

Abstract
Background
Nasopharyngeal carcinoma (NPC) is commonly found in Asia, especially among the Chinese ethnic group. Chromosome rearrangements are common among NPC patients. Although the mechanism underlying the chromosome rearrangements in NPC is unclear, various mechanisms including activation of caspase-activated DNase (CAD) were proposed to contribute to chromosome rearrangements in leukaemia. Activation of CAD can be initiated by multiple agents, including oxidative stress, which is well implicated in carcinogenesis. CAD is the main enzyme that causes DNA fragmentation during apoptosis, and CAD is also implicated in promoting cell differentiation. In view of the role of oxidative stress in carcinogenesis and CAD activation, and since CAD was suggested to contribute to chromosome rearrangement in leukaemia, we hypothesise that oxidative stress-induced CAD activation could be one of the mechanisms that leads to chromosome rearrangements in NPC.
Methods
SUNEI cells were treated with various concentrations of H2O2 for different period of time to ensure that cells undergo H2O2-induced MLL gene cleavage. Transfections with hCAD, mCAD, mutant hCAD, or cotransfection with hCAD and mICAD, and cotransfection with mutant hCAD and mICAD were performed. Gene expression was confirmed by Western blotting and MLL gene cleavage was assessed by inverse polymerase chain reaction (IPCR).
Results
Treatment with H2O2 clearly induces cleavages within the MLL gene which locates at 11q23, a common deletion site in NPC. In order to investigate the role of CAD, CAD was overexpressed in SUNE1 cells, but that did not result in significant changes in H2O2-induced MLL gene cleavage. This could be because CAD requires ICAD for proper folding. Indeed, by overexpressing ICAD alone or co-expressing ICAD with CAD, Western blotting showed that CAD was expressed. In addition, ICAD overexpression also suppressed H2O2-induced MLL gene cleavage, suggesting a possible role of CAD in initiating chromosome cleavage during oxidative stress.
Conclusions
Oxidative stress mediated by H2O2 induces cleavage of the MLL gene, most likely via the caspase-activated DNase, CAD, and CAD expression requires ICAD. Since the MLL gene is located at 11q23, a common deletion site in NPC, thus stress-induced CAD activation may represent one of the mechanisms leading to chromosome rearrangement in NPC.
Keywords: CAD, ICAD, Oxidative stress, Nasopharyngeal carcinoma, MLL, Chromosome breaks
Background
Nasopharyngeal Carcinoma (NPC) is one of the most common cancers in Asia especially in South East Asia, particularly among the Chinese ethnic group [1, 2]. It is well associated with Epstein-Barr virus infection, environmental, dietary, genetic and epigenetic factors [3–7]. The epigenetic effect was demonstrated by promoter methylation of tumour suppressor gene, which was suggested to play a role in the growth and invasion of NPC cells [8]. Furthermore, various chromosome rearrangements are found in NPC, including chromosome gains and losses [9]. A number of studies have identified potential tumour suppressor genes and oncogenes [10]. Nevertheless, the mechanism (s) leading to the non-random chromosome rearrangements in NPC is still elusive. Various mechanisms of chromosome rearrangements have been proposed in other malignancies. These include mechanisms involving Alu repeats [11], V (D) J recombination [12], DNA topoisomerase II [13] and apoptosis [14, 15]. More recently, caspase-activated DNase (CAD) is also implicated in chromosome rearrangement in NPC [16].
Apoptosis is a naturally occurring cell death process that is important in various biological systems [17]. Apoptosis is characterised by a series of distinct morphological and biochemical changes [18]. Most of these morphological changes result from the activity of a class of cysteine proteases, called caspases [19]. A number of caspases have been identified, where about two-thirds of them function in apoptosis [20]. Caspases normally exist as inactive pro-enzymes. When apoptois is triggered, caspases are converted into active enzyme to cleave a subset of proteins [21], either inactivating or activating the target proteins [19]. During apoptosis, while the caspases are activated, the genomic DNA is fragmented into high-molecular-weight (HMW) DNA as well as the smaller fragments known as the internucleosomal DNA ladder [22]. These HMW DNA fragments of 50–300 kb correspond to the DNA-loop structures [23], which interact with the nuclear matrix via the matrix-attachment region or scaffold-associated region (MAR/SAR) sequence [24]. Thus, the HMW DNA formation during apoptosis appears to be DNA loop excision at the MAR/SAR sequence at the base of DNA loop [25]. One of the key enzymes in apoptosis is caspase-activated DNAase (CAD) [26]. Normally CAD exists as an inactive complex with its inhibitor, the Inhibitor of CAD (ICAD). During apoptosis induction, ICAD is cleaved by caspase-3, thus releasing the activated CAD, allowing it to cleave the genomic DNA into HMW DNA as well as internucleosomal DNA ladder [27]. CAD seems to be playing multiple roles. On one hand it is the apoptotic nuclease, one the other hand, it was also found to play a role in chromosome rearrangement commonly found in leukaemia [14, 15, 28]. In addition, CAD was also shown to promote cell differentiation by inducing DNA strand breaks [29].
Apoptotic DNA fragmentation can be induced by a range of stimuli including cytotoxic drugs [15], Fas ligands [30], virus infection [31] and oxidative stress [32]. Oxidative stress induces apoptotic DNA fragmentation in endothelial cells under an ATP rich environment [33] as well as in skeletal muscle myoblasts [34]. It was also found to induce the formation of High Molecular Weight (HMW) DNA fragmentation in leukaemic cells [35]. Oxidative stress produces reactive oxygen species (eg. H2O2, hydroxyl radical and superoxide), that cause injuries on various cellular macromolecules. These damages have been proposed to contribute to the development of cancer [36, 37]. Oxidatively damaged DNA are repaired by the Base Excision Repair pathway (BER) [38] which involves the function of hOGG1 and XRCC1 [39, 40]. Polymorphism of these two genes was shown to be associated with elevated risk of NPC [41], supporting the role of oxidative stress in NPC development.
Knowing that oxidative stress is contributing to NPC, which contains multiple chromosome rearrangements; while oxidative stress also induces apoptotic DNA fragmentation and the apoptotic nuclease, CAD, has been implicated in chromosome rearrangements; thus, we hypothesise that, oxidative stress-induced CAD activation may result in chromosome rearrangement which partly contributes to NPC development. In general, chromosome rearrangement requires chromosome breaks. Since CAD is the major enzyme in apoptotic chromosome fragmentation, we intend to investigate the effect of CAD expression on hydrogen peroxide-induced chromosome breaks within the mixed lineage leukaemia (MLL) gene. The MLL gene is chosen based on the following criteria: (1) it is located at 11q23 [42], a commonly deleted site in NPC [9]; (2) it is commonly translocated in leukaemia [43] and (3) expression of the Epstein-Barr virus latent membrane protein 1 (LMP1) gene induces cleavage of the MLL gene [16].
In this report, we show that oxidative stress induced cleavage of the MLL gene. Expression of CAD alone did not enhance MLL gene cleavage. Interestingly, expression of ICAD not only enhanced CAD expression, but also inhibited MLL gene cleavage, supporting the role of CAD in MLL gene cleavage during oxidative stress.
Results
Hydrogen peroxide (H2O2) induces cleavage of the MLL gene
In order to examine the contribution of H2O2-induced chromosome breaks leading to chromosome rearrangement in NPC, SUNE1 cells were treated with various concentrations of H2O2 for 20 h. Genomic DNA was modified and analysed by nested inverse polymerase chain reaction (IPCR). Based on the primer position, an intact MLL would yield a 2.2 kb band while any band smaller than 2.2 kb represents amplification of the cleaved MLL. As shown in Fig. 1, cells treated with 50 μM, 100 μM and 500 μM of H2O2 resulted in cleavage of the MLL gene as indicated by the presence of distinct bands smaller than 2.2 kb (Lanes 3–5 respectively). Treatment with 1 and 10 mM (Lanes 6 and 7) results in no amplification at all, this is probably due to extensive damage of the template DNA at high H2O2 concentration.
Fig. 1.
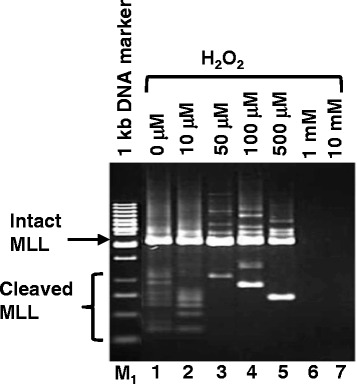
Hydrogen peroxide (H2O2) induces MLL gene cleavage in NPC cells. SUNE1 cells were treated with either 0 μM, 10 μM, 50 μM, 100 μM, 500 μM, 1 mM or 10 mM of H2O2 for 20 h (lanes 1–7 respectively). Genomic DNA was extracted and modified for IPCR as described in Materials and Methods. DNA was digested with Msc I during the modification. Side arrow shows the 2.2 kb band resulting from amplification of the intact MLL gene. Side bracket shows bands smaller than 2.2 kb resulting from amplification of cleaved MLL gene. M1 represents 1 kb DNA marker
Transient transfection with normal and mutant CAD did not enhance H2O2-induced MLL gene cleavage
Since H2O2 clearly induces cleavage of the MLL gene, we would like to investigate if this cleavage is mediated by CAD. Overexpression of CAD in HeLa cells was shown to result in nucleosomal DNA ladder formation [44], thus we chose to transiently transfect both HeLa and and SUNE1 cells, follow by H2O2 treatment, to examine if CAD expression enhances H2O2-induced MLL gene cleavage. As shown in Fig. 2a, the various transfections were successful, with expression of the green fluorescence protein (GFP) clearly shown for each transfection, in the presence or absence of H2O2 Nested IPCR was then performed to detect MLL gene cleavage in transfected cells. To our surprise, comparing with transfection with vector alone (Fig. 2b, lane 6), neither transfection with human CAD nor mouse CAD enhances H2O2 -induced MLL gene cleavage (Fig. 2b, lanes 7 and 8). Similarly, transfection with the mutant human CAD (Fig. 2b, lanes 9) did not show significant difference from transfection with wild-type human or mouse CAD (Fig. 2b, lanes 7 and 8). In the absence of H2O2, the various transfection results in similar background cleavage (Fig. 2b, lanes 1–5). The same transfections were done in HeLa cells and similar results were observed (data not shown).
Fig. 2.
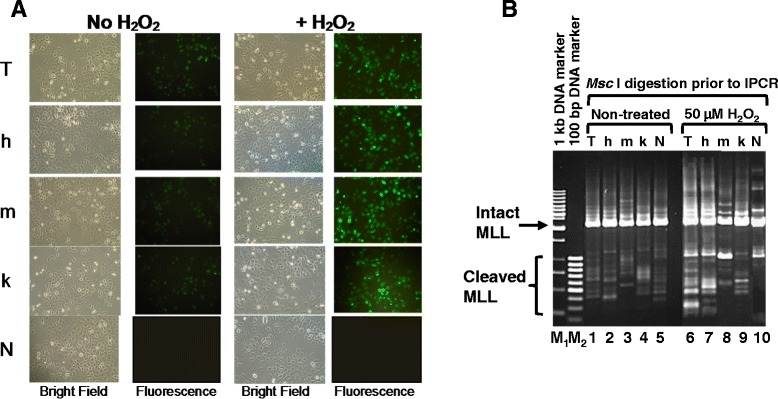
Transient transfection with normal and mutant CAD do not enhance H2O2-induced MLL gene cleavage. a Microscopic morphology of transiently transfected SUNE1 cells. SUNE1 cells were transfected with either the vector pTracer, T; pTracer-hCAD, h; pTracer-mCAD, m; or pTracer-hCAD (k157Q), k. A mock transfection (no DNA), N, was included as control. Transfected cells were either treated with 50 μM of H2O2 for 8 h at 17 h post-transfection, or left untreated. Observation was then carried out with light microscopy and fluorescence microscopy. b Nested IPCR detection of MLL gene cleavage. SUNE1 cells were transfected with either the vector pTracer, T (Lanes1 and 6), pTracer-hCAD, h (Lanes 2 and 7), pTracer-mCAD, m (Lanes 3 and 8) or pTracer-hCAD (k157Q), k (Lanes 4 and 9). A mock transfection (no DNA), N (Lanes 5 and 10) was included as control. The cells were either non-treated (Lanes 1–5) or treated with 50 μM of H2O2 for 8 h at 17 h post-transfection. Genomic DNA extracted was modified for IPCR as described in Materials and Methods. Side arrow shows the 2.2 kb band resulting from amplification of the intact MLL gene. Side bracket shows bands smaller than 2.2 kb resulting from amplification of cleaved MLL gene. M1 represents 1 kb DNA marker. M2 represents 100 bp DNA marker
ICAD expression enhances CAD expression
Although CAD overexpression in HeLa was shown to enhance DNA nucleosomal ladder formation [44], ICAD was also suggested to be the chaperon for CAD [26]. In view of this, co-transfection of CAD and ICAD was carried out in SUNE1 cells. As shown in Fig. 3, human CAD transfection alone in deed did not show enhanced CAD expression (Panel A, lane 4). However, transfection with full-length mouse ICAD alone greatly enhanced CAD expression (Panel A, lane 5). At the same time, co-transfection wtih human CAD and mouse ICAD, or co-transfection with mutant human CAD and mouse ICAD also enhanced CAD expression (Panel A, lanes 6 and 7). Western blot with anti-ICAD clearly shows that ICAD expression was successful (Panel A, lanes 5–7). Expression of GAPDH shows consistent loading in all samples (Panel A, lanes 1–7). To ensure that subsequent H2O2 treatment does not change the expression pattern, transfected cells were also treated with H2O2 at 17 h post-trasfection. The results show that the expression pattern for the different transfection is similar to that in the absence of H2O2 (Panel B, lanes 1–7).
Fig. 3.
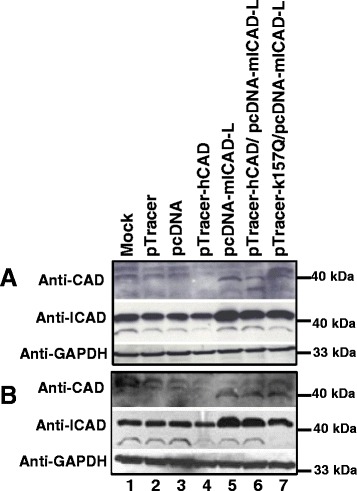
ICAD expression enhances CAD expression. Two sets of SUNE1 cells were transiently transfected with vector pTracer (Lane 2), pcDNA (Lane 3), pTracer-hCAD (Lane 4), pcDNA-mICAD-L (Lane 5), or cotransfection with pTracer-hCAD/pcDNA-mICAD-L (Lane 6) and pTracer-hCAD (K157Q)/pcDNA-mICAD-L (Lane 7). A mock transfection (No DNA) was included as control (Lane 1). Transfected cells were either not treated (Panel a) or treated with 50 μM of H2O2 for 6 h at 17 h post-transfection (Panel b). Protein was extracted as detailed in Materials and Methods. Expression of CAD, ICAD and GAPDH was each analysed by anti-CAD, anti-ICAD and anti-GAPDH respectively
ICAD expression inhibits MLL gene cleavage
Since ICAD expression enhances CAD expression, we intend to investigate this effect on MLL gene cleavage in the presence of H2O2. There are two possibilities, one is ICAD expression enhances CAD expression (as shown in previous section), and thus enhances MLL gene cleavage. Alternatively, due to high level of ICAD overexpressed, it may also inhibit the activity of CAD though CAD’s expression was enhanced. Indeed, ICAD overexpression inhibits cleavage of the MLL gene in the presence of 50 μM H2O2 (Fig. 4, lane 5). Co-transfection with ICAD and human CAD or co-transfection with ICAD and mutant human CAD do not show obvious inhibition of MLL gene cleavage (Fig. 4, lanes 6 and 7 respectively). This could be due to lesser amount of ICAD DNA being used during co-transfection as compared to ICAD transfection alone. Lesser amount of ICAD DNA is required during co-transfection to avoid DNA toxicity during transfection. At the same time, this may also be due to more CAD expressed from the transfected plasmid in addition to endogenous CAD.
Fig. 4.
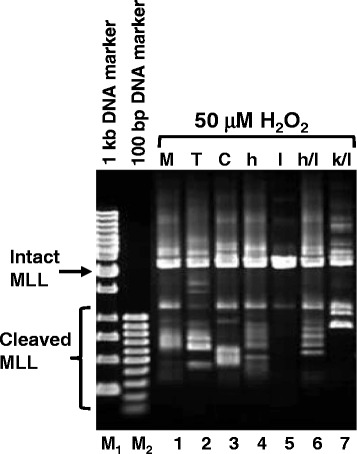
ICAD expression inhibited H2O2-induced MLL gene cleavage. SUNE1 cells were transiently transfected with vector pTracer (Lane 2), pcDNA (Lane 3), pTracer-hCAD (Lane 4), pcDNA-mICAD-L (Lane 5), or cotransfection with pTracer-hCAD/pcDNA-mICAD-L (Lane 6) and pTracer-hCAD (K157Q)/pcDNA-mICAD-L (Lane 7). A mock transfection (No DNA) was included as control (Lane 1). Transfected cells were treated with 50 μM of H2O2 for 6 h at 17 h post-transfection. Genomic DNA was extracted and processed for IPCR as detailed in Materials and Methods. Side arrow shows the 2.2 kb band resulting from amplification of the intact MLL gene. Side bracket shows bands smaller than 2.2 kb resulting from amplification of cleaved MLL gene. M1 represents 1 kb DNA marker. M2 represents 100 bp DNA marker
Discussion
The apoptotic nuclease, caspase activated DNase (CAD) was suggested to play a direct role in mediating chromosome translocation in leukaemia [15, 28]. Although chromosome rearrangements are commonly found in nasoparyngeal carcinoma (NPC), the mechanism leading to these rearrangements is yet unclear. Our earlier data showed that expression of Epstein-Bar virus (EBV) latent membrane protein 1 (LMP1) induced cleavage of the mixed lineage leukemia (MLL) gene at 11q23, which was significantly reduced by caspase inhibitor [16]. Since 11q23 is a commonly deleted site in NPC, thus it was suggested that virus-induced CAD activation could be involved in the initiation of chromosome rearrangement in NPC. Oxidative stress was found to be a contributing factor to NPC [41] and is known to induce apoptotic DNA fragmentation [32], thus, the current study extended to investigate the role of CAD in oxidative stress-induced chromosome breaks within the MLL gene in NPC cells.
Although oxidative stress was suggested to be a contributing factor to NPC, it was unclear of the molecular mechanism. In this current study, we clearly demonstrated that hydrogen peroxide induced chromosome breaks within the MLL gene. Since MLL locates at 11q23, a common deletion site in NPC [9], thus our result suggests that oxidative stress could be initiating chromosome aberration by inducing chromosome breaks at the MLL gene. MLL gene contains MAR/SAR sequences [45] which has been implicated in chromosome rearrangement [46], while oxidative stress has also been shown to poison DNA topoisomerase II, resulting in high molecular weight (HMW) DNA fragmentation [35]. High molecular weight (HMW) DNA fragmentation appears to result from excision of the chromosomal DNA loops [25], whose base is interacting with the nuclear matrix via the MAR/SAR sequence [47]. Although it was suggested that, the loop excision was mediated by DNA topoisomerase II [35], it is also known that DNA topoisomerase II interacts with CAD at the nuclear matrix [48], thus not excluding the role of CAD in the event. This is supported by our result that hydrogen peroxide induces cleavage of the MLL gene near the MAR/SAR region.
Overexpression of CAD in HeLa cells was shown to induce nucleosomal DNA ladder formation [44], thus we chose to express CAD in HeLa and SUNE1 cells and then assess for H2O2-induced chromosome breaks. No obvious difference was observed among the different transfectants as detailed in the result section. This could be due to the reason that ICAD is acting as a chaperone for the proper folding of CAD during its expression [26]. This result seems to contradict with the observation of Mukae et al. [44]. However, Mukae et al. [44] employed both the anti-Fas antibody and actinomycin D and successfully demonstrated that overexpression of CAD alone in HeLa results in nucleosomal DNA ladder formation [44]. Anti-Fas antibody activates the death receptor pathway during apoptosis [49], while actinomycin D targets DNA topoisomerase II, which activates the mitochondrial pathway during apoptosis [50]. In the current study, H2O2 is the sole apoptosis inducer, while H2O2-induced apoptosis is CD-95 independent [51] and it induces apoptosis through the mitochondrial pathway [52]. This subtle difference may explain the different observations in these two studies.
During protein synthesis in a cell-free system, CAD was recovered as a complex with ICAD, suggesting that ICAD acts as a chaperon for CAD [26]. This is further supported by our result that, expression of ICAD enhanced the expression of endogenous and exogenous CAD. In the current study, mouse ICAD and human CAD were used. Thus it seems that the chaperone activity of mouse ICAD can be applied to human CAD. This is supported by the fact that mouse ICAD and human ICAD are almost identical at the N-terminal domain [44], the so called CAD domain [53]. This domain is required for the chaperon activity of ICAD, as well as for CAD/ICAD association during formation of the heterotetrameric structure of (CAD/ICAD) 2 [54]. This kind of chaperone-assisted protein expression is also evidenced in microorganism such as Escherichia coli [55].
Our result shows that ICAD expression enhances CAD expression, while we also hypothesise that CAD is mediating the H2O2-induced MLL gene cleavage. Thus, in order to investigate the final effect of ICAD expression in terms of MLL gene cleavage, the transfected cells were treated with H2O2 and assessed by IPCR. Our result clearly shows that although ICAD expression induced endogenous CAD expression, it has also extensively reduced H2O2-induced MLL gene cleavage. This is obviously explained by the fact that ICAD expression was much higher than CAD as shown by the Western blot result. Thus, the endogenous CAD expressed was inhibited by the overexpressed exogenous CAD. Similarly, ICAD expression also inhibited Fas ligand-induced and staurosporine-induced DNA fragmentation [27]. This supports our hypothesis that CAD could be involved in MLL gene cleavage during oxidative stress. This is also consistent with CAD’s role in chromosome translocation in treatment-related leukemia [28]. In the absence of apoptotic stimuli, CAD is in complex with ICAD and thus stays inactive [26]. Both CAD and ICAD share a homologous N-terminal domain of about 80 amino acids [44]. This domain is known as the CAD domain. The CAD domain of CAD and ICAD interact with each other during CAD/ICAD heterodimeric complex formation, and this interaction is important for the proper folding of CAD [53, 56]. Upon induction of apoptosis by apoptotic stimuli, the caspase cascade is activated, with caspase 3 cleaving ICAD, thus releasing the activated CAD [27]. CAD/ICAD complex is normally mobile in the nucleosol of dividing cells, but during apoptosis induction, CAD becomes progressively immobilised and associates with the nuclear matrix [57]. Nuclear matrix is the anchorage site for the organisation of DNA-loop structure in the cell [47]. The base of DNA loops contain DNA sequences known as the matrix-attachment region or scaffold-associated region (MAR/SAR) and it is through these sequences that DNA is interacting with the nuclear matrix [24]. With CAD being associated with the nuclear matrix during apoptosis [57], it is obviously in close proximity to the MAR/SAR sequence of DNA and potentially cleave the DNA when it is activated. The region of DNA involved in this study falls within the MAR/SAR sequence, thus supporting the role of CAD in mediating MLL gene cleavage during oxidative stress.
Conclusions
From the current findings, we concluded that oxidative stress mediated by H2O2 induces cleavage of the MLL gene, most likely via the activity of CAD, and CAD expression requires ICAD. Since the MLL gene is located at 11q23, a common deletion site in NPC, thus stress-induced CAD activation may represent one of the mechanisms leading to chromosome rearrangement in NPC.
Methods
Cell lines and materials
HeLa cell line was a gift from Dr. Edmund Sim Ui Hang of Universiti Malaysia Sarawak. Nasopharyngeal carcinoma cell line, SUNE1 was generously provided by Professor Dr. Sam Choon Kook previously from Universiti Malaya. Plasmid clones for hCAD, mCAD and mICAD were generous gifts from Professor Dr. Shigekazu Nagata of Osaka Bioscience Institute. DNA Polymerase I, Large (Klenow) fragment, T4 DNA Ligase and all the restriction enzymes were purchased from New England Biolabs (NEB, England). PCR primers were obtained from First Base Laboratories. Phusion DNA Polymerase was purchased from Finnzymes, Finland. Blood and Cell Culture DNA Mini-Prep kit and QIAquick Gel Extraction Kit were purchased from QIAGEN, Germany. All cell culture and trasfection reagents were obtained from GIBCO, Invitrogen. Protease inhibitor cocktail was purchased from Sigma, USA.
Cell culture and H2O2 treatment
HeLa cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM), which contains low glucose (1X), 1 g/L D-glucose, L-glutamine (2 mM) and 100 mg/L sodium pyruvate, supplemented with 10 % heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100 μg/ml). Meanwhile, SUNE1cells were maintained in RPMI 1640 medium supplemented with 10 % heat-inactivated fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml). All cells were grown at 37 °C with 5 % CO2. HeLa and SUNE1 cells in 60 mm dishes were treated with 0, 10, 50, 100, 500, 1000 and 10,000 μM of H2O2 for 20 h. Cells were collected, genomic DNA was extracted using Blood and Cell Culture mini kit.
Nested Inverse polymerase chain reaction (IPCR) detection of H2O2-induced chromosome breaks within the MLL gene
Figure 5 shows a summary of DNA modification as well as the IPCR. Briefly, extracted genomic DNA was digested with BamH I (NEB, USA) at 37 °C overnight, followed by Klenow fill-in with DNA Polymerase I Large Fragment. Cyclisation by T4 DNA Ligase was performed and subsequently linearisation by Msc I. Nested IPCR was carried out with 150 ng of Msc I-digested template DNA, 50 pmol each of the primers, 200 μM each of the dNTP, 0.4 unit of Phusion polymerase and 1X of Phusion HF buffer. PCR cycle condition was: 1 cycle at 98 °C for 30 s, followed by 30 cycles at 98 °C for 10 s, 59 °C for 30 s, 72 °C for 15 s and a final cycle at 72 °C for 10 min. Second round PCR was performed with 2 μl of 5 time-diluted first round PCR products. PCR cycle condition was similar to the first round, except that the annealing and extension steps were carried out at 54 °C for 30 s and 72 °C for 11 s respectively. PCR products were analysed on 1 % agarose gel in 0.5X TBE buffer. The primers used were 5′-GAGAATCGCTTGAACCCAACAG-3′ and 5′-CTTGTGGGTCAGCAATTCCTTC-3′ in the first round, 5′-CCACTCCTTTATATTCCCATAGC-3′ and 5′-TCCTCCACTCACCTGATTC-3′ in the second round. Intact MLL gene will give rise to an amplification product of 2.2 kb while breaks within the region encompassed by the primers will produce amplification products smaller than 2.2 kb.
Fig. 5.
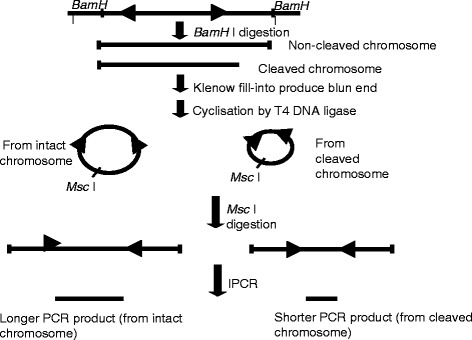
Flow chart showing DNA modification and IPCR. The arrow heads indicate the forward and reverse primers that were designed in opposite direction. BamH I digestion yielded a mixture of intact chromosome and cleaved chromosome. Klenow fill-in produced blunt ended chromosome fragments which were then cyclilsed by T4 DNA ligase. The intact chromosome will become a large circle while the cleaved chromosome will become a smaller circle. Upon cyclisation, the primers are now in correct orientation for amplification. Msc I digestion cleaved both circles outside the amplification region, thus merely linearise the molecule. Amplification from intact MLL gene will produce longer PCR products while amplification from cleaved MLL gene will yield shorter PCR products
Site-directed mutagenesis of human CAD (hCAD)
Site-directed mutagenesis of hCAD was performed using megaprimer site-directed mutagenesis protocol [58]. During the first round of PCR, pTracer-hCAD which carries the wild type human CAD gene was used as the template. Forward primer used was 5′-CGATTTCAGAGCCAGTCTGG-3′, which carries a mutation from Lysine (K), AAG to Glutamine (Q), GAG as highlighted in bold, while the reverse primer used was 5′-CGTCTAGACTGGCGTTTCCG-3′ which carries an Xba I restriction site (in bold). An amplification product of 589 bp was produced and was used as the reverse primer (a megaprimer) in second round of PCR. The forward primer used in second PCR was 5′-CTGGTACCATGCTCCAGAAG-3′ which carries a Kpn I restriction site (in bold). The final amplification product was a 1 kb fragment of mutated hCAD (K157Q) with Xba I and Kpn I restriction sites at the 5′ and 3′ ends respectively. Gel-purified amplification product was subsequently subcloned into pTracer producing pTracer-hCAD (K157Q). The subcloned gene was then confirmed by DNA sequencing.
Transient transfection and co-transfection of NPC cells
SUNE1 cells were transfected with 2 μg each of the following: pTracer-hCAD carrying the wild type human CAD gene; pcDNA-mICAD-L carrying the full length wild type mouse ICAD; pTracer and pcDNA as controls. SUNE1 cells were also co-transfected with the following: pTracer-hCAD and pcDNA-mICAD-L; pTracer-hCAD (k157Q) carrying the mutant human CAD and pcDNA-mICAD-L in a ratio of 2:1 (1.33 μg: 0.67 μg). Mock transfection was also carried out where cells underwent transfection process in the absence of DNA. Transfection was performed with LipofectAMINE™ reagent and PLUS reagent (Invitrogen, Carlsbad, USA) following the manufacturer’s protocol.
SDS PAGE and Western analysis
Attached cells were lysed with 0.5 ml of triple detergent mix (50 mM Tris.Cl, pH8; 150 mM sodium chloride; 0.02 % sodium azide; 0.1 % SDS; 1 % Nonidet P-40 and 0.5 % sodium deoxycholate) containing freshly added protease inhibitor cocktail and PMSF. Lysis was carried out on ice for 5 min, and then transferred to 1.5 ml microcentrifuge tubes. The lysate was further lysed by freezing in liquid nitrogen for 5 min followed by thawing at 37 °C. The lysate was vortexed vigorously for 1 min and the freez-thaw process was repeated for another 4 times. Centrifugation at 20,000x g was carried out for 10 min. Protein concentration in the supernatant was estimated using Bradford Assay. Fifty migrogram of each crude extract was boiled for 10 min in the presence of 2x SDS sample buffer (1X Tris.Cl, pH 6.8, 20 % glycerol, 4 % SDS, 0.2 % 2-merchaptoethanol, 0.001 % bromophenol blue). Samples were centrifuged, and analysed on 10 % SDS-polyacrylamide gel, followed by transfer onto polyvinylidene fluoride (PVDF) membrane (Millipore, Burlington, MA). Immunoblotting was performed with rabbit anti-CAD (Stressgen) at 1:1000 dilution, rabbit anti-ICAD (Stressgen) at 1:4000 dilution and rabbit anti-GAPDH (Santa Cruz, USA) at 1:4000 dilution. Goat anti-rabbit IgG HRP (Santa Cruz, USA) was used as the secondary antibody at 1:10,000 dilution. Chemiluminescent detection was carried out using the WestPico Chemiluminescent Substrate Kit (Pierce, USA) following the manufacturer’s protocol.
Acknowledgements
We wish to express our greatest gratitude to Professor Dr. Shigekazu Nagata of Osaka University Medical School for the hCAD, mCAD and mICAD plasmid clones. We also would like to thank Professor Dr. Sam Choon Kook, previously from Universiti Malaya, for the NPC cell line. We also thank Associate Professor Dr. Edmund Sim Ui Hang of Universiti Malaysia Sarawak for providing us the HeLa cell line. We sincerely thank Associate Professor Dr. William Lim Kiong Seng of Universiti Malaysia Sarawak and Madam Chin Mei Yieng for reagent sharing and technical advice. We also express our gratitude to Universiti Malaysia Sarawak for providing the necessary facilities. This work was supported by research grant from the Ministry of Science, Technology and Innovation, Malaysia (grant number: 02-01-09SF0014).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SSB performed detailed experimental design, conducted experiments and analysed experimental results. SPS contributed to the original idea of the research, the design of the project, data interpretation and manuscript writing. All authors read and approved the final manuscript.
Contributor Information
Siaw Shi Boon, Email: ssboon81@gmail.com.
Sai-Peng Sim, Email: spsim@fmhs.unimas.my.
References
- 1.Fandi A, Altun M, Azli N, Armand JP, Cvitkovic E. Nasopharyngeal cancer: epidemiology, staging, and treatment. Semin Oncol. 1994;21:382–97. [PubMed] [Google Scholar]
- 2.Voravud N. Cancer in the far East. In: Sikora K, HKE, editor. Treatment of cancer. London: Chapman and Hall Medical; 1990. pp. 887–94. [Google Scholar]
- 3.Niedobitek G. Epstein-Barr virus infection in the pathogenesis of nasopharyngeal carcinoma. Mol Pathol. 2000;53:248–54. doi: 10.1136/mp.53.5.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ning JP, Yu MC, Wang QS, Henderson BE. Consumption of salted fish and other risk factors for nasopharyngeal carcinoma (NPC) in Tianjin, a low-risk region for NPC in the People’s Republic of China. J Natl Cancer Inst. 1990;82:291–6. doi: 10.1093/jnci/82.4.291. [DOI] [PubMed] [Google Scholar]
- 5.Chow WH, McLaughlin JK, Hrubec Z, Nam JM, Blot WJ. Tobacco use and nasopharyngeal carcinoma in a cohort of US veterans. Int J Cancer. 1993;55:538–40. doi: 10.1002/ijc.2910550403. [DOI] [PubMed] [Google Scholar]
- 6.Hutajulu SH, Indrasari SR, Indrawati LP, Harijadi A, Duin S, Haryana SM, et al. Epigenetic markers for early detection of nasopharyngeal carcinoma in a high risk population. Mol Cancer. 2011;10:48. doi: 10.1186/1476-4598-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hildesheim A, Wang CP. Genetic predisposition factors and nasopharyngeal carcinoma risk: a review of epidemiological association studies, 2000–2011: Rosetta Stone for NPC: genetics, viral infection, and other environmental factors. Semin Cancer Biol. 2012;22:107–16. doi: 10.1016/j.semcancer.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Li S, Gao JL. Promoter methylation status of the tumor suppressor gene SOX11 is associated with cell growth and invasion in nasopharyngeal carcinoma. Cancer Cell Int. 2013;13:109. doi: 10.1186/1475-2867-13-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien G, Yuen PW, Kwong D, Kwong YL. Comparative genomic hybridization analysis of nasopharygeal carcinoma: consistent patterns of genetic aberrations and clinicopathological correlations. Cancer Genet Cytogenet. 2001;126:63–7. doi: 10.1016/S0165-4608(00)00392-7. [DOI] [PubMed] [Google Scholar]
- 10.Hu C, Wei W, Chen X, Woodman CB, Yao Y, Nicholls JM, et al. A global view of the oncogenic landscape in nasopharyngeal carcinoma: an integrated analysis at the genetic and expression levels. PLoS One. 2012;7:e41055. doi: 10.1371/journal.pone.0041055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y, Alder H, Nakamura T, Schichman SA, Prasad R, Canaani O, et al. Sequence analysis of the breakpoint cluster region in the ALL-1 gene involved in acute leukemia. Cancer Res. 1994;54:2327–30. [PubMed] [Google Scholar]
- 12.Gu Y, Cimino G, Alder H, Nakamura T, Prasad R, Canaani O, et al. The (4;11)(q21;q23) chromosome translocations in acute leukemias involve the VDJ recombinase. Proc Natl Acad Sci U S A. 1992;89:10464–8. doi: 10.1073/pnas.89.21.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strissel PL, Strick R, Rowley JD, Zeleznik L. An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood. 1998;92:3793–803. [PubMed] [Google Scholar]
- 14.Nicholas CP, Sim SP. Etoposide-induced apoptosis results in chromosome breaks within the AF9 gene: Its implication in chromosome rearrangement in leukaemia. Adv Biosci Biotechnol. 2012;3:686–94. doi: 10.4236/abb.2012.326089. [DOI] [Google Scholar]
- 15.Sim SP, Liu LF. Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. J Biol Chem. 2001;276:31590–5. doi: 10.1074/jbc.M103962200. [DOI] [PubMed] [Google Scholar]
- 16.Yee PH-C, Sim SP. High cell density and latent membrane protein 1 expression induce cleavage of the mixed lineage leukemia gene at 11q23 in nasopharyngeal carcinoma cell line. J Biomedical Sci. 2010;17:77. doi: 10.1186/1423-0127-17-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–98. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 18.Israels LG, Israels ED. Apoptosis. Stem Cells. 1999;17:306–13. doi: 10.1002/stem.170306. [DOI] [PubMed] [Google Scholar]
- 19.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 20.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 21.Alenzi FQ, Lotfy M, Wyse R. Swords of cell death: caspase activation and regulation. Asian Pac J Cancer Prev. 2010;11:271–80. [PubMed] [Google Scholar]
- 22.Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, et al. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 1993;12:3679–84. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filipski J, Leblanc J, Youdale T, Sikorska M, Walker PR. Periodicity of DNA folding in higher order chromatin structures. EMBO J. 1990;9:1319–27. doi: 10.1002/j.1460-2075.1990.tb08241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cockerill PN, Garrard WT. Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell. 1986;44:273–82. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- 25.Lagarkova MA, Iarovaia OV, Razin SV. Large-scale fragmentation of mammalian DNA in the course of apoptosis proceeds via excision of chromosomal DNA loops and their oligomers. J Biol Chem. 1995;270:20239–41. doi: 10.1074/jbc.270.35.20239. [DOI] [PubMed] [Google Scholar]
- 26.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 27.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 28.Hars ES, Lyu YL, Lin CP, Liu LF. Role of apoptotic nuclease caspase-activated DNase in etoposide-induced treatment-related acute myelogenous leukemia. Cancer Res. 2006;66:8975–9. doi: 10.1158/0008-5472.CAN-06-1724. [DOI] [PubMed] [Google Scholar]
- 29.Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci U S A. 2010;107:4230–5. doi: 10.1073/pnas.0913089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–8. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 31.Koyama AH. Induction of apoptotic DNA fragmentation by the infection of vesicular stomatitis virus. Virus Res. 1995;37:285–90. doi: 10.1016/0168-1702(95)00026-M. [DOI] [PubMed] [Google Scholar]
- 32.Higuchi Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem Pharmacol. 2003;66:1527–35. doi: 10.1016/S0006-2952(03)00508-2. [DOI] [PubMed] [Google Scholar]
- 33.Lelli JL, Jr, Becks LL, Dabrowska MI, Hinshaw DB. ATP converts necrosis to apoptosis in oxidant-injured endothelial cells. Free Radic Biol Med. 1998;25:694–702. doi: 10.1016/S0891-5849(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 34.Stangel M, Zettl UK, Mix E, Zielasek J, Toyka KV, Hartung HP, et al. H2O2 and nitric oxide-mediated oxidative stress induce apoptosis in rat skeletal muscle myoblasts. J Neuropathol Exp Neurol. 1996;55:36–43. doi: 10.1097/00005072-199601000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Li TK, Chen AY, Yu C, Mao Y, Wang H, Liu LF. Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev. 1999;13:1553–60. doi: 10.1101/gad.13.12.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ames BN. Endogenous oxidative DNA damage, aging, and cancer. Free Radic Res Commun. 1989;7:121–8. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt 1):17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 39.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–66. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 40.Marsin S, Vidal AE, Sossou M, Menissier-de Murcia J, Le Page F, Boiteux S, et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J Biol Chem. 2003;278:44068–74. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 41.Cho EY, Hildesheim A, Chen CJ, Hsu MM, Chen IH, Mittl BF, et al. Nasopharyngeal Carcinoma and Genetic Polymorphisms of DNA Repair Enzymes XRCC1 and hOGG1. Cancer Epidemiol Biomark Prev. 2003;12:1100–4. [PubMed] [Google Scholar]
- 42.Ziemin-van der Poel S, McCabe NR, Gill HJ, Espinosa R, III, Patel Y, Harden A, et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci U S A. 1991;88:10735–9. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowley JD. Rearrangements involving chromosome band 11Q23 in acute leukaemia. Semin Cancer Biol. 1993;4:377–85. [PubMed] [Google Scholar]
- 44.Mukae N, Enari M, Sakahira H, Fukuda Y, Inazawa J, Toh H, et al. Molecular cloning and characterization of human caspase-activated DNase. Proc Natl Acad Sci U S A. 1998;95:9123–8. doi: 10.1073/pnas.95.16.9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broeker PL, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, et al. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood. 1996;87:1912–22. [PubMed] [Google Scholar]
- 46.Strick R, Zhang Y, Emmanuel N, Strissel PL. Common chromatin structures at breakpoint cluster regions may lead to chromosomal translocations found in chronic and acute leukemias. Hum Genet. 2006;119:479–95. doi: 10.1007/s00439-006-0146-9. [DOI] [PubMed] [Google Scholar]
- 47.Mirkovitch J, Gasser SM, Laemmli UK. Scaffold attachment of DNA loops in metaphase chromosomes. J Mol Biol. 1988;200:101–9. doi: 10.1016/0022-2836(88)90336-1. [DOI] [PubMed] [Google Scholar]
- 48.Durrieu F, Samejima K, Fortune JM, Kandels-Lewis S, Osheroff N, Earnshaw WC. DNA topoisomerase IIalpha interacts with CAD nuclease and is involved in chromatin condensation during apoptotic execution. Curr Biol. 2000;10:923–6. doi: 10.1016/S0960-9822(00)00620-5. [DOI] [PubMed] [Google Scholar]
- 49.Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 2000;102:1–4. doi: 10.1016/S0092-8674(00)00003-9. [DOI] [PubMed] [Google Scholar]
- 50.Alexander RB, Nelson WG, Coffey DS. Synergistic enhancement by tumor necrosis factor of in vitro cytotoxicity from chemotherapeutic drugs targeted at DNA topoisomerase II. Cancer Res. 1987;47:2403–6. [PubMed] [Google Scholar]
- 51.Dumont A, Hehner SP, Hofmann TG, Ueffing M, Droge W, Schmitz ML. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-kappaB. Oncogene. 1999;18:747–57. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- 52.Singh M, Singh N. Induction of apoptosis by hydrogen peroxide in HPV 16 positive human cervical cancer cells: involvement of mitochondrial pathway. Mol Cell Biochem. 2008;310:57–65. doi: 10.1007/s11010-007-9665-5. [DOI] [PubMed] [Google Scholar]
- 53.Uegaki K, Otomo T, Sakahira H, Shimizu M, Yumoto N, Kyogoku Y, et al. Structure of the CAD domain of caspase-activated DNase and interaction with the CAD domain of its inhibitor. J Mol Biol. 2000;297:1121–8. doi: 10.1006/jmbi.2000.3643. [DOI] [PubMed] [Google Scholar]
- 54.Lechardeur D, Dougaparsad S, Nemes C, Lukacs GL. Oligomerization state of the DNA fragmentation factor in normal and apoptotic cells. J Biol Chem. 2005;280:40216–25. doi: 10.1074/jbc.M502220200. [DOI] [PubMed] [Google Scholar]
- 55.de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007;7:32. doi: 10.1186/1472-6750-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otomo T, Sakahira H, Uegaki K, Nagata S, Yamazaki T. Structure of the heterodimeric complex between CAD domains of CAD and ICAD. Nat Struct Biol. 2000;7:658–62. doi: 10.1038/77957. [DOI] [PubMed] [Google Scholar]
- 57.Lechardeur D, Xu M, Lukacs GL. Contrasting nuclear dynamics of the caspase-activated DNase (CAD) in dividing and apoptotic cells. J Cell Biol. 2004;167:851–62. doi: 10.1083/jcb.200404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barik S. Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol Biol. 1996;57:203–15. doi: 10.1385/0-89603-332-5:203. [DOI] [PubMed] [Google Scholar]