ∼100 | ||
| Human (N = 104) | ∼20,000 | 100 | 1 Mb [20 genes] | ≤1.00 | 1/(genome) | ∼0.1 |
| 1 | 100 kb [3 genes] | ≤0.10 | 1/(100 genes) | ∼3 | ||
| 1/(30 genes) | ∼10 | |||||
| 1/(10 genes) | ∼30 |
Due to variation in functional density across genomes and organisms, we frame our estimates in terms of coding genes and their densities, using the Drosophila and human reference genomes (SI Text, section 3). Column information is as follows: column 1 indicates the species and population size considered, column 2 indicates the number of coding genes in a haploid set of autosomes, column 3 indicates the recessive deleterious mutation effect size of interest, column 4 indicates the zone around this recessive deleterious mutation within which adaptation is suppressed (SI Text, sections 3.1 and 3.2), where the bracketed information is the number of coding genes that typically appear in a region of this size (centered on a coding gene, SI Text, section 3.3), column 5 indicates the beneficial mutation effect size of interest (where we consider all beneficial mutations which fall within the regime because they behave similarly and are greatly impacted), column 6 indicates the densities of recessive deleterious mutations as obtained from refs. 20, 24, and 54, where we use a range of possible densities for mildly deleterious mutations in humans due to a lack of information, and column 7 indicates the proportion of new adaptive mutations of the given effect size impacted (i.e., the proportion of coding genes in a genome within which adaptation is suppressed), which can be substantial.
Data on the abundance of recessive lethals (%) are perhaps the most unambiguous. They are thought to occur at a rate of per genome in both humans and Drosophila (24, 54), which translates to 1 per ∼20,000 coding genes in humans and 1 per ∼12,000 coding genes in Drosophila. The density of mildly deleterious recessive mutations is less well characterized; however, they are thought to be much more abundant (14). One estimate in Drosophila places the number of mildly deleterious recessive mutations (sd ∼1%) at about 200 per autosome (20), which is 1 per ∼30 coding genes. There are no data like these for humans yet; thus we use the Drosophila density as a starting point for a range of possible densities. The size of the zones around these deleterious mutations depends on multiple parameters (N, , , r), where, for example, a recessive lethal in a natural Drosophila population impacts a region of ∼100 kb, but in humans, the size of this zone is ∼1 Mb (Table 1, column 4).
The overall impact of recessive deleterious mutations on adaptation will depend not only on their densities but also on the density of functional regions (in this case, coding genes) around each deleterious mutation. Estimates of gene density are currently quoted at about 1 gene per ∼100 kb in humans and 1 gene per ∼10 kb in Drosophila (55, 56), yet a significant proportion of genes are clustered in both organisms, especially in humans (57, 58). Thus, we frame our estimates in terms of coding gene densities within a given zone size, where we used the Drosophila and human reference genomes to estimate the gene density around every coding gene (column 4 brackets in Table 1; method in SI Text, section 3). For example, a recessive lethal in a human population is likely to suppress adaptation for ∼1 Mb around itself, which contains on average ∼20 other coding genes (SI Text, section 3.3). Given the density of recessive lethals, this translates to only about 0.1% of human coding genes within which adaptation is suppressed (Table 1).
Although the impact of recessive lethals is unlikely to be very dramatic, mildly deleterious mutations paint a very different picture (Table 1). For example, a mildly deleterious mutation in a wild Drosophila population affects about 10 kb around itself, within which there are typically three other coding genes (SI Text, section 3.3). This translates to of coding genes within Drosophila that are impacted. These effects become exacerbated in the context of smaller population sizes, as might be seen in experimental populations. For example, in a Drosophila population of N = 1,000 flies, beneficial mutations with effect size will have suppressed fixation probabilities in the entirety of the Drosophila genome (Table 1). If we consider the beneficial mutations that do fix (SI Text, section 3.5), we find that beneficial mutations with comparable selective effect to the deleterious mutation (sb ≈ 1%) will be subject to staggered phases within the majority of the genome ( of coding genes). This is interesting in light of a number of experimental evolutions in small populations of Drosophila (N ≈ 102−103), where studies have often shown a number of alleles that initially increase in frequency with a rate and direction suggestive of selection but then do not finish the sweep to fixation (59, 60) [older experiments also exhibited similar patterns (61–65)]. Possible explanations [including individually overdominant loci or selection on polygenic traits (66)] are still under investigation; however, it is plausible that this behavior could in part be due to linked recessive deleterious alleles in small populations causing staggered sweeps.
For humans, there are no current estimates for the densities of weakly deleterious recessive mutations; however, if we consider a range of densities from 1 mildly deleterious mutation every 10 coding genes to every 100 coding genes, we find that anywhere from 3% to 30% of coding genes may be subject to a reduced rate of adaptation (Table 1). This result strongly emphasizes the need for more information regarding the actual numbers of weakly deleterious recessive mutations segregating in human populations, as their combined effect could potentially result in a significantly suppressed rate of fixation of weakly adaptive mutations, particularly in small populations. We note that in classical population genetics, the term is generally required to be satisfied for adaptation to proceed. However, in a population with densely distributed recessive mutations, the requirement becomes , a factor smaller. If genomes are indeed rich in recessive deleterious variation, it may be the case that beneficial mutations must have substantially larger fitness effects ( instead of ) to spread through a population.
Altered Signatures of Selection.
A beneficial mutation that begins its selective sweep with a linked recessive deleterious hitchhiker may be expected to leave an altered genomic signature of selection upon reaching fixation compared with a hard sweep. Under the classic hard sweep model of adaptation, a single de novo adaptive mutation occurs on a single haplotype and drives it to fixation. As a result, diversity in the immediate vicinity of the adaptive site is expected to be greatly reduced at the completion of the sweep and should recover to background levels farther away in the genome (67). In contrast, during a successful staggered sweep in which recombination must unlink a recessive deleterious hitchhiker, the haplotype that begins the sweep will always be distinct from the haplotype that finishes the sweep. It is thus possible in this scenario for both haplotypes to persist in the population after fixation occurs, producing higher levels of haplotype diversity around the beneficial mutation.
To illustrate this point, we simulated staggered sweeps, hard sweeps, and soft sweeps with linked neutral diversity using SLiM, a program for forward population genetic simulations of linked loci (68) (soft sweep simulations used a high beneficial mutation rate, such that beneficial mutations occurred on multiple haplotypes and swept concurrently). Upon fixation of the beneficial mutation, we plotted patterns of heterozygosity around the adaptive site, and found that staggered sweeps leave distinct signatures (Fig. 5; additional statistics in SI Text, section 5). At the conclusion of a staggered sweep, there can be multiple haplotypes present in the population at substantial frequencies (Fig. 5 A vs. B), a signature that is qualitatively similar to that generated by a soft or partial sweep. Additionally, compared with both hard and soft sweeps, staggered sweeps consistently leave higher levels of heterozygosity above the region where the recessive deleterious hitchhiker was unlinked (Fig. 5C). This asymmetry is caused by the requirement that, in order for fixation to occur, at least two haplotypes must participate in the selective sweep on the side of the beneficial mutation that contained the deleterious hitchhiker. These staggered sweep signatures are interesting in light of the many studies that have found partial sweeps (69–71), a signature characterized by a single beneficial mutation (or haplotype, if the mutation is not yet identified) that has spread through a population but not yet reached fixation. Our model suggests that linked recessive deleterious variation could potentially be another factor contributing to such signatures observed in natural populations.
Fig. 5.
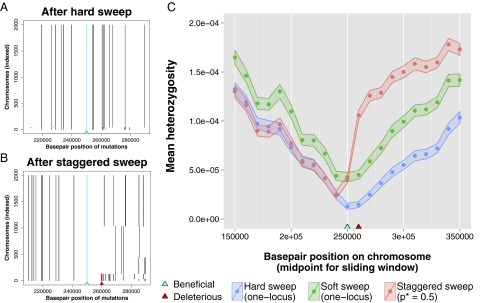
Altered signatures of selection in the genome after a staggered sweep. Simulations were performed using SliM (68) to generate and track neutral diversity around the adaptive site, where simulations used and N = 1,000 diploids. (A) Haplotypes present in a population at the conclusion of single simulation of a hard sweep in which a beneficial mutation on a haplotype containing only neutral mutations was seeded at establishment frequency; note that a single haplotype dominates the population. (B) Haplotypes present in a population at the conclusion of single simulation of a staggered sweep in which a beneficial mutation on a haplotype containing both neutral mutations and a single recessive deleterious mutation () 10 kb away (where ) was seeded at establishment frequency. Note that recombination has unlinked the beneficial and recessive deleterious mutations, such that multiple haplotypes are at high frequency in the population after fixation of the beneficial mutation. (C) Mean heterozygosity across 200 simulations calculated in sliding windows of length 30 kb with step size 10 kb, where the ribbon around data points indicates the SEM. Results are plotted for hard sweep simulations in which a new adaptive mutation occurs on a single haplotype, soft sweep simulations in which a new adaptive mutation occurs on multiple haplotypes (Nub ≈ 1), and staggered sweep simulations in which an adaptive mutation occurs on a single haplotype background containing a recessive deleterious mutation ().
Possible Extensions to Our Model.
Our model is unique in that our assumption of recessivity allows for the consideration of deleterious mutations with appreciable selective effects, a class of deleterious mutations that are observed in natural populations and yet are generally precluded from hitchhiking models that assume codominance (due to ). Thus, for a deleterious mutation with a given effect size , a linked beneficial mutation should have a higher probability of fixation if that deleterious hitchhiker is recessive. This is due to the fact that the haplotype can potentially establish (as in the regime) or drift at low frequencies (as in the regime), which, in either case, allows the beneficial mutation to persist for longer times in the population (and thus have more chances at escape) than if its hitchhiker had codominant effects. One possible extension would be to require the deleterious mutation to be weaker (), which would not cause temporary balancing selection, as in our model, but may allow the beneficial mutation on the haplotype to initially establish with a higher probability than if the hitchhiker had codominant effects (i.e., instead of ). Another interesting extension would be to build a model with an arbitrary number, n, of deleterious sites, in which the deleterious effect is correlated with the dominance coefficient. This would better reflect what is observed in nature, and additionally would allow for the interesting case where many neighboring weakly deleterious recessive mutations can cumulatively cause a balanced state (if ), and which would also require longer timescales to be unlinked. In general, the ability of recessive deleterious mutations to hitchhike to appreciable frequencies suggests they could play a role in the dynamics of rapid adaptation and potentially in the maintenance of genetic variation. Our model of a single beneficial and deleterious mutation is appropriate if mutation rates are small (i.e., ). An interesting extension would thus be to incorporate arbitrary rates of beneficial and deleterious mutations (similar to ref. 30), allowing for beneficial mutations to be so frequent that when a new adaptive event occurs the effects of the previous one may not have yet been “forgotten.”
Conclusions.
Studies of linkage interference due to deleterious hitchhikers have largely been concerned with mutations of weak effects, as these are able to hitchhike to fixation with a beneficial mutation of larger effect. However, we have shown here that recessive mutations, which behave like weakly deleterious mutations at low frequencies but like strongly deleterious mutations at high frequencies, can significantly interfere with the rate and dynamics of adaptation. We find that a single recessive deleterious mutation will inhibit adaptation for large genomic distances around itself, and linked adaptive mutations sweeping through a population may stagger at an intermediate frequency for an extended time before reaching fixation. The consequences of recessive deleterious variation for adaptation are amplified in small populations, for closely linked sites, for weakly adaptive events, and for populations that harbor substantial recessive deleterious load. Although definitive experimental data for staggered sweeps are yet to be acquired, the evidence for abundant recessive deleterious variation from both natural populations (inbreeding depression data) and de novo mutations (mutation accumulation and mutagenesis experiments) suggests that staggered sweeps may be important during adaptation.
Supplementary Material
Acknowledgments
The authors would like to thank Daniel Fisher for helpful discussions. We would also like to thank Gavin Sherlock, Hua Tang, Carlos Bustamante, Philipp Messer, Mike McLaren, and all members of the D.A.P. laboratory, as well as Daniel Weissman, Michael Desai, Matthew Hartfield, Deborah Charlesworth, Brian Charlesworth, and Nick Barton for useful feedback. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant DGE-114747 (to Z.J.A.) and by National Institutes of Health Grants RO1GM100366 and RO1GM097415 (to D.A.P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424949112/-/DCSupplemental.
References
- 1.Haldane JBS. A mathematical theory of natural and artificial selection, part V: Selection and mutation. Math Proc Cambridge Philos Soc. 1927;23(7):838−844. [Google Scholar]
- 2.Charlesworth B. Adaptive evolution: The struggle for dominance. Curr Biol. 1998;8(14):R502–R504. doi: 10.1016/s0960-9822(98)70318-5. [DOI] [PubMed] [Google Scholar]
- 3.Fisher RA. The possible modification of the response of the wild type to recurrent mutations. Am Nat. 1928;62(1):115–116. [Google Scholar]
- 4.Wright S. Physiological and evolutionary theories of dominance. Am Nat. 1934;68(714):24–53. [Google Scholar]
- 5.Kacser H, Burns JA. The molecular basis of dominance. Genetics. 1981;97(3-4):639–666. doi: 10.1093/genetics/97.3-4.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manna F, Martin G, Lenormand T. Fitness landscapes: An alternative theory for the dominance of mutation. Genetics. 2011;189(3):923–937. doi: 10.1534/genetics.111.132944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukai T, Chigusa S, Yoshikawa I. The genetic structure of natural populations of Drosophila melanogaster. 3. Dominance effect of spontaneous mutant polygenes controlling viability in heterozygous genetic backgrounds. Genetics. 1965;52(3):493–501. doi: 10.1093/genetics/52.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houle D, Hughes KA, Assimacopoulos S, Charlesworth B. The effects of spontaneous mutation on quantitative traits. II. Dominance of mutations with effects on life-history traits. Genet Res. 1997;70(1):27–34. doi: 10.1017/s001667239700284x. [DOI] [PubMed] [Google Scholar]
- 9.García-Dorado A, Caballero A. On the average coefficient of dominance of deleterious spontaneous mutations. Genetics. 2000;155(4):1991–2001. doi: 10.1093/genetics/155.4.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chavarrías D, López-Fanjul C, García-Dorado A. The rate of mutation and the homozygous and heterozygous mutational effects for competitive viability: A long-term experiment with Drosophila melanogaster. Genetics. 2001;158(2):681–693. doi: 10.1093/genetics/158.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fry JD, Nuzhdin SV. Dominance of mutations affecting viability in Drosophila melanogaster. Genetics. 2003;163(4):1357–1364. doi: 10.1093/genetics/163.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters AD, Halligan DL, Whitlock MC, Keightley PD. Dominance and overdominance of mildly deleterious induced mutations for fitness traits in Caenorhabditis elegans. Genetics. 2003;165(2):589–599. doi: 10.1093/genetics/165.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szafraniec K, Wloch DM, Sliwa P, Borts RH, Korona R. Small fitness effects and weak genetic interactions between deleterious mutations in heterozygous loci of the yeast Saccharomyces cerevisiae. Genet Res. 2003;82(1):19–31. doi: 10.1017/s001667230300630x. [DOI] [PubMed] [Google Scholar]
- 14.Charlesworth D, Willis JH. The genetics of inbreeding depression. Nat Rev Genet. 2009;10(11):783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 15.Ives PT. The genetic structure of American populations of Drosophila melanogaster. Genetics. 1945;30(2):167–196. doi: 10.1093/genetics/30.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Band HT, Ives PT. Comparison of lethal + semilethal frequencies in second and third chromosomes from a natural population of Drosophila melanogaster. Can J Genet Cytol. 1963;14:351–357. doi: 10.1139/g63-047. [DOI] [PubMed] [Google Scholar]
- 17.Mukai T, Cardellino RA, Watanabe TK, Crow JF. The genetic variance for viability and its components in a local population of Drosophila melanogaster. Genetics. 1974;78(4):1195–1208. doi: 10.1093/genetics/78.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukai T, Yamaguchi O. The genetic structure of natural populations of Drosophila melanogaster. XI. Genetic variability in a local population. Genetics. 1974;76(2):339–366. doi: 10.1093/genetics/76.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmons MJ, Crow JF. Mutations affecting fitness in Drosophila populations. Annu Rev Genet. 1977;11:49–78. doi: 10.1146/annurev.ge.11.120177.000405. [DOI] [PubMed] [Google Scholar]
- 20.Latter BD. Mutant alleles of small effect are primarily responsible for the loss of fitness with slow inbreeding in Drosophila melanogaster. Genetics. 1998;148(3):1143–1158. doi: 10.1093/genetics/148.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sved JA. An estimate of heterosis in Drosophila melanogaster. Genet Res. 1971;18(1):97–105. doi: 10.1017/s0016672300012453. [DOI] [PubMed] [Google Scholar]
- 22.Sved JA. Fitness of third chromosome homozygotes in Drosophila melanogaster. Genet Res. 1975;25(2):197–200. doi: 10.1017/s0016672300015603. [DOI] [PubMed] [Google Scholar]
- 23.Wilton AN, Sved JA. X-chromosomal heterosis in Drosophila melanogaster. Genet Res. 1979;34(3):303–315. doi: 10.1017/s0016672300019534. [DOI] [PubMed] [Google Scholar]
- 24.Kusakabe S, Yamaguchi Y, Baba H, Mukai T. The genetic structure of the Raleigh natural population of Drosophila melanogaster revisited. Genetics. 2000;154(2):679–685. doi: 10.1093/genetics/154.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8(3):269–294. [PubMed] [Google Scholar]
- 26.Birky CW, Jr, Walsh JB. Effects of linkage on rates of molecular evolution. Proc Natl Acad Sci USA. 1988;85(17):6414–6418. doi: 10.1073/pnas.85.17.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto SP, Barton NH. The evolution of recombination: removing the limits to natural selection. Genetics. 1997;147(2):879–906. doi: 10.1093/genetics/147.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barton NH. Genetic linkage and natural selection. Philos Trans R Soc Lond B Biol Sci. 2010;365(1552):2559–2569. doi: 10.1098/rstb.2010.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neher RA, Shraiman BI, Fisher DS. Rate of adaptation in large sexual populations. Genetics. 2010;184(2):467–481. doi: 10.1534/genetics.109.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Good BH, Desai MM. Deleterious passengers in adapting populations. Genetics. 2014;198(3):1183–1208. doi: 10.1534/genetics.114.170233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78(2):737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peck JR. A ruby in the rubbish: Beneficial mutations, deleterious mutations and the evolution of sex. Genetics. 1994;137(2):597–606. doi: 10.1093/genetics/137.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barton NH. A general model for the evolution of recombination. Genet Res. 1995;65(2):123–145. doi: 10.1017/s0016672300033140. [DOI] [PubMed] [Google Scholar]
- 34.Roze D, Barton NH. The Hill-Robertson effect and the evolution of recombination. Genetics. 2006;173(3):1793–1811. doi: 10.1534/genetics.106.058586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartfield M, Keightley PD. Current hypotheses for the evolution of sex and recombination. Integr Zool. 2012;7(2):192–209. doi: 10.1111/j.1749-4877.2012.00284.x. [DOI] [PubMed] [Google Scholar]
- 36.Hadany L, Feldman MW. Evolutionary traction: The cost of adaptation and the evolution of sex. J Evol Biol. 2005;18(2):309–314. doi: 10.1111/j.1420-9101.2004.00858.x. [DOI] [PubMed] [Google Scholar]
- 37.Hartfield M, Otto SP. Recombination and hitchhiking of deleterious alleles. Evolution. 2011;65(9):2421–2434. doi: 10.1111/j.1558-5646.2011.01311.x. [DOI] [PubMed] [Google Scholar]
- 38.Luksza M, Lässig M. A predictive fitness model for influenza. Nature. 2014;507(7490):57–61. doi: 10.1038/nature13087. [DOI] [PubMed] [Google Scholar]
- 39.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci USA. 2013;110(8):2910–2915. doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lang GI, et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500(7464):571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elena SF, Lenski RE. Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4(6):457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- 42.Chun S, Fay JC. Evidence for hitchhiking of deleterious mutations within the human genome. PLoS Genet. 2011;7(8):e1002240. doi: 10.1371/journal.pgen.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williamson SH, et al. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3(6):e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huff CD, et al. Crohn’s disease and genetic hitchhiking at IBD5. Mol Biol Evol. 2012;29(1):101–111. doi: 10.1093/molbev/msr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fay JC. Disease consequences of human adaptation. Appl Transl Genom. 2013;2:42–47. doi: 10.1016/j.atg.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cruz F, Vilà C, Webster MT. The legacy of domestication: Accumulation of deleterious mutations in the dog genome. Mol Biol Evol. 2008;25(11):2331–2336. doi: 10.1093/molbev/msn177. [DOI] [PubMed] [Google Scholar]
- 47.Lu J, et al. The accumulation of deleterious mutations in rice genomes: A hypothesis on the cost of domestication. Trends Genet. 2006;22(3):126–131. doi: 10.1016/j.tig.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 48.Hartfield M, Glémin S. Hitchhiking of deleterious alleles and the cost of adaptation in partially selfing species. Genetics. 2014;196(1):281–293. doi: 10.1534/genetics.113.158196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clegg MT. Dynamics of correlated genetic systems. II. Simulation studies of chromosomal segments under selection. Theor Popul Biol. 1978;13(1):1–23. doi: 10.1016/0040-5809(78)90033-3. [DOI] [PubMed] [Google Scholar]
- 50.Ohta T, Kimura M. Development of associative overdominance through linkage disequilibrium in finite populations. Genet Res. 1970;16(2):165–177. doi: 10.1017/s0016672300002391. [DOI] [PubMed] [Google Scholar]
- 51.Robertson A. Selection for heterozygotes in small populations. Genetics. 1962;47:1291–1300. doi: 10.1093/genetics/47.9.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ewens W, Thomson G. Heterozygote selective advantage. Ann Hum Genet. 1970;33(4):365–376. [Google Scholar]
- 53.Nei M, Roychoudhury AK. Probability of fixation and mean fixation time of an overdominant mutation. Genetics. 1973;74(2):371–380. doi: 10.1093/genetics/74.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao Z, Waggoner D, Stephens M, Ober C, Przeworski M. An estimate of the average number of recessive lethal mutations carried by humans. Genetics. 2015;199(4):1243–1254. doi: 10.1534/genetics.114.173351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lander ES, et al. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 56.Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 57.Adachi N, Lieber MR. Bidirectional gene organization: A common architectural feature of the human genome. Cell. 2002;109(7):807–809. doi: 10.1016/s0092-8674(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 58.Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1(1):5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orozco-terWengel P, et al. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Mol Ecol. 2012;21(20):4931–4941. doi: 10.1111/j.1365-294X.2012.05673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burke MK, et al. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467(7315):587–590. doi: 10.1038/nature09352. [DOI] [PubMed] [Google Scholar]
- 61.Clegg MT, Kidwell JF, Kidwell MG, Daniel NJ. Dynamics of correlated genetic systems. I. Selection in the region of the Glued locus of Drosophila melanogaster. Genetics. 1976;83(4):793–810. doi: 10.1093/genetics/83.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoo BH. Long-term selection for a quantitative character in large replicate populations of Drosophila melanogaster: Part 1. Response to selection. Genet Res. 1980;35(1):1–17. doi: 10.1007/BF00276006. [DOI] [PubMed] [Google Scholar]
- 63.Yoo BH. Long-term selection for a quantitative character in large replicate populations of Drosophila melanogaster: Part 2. Lethals and visible mutants with large effects. Genet Res. 1980;35(1):19–31. [Google Scholar]
- 64.Robertson FW, Reeve E. Studies in quantitative inheritance I. The effects of selection of wing and thorax length in Drosophila melanogaster. J Genet. 1952;50(3):414–448. [Google Scholar]
- 65.Frankham R, Jones LP, Barker JSF. The effects of population size and selection intensity in selection for a quantitative character in Drosophila. 3. Analyses of the lines. Genet Res. 1968;12(3):267–283. doi: 10.1017/s0016672300011861. [DOI] [PubMed] [Google Scholar]
- 66.Chevin LM, Hospital F. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics. 2008;180(3):1645–1660. doi: 10.1534/genetics.108.093351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 2007;89(5-6):391–403. doi: 10.1017/S0016672308009579. [DOI] [PubMed] [Google Scholar]
- 68.Messer PW. SLiM: Simulating evolution with selection and linkage. Genetics. 2013;194(4):1037–1039. doi: 10.1534/genetics.113.152181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc Natl Acad Sci USA. 2006;103(1):135–140. doi: 10.1073/pnas.0509691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol. 2014;31(5):1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.