

Abstract
PTEN mutation is a frequent feature across a plethora of human cancers, the hot-spot being its C-terminus (PTEN-CT) regulatory domain resulting in a much diminished protein expression. In this study, the presence of C-terminus mutations was confirmed through sequencing of different human tumor samples. The kinase CKII-mediated phosphorylation of PTEN at these sites makes it a loopy structure competing with the E3 ligases for binding to its lipid anchoring C2 domain. Accordingly, it was found that PTEN-CT expressing stable cell lines could inhibit tumorigenesis in syngenic breast tumor models. Therefore, we designed a novel exosome-mediated delivery of the intrinsic PTEN domain, PTEN-CT into different cancer cells and observed reduced proliferation, migration, and colony forming ability. The delivery of exosome containing PTEN-CT to breast tumor mice model was found to result in significant regression in tumor size with the tumor sections showing increased apoptosis. Here, we also report for the first time an active PTEN when its C2 domain is bound by PTEN-CT, probably rendering its anti-tumorigenic activities through the protein phosphatase activity. Therefore, therapeutic interventions that focus on PTEN E3 ligase inhibition through exosome-mediated PTEN-CT delivery can be a probable route in treating cancers with low PTEN expression.
Introduction
PTEN since its identification has been in large associated with the control of human malignancies.1,2 The dominance of PTEN over the PI3K signaling cascade is mainly responsible for rendering it the tumor suppressive role that results in the control of a wide array of physiological processes; growth, apoptosis, proliferation, and migration.3,4 However, in recent years PTEN has expanded its TENtacles in regulating processes such as stem cell population maintenance, genomic stability, and activation of ovarian follicle.5,6,7 Meanwhile, PTEN protein phosphatase activity has gained significant importance and is even capable of inhibiting cell migration irrespective of the Akt pathway.8 As a protein phosphatase, it targets among others SRC kinases, CREB in the nucleus thereby regulating migration of glioma cells. PTEN can even antagonize the expression of epidermal growth factor receptor mutation type three (EGFR νIII).9,10,11 Crucial for PTEN to be able to render its functions properly is the ability to translocate to different sub-cellular compartments of the cell and even out of it through exosomes while retaining its phosphatase activity in the cells where it is internalized.12,13,14 Justifiably, PTEN has fast attained the stature of one of the prime regulators of the cell and therefore it needs to be subjected to intricate surveillance. Although, PTEN like other major players of the cell shows regulation at the transcriptional level as well as through epigenetic silencing, post-translational modifications of PTEN and the associated localization-based alterations in its activities have been the focal point of research on PTEN regulatory events.15,16
PTEN is a relatively stable protein with a half-life of nearly 9 hours. However, a number of human cancer cases have revealed that its stability is under significant threat, which contributes to their aggressive nature in terms of proliferation and migration. Quite a number of such PTEN-associated cancers yielded the revelation of a C-terminus curtailed PTEN (351–403 residues).17,18 Indeed, the C-terminus PTEN truncation resulted in a much lower expression when compared with the wild-type (WT) protein because of its accelerated degradation.19 Now, the C-terminus of PTEN has potential stabilizing factor(s): a PDZ domain that participates in protein–protein association with other PDZ containing proteins resulting in large scaffold structures, which has the capability to prevent its degradation. Interactions with the members of the membrane guanylate-kinase inverted family, MAGI2 through this domain result in its stability, which again is modulated by the cytoskeletal protein Vinculin.20,21 As a matter of fact, knock out of vinculin was reported with negligible PTEN expression even with normal transcript levels.21 Phosphorylation of the C-terminus PTEN tail (S380, T382, and T383) primarily through the kinase CKII forms a closed (inactive lipid phosphatase) loop like structure that is involved in its stabilization.22,23 This PTEN tail phosphorylation is regulated delicately by the C-terminus PDZ-interacting proteins, protein interacting with carboxyl terminus 1, syntrophin-associated serine/threonine kinase.24,25
The E3 ubiquitin ligase-mediated regulation of PTEN is a crucial factor in determining its cellular level. Nedd4-1 was the first E3 ligase reported in connection with PTEN that targeted its C-terminus curtailed mutant form with better efficiency. However, the exact mechanism for such preference was not detailed.26,27 Interestingly though, it was reported that Nedd4-1 was not indispensable for regulating PTEN stability. As such, other E3 ligases have been discovered with the ability to control PTEN turnover.28,29,30 Recently, C-terminus of Hsc-interacting protein (CHIP) has been identified as a prominent E3 ligase that takes part in controlling PTEN stability.31
An important turn of events took place with respect to PTEN research when it was reported that PTEN normally a resident of cytoplasm and nucleus is secreted into exosomes. Such PTEN containing exosomes are internalized by other cells with intact lipid phosphatase activity of the protein.14 Exosomes are 30–100 nm diameter sized vesicular structures with multivesicular body and late endosomal origin. The exosomes that have been observed in a variety of cell types such as neurons, lymphocytes, and a number of mammalian cells are secreted into the extracellular environment, an event regarded as highly controlled.32 Recently, it has been suggested that this mechanism can be exploited for the targeted delivery of siRNA or other factors in the regulation of various diseases.33
Here, we show that the PTEN-CT packaged exosomes elicit anti-proliferative and anti-tumorigenic responses when introduced into different cancer cell lines. In order to increase the PTEN-CT amount in the exosomes, an artificial exosomal delivery system was generated to incorporate purified recombinant PTEN-CT into the exosomes. These exosomes when injected into mice breast tumor models could significantly inhibit tumorigenesis through stabilized PTEN-induced apoptosis. The major apprehension about the system was whether the stabilized PTEN would remain functionally active. A number of previous in vitro results have suggested otherwise, which focused mainly on the PTEN lipid phosphatase activity. However, the results of our study reveal for the first time that C2 bound PTEN is functionally active with anti-tumorigenic potential. Upon further analysis, it was found that such an effect might be produced by the lipid independent protein phosphatase activity of PTEN. The lipid phosphatase-independent cancer inhibitory role of PTEN protein phosphatase activity has increasingly been highlighted in recent times.8 In this study, we report a novel exosome-mediated delivery of the intrinsic PTEN-stabilizing factor PTEN-CT that shows the potential to be exploited as a therapeutic intervention in the treatment of cancer.
Results
The C-terminus lacking PTEN undergoes accelerated degradation
PTEN deletion or truncation is a frequent event in quite a number of human cancers. As a matter of fact when these PTEN mutants are transfected in cells, they are expected to be expressed at a much diminished level as compared to their WT counterpart suggestive of their probable rapid degradation.19 In order to investigate this possibility, the half-lives of WT and C-terminus truncated PTEN (ΔCT) were analyzed through cycloheximide chase experiment. The half-life of ΔCT was less than 3 hours way below the 9-hour half-life of the WT form (Figure 1a). The mutant form also seemed to respond better to one of the E3 ligases (CHIP) of PTEN (Supplementary Figure S1a). Now, it needed to be confirmed whether this difference in expression of the two PTEN forms was due to the proteasome-mediated degradation. Upon treatment of the HEK293 cells expressing either WT- or ΔCT-PTEN with the proteasome inhibitor MG132, it was observed that the recovery of ΔCT was much more pronounced in presence of the inhibitor (Figure 1b), the observations being corroborated with the use of CHIP siRNA (Supplementary Figure S1b). These findings were substantiated by both in vivo and in vitro ubiquitination assays, wherein ΔCT was found to form higher molecular weight ubiquitinated adducts whether in absence (Figure 1c) or presence of an E3 ligase (Figure 1d).
Figure 1.
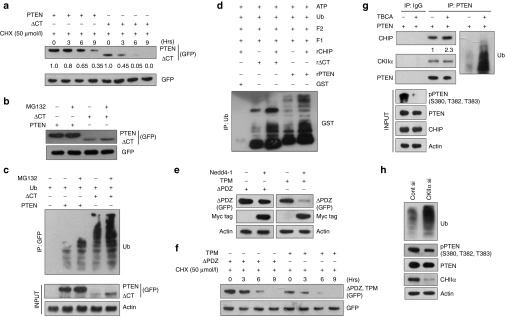
The C-terminus regulatory region lacking PTEN is a better E3 ligase substrate. Cells transfected with: (a) 3.0 µg GFP-PTEN or GFP-ΔCT along with 1.5 µg of pGZ21dx-GFP empty vector (EV). Whole cell lysates (WCLs) were prepared from the cells after treatment with 50 µmol/l cycloheximide (CHX) at the time points (in hours) as mentioned in the figure and immunoblotted against PTEN and ΔCT (using anti-GFP antibody). Empty GFP acted as transfection control. The numbers below the blot represents the densitometric values. (b) 3.0 µg GFP-PTEN or GFP-ΔCT and treated with 50 µmol/l of the proteasomal inhibitor MG132 for 4 hours. Immunoblots were used to detect PTEN, ΔCT (anti-GFP), and GFP. (c) Ub (2.0 µg) along with GFP-PTEN or ΔCT (3.0 µg) and treated with DMSO or 50 µmol/l MG132 for 4 hours. The resultant lysates were immunoprecipitated with anti-GFP antibody, immunoblots show Ub-adducts. The expression levels of all the transfected genes were checked as input (5%). (d) Bacterially expressed and purified, 1 µg each of GST, GST-PTEN (rPTEN), or GST-ΔCT (rΔCT) were incubated with Ub, ATP, “F1 and F2” in presence or absence of 0.5 µg GST-CHIP (rCHIP) for 4 hours at room temperature (RT). Mixtures were then immunoprecipitated with anti-Ub antibody and detected for GST. (e) GFP-ΔPDZ or GFP-TPM (3.0 µg) with or without Myc/His-Nedd4-1 (2.0 µg) and the resultant lysates probed for ΔPDZ, TPM (GFP), Nedd4-1 (Myc tag), and Actin. (f) 3.0 µg GFP-ΔPDZ or GFP-TPM with pGZ21dx-GFP (1.5 µg), treated with 50 µmol/l CHX at the time points (in hours) as mentioned in the figure and probed for ΔPDZ, TPM (GFP). Empty GFP served as transfection control. (g) 3.0 µg GFP-PTEN and treated with or without 20 µmol/l TBCA for 4 hours. WCLs were immunoprecipitated using anti-PTEN antibody and immunoblotted against CHIP, PTEN, and Ub. Input (5%) was run for checking PTEN, pPTEN (S380, T382, T383), CHIP, and Actin. (h) 30 nmol/l Control siRNA (Cont si) or CKIIα siRNA (CKIIα si), WCLs prepared, immunoprecipitated with PTEN, and checked for Ub. Input (5%) was used to detect PTEN, pPTEN (S380, T382, T383), CKIIα, and Actin. The transfected cells were incubated for 36 hours in all the cases. All the experiments were performed in HEK293 cells with at least three repeats.
Thus, the C-terminus of PTEN possessed some protective function for the protein. Available literatures suggest that it could be either of the PDZ domain or a stretch of sites (S380, T382, and T383) phosphorylated by CKII. Both the possibilities were contemplated through the generation of PTEN mutants, ΔPDZ and TPM (triple phosphorylation mutant) and checking their relative stability in presence of the E3 ligase Nedd4-1. It was quite obvious that TPM was highly unstable in presence of the E3 ligase as compared to ΔPDZ (Figure 1e) with a much reduced half-life (Figure 1f). This was not surprising with the earlier observation that the in vitro ubiquitinated adducts of ΔCT- and WT-PTEN showed little difference as compared to the in vivo adducts (Figure 1c,d) due to the unavailability of the distinguishing factor under the in vitro condition, the phosphorylation of the C-terminus PTEN sites (Supplementary Figure S1c). Since, these sites were mainly phosphorylated by the kinase CKII, its role in this process was explored. Accordingly, the cells were treated with the potent CKII inhibitor TBCA. Immunoprecipitation (IP) performed with the resultant lysates confirmed for a better interaction of PTEN with the E3 ligase CHIP along with a much enhanced ubiquitination (Figure 1g). The same result was obtained when CKIIα was knocked down (Figure 1h). From these results, it can be concluded that PTEN without the C-terminus regulatory domain is susceptible to rapid proteasomal degradation owing to the lack of CKII-mediated stabilizing phosphorylation.
Differential pattern of PTEN expression in multiple human cancer types
The expression of PTEN has been found to be highly compromised across different types of human tumors. The PTEN protein level was found to be varying considerably among multiple human cancer cell lines including brain, prostate, breast, and colon with concomitant variation in the levels of the different E3 ligases regulating it; CHIP, XIAP, Nedd4-1, and WWP2 (Figure 2a). Under such a scenario, the expression of PTEN was checked in different human tumor samples. Glioma raw patient samples (n = 30) were used to extract proteins and the level of PTEN analyzed through western blotting. There was a great degree of variation in the PTEN level in those samples (Figure 2b). Paraffinized sections of the patient samples were received for the rest of the cancer types (breast, n = 20; colon, n = 14; and prostate, n = 9). Immunohistochemical analyses followed by H-scoring for PTEN confirmed for very low to high PTEN levels in the breast (Figure 2c,d), colon (Figure 2e,f), and prostate (Figure 2g,h) samples as well. Henceforth, it was concluded from the available tumor samples that the level of PTEN varied considerably among them.
Figure 2.

PTEN expression pattern in different human cancer samples and cell lines. (a) Whole cell lysates (WCLs) prepared from the human mammalian cancer cell lines: T98G, DBT-RG (glioma); DU145, PC3 (prostate); MCF7 (breast); HCT116 (colon); the human embryonic kidney cell line HEK293 and immunoblotted against PTEN, CHIP, XIAP, Nedd4-1, and WWP2. Actin served as the loading control. (b) Proteins isolated from raw human glioma samples (n = 30) using TRIzol method and immunoblotted against PTEN and Hsp90, which served as the loading control. Densitometric analysis of the immunoblots performed with ImageJ software and the control normalized PTEN bands plotted (relative intensity represent arbitrary units). (c) Immunohistochemistry (IHC) performed with sections of formalin fixed paraffinized human breast cancer tissue samples (n = 20) against PTEN, H-scores calculated and plotted for the different patient samples. (d) Figure representing high and low PTEN levels as found by IHC of the breast samples. (e) Human colon sample (n = 14) paraffinized sections used for immunohistochemical staining against PTEN, H-scores calculated and plotted for the different patient samples. (f) Figure representing high and low PTEN levels as found by IHC of the colon samples. (g) IHC and H-scoring against PTEN performed with sections of formalin fixed paraffinized human prostate cancer tissue samples (n = 9). (h) Figure representing high and low PTEN levels as found by IHC of the prostate samples. Images were acquired at 10× original magnification. All the experiments performed were repeated thrice.
Relationship between PTEN expression and its C-terminus mutations
The reason behind the differential expression pattern of PTEN needed to be addressed given the significance of PTEN in inhibiting tumor progression. In order to find a probable clue for these observations, the tumor samples from all the different cancer types were used for RNA extraction followed by cDNA preparation for the potential PTEN mutational hot-spot region (1051–1212 bp). This region of PTEN was sequenced to check for the presence of any mutation. Interestingly, 3 of the 30 glioma samples (11, 13, and 30) were found to harbor a PTEN point mutation (A1147→C, T383P) affecting the CKII-mediated phosphorylation site T383 while in a fourth sample (21), a silent mutation (T1140→G) was detected (Figure 3a). The PTEN expression in those three samples was found to be negligible as compared to the other randomly selected samples (Figure 3b). However, the silent mutation seemed to cause no effect on PTEN expression. In case of breast cancer, 5 of the 20 samples screened were observed to carry mutations; 3 of the samples showed (A1147→C, T383P) mutations while the rest 2 possessed the silent mutation (T1140→G) (Figure 3c). Here, again the samples carrying the mutations were compared with randomly selected other samples for PTEN expression and the observations corroborated the previous findings with the glioma samples. Representative immunohistochemical images comparing sample 6 (mutation) and sample 4 (silent mutation) with another sample 3 (no mutation) clearly show the difference in PTEN levels between the three samples (Figure 3d). Chromatograms showing the mutated and normal sites from both the sets (glioma and breast) have been provided (Supplementary Figure S2a,b). Therefore, the findings point toward a critical relationship between PTEN mutations at the phosphorylation sites and its diminished expression pattern.
Figure 3.
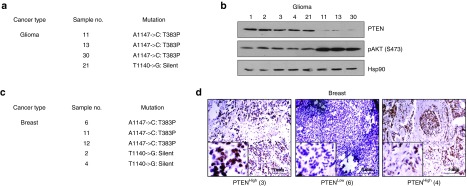
Influence of PTEN C-terminus mutation on its expression. (a) Table represents the number and types of PTEN C-terminus mutations detected in the sequenced raw glioma samples (n = 30). (b) Isolated proteins from the glioma samples carrying mutations (11, 13, 21, and 30) and other randomly selected samples (1, 2, 3, and 4) were immunoblotted against PTEN and pAKT (S473). Hsp90 used as the loading control. (c) Table represents the number and types of PTEN C-terminus mutations detected in the sequenced paraffinized breast cancer samples (n = 20). (d) A comparative representative figure of PTEN staining in sample 6 (mutated) and sample 4 (silent) against sample 3 (non-mutated) as detected through immunohistochemistry (IHC). Images were acquired at 10× original magnification. All the experiments were performed with three repeats.
PTEN-CT protects PTEN from E3 ligase-mediated proteasomal degradation
The phosphorylation of PTEN at its C-terminus tail by CKII results in its folding back to interact with the lipid anchoring C2 domain, thereby conferring stability to the protein. PTEN can be stabilized either by increasing the level of CKII, a proposition that may cause significant turmoil within the cell given the tumorigenic potential of the kinase and hence cannot be taken into account. The other option is to introduce an exogenous PTEN-CT that would not depend on its phosphorylation status and would readily be available to interact with the C2 domain of PTEN. Since, the two domains (CT and C2) interact with each other, it was important to decipher the PTEN domain that interacts with the E3 ligases. Available information in the literature indicated that PTEN might interact with CHIP and Nedd4-1 through its C2 domain.27,31 We also found similar indications for the E3 ligase XIAP (Supplementary Figure S3). Subsequently, in an IP experiment, it was found that these PTEN regulating E3 ligases; CHIP, Nedd4-1, and XIAP interact with its C2 domain (Figure 4a,b). The interactions seem to decrease with the full-length PTEN (Figure 4c) that again suggests for the protective role of PTEN-CT for the tumor suppressor against the E3 ligases. Subsequently, a competitive IP assay was performed to check the interacting potential of the E3 ligases; CHIP, Nedd4-1, and XIAP with PTEN in presence of increasing concentration of PTEN-CT. The presence of PTEN-CT greatly influenced the interaction of PTEN with either of CHIP, Nedd4-1, or XIAP (Figure 4d), the same trend being observed in the ubiquitination pattern of PTEN (Figure 4e). These findings were corroborated by an in vitro competitive binding assay, wherein PTEN-CT inhibited the binding of CHIP with PTEN (Figure 4f). In order to confirm the protective function of PTEN-CT for PTEN under in vivo conditions, the mice mammary carcinoma cell line 4T1 was transfected with either PTEN-CT or empty vector. The cells were selected based on their puromycin resistance at 10 µg/ml final concentration and verified for the presence of PTEN-CT through western blotting (Supplementary Figure S4). These cells were then injected into the mammary fat pad of female BALB/c mice for tumor development. The animals were sacrificed post 4 weeks of injection and the tumor volumes were measured when a significantly reduced tumor volume was observed with the PTEN-CT stable cell-injected animals (n = 6) as compared to the control sets (n = 6) (Figure 4g). Immunohistochemical analyses with the sections of these tumor sets suggested for PTEN stabilization with a concomitant reduction in pCREB (S133) levels in the PTEN-CT set as compared to the control (Figure 4h). However, not much of a difference could be detected in the levels of pAKT (S473). This point toward the fact that may be the protein phosphatase activity of PTEN is behind its anti-tumorigenic potential here. Moreover, another two sets of animals were injected with either WT (n = 6)—or TPM (n = 6)—PTEN expressing stable 4T1 cells. Compared to WT-PTEN, TPM-PTEN possessed very lesser ability of tumor regression (Supplementary Figure S5). Hence, it is now confirmed that PTEN-CT could antagonize the interaction of PTEN with the E3 ligases, thereby stabilize it and inhibit tumor progression.
Figure 4.

PTEN is protected by PTEN-CT from E3 ligase-mediated proteasomal degradation. (a) Schematic representation of PTEN-WT and its truncated mutants. Cells transfected with: (b) 2.5 µg GFP-C2, whole cell lysates (WCLs) prepared, immunoprecipitated with GFP or IgG and immunoblotted against CHIP, Nedd4-1, XIAP, and GFP with 5% input. (c) 2.5 µg GFP-PTEN or GFP-C2, WCLs prepared, immunoprecipitated with GFP or IgG, and immunoblotted against CHIP, XIAP, Nedd4-1, and GFP with 5% input (lower panel). (d) 2.0 µg GFP-PTEN with or without 1.5 µg or 3.0 µg GFP-PTEN-CT, WCLs prepared, immunoprecipitated with PTEN or IgG, and immunoblotted against CHIP, Nedd4-1, XIAP, PTEN-CT (GFP), PTEN, and with 5% input (lower panel). (e) The experiment performed as in d and immunoblotted against Ub. Input (5%) was run to check the levels of PTEN, PTEN-CT (GFP), pAKT (S473), pCREB (S133), and Actin (lower panel). The experiments upto e were performed in HEK293 cells. (f) One hundred microgram of GST-PTEN or GST were immobilized on GSH resin. Fifty microgram of FLAG-CHIP and/or GFP-CT were allowed to bind with them at RT for 2 hours. The resultant immunoblots were checked for PTEN, CHIP, PTEN-CT (GFP), and GST. (g) Two sets of 4–6 weeks old female BALB/c mice (n = 6) injected with pEGFP or pEGFP-PTEN-CT expressing stable 4T1 cells, sacrificed in the 4th week, tumors excised, measured with vernier calipers, and the calculated corresponding volumes represented graphically. Data are represented as mean ± SEM. (h) Paraffinized sections of the tumors in g were used for immunohistochemical analysis against PTEN, pAKT (S473), and pCREB (S133). Images acquired at 20× original magnification. All the experiments were repeated three times.
The PTEN-CT-exosome delivery stabilizes PTEN to inhibit cellular proliferation
It was now established that PTEN-CT-mediated PTEN stabilization could inhibit tumorigenesis. The next goal was to devise a novel mechanism to deliver PTEN-CT into the cancer cells. Exosomes have been previously suggested for the delivery of siRNAs both under in vivo and in vitro conditions.33 Since, PTEN could be trafficked through the exosomes,14 these vesicles seemed to be the perfect natural delivery system for PTEN-CT into the cells and render its effects. HEK293 cells were stained with PKH26 (a lipophilic vital dye) that is used for membrane labeling and transfected with either pEGFP or pEGFP-CT (a near 90% transfection efficiency was observed) (Figure 5a). These cells were then used for the isolation of exosomes. The exosomes carrying PTEN-CT were confirmed through western blotting (Figure 5b). The internalization of PTEN-CT in the exosomes was confirmed when it was found that the internalized PTEN-CT could be protected from trypsin digestion as shown in the lower panel of Figure 5b. Before proceeding further with the exosomes for the study, vesicles needed to be confirmed for being exosomes. The characterization of exosomes was carried out through atomic force microscopy (AFM) where the average size of the vesicles was found to be ~50 nm in diameter, a key feature of the exosomes (Figure 5c,d). A representative three-dimensional AFM image of the exosomes suggest for the consistency of the size (Supplementary Figure S6). The PTEN-CT-exosomes were then applied on HeLa cells and observed for their uptake up to 72 hours. It was found that the uptake of the exosomes carrying PTEN-CT increased with time as was evident through the increase in intensity of both GFP and PKH26 (Figure 5e). Since, PKH26 stained the exosome membranes; its presence could be used as an indication for the uptake of exosomes. In order to increase the amount of PTEN-CT in the exosomes, PTEN-CT was purified (Supplementary Figure S7) and packaged into exosomes through electroporation. Internalization of PTEN-CT into exosomes was confirmed through trypsin digestion (Supplementary Figure S8a). The integrity of the exosomes was also found to be quite consistent after electroporation as analyzed through AFM (Supplementary Figure S8b). The exosomes were further used to treat the breast cancer cell line MDA-MB-231 for 48 hours and the PTEN-CT internalization was further confirmed through western blotting (Figure 5f). The mice cell line 4T1 when treated with PTEN-CT-exosomes for 48 hours were found to have significantly reduced colony-forming ability (Figure 5g). The treatment also resulted in significant compromise in the viability of the cells (Figure 5h). A similar experiment was designed to check the anti-proliferative effects of PTEN-CT in the prostate cancer cell lines PC3 (PTEN null) and DU145 (PTEN WT). However, PTEN-CT alone could not produce such effects in absence of PTEN (Supplementary Figure S9a,b). In summary, it can be clearly said that the exosome packaged PTEN-CT is readily taken up by the cancer cells that in turn induces PTEN-mediated inhibition of cellular proliferation.
Figure 5.

Exosome mediated CT delivery inhibits cancer cell proliferation. (a) HEK293 cells stained with PKH26, seeded on 150 mm culture plates and transfected with 10 µg pEGFP or pEGFP-PTEN-CT. A small part of these cells were reseeded in 35 mm culture plates and visualized with Olympus BX61 microscope for PTEN-CT (GFP, green), PKH26 (red), and DAPI (blue) at 60× original magnification. The images were then merged with ImageJ software. (b) Exosomes isolated from the cells in a, lysates prepared and immunoblotted against PTEN-CT (GFP). Alix and TSG101 are controls for exosome (exo). Lower panel: PTEN-CT (in WCL) and exosome packaged PTEN-CT (through electroporation) were treated with trypsin (Sigma Aldrich). A 1:50 enzyme to substrate ratio (w/w) was maintained and the samples were incubated at 37 °C for 3 hours. Exosomes were lysed and both the samples were then checked for the presence of PTEN-CT. (c) Freshly isolated exosome were diluted 1:10 in PBS, 10 µl sample put on muscovite Ruby mica sheet and analyzed in AFM to get the real topographic image and surface profile extraction (lower panel). (d) The same sample was used to make the amplitude image and size measurement analysis. The average size of the exosomes was found to be around 50 nm. (e) HeLa cells treated with 10 µg CT-exosomes and observed for their uptake at the time points as specified in the figure. Here, PKH26 (red) is checked for exosome internalization and GFP (green) for PTEN-CT. The images were then merged with ImageJ software to look for co-localization. Images were acquired at 60× original magnification. (f) MDA-MB-231 cells treated with 10 µg empty or PTEN-CT packaged exosomes, incubated for 48 hours, cells lysed and exosome internalization was checked by immunoblotting against PTEN-CT (GFP). The immunoblots were also checked for PTEN, pAKT (S473), pCREB (S133), and Actin. (g) 4T1 cells treated with 10 µg empty or PTEN-CT packaged exosomes, incubated for 48 hours, and checked for their colony forming abilities. After 2 weeks, the colonies were counted and size measured in three different microscopic fields and average values of two biological repeats in duplicates were plotted. The images were captured with Olympus IX81 inverted microscope at 20× original magnification after staining the colonies with crystal violet overnight at 4 °C. The graph represents the number of colonies and relative size of the colonies formed by the PTEN-CT exosome treated and untreated cells. Data represented mean ± SEM. (h) 5 or 10 µg empty or PTEN-CT packaged exosomes were used to treat 4T1 cells and checked for their viability through MTT assay. The reading was taken at 550 nm in a Multimode Varioskan Reader and the average values of two biological repeats in duplicates were plotted. Data represented mean ± SEM. All other experiments were performed at least three times.
PTEN-CT induces PTEN-mediated apoptosis and ablates tumorigenesis
PTEN-CT was able to inhibit cellular proliferation through PTEN stabilization. It was observed earlier that the treatment of 4T1 cells with PTEN-CT significantly reduced the cell viability (Figure 5h). It was therefore, interesting to check whether the stabilization of PTEN by PTEN-CT could promote PTEN-mediated apoptosis. Indeed that turned out to be the case when the HCT116 colon cancer cells treated with 2.5, 5.0, and 10 µg of PTEN-CT-exosomes showed increased Sub G0 cell population (2.1%, 5.3%, and 7.9%, respectively) in a concentration gradient manner (Figure 6a). This is suggestive of the fact that the cells might have undergone apoptosis and was confirmed by Annexin V binding with the percentage of cells undergoing apoptosis increased from 4.1 to 39.4 for empty exosome to PTEN-CT-exosome treatment at 10 µg (Figure 6b). In addition, PTEN-CT treatment of the breast cancer cells MCF7 seemed to inhibit cell migration as was observed through scratch assay post 36 hours (Figure 6c). Next, PKH26 stained PTEN-CT-exosomes (150 µg) or equivalent amount of empty exosomes were injected into BALB/c mice carrying size matched tumors (post 2 weeks of 4T1 cell injection) either at the tail vein or site of the tumor. Under both circumstances, the exosomes seemed to enter and diffuse within the tissues of the mice as suggested through whole body imaging of the mice (Figure 6d, Supplementary Figure S10). However, an organ specific uptake and distribution could be observed only in case of the tail vein-injected mice upon fluorescence imaging of the cryo-sectioned tissues (from heart, lung, liver, kidney, and spleen) (Figure 6e). The cryo-sectioned tissues showed the presence of PKH26 to the maximum at 24 hours while the internalization seemed to have initiated at around the 1st hour (Supplementary Figure S11). From the 3rd day onwards, the major accumulation was within the liver and to some extent in the lungs. The mice were sacrificed after 4 weeks and a significant tumor volume reduction in the PTEN-CT-exosome-injected mice sets (n = 6 each) in both cases (tumor site or tail-vein injection) was observed as compared to the control sets (n = 6 each) (Figure 6f, Supplementary Figure S12). However, PTEN-CT (n = 6) or empty exosome (n = 6) alone could not elicit the same tumor regressing effect as PTEN-CT packaged in exosomes (n = 6) (Supplementary Figure S13). Furthermore, the PTEN-CT treated tissue sections revealed significantly higher percentage of apoptotic cells when in situ apoptosis detection for the two sets of tissues were performed (Figure 6g). Similar results were observed from the immunohistochemical analysis of the tissues when checked for the expressions of proliferation marker (Ki67) and apoptosis marker (Caspase 3) (Figure 6h). From these findings, it can be concluded that exosome packaged PTEN-CT can even be successfully delivered into mice tumor models resulting in significant inhibition of tumor progression through stabilized PTEN-induced apoptosis.
Figure 6.

PTEN-CT induces PTEN-mediated apoptosis and ablates tumorigenesis in vivo. (a) HCT116 cells treated with empty (EV) or PTEN-CT packaged (2.5 µg, CT1; 5.0 µg, CT2; 10.0 µg, CT3) exosomes and cultured for 48 hours. The cells were processed; PI stained and proceeded for cell cycle analysis with a focus on the Sub G0 population. The data from four experimental repeats represented as mean ± SEM. (b) A different set of cells with similar treatment were used for checking the apoptotic cell population with FITC Annexin V/PI staining. Here, Cont: control (empty exosomes); CT2: 5.0 µg CT packaged exosomes; CT3: 10.0 µg PTEN-CT packaged exosomes. The experiment was done with three biological repeats. (c) MCF7 cells treated with 10.0 µg empty or PTEN-CT packaged exosomes, incubated for 48 hours and reseeded onto 35 mm culture dishes. Transverse scratches are made on the plates with 200 µl sterile tips and the images captured with Olympus IX81 inverted microscope at 10× original magnification at the specified time points as mentioned in the figure. (d) BALB/c mice (n = 6) carrying tumors were injected with PTEN-CT-exosomes (PKH26 labeled) or empty exosomes at the tail vein. Whole body imaging was performed on the 3rd and 10th day after exosome injection in Xenogen IVIS 200 biphotonic imager. For a similarly sized region, luminescence obtained is represented as photons/sec/region of interest (ROI)-luminescence of the background. (e) The cryo-sectioned tissues (20 µm) from the various organs (heart, lung, liver, kidney and spleen) of the mice (injected at tail vein) were imaged under fluorescence microscope. The organs sectioned were from the 3 and 10 days post-injected mice. The images were acquired at 10× original magnification. (f) The tumors from the mice treated with empty or PTEN-CT packaged exosomes were excised and size measured with vernier calipers and three representative tumors from the control and PTEN-CT treated mice sets are provided. Graph represents tumor volumes from the tumor site injected mice. Volume was calculated and data represented as mean ± SEM. (g) The tumor sections of the mice treated with empty exosomes or PTEN-CT-exosomes used for in situ apoptosis detection. H-scores were calculated and plotted. (h) The adjacent sections were used for the immunohistological detection of Caspase 3 and Ki67. Images were acquired at 20× original magnification with Olympus BX61 microscope. The data represented mean ± SEM.
Discussion
The C-terminus of PTEN, the mutational hot-spot of the tumor suppressor in a wide variety of human cancers is an important part of the protein with respect to its stability and functions.19 Here, we establish a novel exosome-mediated delivery of PTEN-CT, the intrinsic regulatory domain of PTEN for its stabilization and inhibition of tumor progression. It was found that PTEN without its C-terminus domain was rapidly targeted for E3 ligase-mediated proteasomal degradation. The PTEN C-terminus possesses two potential stabilizing factors that could have been involved in this process. A PDZ domain at the extreme end of the protein could interact with other PDZ domain containing proteins, form large scaffold structures, and stabilize PTEN. PTEN also consists of a stretch of phosphorylation sites (S380, T382, and T383) at the C-terminus end phosphorylated mainly by CKII. In presence of the E3 ligase Nedd4-1, it was demonstrated that the PTEN mutant TPM is more susceptible to the E3 ligase as compared to ΔPDZ. Also, the treatment of the cells with the CKII inhibitor TBCA followed by IP suggested for increased interaction of PTEN with the E3 ligase CHIP. Thus, it was confirmed that the lack of phosphorylation at the C-terminus could result in accelerated degradation of the tumor suppressor. Previous reports have suggested for an intramolecular rearrangement of PTEN following phosphorylation at its C-terminus tail when it forms a loop-like structure by interacting with the C2 lipid anchoring domain.34 This form of PTEN was not only found to be more stable but also suggested to be inactive since its lipid binding ability was severely compromised. Although a negative feedback response of PTEN to its E3 ligase Nedd4-1 was reported by one of the studies, no detailed mechanism was provided for the enhanced stability of the PTEN loop-like structure.35 In this study, we found that most of the E3 ligases interact with PTEN through its C2 domain. It was also observed that with increasing amounts of PTEN-CT, the interaction of PTEN with the E3 ligases was severely hampered. Therefore, a competition exists between the E3 ligases and PTEN-CT for binding to the PTEN-C2 (Figure 7). It was observed in the available human cancer patient samples that the PTEN expression varied considerably. With the increasing evidences for the significance of CKII-mediated phosphorylation of PTEN and its stability, we screened for the existence of any mutation that might be present at those PTEN phosphorylation sites in different human cancer samples. It has been previously suggested that cDNA sequencing improves the detection of mutation.36 Accordingly, the cDNA of the PTEN target region (1,050–1,212 bp) was PCR amplified and sequenced. We found three mutations each in the glioma and breast cancer samples that affected the T383 phosphorylation site along with some silent mutations (T1140→G). Although the PTEN expression was not affected in the samples carrying silent mutations, it will be highly interesting to delineate the role of these mutations. The expression of PTEN was greatly reduced in the samples showing (A1147→C) point mutations, thereby further strengthening the importance of the relationship between PTEN C-terminus tail phosphorylation and its stability. Intriguingly, CKII also provides the priming phosphorylation of PTEN at S370 for GSK3β-induced phosphorylation at S366, which makes PTEN unstable.37 So, what is the implication of CKII-mediated PTEN phosphorylation and stabilization? The prime reason of PTEN phosphorylation by the pro-oncogenic kinase CKII could be to render it into an inactive form. However, it cannot be denied that the CT–C2 interaction in PTEN can result in its significant stabilization. Therefore, strategies exploiting this fact can be utilized to stabilize PTEN to interfere with tumor development and progression provided the stabilized PTEN remains in an active form. It was revealed through our findings that PTEN-CT could ablate tumorigenesis under in vivo conditions. Immunohistochemical analyses of the paraffinized sections of the mice tumor samples suggested increased levels of PTEN with a concomitant decrease in the pCREB (S133) levels in the tumors derived from the PTEN-CT stable line injected mice. Nothing significant was noticed in the level of pAKT (S473). Thus, it could be assumed that stabilized PTEN here was able to render anti-tumorigenic activities through its protein phosphatase activity independent of the lipid phosphatase activity. This is the first ever report to suggest for this stable PTEN conformation in an active form. In recent times, it has been found that the protein phosphatase activity of PTEN alone can inhibit tumorigenesis.8 Since, it was now confirmed that exogenous introduction of PTEN-CT can stabilize PTEN with its intact anti-oncogenic properties, designing of an appropriate PTEN-CT natural delivery system was the need of the hour that can easily evade the degrading system of the host. Here, we have devised a novel exosome-mediated delivery of PTEN-CT and observed that this system could elicit the desired effects under both in vitro and in vivo conditions. PTEN-CT was overexpressed in HEK293 cells and the exosomes containing PTEN-CT were isolated. The PTEN-CT-exosomes when treated to other cells were readily taken up. Earlier reports have suggested for the delivery of siRNA with the aid of exosomes.33 Alternatively, purified PTEN-CT was packaged into the exosomes. These, PTEN-CT-exosomes when delivered into the cancer cells could induce PTEN-mediated inhibition of cell proliferation, migration, and colony formation as well as promote apoptosis. We could monitor the entry of the PTEN-CT carrying exosomes (PKH26 stained) into animal breast tumor models, thereby suggesting for the possible uptake through fusion of the exosome membrane and release of PTEN-CT. This was confirmed by the significant reduction in tumor size in the animal sets injected with PTEN-CT-exosomes. In addition, PTEN-CT treated tissue sections revealed significantly higher percentage of apoptotic cells as compared to the control tumors. Hence, an intrinsic part of a protein packaged in a natural membrane system can be readily taken up by the cells, established within and not regarded foreign.
Figure 7.

A model for the E3 ligase-PTEN-CT competition for binding to the C2 domain of PTEN. When the PTEN tail (S380, T382 and T383) is unphosphorylated, it stays in a linear active conformation that may be subjected to E3 ligase induced proteasomal degradation. Phosphorylation of these sites mainly by CKII leads to the formation of a closed loop like structure within the C2 and PTEN-CT domains of PTEN inhibiting the accessibility of the E3 ligases to the C2 domain thereby resulting in a stable state. Many cancer cases show C-terminus truncation or point mutations of PTEN when it is destined for accelerated degradation ending up with very low or negligible PTEN expression. The introduction of exogenous PTEN-CT into the system results in a functionally active stable PTEN.
With the kind of difference the presence or absence and the level of PTEN expression makes to the cancer scenario, this finding might be beneficial in the prevention of untimely degradation or low/negligible PTEN protein levels. A very recent report has connected PTEN mutations with poor prognosis in glioma.38 Therefore, this novel technique of delivering an intrinsic domain of the critical tumor suppressor PTEN can be a crucial lead in the future development of pharmacological interventions in the inhibition and control of cancer progression.
Materials and Methods
Plasmids. The constructs; pIRES-CHIP (FLAG-CHIP) and pGEX4T1-CHIP were generated as described earlier31 while pGZ21dx-GFP-PTEN-WT (GFP-PTEN) was a kind gift from K.M. Yamada. PTEN-WT construct was subsequently used to generate the mutants; pEGFP-PTEN-WT, pGZ21dx-GFP-ΔPDZ, pGZ21dx-GFP-ΔCT, pGZ21dx-GFP-CT (GFP-CT), pGZ21dx-GFP-C2 (GFP-C2), pcDNA3.1-Myc/His-CT, pEGFP-CT, pGZ21dx-GFP-TPM (triple phosphorylation mutant, S380A, T382A, and T383A), and pEGFP-TPM. The plasmids; pRK5-HA-ubiquitin-WT, pCI-HA-NEDD4-1, and pGEX-Xiap were obtained from Addgene. Nedd4-1 and pGEX-Xiap were subsequently cloned into pcDNA3.1-Myc/His.
Mammalian cell maintenance and lysate preparation. The human cell lines including HEK293 (human embryonic kidney), DBTRG, T98G (glioma), DU145, PC3 (prostate cancer), MDA-MB-231, MCF7 (breast cancer), HCT116 (colon cancer), HeLa (cervical cancer) as well as the mice mammary carcinoma cell line, 4T1 were obtained from ATCC (American Type Culture Collection). All the cells except MCF7 and 4T1 (that were maintained in RPMI) were cultured in DMEM supplemented with 10% FBS, 2,000 units/l penicillin, 2 mg/l streptomycin, and 3 mg/l gentamycin solution (Invitrogen, Grand Island, NY) and maintained at 37 °C with 5% CO2. Tris lysis buffer (50 mmol/l Tris pH 7.4, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% NP40, 10% glycerol, and Protease Inhibitor Cocktail Set V, Calbiochem, Darmstadt, Germany) and IP lysis buffer (50 mmol/l HEPES pH 7.2, 250 mmol/l NaCl, 10% glycerol, 1% NP40, 1.0 mmol/l EDTA, 0.5 mmol/l DTT, 10 mmol/l PMSF, and Protease Inhibitor Cocktail Set V) were used for the preparation of whole cell lysates for western blotting and IP respectively as per previous protocol.39 Lysates from raw glioma tissues were prepared after TRIzol RNA extraction following established protocol.40 The patient samples were acquired and proceeded for experiments following the Human Ethics Guidelines at CSIR-Indian Institute of Chemical Biology and Park Clinic (source: Center for the samples), Kolkata. Consents were taken from the patients after explaining the nature and possible consequences of the experiments and publication of the observations.
Preparation of stable cell lines. 4T1 cells were transfected with pEGFP, pEGFP-CT, pEGFP-PTEN, or pEGFP-TPM using Lipofectamine 2000 (Invitrogen) following previous protocol.41 The cells were selected based on their puromycin resistance at 10 µg/ml final concentration. A number of colonies were picked for the expression of the desired proteins. Colonies with the highest expression of the proteins of interest were used for injection into the mammary fat pad of the BALB/c mice.
Antibodies and chemicals. The following antibodies were used in the study: PTEN, CHIP, Hsp90, Nedd4-1, XIAP, Alix, pPTEN (S380, T382, T383), pAKT (S473), pCREB (S133), GST, GFP, Ub, Myc tag, Anti-rabbit IgG, Anti-mouse IgG (Cell Signaling Technology, Danvers, MA); CKIIα, PTEN, pAKT (S473), TSG101 (Abcam, Cambridge, MA); PTEN, β-Actin, normal mouse serum (Santa Cruz Biotechnology, Santa Cruz, CA); and anti-goat IgG (Sigma Aldrich, St Louis, MO). The chemicals used included TBCA, MG132, and cycloheximide (Calbiochem). Cycloheximide was dissolved in water while MG132 and TBCA were dissolved in DMSO. The cycloheximide and MG132 treatments at 50 µmol/l final concentrations were used for 4 hours while TBCA was treated at 20 µmol/l final concentration for the same time period.
Small interfering RNA and transfections. CHIP, CKIIα, and scrambled siRNAs were purchased from Santa Cruz Biotechnology. The siRNA (30 nmol/l final concentration) transfection was performed with RNAiMAX (Invitrogen) while transfections of the cancer cell lines were through Lipofectamine-2000 (Invitrogen). All other transfections were performed with calcium phosphate method following previous protocol.41
Immunoprecipitation and immunoblotting. A total protein amount of 1 mg was used in all IP experiments (unless otherwise mentioned) using desired antibodies and pulled down with Protein A Sepharose beads (Amersham Biosciences, Piscataway, NJ). Both IP and immunoblotting were done following established protocol.31
Recombinant protein purification. GST-ΔCT (rΔCT), GST-PTEN (rPTEN), GST-CHIP (rCHIP), GST-PTEN-CT (rPTEN-CT), and GST (control) vectors were transformed and expressed in BL21DE3 cells for purification of recombinant proteins. Freshly grown cultures were induced with 1 mmol/l IPTG at 22 °C for 4 hours. The cells were then harvested and the proteins purified and concentrated following previous protocol.31 The purity of the proteins was checked through SDS–PAGE and Coomassie staining. The GST tag was cleaved from PTEN-CT with human thrombin (Calbiochem) following established protocols42 before its loading into exosomes.
Ubiquitination assay. Whole cell lysates prepared from 48 hours post-transfected cells were used for the in vivo ubiquitination assay using the co-IP protocol as described earlier. The recombinant proteins; rPTEN, rΔCT, rCHIP, and GST were purified and used for in vitro ubiquitination assay with the Ubiquitin Protein Conjugation Kit (Calbiochem) following previous protocol.31
GST pull-down assay. One hundred microgram of GST or GST-PTEN were incubated with 50 µl (50% slurry) of Glutathione HiCap Matrix (Qiagen, Valencia, CA). After proper washing of the beads, 50 µg of FLAG-CHIP and/or GFP-PTEN-CT (overexpressed in HEK293 cells) were allowed to bind to the resin for 2 hours at room temperature following manufacturer's protocol after which the beads were washed thrice with wash buffer and eluted with 2X SDS loading buffer.
Labeling of cells with PKH26. HEK293 cells were labeled with PKH26 using the PKH26 Fluorescent Cell Linker Kit (Sigma Aldrich) following manufacturer's protocol. Briefly, the cells were cultured in 150 mm dishes (2 × 107 cells), trypsinized, washed once with serum free medium, and centrifuged into a loose pellet at 400g for 5 minutes. A 2X cell suspension and a 2X dye suspension were prepared, mixed well, and allowed to stand for 2 minutes. The reaction was blocked by adding an equal volume of FBS, washed twice, and the cells were plated in 150 mm dishes to be used for the isolation of PKH26 stained exosomes.
Exosome isolation and loading with PTEN-CT. PKH26 labeled HEK293 cells were seeded on 150 mm dishes and used for the isolation of exosomes. The exosomes were isolated from 4T1 cells for all the in vivo experiments. Alternatively, the cells were transfected with pEGFP (EV) or pEGFP-PTEN-CT, kept for 48 hours and used for the purpose. The conditioned medium was collected in 50 ml polypropylene tubes and centrifuged at 200g at 4 °C for 10 minutes. The supernatant was centrifuged at 20,000g at 4 °C for 20 minutes. The resultant supernatant was used for the recovery of exosomes by ultracentrifugation at 100,000g for 1 hour at 4 °C. The pellet was washed with 1X PBS and the centrifugation repeated. The exosome pellet was resuspended in PBS for treating the cancer cells. The empty exosomes were used for loading of PTEN-CT following existing protocol.33 A mixture of equal amounts of purified PTEN-CT and exosomes in 1:1 (wt/wt) ratio with the final amount of exosomes not exceeding 0.5 µg in electroporation buffer (400 µl max.) was used for the purpose. The PTEN-CT loaded exosomes (150 µg) were injected either into the tail vein or tumor site of the mice making sure that the whole resuspension entered the site of injection.
Atomic force microscopy. The isolated exosomes were diluted 10 times in PBS and 10 µl of it was applied onto freshly cleaved muscovite Ruby mica sheet (ASTM V1 Grade Ruby Mica from MICAFAB, Chennai, Tamil Nadu, India). The sample was then dried with a vacuum dryer for 15–30 minutes. The loosely adhered molecules were washed with 0.5 ml MilliQ water and the sample was dried again. Acoustic Alternative Current mode AFM was performed using a Pico plus 5500 ILM AFM (Agilent Technologies, Santa Clara, CA) with a piezoscanner maximum range of 9 µm. Micro fabricated silicon cantilever nanosensors (NanoAndMore, Lady's Island, SC) of 225 µm in length with a nominal spring force constant of 21–98 N/m were used (Nano sensors). Cantilever oscillation frequency was tuned into resonance frequency (250–300 kHz). A scan size (between 0.5 and 5 µm) with a scan speed rate (0.5 lines/second) were used to acquire the images. The image processing was performed with Pico view 1.12 software and Pico Image Advanced version software (Agilent Technologies).
Immunocytochemistry (ICC). The cells were treated as follows: 4% paraformaldehyde fixation, 0.5% Triton-X-100 permeabilization, and blocking with 2.5% BSA in PBS. The rest of the procedure followed established protocols.43 The images were acquired with Olympus BX61 motorized fluorescence microscope using Image ProPlus software at the desired magnification. The images for the uptake of exosomes carrying PTEN-CT were captured directly with Olympus IX81 inverted microscope at the desired time points.
Preparation of cDNA and sequencing. TRIzol (Invitrogen) method was used to isolate RNA from the raw glioma tissues following manufacturer's protocol. RNA from the paraffinized tissue sections was isolated with High Pure RNA Tissue Kit (Roche, Mannheim, Germany). Following manufacturer's protocol, cDNA was prepared from the isolated RNA using the Revert Aid H Minus First Strand cDNA Synthesis Kit (Fermentas, Glen Burnie, MD). The primers used for the PCR amplification of PTEN-CT cDNA sequences were: 5′-GTAGAGGAGCCGTCAAAT-3′ (sense) and 5′-CGACTTTTGTAATTTGTG-3′ (antisense). PCR products were amplified and then sequenced using the dideoxy termination method with 3130X Genetic Analyzer (Applied Biosystems, Carlsbad, CA). The sequences were extracted and chromatograms analyzed using Chromas Lite 2.1 (Technelysium, South Brisbane, Queensland, Australia). PTEN sequence was blasted with the extracted sequences making use of EMBOSS tools for sequence analysis (EMBL-EBI, Hinxton, Cambridge, UK). ExPASy translate tool was used for the detection of the putative amino acid changes for the mutated sequences. The patient samples were acquired and proceeded for experiments following the Human Ethics Guidelines at CSIR-Indian Institute of Chemical Biology and Park Clinic (source: Center for the samples), Kolkata. Consents were taken from the patients after explaining the nature and possible consequences of the experiments.
Cell viability. The viability of the cells were assayed with MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide] following established protocol.44 4T1 cells were seeded on 96-well plates and treated with 10 µg PTEN-CT-exosomes and incubated for 48 hours. The cells were then kept in dark in freshly prepared MTT (0.5 mg/ml) in RPMI medium for 3 hours at 37 °C following which the media were sucked from the wells. One hundred microliter of DMSO was added to each well and kept for 30 minutes to solubilize the resultant formazan. The spectrophotometric absorbance of the solubilized colored product was read at 550 nm by Varioskan Flash Multimode Reader (Thermo Scientific, Waltham, MA).
Flow cytometry. HCT116 cells were treated with empty exosomes, 2.5 µg, 5.0 µg, or 10.0 µg PTEN-CT-exosomes and kept for 48 hours. Cells (1 × 106) were then trypsinized, washed twice with PBS, ethanol (70%) fixed and washed twice with PBS. Five hundred microliter of RNase/PI staining buffer (BD Biosciences, San Jose, CA) was then added to the cells and incubated for 10 minutes in dark and proceeded for flow cytometric analysis with LSRFortessa cell analyzer (BD Biosciences). Alternatively, cells were used for apoptosis detection following manufacturer's protocol using FITC Annexin V Apoptosis Detection Kit II (BD Biosciences) and analyzed in LSRFortessa cell analyzer (BD Biosciences).
Soft agar colony formation. The 4T1 cells were seeded on 6-well plates, treated with 10 µg of empty or exosomes loaded with PTEN-CT and incubated at 37 °C for 48 hours. The cells were then trypsinized, counted with a hemocytometer, 3,000 cells/well mixed in 0.35% agar-RPMI and seeded on the top of 1% solidified agar-RPMI. Colonies were counted (in three different microscopic fields) after incubating the plates at 37 °C in 5% CO2 for 2 weeks. Images of the visible colonies were acquired with IX81 inverted microscope after staining them with crystal violet overnight at 4 °C.
Scratch assay. MCF7 cells were seeded onto 6-well plates and treated with 10 µg of empty exosomes or exosomes carrying PTEN-CT for 48 hours. The cells were then reseeded at ~30% confluency onto 6-well plates. Two transverse scratches were made on the monolayer with a 200 µl sterile pipette tip when the cells reached 50% confluency, washed with PBS and images captured in IX81 inverted microscope. The images were reacquired after 36 hours to monitor the migration of the cells.
In vivo animal studies. 4T1 cells were seeded at 40% confluency and maintained for 24 hours. Cells were removed with trypsin, washed twice with PBS and resuspended in RPMI. Aliquots of 100 µl were prepared to contain 1 × 107 cells and kept in ice. The viability of the cells was checked with Trypan Blue exclusion and cell suspensions showing ~97% viability were kept ready for injection. With the help of a 26-gauge needle, 4–6 weeks old female BALB/c mice were injected subcutaneously into the mammary fat pads with prepared aliquots of cells. The onset of tumor development in the animals was monitored daily by palpating the injection area and on every 3–4 days tumor diameters were measured with vernier calipers. The mice were sacrificed in the 4th week after cell injection, tumors excised and measured for volume. All the animal experiments performed were in compliance with the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA) guidelines and approved by the Animal Ethics Committee at CSIR-Indian Institute of Chemical Biology.
Whole body imaging. The exosomes labeled with PKH26 (Sigma Aldrich) were injected into tumor carrying mice post 1 week of injection of the 4T1 cells. PTEN-CT loaded or empty exosomes (150 µg) were injected into the animals either at the tumor site (n = 6 each) or tail vein (n = 6 each). The animals were anesthetized (2.5% isofluorane) and imaged (on the 3rd and 10th day after the exosomes were injected) in a Xenogen IVIS 200 biphotonic imager. For a similarly sized region, luminescence obtained is represented as photons/sec/region of interest (ROI)-luminescence of the background.
Perfusion and Cryo-sectioning. The animals were injected with chloral hydrate (3.6%) intraperitoneally and perfused through a cardiac peristaltic pump. After perfusion, the organs (heart, lung, liver, kidney and spleen) were taken out and kept in 4% paraformaldehyde for 24 hours followed by incubation in 30% sucrose for 48 hours at 4°C. The tissues were then cryo-sectioned (20 µm thick) using Cryotome (Thermo Scientific), spread on glass slides pre-coated with Poly-L-lysine (Sigma Aldrich) and visualized under fluorescence microscope.
Immunohistochemistry. Paraffinized human cancer samples were obtained from Park Clinic, Kolkata. For the animal tumors, the formalin preserved tissues were embedded in paraffin blocks following standard protocol.44 Using a Leica microtome system, 5 µm thick tissue sections were prepared and spread on glass slides pre-coated with Poly-l-lysine. Immunohistochemistry was performed with the sections following previous protocol.31 The images were captured with BX61 microscope at the desired magnification and H-scoring performed where applicable.
In situ apoptosis detection. Paraffinized animal tumor tissue sections from PTEN-CT treated and control sets were tested for apoptosis with TACS TdT DAB In Situ Apoptosis Detection Kit (R&D Systems) following manufacturer's protocol. The images were acquired in BX61 microscope at 20× original magnification and proceeded for H-scoring.
Statistical analysis. GraphPad (QuickCalcs, http://www.graphpad.com/quickcalcs/index.cfm) calculator was used for all the statistical calculations to create the graphs. In order to ascertain statistical significance, Student's t-test (unpaired) analyses were performed. The P-values <0.05 were considered statistically significant. The error bars present in the figures represent ± SEM.
SUPPLEMENTARY MATERIAL Figure S1. ΔCT is more susceptible to E3 ligase-mediated proteasomal degradation. Figure S2. Comparative chromatograms of normal and mutated PTEN cancer samples. Figure S3. XIAP interacts more efficiently with ΔCT as compared to wild-type PTEN. Figure S4. PTEN-CT expression in 4T1 stable lines. Figure S5. TPM-PTEN is inefficient in inhibiting tumor progression. Figure S6. 3D topographic view of the isolated exosomes. Figure S7. Purified PTEN-CT samples. Figure S8. Consistency of the PTEN-CT internalized exosomes. Figure S9. PTEN-CT cannot induce anti-proliferative effects in absence of PTEN. Figure S10. Mice injected with exosomes at tumor site. Figure S11. Initial internalization of tail vein injected exosomes. Figure S12. PTEN-CT exosome induced reduction in tumor volume. Figure S13. PTEN-CT alone cannot elicit tumor regressing effects.
Acknowledgments
We thank K.M. Yamada (Chief, Laboratory of Cell and Developmental Biology, National Institute of Health/NIDCR, Building 30, Room 426, 30 Convent DR MSC 4370, Bethesda, MD 20892-4370) for kindly gifting us the PTEN constructs. We would also like to convey our thanks to T. Muruganandan for analysis and assistance with the AFM; A. Gangopadhyay and A. Ghosh for assistance with the FACS analysis; and T. Chowdhury for the animal whole body imaging. This work was supported by grants provided by Council of Scientific and Industrial Research (CSIR): EMPOWER (OLP-002), MEDCHEM (BSC0108), mind-0115, and DST (SR/SO/HS150/2010) to M.K.G. All the authors provided critical inputs during the manuscript preparation and approved its final version. The authors declare no conflict of interest.
Supplementary Material
ΔCT is more susceptible to E3 ligase-mediated proteasomal degradation.
Comparative chromatograms of normal and mutated PTEN cancer samples.
XIAP interacts more efficiently with ΔCT as compared to wild-type PTEN.
PTEN-CT expression in 4T1 stable lines.
TPM-PTEN is inefficient in inhibiting tumor progression.
3D topographic view of the isolated exosomes.
Purified PTEN-CT samples.
Consistency of the PTEN-CT internalized exosomes.
PTEN-CT cannot induce anti-proliferative effects in absence of PTEN.
Mice injected with exosomes at tumor site.
Initial internalization of tail vein injected exosomes.
PTEN-CT exosome induced reduction in tumor volume.
PTEN-CT alone cannot elicit tumor regressing effects.
References
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–613. doi: 10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, et al. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal. 2012;5:ra18. doi: 10.1126/scisignal.2002138. [DOI] [PubMed] [Google Scholar]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, et al. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68:1862–1871. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011;71:2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai XM, Tao BB, Wang LY, Liang YL, Jin JW, Yang Y, et al. Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int J Cancer. 2005;117:905–912. doi: 10.1002/ijc.21251. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hoell P, Ahlemeyer B, Krieglstein J. PTEN: a crucial mediator of mitochondria-dependent apoptosis. Apoptosis. 2006;11:197–207. doi: 10.1007/s10495-006-3714-5. [DOI] [PubMed] [Google Scholar]
- Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal. 2012;5:ra70. doi: 10.1126/scisignal.2003084. [DOI] [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;157:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- Teng DH, Hu R, Lin H, Davis T, Iliev D, Frye C, et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 1997;57:5221–5225. [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci USA. 1999;96:10182–10187. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci USA. 2000;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste MC, Nalbant P, Adamson ED, Hahn KM. Vinculin controls PTEN protein level by maintaining the interaction of the adherens junction protein beta-catenin with the scaffolding protein MAGI-2. J Biol Chem. 2005;280:5676–5681. doi: 10.1074/jbc.M405561200. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- Okahara F, Ikawa H, Kanaho Y, Maehama T. Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J Biol Chem. 2004;279:45300–45303. doi: 10.1074/jbc.C400377200. [DOI] [PubMed] [Google Scholar]
- Valiente M, Andrés-Pons A, Gomar B, Torres J, Gil A, Tapparel C, et al. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem. 2005;280:28936–28943. doi: 10.1074/jbc.M504761200. [DOI] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Shi Y, Wang J, Huang G, Jiang X. Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem J. 2008;414:221–229. doi: 10.1042/BJ20080674. [DOI] [PubMed] [Google Scholar]
- Fouladkou F, Landry T, Kawabe H, Neeb A, Lu C, Brose N, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci USA. 2008;105:8585–8590. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Themsche C, Leblanc V, Parent S, Asselin E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J Biol Chem. 2009;284:20462–20466. doi: 10.1074/jbc.C109.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN, et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol. 2011;13:728–733. doi: 10.1038/ncb2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, et al. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem. 2012;287:15996–16006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012;7:2112–2126. doi: 10.1038/nprot.2012.131. [DOI] [PubMed] [Google Scholar]
- Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn Y, Hwang CY, Lee SR, Kwon KS, Lee C. The tumour suppressor PTEN mediates a negative regulation of the E3 ubiquitin-protein ligase Nedd4. Biochem J. 2008;412:331–338. doi: 10.1042/BJ20071403. [DOI] [PubMed] [Google Scholar]
- Szybka M, Zakrzewska M, Rieske P, Pasz-Walczak G, Kulczycka-Wojdala D, Zawlik I, et al. cDNA sequencing improves the detection of P53 missense mutations in colorectal cancer. BMC Cancer. 2009;9:278. doi: 10.1186/1471-2407-9-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280:35195–35202. doi: 10.1074/jbc.M503045200. [DOI] [PubMed] [Google Scholar]
- Xiao WZ, Han DH, Wang F, Wang YQ, Zhu YH, Wu YF, et al. Relationships between PTEN gene mutations and prognosis in glioma: a meta-analysis. Tumour Biol. 2014;35:6687–6693. doi: 10.1007/s13277-014-1885-1. [DOI] [PubMed] [Google Scholar]
- Ghosh MK, Harter ML. A viral mechanism for remodeling chromatin structure in G0 cells. Mol Cell. 2003;12:255–260. doi: 10.1016/s1097-2765(03)00225-9. [DOI] [PubMed] [Google Scholar]
- Simões AE, Pereira DM, Amaral JD, Nunes AF, Gomes SE, Rodrigues PM, et al. Efficient recovery of proteins from multiple source samples after TRIzol(®) or TRIzol(®)LS RNA extraction and long-term storage. BMC Genomics. 2013;14:181. doi: 10.1186/1471-2164-14-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh MK, Sharma P, Harbor PC, Rahaman SO, Haque SJ. PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene. 2005;24:7290–7300. doi: 10.1038/sj.onc.1208894. [DOI] [PubMed] [Google Scholar]
- LaVallie ER, McCoy JM, Smith DB, Riggs P. Enzymatic and chemical cleavage of fusion proteins. Curr Protoc Mol Biol. 2001;Ch. 16:Unit16.4B. doi: 10.1002/0471142727.mb1604bs28. [DOI] [PubMed] [Google Scholar]
- Sha J, Ghosh MK, Zhang K, Harter ML. E1A interacts with two opposing transcriptional pathways to induce quiescent cells into S phase. J Virol. 2010;84:4050–4059. doi: 10.1128/JVI.02131-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmik A, Das N, Pal U, Mandal M, Bhattacharya S, Sarkar M, et al. 2,2′-diphenyl-3,3′-diindolylmethane: a potent compound induces apoptosis in breast cancer cells by inhibiting EGFR pathway. PLoS One. 2013;8:e59798. doi: 10.1371/journal.pone.0059798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ΔCT is more susceptible to E3 ligase-mediated proteasomal degradation.
Comparative chromatograms of normal and mutated PTEN cancer samples.
XIAP interacts more efficiently with ΔCT as compared to wild-type PTEN.
PTEN-CT expression in 4T1 stable lines.
TPM-PTEN is inefficient in inhibiting tumor progression.
3D topographic view of the isolated exosomes.
Purified PTEN-CT samples.
Consistency of the PTEN-CT internalized exosomes.
PTEN-CT cannot induce anti-proliferative effects in absence of PTEN.
Mice injected with exosomes at tumor site.
Initial internalization of tail vein injected exosomes.
PTEN-CT exosome induced reduction in tumor volume.
PTEN-CT alone cannot elicit tumor regressing effects.