

Editor’s Note:
Numerous factors make us react to situations differently: age, gender, education, relationships, socioeconomic status, environment, cultural background, life experience. But as our author describes, biological bases, such as the way genetics and neurochemicals affect our brains, are providing insight into addiction, posttraumatic stress disorder, and other stresses that he calls “an intimate part of modern life.”
Stress is everywhere. It is an intimate part of modern life. But what is stress? How does the brain process the feeling as a “stress system”? What chemicals in our brains mediate the stress response, and, most important, can we control it? Moreover, what conveys individual differences in stress responsivity that leave some of us vulnerable to stress disorders and others resilient? When does stress go rogue and produce psychopathology? And why do I think of it as the “dark side” of reward pathways in the brain?
My hypotheses are that individual differences in stress vulnerability and resilience are key determinants of the development of posttraumatic stress disorder (PTSD) and addiction, and these differences derive from the neurocircuitry of our emotional dark side. I’ll take you through this neurocircuitry to explain what I mean.
What is Stress?
Stress can be classically defined as “the nonspecific (common) result of any demand upon the body”1 or, from a more psychological perspective, “anything which causes an alteration of psychological homeostatic processes.”2 Historically, the physiological response that is most associated with a state of stress is an elevation of chemicals called glucocorticoids that help control inflammation. Glucocorticoids are derived from the adrenal cortex, a gland situated above the kidneys, and glucocorticoid elevations were thought to be controlled by the brain’s hypothalamus, a region that is associated with emotion. Maintaining psychological homeostasis, therefore, involves responses among the nervous, endocrine, and immune systems. This nexus is referred to as the hypothalamic-pituitary-adrenal axis (HPA).
Efforts to identify processes involved in disrupting psychological homeostasis began while I was a staff scientist at the Arthur Vining Davis Center for Behavioral Neurobiology at the Salk Institute in California. My colleagues Wylie Vale, Catherine Rivier, Jean Rivier, and Joachim Spiess first demonstrated that a peptide called corticotropin-releasing factor (CRF) initiates the HPA axis’s neuroendocrine stress response. Research showed that CRF emanated from a part of the hypothalamus called the paraventricular nucleus, which is the primary controller of the hypothalamic-pituitary-adrenal axis. When the hypothalamus releases CRF, it travels through blood vessels to the pituitary gland, located at the base of the brain. There, CRF binds to receptors located in the anterior part of this gland to release adrenocorticotropic hormone (ACTH) into the blood stream.3
ACTH in turn travels to the cortex of the adrenal gland to release glucocorticoids. Glucocorticoids, in turn, synthesize glucose to increase energy used by the brain, and glucocorticoids also decrease immune function by blocking “proinflammatory” proteins that ordinarily produce inflammation. Together these responses facilitate the body’s mobilization in response to acute stressors. Indeed, acute and chronic glucocorticoid responses differentially affect brain function, with acute high-dose glucocortoids imparting a protective effect.4
Fight or Flight?
When faced with stressors, what determines whether we fight or flee? The human brain’s “extended amygdala” processes fear, threats, and anxiety (which cause fight or flight responses in animals)5,6 and encodes negative emotional states. Located in the lower area of the brain called the basal forebrain, the extended amygdala is composed of several parts, including the amygdala and nucleus accumbens.7 This system receives signals from parts of the brain that are involved in emotion, including the hypothalamus and, most important for this examination, the prefrontal cortex. Extended amygdala neurons send axons or connections heavily to the hypothalamus and other midbrain structures that are involved in the expression of emotional responses.7,8
In psychopathology, dysregulation of the extended amygdala has been considered important in disorders related to stress and negative emotional states. These disorders include PTSD, general anxiety disorder, phobias, affective disorders, and addiction.9,10 For example, animals exposed to a stressor will show an enhanced freezing response to a conditioned fear stimulus, an enhanced startle response to a startle stimulus, and avoidance of open areas, all of which are typical responses to an aversive stimulus and are mediated in part by the extended amygdala.
The Neurochemical Mediators
Why then do individual responses to stress differ? Two important neurochemical systems are involved and help answer this question. The first one is CRF, the neurochemical system mentioned above. It turned out CRF is also a major component of the extended amygdala and works to effect behavioral changes.
While the glucocorticoid response mobilizes the body for physiological responses to stressors, CRF mobilizes the body’s behavioral response to stressors via brain circuits outside the hypothalamus. One of my first eureka moments was when my laboratory helped demonstrate initially that CRF mediates not only physiological and hormonal responses to stressors but also behavioral responses.
In our first study, I injected the newly discovered CRF peptide into the brain in rats and observed very peculiar behavioral hyperactivity. The rats climbed all over the wire-mesh testing cages, including the walls. I called Wylie Vale over to observe the animals because they seemed to be levitating. We subsequently showed that injecting CRF into the rats’ brains produced a pronounced hyperarousal in a familiar environment but a pronounced freezing-like response in a novel stressful environment.11 Subsequent work showed that the extended amygdala mediates such responses to CRF and fear and anxiety in general. When agents were used to block CRF receptors from binding CRF, anti-stress effects occurred, confirming that the release of naturally produced CRF is central in behavioral responses to stressors.12 Equally intriguing, in chronic prolonged stress, glucocorticoids stimulate CRF production in the amygdala while inhibiting it in the hypothalamus, suggesting a means of protecting the body from high chronic exposure to glucocorticoids by shutting off the HPA axis but driving the extrahypothalamic CRF stress system.
The other key neurotransmitter system involved in individual differences in stress responsiveness is called the dynorphin-kappa opioid system (also located in the extended amygdala). This system is implicated in effecting negative emotional states by producing aversive dysphoric-like effects in animals and humans.13 Dysphoria is a negative mood state, the opposite of euphoria. Dynorphins are widely distributed in the central nervous system.14 They have a role in regulating a host of functions, including neuroendocrine and motor activity, pain, temperature, cardiovascular function, respiration, feeding behavior, and stress responsivity.15
In addition to these two neurochemical systems, we now know that other neurochemical systems interact with the extended amygdala to mediate behavioral responses to stressors. They include norepinephrine, vasopressin, hypocretin (orexin), substance P, and proinflammatory cytokines. Conversely, some neurochemical systems act in opposition to the brain stress systems. Among these are neuropeptide Y, nociceptin, and endocannabinoids. A combination of these chemical systems sets the tone for the modulation of emotional expression, particularly negative emotional states, via the extended amygdala.16
Psychopathology and Stress Systems
How are stress systems involved in PTSD? PTSD is characterized by extreme hyperarousal and hyperstress responsiveness. These states contribute greatly to the classic PTSD symptom clusters of re-experiencing, avoidance, and arousal. Perhaps more insidious, about 40 percent of people who experience PTSD ultimately develop drug and alcohol use disorders. Data suggest that the prevalence of an alcohol use disorder in people with PTSD may be as high as 30 percent.17 The major model of PTSD neurocircuitry evolved from early animal work on fear circuits,18 which suggested that brain stress systems are profoundly activated in the extended amygdala.
PTSD patients exhibit abnormally high glucocorticoid receptor sensitivity. This hypersensitivity results in excessive suppression of the HPA axis through corticosteroid negative feedback.19 Research has found that military participants who developed high levels of PTSD symptoms after deployment tended to be those who had significantly higher glucocorticoid receptor expression levels before deployment.20 Another key preclinical study showed that strong activation of CRF receptor signaling in animal models can induce severe anxiety-like and startle hyperreactivity that corresponds to the severe anxiety and startle reactivity seen in patients with PTSD.21 Research also has demonstrated that patients with severe PTSD exhibit overly active brain CRF neurotransmission, measured by increases in CRF in their cerebrospinal fluid.22
While data on PTSD and the dynorphin-kappa system are limited, significant data suggest that brain kappa-opioid receptors play an important role in mediating stress-like responses and encoding the aversive effects of stress.13 An exciting recent imaging study with a kappa-opioid tracer showed decreased kappa-opioid binding in the brain in PTSD patients. This finding suggests increased dynorphin release in patients who are clinically diagnosed with PTSD.23
From a neurocircuitry perspective, functional imaging studies of patients with PTSD show that the amygdala is hyperactive while the ventromedial prefrontal cortex (PFC) and inferior frontal gyrus area show reduced activity.24 These findings suggest that the ventromedial PFC no longer inhibits the amygdala. This loss of inhibition in turn drives increased responses to fear, greater attention to threatening stimuli, delayed or decreased extinction of traumatic memories, and emotional dysregulation.25
One attractive hypothesis for the functional neurocircuitry changes that occur in PTSD suggests a brain-state shift from mild stress (in which the PFC inhibits the amygdala) to extreme stress (in which the PFC goes offline and the amygdala dominates; see figure 1).26 Under this paradigm (rubric means “a standard of performance for a defined population”), relative dominance by the cerebral cortex conveys resilience, and relative dominance by the amygdala conveys vulnerability.26 Delving further into the effects of prefrontal control, two related studies showed that ventromedial PFC activation correlates with the extinction of fear, whereas amygdala activation by the dorsal anterior cingulate cortex (ACC) correlates with a failure to eliminate fear.27,28
Figure 1.
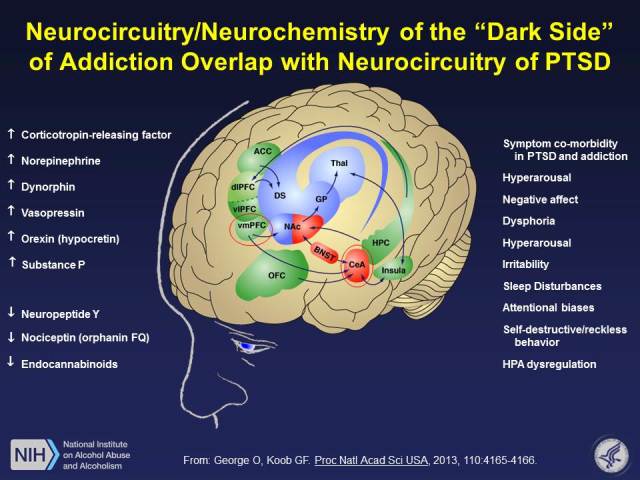
Common neurocircuitry in addiction and posttraumatic stress disorder (PTSD) with a focus of prefrontal cortex (PFC) control over the extended amygdala. The medial PFC inhibits activity in the extended amygdala, where key stress neurotransmitters mediate behavioral responses to stressors and negative emotional states. Key neurotransmitters include corticotropin-releasing factor (CRF) and dynorphin but also other stress and antistress modulators. Notice a significant overlap in the symptoms of PTSD and the withdrawal/negative affect stage of the addiction cycle.
The Paradoxical ‘Darkness Within’
I often tell people that I spent the first fifteen years of my career studying why we feel good and the most recent fifteen years studying why we feel bad. However, these two emotional states are intimately linked, which raises the seemingly contradictory possibility that excessive activation of the reward system can lead to stress-like states that, in their severest form, resemble PTSD. So how did I get to the “dark side”? Well, by first studying the “light side,” or how drugs produce their rewarding effects.
My research team and others hypothesized that addiction involves three stages that incorporate separate but overlapping neurocircuits and relevant neurotransmitter systems: binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation or “craving.”29,30 The binge/intoxication stage involves the facilitation of incentive salience (the linking of previously neutral stimuli in the environment to rewards to give those stimuli incentive properties), mediated largely by neurocircuitry in the basal ganglia. The focus is on activation of the “reward” neurotransmitters dopamine and opioid peptides that bind to mu-opioid receptors in the brain. Early work in the addiction field showed that the nucleus accumbens was a key part of this neurocircuitry that mediates the rewarding properties of abused drugs.
Franco Vaccarino and I showed that we could block heroin self-administration when we injected minute amounts of methylnaloxonium, which blocks opioid receptors, into animals’ nucleus accumbens.31 Subsequently, several classic human imaging studies showed that intoxicating doses of alcohol result in the release of dopamine and opioid peptides in the nucleus accumbens.32,33 We now know that activation of the nucleus accumbens leads to the recruitment of basal ganglia circuits that engage the formation and strengthening of habits. This process is hypothesized to reflect the beginning of compulsive-like responding for drugs—in other words, addiction.
An experiment that turned out exactly the opposite of what I had predicted is the second reason I landed on addiction’s dark side. Tamara Wall, Floyd Bloom, and I set out to identify which regions of the brain mediate physical withdrawal from opiates. We began by training opiate-dependent rats to work for food. Then we disrupted their food-seeking behavior by injecting them with naloxone. This drug precipitated withdrawal, producing a malaise- and dysphoric-like state; as a result, the rats stopped pressing the lever. Thus far, we had successfully replicated original findings.34 We then set out to inject methylnaloxonium, a drug that blocks opioid receptors in brain areas previously implicated in physical withdrawal from opiates. We injected this drug because it was a naloxone analog that would spread less in the brain and precipitate “local” withdrawal as measured by a decrease in lever pressing for food.
We speculated that the most sensitive brain areas to produce a decrease in lever pressing would be the periaqueductal gray and medial thalamus because they had been shown to mediate physical withdrawal from opiates. However, injections into the periaqueductal gray and medial thalamus were ineffective in decreasing lever pressing for food. Instead, injections into the nucleus accumbens proved effective—so effective that we had to drop the dose. Even at a very low dose, we saw some modest effect in decreasing lever pressing for food.35 It then dawned on me that the same brain region responsible for making you feel good also made you feel bad when you became dependent (addicted). This epiphany led me to devote the rest of my career to trying to understand exactly how such opposite reactions that occur during withdrawal, termed opponent processes, are mediated.
This observation led me to a completely new conceptualization of the withdrawal/negative affect stage of addiction. I concluded that this stage is characterized not only by drug-induced specific “physical withdrawal” but also common drug-induced “motivational” withdrawal, characterized by dysphoria, malaise, irritability, sleep disturbances, and hypersensitivity to pain. (These symptoms are virtually identical to the hyperarousal/stress symptoms seen in PTSD; see figure 1).
Two processes were subsequently hypothesized to form the neurobiological basis for the withdrawal/negative affect stage. One is the loss of function in the reward systems in the medial part of the nucleus accumbens of the extended amygdala. This reward system loss is mediated by a loss of function in dopamine systems. The other process is the recruitment of brain stress systems in other parts of the extended amygdala (notably, the central nucleus of the amygdala), including recruitment of the neurochemical systems CRF and dynorphin.36,37 The combination of decreases in reward neurotransmitter function and recruitment of brain stress systems provides a powerful motivation for reengaging in drug taking and drug seeking.
Yet another breakthrough came when my laboratory first realized the dramatic role of CRF in compulsive alcohol seeking, via the amelioration of anxiety-like responses when a CRF receptor antagonist or receptor blocker was used to block the anxiety-like responses of alcohol withdrawal.38 Subsequently, we showed that acute alcohol withdrawal activates CRF systems in the central nucleus of the amygdala.39 Moreover, in animals we found that site-specific injections of CRF receptor antagonists into the central nucleus of the amygdala or systemic injections of small-molecule CRF antagonists reduced the animals’ anxiety-like behavior and excessive self-administration of addictive substances during acute withdrawal.12,40 Perhaps equally compelling, Leandro Vendruscolo and I recently showed that a glucocortoid receptor antagonist could also block the excessive drinking during acute alcohol withdrawal, linking sensitization of the CRF system in the amygdala to chronic activation of the HPA glucocorticoid response.41
But how is excessive activation of the reward system linked to activation of the brain stress systems? Seminal work by Bill Carlezon and Eric Nestler showed that the activation of dopamine receptors that are plentiful in the shell of the nucleus accumbens stimulates a cascade of events that ultimately lead to changes in the rate of DNA transcription initiation and alterations in gene expression. Ultimately, the most notable alteration is activation of dynorphin systems. This dynorphin system activation then feeds back to decrease dopamine release.37 Recent evidence from my laboratory and that of Brendan Walker suggests that the dynorphin-kappa opioid system also mediates compulsive-like drug responses (to methamphetamine, heroin, nicotine, and alcohol); this response is observed in rat models during the transition to addiction. Here, a small-molecule kappa-opioid receptor antagonist selectively blocked the animals’ development of compulsive drug self-administration.42–45 Given that the activation of kappa receptors produces profound dysphoric effects, this plasticity within the extended amygdala may also contribute to the dysphoric syndrome associated with drug withdrawal that is thought to drive the compulsive responses mediated by negative reinforcement.46
Yet another pleasant surprise was the realization that the preoccupation/anticipation, or “craving,” stage in alcoholism mediates the dysregulation of executive control via prefrontal cortex circuits. Importantly, these circuits can become a focal point for individual differences in vulnerability and resilience. Many researchers have conceptualized two generally opposing systems, a “Go” system and a “Stop” system, where the Go system engages habitual and emotional responses and the Stop system brakes habitual and emotional responses. The Go system circuit consists of the anterior cingulate cortex and dorsolateral PFC, and it engages habit formation via the basal ganglia. The Stop system circuit consists of the ventromedial PFC and ventral anterior cingulate cortex and inhibits basal ganglia habit formation, as well as the extended amygdala stress system. People with drug or alcohol addiction experience disruptions of decision making, impairments in the maintenance of spatial information, impairments in behavioral inhibition, and enhanced stress responsivity, all of which can drive craving. More important, this Stop system controls the “dark side” of addiction and the stress reactivity observed in PTSD.
This realization was brought home to me when my colleague Olivier George and I showed that, even in rats that simply engaged in the equivalent of binge drinking, there was a disconnection of the frontal cortex’s control over the amygdala but not nucleus accumbens.47 These results suggest that early in excessive alcohol consumption, a disconnect occurs in the pathway between the PFC and the central nucleus of the amygdala, and this disconnect may be key to impaired executive control over emotional behavior.
Evidence for a Genetic/Epigenetic Mechanism
I suspect that the neurocircuitry focus on the frontal cortex and amygdala in the development of PTSD and addiction will reveal targets for individual differences in vulnerability and resilience. Human imaging studies have established that reduced functioning of the ventromedial PFC and anterior cingulate cortex and increased functioning of the amygdala are reliable findings in PTSD.26. Similarly, drug addiction also has been associated with general reduced function of the ventromedial PFC.48 So what is the contribution of the ventralmedial PFC and anterior cingulate cortex in stress and negative emotional states associated with craving, particularly given what we already know in PTSD? Considering the high co-occurrence of substance abuse and PTSD and the key role of the PFC in controlling the stress systems, the dysregulation of specific subregions of the PFC may be involved in both disorders.
Converging evidence in humans suggests major individual differences in the response of the extended amygdala to emotional stimuli, particularly those considered stressful, and in vulnerability to PTSD and addiction. Research has demonstrated that the central nucleus of the amygdala (the dorsal amygdala in humans) is involved in the conscious processing of fearful faces in healthy volunteers and, more important, that individual differences in trait anxiety predicted the response of a key input to the central nucleus of the amygdala, the basolateral amygdala, to unconsciously processed fearful faces.49 Moreover, a landmark study that used positron emission tomography showed that the amygdala is activated in cocaine-addicted individuals during drug craving but not during exposure to non-drug-related cues.50
Similarly, changes in frontal cortex function can convey individual differences in vulnerability and resilience. In one prospective study that was conducted following the 9.0 Tohoku earthquake in Japan in 2011, participants who had higher gray matter volume in the right ventral anterior cingulate cortex were less likely to have developed PTSD-like symptoms.51 The degree of improvement in symptoms after cognitive behavior therapy was positively correlated with increases in anterior cingulate cortex activation.52 In contrast, other studies have shown that people with PTSD and their high-risk twins show greater resting brain metabolic activity in the dorsal anterior cingulate cortex compared with trauma-exposed individuals without PTSD, suggesting that increased dorsal anterior cingulate cortex activity may be a risk factor for developing PTSD.53
But what molecular neurobiological changes drive these circuit changes? Genetic studies have shown that 30 to 72 percent of the vulnerability to PTSD and 55 percent of the vulnerability to alcoholism can be attributed to heritability. Most would argue that the genetic influences of both disorders stem from multiple genes, and the candidate-gene approach has not yet identified major genetic variants that convey vulnerability to PTSD. However, in two scholarly reviews, at least seventeen gene variants were associated with PTSD and many others with alcoholism.26 Overlapping genes that have been identified in both disorders include gamma-aminobutyric acid, dopamine, norepinephrine, serotonin, CRF, neuropeptide Y, and neurotrophic factors, all of which are relevant to the present hypothesis.
From an epigenetic perspective, some genes may be expressed only under conditions of trauma or stress, and these environmental challenges can modify genetic expression via DNA methylation or acetylation. Both PTSD and alcoholism show epigenetic changes that suggest an increased regulation in genes related to the stress system.54,55 For PTSD, one gene that has been implicated in epigenetic modulation is SLC6A4, which regulates synaptic serotonin reuptake and appears to have a central role in protecting individuals who experience traumatic events from developing PTSD via high methylation activity.56 For alcoholism, histone deacetylase (HDAC) has been implicated in an epigenetic modulation. This gene is involved in the activity-dependent regulation of brain-derived neurotrophic factor (BDNF) expression in neurons. Alcohol-preferring rats with innate higher anxiety-like responses showed higher HDAC activity in the central nucleus of the amygdala. Knockdown of a specific HDAC called HDAC2 in the central nucleus of the amygdala increased BDNF activity and reduced anxiety-like behavior and voluntary alcohol consumption in a selected line of rats that were bred for high alcohol preference.57
Thus, altogether, my hypothesis is that individual differences in stress vulnerability and resilience, which are key determinants of the development of PTSD and addiction, derive from the neurocircuitry of our emotional “dark side.” The origins of activation of the dark side involve both hyperactivity of the extended amygdala (dynorphin and CRF driven by excessive drug use) and reduced activity of the medial PFC (driven by excessive drug use and brain trauma). New advances in our understanding of the neurocircuitry of the dark side and identification of epigenetic factors that weight the function of these circuits will be the key to precision medicine for the diagnosis and treatment of these disorders.
Bio
George F. Koob, Ph.D., is director of the U.S. National Institute on Alcohol Abuse and Alcoholism at the National Institutes of Health. He is on a leave of absence from the Committee on the Neurobiology of Addictive Disorders at The Scripps Research Institute and the Departments of Psychology and Psychiatry and Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California, San Diego. Koob, an authority on drug addiction and stress, has contributed to the understanding of the neurocircuitry associated with the acute reinforcing effects of drugs of abuse and the neuro-adaptations of the reward and stress circuits associated with the transition to dependence. He is co-author of Drugs, Addiction, and the Brain, and he earned his Ph.D. in behavioral physiology at Johns Hopkins University.
References
- 1.Selye H. “Selye’s Guide to Stress Research,”. Van Nostrand Reinhold; New York: 1990. [Google Scholar]
- 2.Burchfield SR. “The stress response: a new perspective,”. Psychosomatic Medicine. 1979;41:661–672. doi: 10.1097/00006842-197912000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Vale W, Spiess J, Rivier C, Rivier J. “Characterization of a 41-residue ovine hypothalamic peptide that stimulates the secretion of corticotropin and β-endorphin,”. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 4.Rao RP, Anilkumar S, McEwen BS, Chattarji S. “Glucocorticoids protect against the delayed behavioral and cellular effects of acute stress on the amygdala.”. Biological Psychiatry. 2012;72:466–475. doi: 10.1016/j.biopsych.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koob GF, Le Moal M. “Addiction and the brain antireward system,”. Annual Review of Psychology. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- 6.LeDoux JE. “Emotion circuits in the brain,”. Annual Review of Neuroscience. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 7.Alheid GF, Heimer L. “New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: the striatopallidal, amygdaloid, and corticopetal components of substantia innominata,”. Neuroscience. 1988;27:1–39. doi: 10.1016/0306-4522(88)90217-5. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds SM, Geisler S, Bérod A, Zahm DS. “Neurotensin antagonist acutely and robustly attenuates locomotion that accompanies stimulation of a neurotensin-containing pathway from rostrobasal forebrain to the ventral tegmental area.”. European Journal of Neuroscience. 2006;24:188–196. doi: 10.1111/j.1460-9568.2006.04791.x. [DOI] [PubMed] [Google Scholar]
- 9.Shin LM, Liberzon I. “The neurocircuitry of fear, stress, and anxiety disorders,”. Neuropsychopharmacology. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koob GF. “Neuroadaptive mechanisms of addiction: studies on the extended amygdala,”. European Neuropsychopharmacology. 2003;13:442–452. doi: 10.1016/j.euroneuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Sutton RE, Koob GF, Le Moal M, Rivier J, Vale W. “Corticotropin releasing factor produces behavioural activation in rats,”. Nature. 1982;297:331–333. doi: 10.1038/297331a0. [DOI] [PubMed] [Google Scholar]
- 12.Koob GF, Zorrilla EP. “Neurobiological mechanisms of addiction: focus on corticotropin-releasing factor,”. Current Opinion in Investigational Drugs. 2010;11:63–71. [PMC free article] [PubMed] [Google Scholar]
- 13.Van’t Veer A A, Carlezon WA., Jr “Role of kappa-opioid receptors in stress and anxiety-related behavior,”. Psychopharmacology. 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watson SJ, Khachaturian H, Akil H, Coy DH, Goldstein A. “Comparison of the distribution of dynorphin systems and enkephalin systems in brain,”. Science. 1982;218:1134–1136. doi: 10.1126/science.6128790. [DOI] [PubMed] [Google Scholar]
- 15.Fallon JH, Leslie FM. “Distribution of dynorphin and enkephalin peptides in the rat brain,”. Journal of Comparative Neurology. 1986;249:293–336. doi: 10.1002/cne.902490302. [DOI] [PubMed] [Google Scholar]
- 16.Koob GF. “The dark side of emotion: the addiction perspective,”. European Journal of Pharmacology. 2014 doi: 10.1016/j.ejphar.2014.11.044. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ouimette P, Read J, Brown PJ. “Consistency of retrospective reports of DSM-IV Criterion A traumatic stressors among substance use disorder patients,”. Journal of Traumatic Stress. 2005;18:43–51. doi: 10.1002/jts.20009. [DOI] [PubMed] [Google Scholar]
- 18.Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Luthi A. “Neuronal circuits of fear extinction,”. European Journal of Neuroscience. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 19.Yehuda R. “Status of glucocorticoid alterations in post-traumatic stress disorder,”. Annals of the New York Academy of Sciences. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- 20.Van Zuiden M, Geuze E, Willermen HL, Vermetten E, Maas M, Heijnen CJ, Kavelaars A. “Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment,”. American Journal of Psychiatry. 2011;168 doi: 10.1176/appi.ajp.2010.10050706. 89e96. [DOI] [PubMed] [Google Scholar]
- 21.Risbrough VB, Stein MB. “Role of corticotropin releasing factor in anxiety disorders: a translational research perspective,”. Hormones and Behavior. 2006;50:550–561. doi: 10.1016/j.yhbeh.2006.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD. “Serial CSF CRH levels and adrenocortical activity in combat veterans with posttraumatic stress disorder,”. American Journal of Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- 23.Pietrzak RH, Naganawa M, Huang Y, Corsi-Travali S, Zheng MQ, Stein MB, Henry S, Lim K, Ropchan J, Lin SF, Carson RE, Neumeister A. “Association of in vivo κ-opioid receptor availability and the transdiagnostic dimensional expression of trauma-related psychopathology,”. JAMA Psychiatry. 2014;71:1262–1270. doi: 10.1001/jamapsychiatry.2014.1221. [DOI] [PubMed] [Google Scholar]
- 24.Hayes JP, Hayes SM, Mikedis AM. “Quantitative meta-analysis of neural activity in posttraumatic stress disorder,”. Biology of Mood and Anxiety Disorders. 2012;2:9. doi: 10.1186/2045-5380-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauch SL, Shin LM, Phelps EA. “Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research: past, present, and future,”. Biological Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. “Biological studies of post-traumatic stress disorder,”. Nature Reviews Neuroscience. 2012;13:769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL. “A role for the human dorsal anterior cingulate cortex in fear expression,”. Biological Psychiatry. 2007a;62:1191–1194. doi: 10.1016/j.biopsych.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 28.Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. “Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert,”. Biological Psychiatry. 2007b;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Koob GF, Le Moal M. “Drug abuse: hedonic homeostatic dysregulation,”. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 30.Koob GF, Volkow ND. “Neurocircuitry of addiction,”. Neuropsychopharmacology Reviews. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaccarino FJ, Pettit HO, Bloom FE, Koob GF. “Effects of intracerebroventricular administration of methyl naloxonium chloride on heroin self-administration in the rat.”. Pharmacology Biochemistry and Behavior. 1985;23:495–498. doi: 10.1016/0091-3057(85)90027-9. [DOI] [PubMed] [Google Scholar]
- 32.Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M, Ma Y, Pradhan K, Wong C. “Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement,”. Journal of Neuroscience. 2007;27:12700–12706. doi: 10.1523/JNEUROSCI.3371-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell JM, O’Neil JP, Janabi M, Marks SM, Jagust WJ, Fields HL. “Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens,”. Science Translational Medicine. 2012;4 doi: 10.1126/scitranslmed.3002902. 116ra6. [DOI] [PubMed] [Google Scholar]
- 34.Sparber SB, Meyer DR. “Clonidine antagonizes naloxone-induced suppression of conditioned behavior and body weight loss in morphine-dependent rats,”. Pharmacology Biochemistry and Behavior. 1978;9:319–325. doi: 10.1016/0091-3057(78)90292-7. [DOI] [PubMed] [Google Scholar]
- 35.Koob GF, Wall TL, Bloom FE. “Nucleus accumbens as a substrate for the aversive stimulus effects of opiate withdrawal,”. Psychopharmacology. 1989;98:530–534. doi: 10.1007/BF00441954. [DOI] [PubMed] [Google Scholar]
- 36.Koob GF, Le Moal M. “Drug addiction, dysregulation of reward, and allostasis,”. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 37.Carlezon WA, Jr, Nestler EJ, Neve RL. “Herpes simplex virus-mediated gene transfer as a tool for neuropsychiatric research,”. Critical Reviews in Neurobiology. 2000;14:47–67. doi: 10.1080/08913810008443546. [DOI] [PubMed] [Google Scholar]
- 38.Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. “CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat,”. Psychopharmacology. 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- 39.Merlo-Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. “Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis,”. Journal of Neuroscience. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Funk CK, O’Dell LE, Crawford EF, Koob GF. “Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats,”. Journal of Neuroscience. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield T, Jr, Logrip ML, Rivier CL, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF. “Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats.”. Journal of Neuroscience. 2012;32:7563–7571. doi: 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker BM, Zorrilla EP, Koob GF. “Systemic κ-opioid receptor antagonism by norbinaltorphimine reduces dependence-induced excessive alcohol self-administration in rats,”. Addiction Biology. 2010;16:116–119. doi: 10.1111/j.1369-1600.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wee S, Orio L, Ghirmai S, Cashman JR, Koob GF. “Inhibition of kappa opioid receptors attenuated increased cocaine intake in rats with extended access to cocaine,”. Psychopharmacology. 2009;205:565–575. doi: 10.1007/s00213-009-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitfield TW, Jr, Schlosburg J, Wee S, Vendruscolo L, Gould A, George O, Grant Y, Edwards S, Crawford E, Koob G. “Kappa opioid receptors in the nucleus accumbens shell mediate escalation of methamphetamine intake,”. Journal of Neuroscience. 2014 doi: 10.1523/JNEUROSCI.1978-13.2015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlosburg JE, Whitfield TW, Jr, Park PE, Crawford EF, George O, Vendruscolo LF, Koob GF. “Long-term antagonism of κ opioid receptors prevents escalation of and increased motivation for heroin intake.”. Journal of Neuroscience. 2013;33:19384–19392. doi: 10.1523/JNEUROSCI.1979-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koob GF. “Negative reinforcement in drug addiction: the darkness within,”. Current Opinion in Neurobiology. 2013;23:559–563. doi: 10.1016/j.conb.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 47.George O, Sanders C, Freiling J, Grigoryan E, Vu CD, Allen S, Crawford E, Mandyam CD, Koob GF. “Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking,”. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry JL, Joseph JE, Jiang Y, Zimmerman RS, Kelly TH, Darna M, Huettl P, Dwoskin LP, Bardo MT. “Prefrontal cortex and drug abuse vulnerability: translation to prevention and treatment interventions.”. Brain Research Reviews. 2011;65:124–149. doi: 10.1016/j.brainresrev.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Etkin A, Klemenhagen KC, Dudman JT, Rogan MT, Hen R, Kandel ER, Hirsch J. “Individual differences in trait anxiety predict the response of the basolateral amygdala to unconsciously processed fearful faces,”. Neuron. 2004;44:1043–1055. doi: 10.1016/j.neuron.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. “Limbic activation during cue-induced cocaine craving,”. American Journal of Psychiatry. 1999;156:11–18. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sekiguchi A, Sugiura M, Taki Y, Kotozaki Y, Nouchi R, Takeuchi H, Araki T, Hanawa S, Nakagawa S, Miyauchi CM, Sakuma A, Kawashima R. “Brain structural changes as vulnerability factors and acquired signs of post-earthquake stress,”. Molecular Psychiatry. 2013;18:618–623. doi: 10.1038/mp.2012.51. [DOI] [PubMed] [Google Scholar]
- 52.Felmingham K, Kemp A, Williams L, Das P, Hughes G, Peduto A, Bryant R. “Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder,”. Psychological Science. 2007;18:127–129. doi: 10.1111/j.1467-9280.2007.01860.x. [DOI] [PubMed] [Google Scholar]
- 53.Shin LM, Lasko NB, Macklin ML, Karpf RD, Milad MR, Orr SP, Goetz JM, Fischman AJ, Rauch SL, Pitman RK. “Resting metabolic activity in the cingulate cortex and vulnerability to posttraumatic stress disorder,”. Archives of General Psychiatry. 2009;66:1099–1107. doi: 10.1001/archgenpsychiatry.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, Goldmann E, Galea S. “Epigenetic and immune function profiles associated with posttraumatic stress disorder,”. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9470–9475. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. “Brain chromatin remodeling: a novel mechanism of alcoholism,”. Journal of Neuroscience. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koenen KC, Uddin M, Chang SC, Aiello AE, Wildman DE, Goldmann E, Galea S. “SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder,”. Depression and Anxiety. 2011;28:639–647. doi: 10.1002/da.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. “Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism,”. Biological Psychiatry. 2013;73:763–773. doi: 10.1016/j.biopsych.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]