


Abstract
In mouse embryonic cells, ligand-activated retinoic acid receptors (RARs) play a key role in inhibiting pluripotency-maintaining genes and activating some major actors of cell differentiation.
To investigate the mechanism underlying this dual regulation, we performed joint RAR/RXR ChIP-seq and mRNA-seq time series during the first 48 h of the RA-induced Primitive Endoderm (PrE) differentiation process in F9 embryonal carcinoma (EC) cells. We show here that this dual regulation is associated with RAR/RXR genomic redistribution during the differentiation process. In-depth analysis of RAR/RXR binding sites occupancy dynamics and composition show that in undifferentiated cells, RAR/RXR interact with genomic regions characterized by binding of pluripotency-associated factors and high prevalence of the non-canonical DR0-containing RA response element. By contrast, in differentiated cells, RAR/RXR bound regions are enriched in functional Sox17 binding sites and are characterized with a higher frequency of the canonical DR5 motif. Our data offer an unprecedentedly detailed view on the action of RA in triggering pluripotent cell differentiation and demonstrate that RAR/RXR action is mediated via two different sets of regulatory regions tightly associated with cell differentiation status.
INTRODUCTION
Retinoic acid (RA), the main active vitamin A metabolite, is a well-known regulator of embryonic development as well as adult physiology (1). The highly pleiotropic organismal and cellular effects of RA are mainly mediated by the combinatorial action of six nuclear receptors [retinoic acid receptors NR1B (RARA, RARB and RARG) and retinoid X receptors NR2B (RXRA, RXRB and RXRG)], which form heterodimers and act as RA-modulated transcription factors.
At the cellular level, RA stimulation triggers fundamental biological processes, such as growth arrest, differentiation and apoptosis. Long before the elucidation of the molecular mechanisms supporting their action, retinoids have been recognized as mediators of cell differentiation both in vivo and in vitro (2–5). Among the various RA sensitive tissues and cell types, embryonal carcinoma (EC) and later, embryonic stem (ES) cells were shown to undergo differentiation upon RA stimulation (6). This ever since remained the treatment of choice to induce in vitro differentiation of mouse and human ES cells. ES and EC cells are characterized by their self-renewal capacity as well as their ability to differentiate into various cell lineages, thus providing invaluable biological models to study early developmental processes. Molecular regulators of pluripotency and self-renewal maintenance have been gradually elucidated and rely on a core transcription factor triumvirate composed of SOX2, NANOG and POU5F1 that acts in a concerted manner to maintain a proliferating and undifferentiated state while preventing lineage specific differentiation (7). Importantly, the elucidation of pluripotency mechanisms provided molecular basis for somatic cells reprogramming into ES-like cells also referred to as induced pluripotent stem cells (iPSc), by the ectopic overexpression of defined transcription factors (8). More recently, additional regulators of gene expression were shown to collaborate with POU5F1, SOX2 and NANOG to control the ESc gene expression program and/or to improve somatic cells reprogramming. These factors exert various transcription-related function ranging from transcriptional factors (e.g. MYC, MYCN, KLF4, STAT3, SMAD1, TCF3) and cofactors (e.g. EP300, Mediator complex subunits) to chromatin modifiers (e.g. PcG). Interestingly, several nuclear receptors emerged as important players in the maintenance of pluripotency and somatic cell reprogramming (i.e. ESRRB, NR5A1, NR5A2, NR0B1) as well as in pluripotent cell differentiation induction (i.e. NR2F1, NR2F2, NR6A1). Paradoxically, RA-activated RARG was also shown to be involved in promoting somatic cell reprogramming toward iPSc (9), in contradiction with its well-documented in vitro differentiating effect of ES/EC cells.
The nuclear hormone receptor family consists of 48/49 (human/mouse) evolutionary-conserved ligand-dependent transcription factors sharing important structural and functional features. As such, they are characterized by the presence of two conserved domains, the central DNA binding domain (DBD) which interacts with the core motif 5′-RGKTSA-3′ (10–12), and the C-terminal ligand-binding domain (LBD) which largely determines nuclear receptor dimerization properties (13). Monomeric NRs recognize a single core motif, while dimeric NR complexes interact with repeated occurrences of this core motif. The spacer size and relative orientation (i.e. Direct Repeat(DR), Inverted Repeat (IR) or Everted Repeat (ER)) of the repeated core enable the selective recognition of the so-called HRE (Hormone Response Element) by specific nuclear receptor dimers (12,14). Accordingly, RAR/RXR specific response elements (RARE) were originally described as direct repeats (DR) of the core consensus sequence separated by 1, 2, or 5 nucleotides (15,16). Additionally, a few instances of non-canonical RAREs were also characterized. They exhibit degenerate core half-site and/or non-classical spacer like DR8 (17), DR3 (18), ER8 (19) and IR0 (20). More recently, whole genome analyses have drawn a more precise RAR/RXR binding picture in various cell types (21–25). Notably, our study of RAR/RXR binding motif repertoire in undifferentiated embryonal carcinoma (EC) and embryonic stem (ES) cells (25) highlighted the underestimated variety of RARE motifs and allows the identification of a new bona fide RARE consisting of a direct repetition of the core motif without spacer (DR0). This motif is already known as a bona fide DNA binding motif for pro-differentiating nuclear receptors like NR6A1 (GCNF), NR2F1 (COUP-TFI) and NR2F2 (COUP-TFII); it is also strongly reminiscent of the monomeric binding motif recognized by ESRRB (26) and NR5A2 (LRH-1) (27) which are both involved in pluripotency maintenance. Hence, the DR0 motif appears as a peculiar DNA motif able to interact with either pro-pluripotency or pro-differentiating nuclear receptors. However, while DR0 or DR0-like motifs allow specific recruitment of GCNF, COUP-TFs, ESRRB and NR5A2 and the transcriptional modulation of neighboring gene, DR0-bound RAR/RXR dimers do not modulate gene expression in reporter in vitro assay (25). Noticeably, by contrast with a previous report which identified the DR5 motif as the most prevalent motif in RAR/RXR occupied DNA regions in MCF7 cells (21), the DR0 motif was found as the most significantly enriched hormone response element in RAR/RXR targeted regions in mES/mEC cells (25), thus suggesting that the recruitment of RAR/RXR on genomic loci encompassing a DR0 motif is favored in pluripotent embryonic cells.
These observations prompted us to hypothesize that the differentiation process is coupled with a drastic reorganization of the RAR/RXR binding repertoire. To get a clear picture of this change and of its transcriptional consequences, we performed a time-course analysis of whole-genome RAR/RXR binding as well as global gene expression profiling, in RA-induced differentiating F9 embryonal carcinoma cells.
Our study highlights the dynamic binding pattern of RAR/RXR during the differentiation process and the diversity and composite nature of recognized DNA motifs. We establish that the dynamic occupation patterns observed result from multiple mechanisms associated with (i) the ability of RAR/RXR heterodimers to interact with DNA, and (ii) region-specific features (e.g. RARE motif, specific transcription factor binding and DNA accessibility). By combining these results with gene expression data, we identified dynamical transcriptional patterns that can be reliably associated with specific cis-regulatory events including RAR/RXR binding dynamics. Altogether, our results enable us to propose an integrated model of RAR/RXR binding dynamics throughout the differentiation process of pluripotent cells.
MATERIALS AND METHODS
Cell culture
Wild-type and RAR paralog specific knockout mouse embryonal carcinoma F9 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 4.5% glucose, L-glutamine and sodium pyruvate (Gibco®, Life Technologies™) supplemented with 5% foetal bovine serum (Gibco®, Life Technologies™), 0.1 mg/ml streptomycin and 100 U/ml penicillin (Gibco®, Life Technologies™) and cultured at 37°C in a humidified 5% CO2 atmosphere. 3.5 × 105 cells were seeded in gelatin-coated 100 mm tissue culture plates (0.1%) 16 h prior to drug treatment. All-trans retinoic acid (RA) (Sigma-Aldrich) was dissolved in 100% DMSO, and diluted in cell culture medium to a final concentration of 1 × 10−7 M. Control samples were treated with a dose of vehicle (DMSO) equivalent to the dose used in RA-treated samples.
Generation of stable knocked-down F9 cells
Esrrb KD, Sox17 KD, shCtrl F9 cells were generated by stable transfection of specific shRNA expression plasmids (Suresilencing, Qiagen) (shCtrl: GGAATCTCATTCGATGCATAC, shEsrrb: CCTGACCACTCTCTGTGAATT, shSox17: CACGGAATTCGAACAGTATCT). The shRNA expression plasmids were transfected with Lipofectamine 2000 transfection reagent (Invitrogen) following manufacturer instructions. The cells were trypsinized 48 h after transfection and seeded into selective medium. F9 cells stably expressing shRNA were established by culture in selection medium containing Neomycin (800μg/ml) (Sigma). The medium was renewed every 3 days. After 2 weeks resistant colonies were screened by RT-qPCR to identified stable clones exhibiting >70% reduced expression level of the targeted mRNA. Selected clones were cultured in complete medium containing maintenance concentration of neomycin (300μg/ml).
Chromatin immunoprecipitation sequencing (ChIP-seq)
ChIP experiments were performed as previously described (28). Briefly, cells were cross-linked by a two-step procedure, with 2mM disuccinimidyl glutarate (Pierce) for 30 min at room temperature and then with 1% formaldehyde (Sigma-Aldrich) for 10 min at 37°C. After lysis, chromatin extract were sonicated to reduce the length of DNA fragments to 200–400 bp using a Diagenode Bioruptor. Antibodies anti-panRAR (Sc-773, Santa Cruz Biotechnology) and anti-panRXR (Sc-553, Santa Cruz Biotechnology) were used to immunoprecipitate RAR- and RXR-bound DNA fragments, respectively. Immunoprecipitated chromatin were both analyzed by quantitative real-time PCR and subjected to next-generation sequencing. For ChIP-seq, nine individual ChIP samples were pooled and concentrated under vacuum. The ChIP templates were sequenced using a Solexa Gene Analyzer II platform (Illumina) at 36 bp read length using standard manufacturer protocols at the CERBM Facility in Illkirch, France. For the validation of ChIP-seq data, selected genomic regions containing RAR and/or RXR were analyzed by ChIP-qPCR on independent biological replicates using Qiagen SYBR Green qPCR Master Mix and genomic primers listed in Supplementary Table S4. The qPCR reaction were performed with an Mx3000 qPCR System (Agilent) using the following amplification profile: 10 min at 95°C, followed by 40 cycles of 30 s at 94°C, 30 s at 55°C and 30 s at 72°C. The results were normalized with respect to input. All data shown correspond to the mean ± SD from at least three biological replicates.
Determination of RAR/RXR binding regions from ChIP-seq data
Reads from ChIP-seq samples were aligned using Bowtie (29) version 0.12.9 with options −n 3, −e 70 and −m 1. We used the UCSC Genome Browser (30) to produce visualizations of the aligned reads along the chromosomes and gene annotations. We performed peak calling using a dedicated procedure in order to deal with the ChIP- seq time series data: (i) call peaks independently for each sample, with a really permissive threshold; (ii) consider the set of positions on the genome which are the summit of a peak in at least one condition; (iii) define a link between any two summits that are closer than 100bp; (iv) compute connected components of this graph; (v) define, for each connected component, a binding region, as a location of 500 bp wide centered at the mean of the summits in the connected component. We thus obtained a fixed set of genomic regions that do not depend on one particular condition anymore. For the first step, we used MACS (31) version 1.4.2 with default options, except the P-value threshold (P-value), which was set to 10−3. For all ChIP samples, we took the whole-cell extract sample in untreated condition as a control. The obtained regions were reduced using two independent filters.
The first filter used the Poisson Margin Test (32) to detect regions displaying a significant difference between RAR binding and control signals for at least one time point (i.e. Benjamini-Hochberg adjusted P-value smaller than 0.001). The second filter was based on the proportion among RAR-positive candidate regions of RXR-positive regions as a function of the unadjusted P-value threshold (see Supplementary Figure S1). We finally kept regions with at most 10−7 unadjusted P-value, provided that they were not located on a mitochondrial chromosome; and they did not belong to the blacklist published in the context of the ENCODE project (33) available at https://sites.google.com/site/anshulkundaje/projects/blacklists. RAR (resp. RXR) was considered present in a region at a given time point if the corresponding Poisson Margin Test yielded a P-value inferior to 10−7; it was considered absent for a P-value superior to 10−3 (negativity threshold in the results section).
Motif discovery and scanning
We used the online version of MEME Suite (34) for the de novo search of enriched motifs in RAR/RXR binding regions, and more precisely the tools MEME ChIP and DREME with standard options except "Scan both DNA strands" set. Additionally, the motif defined by Balmer et al. (15) was used to compute Receiver Operating Characteristic (ROC) curves of direct repeat (DR), inverted repeat (IR) and everted repeat (ER) motif with spacer ranging from 0 to 9 nucleotides, taking as a negative set a control consisting of random DNA sequences with the same length and GC distribution as our RAR/RXR binding regions. For motif occurrences prediction, we chose an alignment score cut-off corresponding to a false positive rate of 0.1 based on the ROC curve.
Clustering of RAR/RXR binding regions
RAR/RXR binding regions were filtered to keep only regions exhibiting a significant difference in binding intensity relative to the initial time point (p < 10−6 with the Poisson Margin Test (32)). Those regions, represented as their RPKM level at each time point, were clustered using Cluster 3 (options: normalization on, k-means algorithm, 100 runs, uncentered Pearson correlation as a distance) (35). The number of clusters was chosen empirically, by visual inspection.
Transcription factor occupancy data sets
We used the occupancy maps provided in publications on mouse ES (26–27,36–39) and F9 cells (40). All the data sets were collected through the GEO database (41). We reused the called peaks distributed with each data set. A list of the accession numbers is provided in Supplementary Table S5.
Assessing the correlation between RAR/RXR and other TFs occupancy
We collected the peak summits from the previously described ChIP-seq data sets as well as ours, expanded them by 50bp in both directions and merged overlapping peaks. For each TF, we computed a 0–1 vector indexed by this set of regions, indicating its presence in each region. We then reported the correlation of each TF vector with the vector for RAR.
Association with RAR/RXR binding clusters
Each of our RAR binding regions was annotated as having or not a detected peak for other TFs less than 1 kb away. We computed the association between a cluster and a specific transcription factor by performing a Fisher's exact test: each region can be a member of the cluster or not, and it can have a detected peak for the indicated transcription factor or not. The contingency table is passed to R's fisher.test and we keep the log odds-ratio, its 95% confidence interval and the P-value resulting from the independence test.
Formaldehyde assisted isolation of regulatory elements (FAIRE)
FAIRE experiments were performed as previously described (42). Briefly, cells were cultured and treated with RA as described above. Following treatment, cells were cross-linked at the indicated time point, by the addition of formaldehyde to 1% for 10 min at 37°C. Following repeated phosphate-buffered saline (PBS) washing, cell pellets were lysed in FAIRE lysis buffer (2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1X Roche protease inhibitor cocktail) and sonicated with a Bioruptor (Diagenode) for 15 min at high intensity 30s ON, 30s OFF. The chromatin containing lysates were then spun down at 15 000 × g for 5 min at 4°C to remove cellular debris. Supernatant was extracted twice with phenol/chloroform and once with chloroform. DNA was precipitated by the addition of 1/10 volume sodium acetate (pH 5.3) and 2 volumes ethanol. DNA pellet was dissolved in water and incubated with 1 μg RNAse A for 2 h at 37°C. Input DNA was reverse cross-linked and purified using a Qiagen PCR purification kit. Purified DNA was analyzed by qPCR using the appropriate primer sets (Supplementary Table S4) as described above.
RNA isolation, library preparation and high-throughput mRNA sequencing (RNA-seq)
Total RNA was extracted with TRIzol (Invitrogen), and treated with RNAse-free DNase I. The concentration of samples was measured using the NanoDrop (Thermo Scientific) and their integrity was determined using an Agilent 2100 Bioanalyzer. RNA-seq libraries were prepared from samples with an RNA Integrity Number (RIN) > = 8.0. RNA-seq libraries were prepared with the True Seq V2 Kit (Illumina) according to the manufacturer's instructions and sequenced using the Solexa Gene Analyzer II or the HiSeq 2500 platform (Illumina).
Detection of differentially expressed genes with RNA-seq data
The reads from mRNA-seq samples were aligned using Tophat (43) version 2.0.7 associated with Bowtie (29) version 0.12.9. Tophat was run with default options. The aligned reads were assigned to genes by htseq-count (44), run with default options on the version 63 of the Ensembl mouse annotation. The tests for differential expression were performed using DESeq (45) version 1.12.0. A gene was declared modulated if it displayed a significant difference between any two time-points (the cut-off was fixed at 5.10−2 of (Bonferroni-Hochberg) adjusted P-value) and expressed if it had a non-zero estimated level (baseMean in DESeq) in some condition.
Gene clustering
Each gene was described as a vector of log2 fold change in estimated expression level (baseMean in DESeq) with respect to the untreated condition. To avoid indefinite values when a gene had no expression in this condition, all baseMean values were added the smallest detected expression level in the data set (this procedure is akin to the use of pseudo-counts in probabilistic models). Only modulated genes were included for the cluster analysis, which was performed by Cluster 3.0 (35). The number of classes in the clustering was chosen by visual inspection of the clusters.
Validation of differentially expressed genes by RT-qPCR
The validation of differentially expressed genes was performed by RT-qPCR analysis using Biomark™ System (Fluidigm®) according to manufacturer's instructions. Briefly, the purified total RNA of individual samples was reverse-transcribed using the SuperScript® III Reverse Transcriptase Kit (Invitrogen). cDNAs were pre-amplified using TaqMan® PreAmp Master Mix (Applied Biosystems) and an equimolar mixture of gene-specific primer couples. The pre-amplified cDNAs were treated with exonuclease1 (New England BioLabs) and amplified using SsoFast™EvaGreen® Supermix (Bio-Rad Laboratories) on a BioMark HD System (Fluidigm). Primers sequences are provided in Supplemental Table S4.
Gel retardation assays
Mouse RARA and RXRA proteins were synthesized in vitro using the TNT-coupled transcription/translation system (Promega). DNA-protein binding assays were performed as previously described (46). The radiolabeled oligonucleotide sequences are provided in Supplementary Table S4. DNA-protein complexes were separated by electrophoresis on a 5% polyacrylamide gel. Dried gels were exposed with Fuji-screen.
Gene Ontology (GO) analysis
GO analysis was performed using GOToolBox (http://genome.crg.es/GOToolBox), using the Mouse Genome Informatics identities of the list of differentially expressed genes. Gene lists and GO term results are provided in Supplementary Table S3.
RESULTS
Genome-wide mapping and characterization of RAR and RXR binding sites during RA-induced differentiation of F9 cells
To obtain a dynamic map of RAR/RXR binding sites during the RA-induced primitive endoderm (PrE) differentiation process, we sampled chromatin at 0, 2, 24 and 48 h after RA stimulation and performed high-resolution ChIP-seq analysis on each chromatin sample. By contrast with previously published studies that focused on RARG paralogs binding (24), we used ChIP validated antibodies recognizing either the three RAR or the three RXR paralogs. To collect a sufficient amount of material, several individual chromatin immunoprecipitation (ChIP) experiments were performed at each time point. Individual samples were validated by ChIP-qPCR analysis of positive and negative genomic regions and pooled before library preparation. Individual libraries were then sequenced on Illumina GAII. In addition, total genomic DNA was sequenced to obtain a reference input profile. Sequencing data, ChIP-qPCR validation data (Supplementary Figure S1) and detailed bioinformatics analyses can be found in Supplementary Experimental Procedure.
An example of the binding profiles for RAR and RXR around the Hoxa locus is shown in Figure 1A. We performed peak calling on each ChIP-seq sample and merged the results so as to determine a fixed set of genomic regions across all tested conditions (see Supplementary Experimental Procedure). The defined binding regions, considered as a genomic feature, do not depend on one particular condition anymore, and we analyzed their occupation status along the time series.
Figure 1.
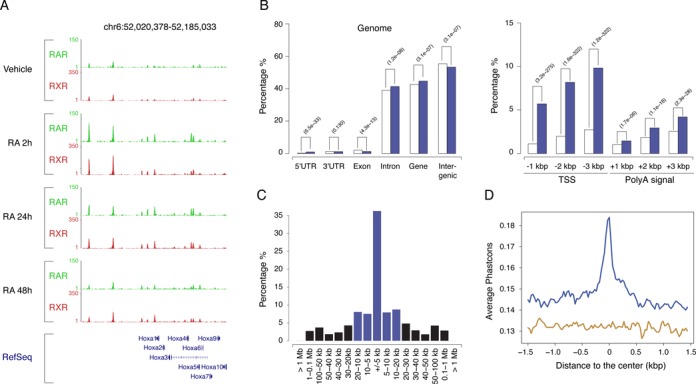
RAR/RXR heterodimers binding regions. (A) Time course analysis of RAR (green) and RXR (red) binding profiles in the Hoxa locus after RA stimulation of F9 EC cells. A screenshot (http://genome.ucsc.edu) of the RAR and RXR binding signal at the different time points after RA stimulation, is shown. (B) Characterization of RAR/RXR binding region relative to specific genomics features. Open and blue bars show the proportion of the indicated genomics features in the genome and ChIP regions, respectively. (C) Distribution of the distance to the first annotated transcription start site (TSS) (blue bars: absolute distance < 20 kb, black bars: absolute distance > 20 kb). (D) Multiple species alignment scores (Phastcons) for RAR/RXR binding regions (blue curve) and randomly selected genome regions (brown curve).
To be able to compare occupancy of the identified binding sites in the various sampled conditions, statistical tests were performed to detect significantly occupied regions relative to the input control. More specifically, we used the Poisson Margin significance Test (PMT) (32) which is robust to imbalances in sample sequencing depth. A region was declared occupied at a given condition for an adjusted P-value (FDR) < 0.01.
We further strengthened this procedure by requiring that an RAR/RXR binding region should be simultaneously occupied both by RAR and RXR in at least one time-point. We finally fixed a P-value threshold for which more than 90% of the RAR binding regions were also occupied by RXR (Supplementary Figure S2). At this threshold, RAR and RXR are respectively bound to a total of 16222 and 34406 genomic loci. Among these, we identified a total of 13791 genomic regions simultaneously bound by RAR and RXR at least at one time point during the PrE differentiation process. These regions will be referred to as our RAR/RXR data set in the remainder of the text (Supplementary Table S1). The difference between the total number of RAR and RAR/RXR bound regions is likely due to a threshold effect and should not be interpreted as the existence of RXR-independent RAR binding events. On the opposite, our study failed to identify RAR positive/RXR negative loci after the definition of a negativity threshold (Supplementary Experimental Procedure) above which one may safely consider that a genomic region is not occupied by a given TF. By contrast, RAR-independent binding of RXR is frequent and mirrors its ability to form homodimers or heterodimers with numerous nuclear receptor superfamily members (13).
The localization of identified RAR/RXR-binding regions using cis-regulatory Element Annotation System (47) (Figure 1B), showed a significant enrichment in promoters as well as in regions localized immediately downstream of polyadenylation signal (Figure 1B, right panel). Furthermore, the analysis of the position of transcription start sites (TSS) relative to RAR/RXR binding regions showed that 68% of RAR/RXR-binding regions were located at less than 20 kb from an annotated TSS (Figure 1C).
A way to assess the potential relevance of our binding regions is to determine whether they are phylogenetically conserved. Indeed, relevant binding regions are expected to be under selective constraints and thus to exhibit better conservation between species (48). We therefore analyzed the evolutionary conservation of genomic regions exhibiting significant interaction with RAR/RXR using the vertebrate PhastCons scores (49). Compared to randomly chosen DNA loci (brown curve), RAR/RXR bound regions (blue curve) exhibit a better evolutionary conservation score, indicating that a significant subset of the identified regions is conserved within vertebrates (Figure 1D). Noticeably, maximum conservation is observed at the center of the analyzed genomic regions, indicating that recognized DNA motifs are globally more conserved than the neighboring sequences.
Dynamic analysis of RAR/RXR binding region occupancy
The crude observation of the number of RAR/RXR bound regions at the various tested time points indicates that the absolute number of RXR bound loci is more stable than the number of RAR bound loci. Furthermore, the observed variations do not occur simultaneously during RA-induced differentiation process (Figure 2A) indicating that RAR and RXR binding dynamics significantly differ. Figure 2B displays the proportion of regions for each qualitative (bound/unbound) occupancy profile for RAR, RAR/RXR and RXR respectively (Figure 2B). Interestingly, 66% of the regions interacting with the heterodimeric complex in absence of RA remain bound over time (2142 out of 3247). Upon RA stimulation, the number of RAR/RXR bound regions increases massively but transiently with 30% and 15.5% of RAR/RXR bound regions meeting the defined criteria only after 2 h and at all times, respectively (Figure 2B central panel). Additionally, occupancy profiles are very similarly distributed in RAR and RAR/RXR bound regions, which strongly suggests that variation in the number of heterodimer bound regions over time is mostly due to the variation in RAR subunit recruitment. In comparison, RXR subunit binding is significantly more stable with 60.3% of all RAR/RXR bound regions being continuously bound by RXR (Figure 2B right panel). To validate this observation and to exclude that it is not only reflecting differences in bound DNA recoveries between the RAR and RXR antibodies, we used the previously defined negativity threshold to quantify the proportion of identified site exhibiting RAR-independent binding of RXR in absence of ligand. We found that 18% of all RAR/RXR binding regions (2464 out of 13791 regions) are occupied by RXR but not by RAR in untreated F9 cells. Furthermore, as shown in Figure 2C, the occupancy level distribution of these regions over time point shows that RA treatment affects RAR binding more drastically than RXR binding (Figure 2C).
Figure 2.
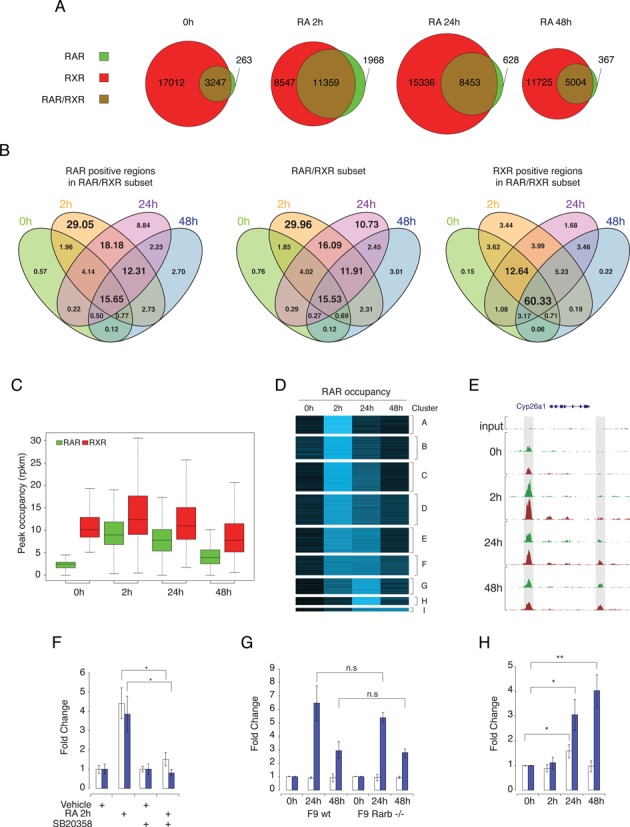
RAR and RXR binding dynamics in differentiating F9 EC cells. (A) The number of RAR- and RXR-binding sites detected by ChIP-seq during PrE differentiation. Results are shown as Venn diagrams representing the number of binding sites during differentiation for RAR only (green), RXR only (red) and shared RAR/RXR (brown). Circle sizes are representative of the number of binding sites at the indicated time point. (B) Intersection of RAR, RXR and RAR/RXR binding regions occupation throughout the PrE differentiation process. Results are shown as four-way Venn diagrams representing the proportion (%) of binding sites assigned to specific temporal behavior in each sub-category. (C) Distribution of the occupancy level over time for binding sites exhibiting no binding of RAR (green) and significant binding of RXR (red) in untreated condition. Occupancy levels are expressed in read per region and per million mapped reads. (D) K-means clustering of RAR/RXR dynamic regions based on their changes in RAR occupancy during PRE differentiation. Results are shown as heat map representing the normalized RAR binding region coverage intensity over time. (E) RAR (green) and RXR (red) binding profiles variation in the Cyp26a1 locus after RA stimulation of F9 EC cells. Upstream and downstream binding region are highlighted by gray box. (F) qPCR quantification of RAR binding intensity at the Cyp26a1 upstream (open bars) and downstream (blue bars) binding sites. RAR binding intensity was normalized on binding level in untreated cells for each binding region. The data shown represent mean ± SD of triplicate experiments. Student's t-test was applied to assess statistical difference of the mean (*: P < 0.05; **: P < 0.01). (G) qPCR quantification for temporal binding pattern of RAR at Cyp26a1 upstream (open bars) and downstream (blue bars) binding sites in wild-type (WT) and Rarb -/- F9 cells. Statistical analysis similar to (F). (H) qPCR quantification of temporal open chromatin enrichment at Cyp26a1 upstream (open bars) and downstream (blue bars) RAR/RXR binding sites. Results show open chromatin enrichment in RA-stimulated relative to untreated F9 EC cells. Statistical analysis similar to (F).
Collectively, these data suggest that RXR and RAR subunits binding are loosely related in absence of RA stimulation and that in this condition, RXR subunit occupies a large subset of the identified regions independently of RAR subunit. Whether RXR interacts with these regions as homodimers or as heterodimers with other partners remains unknown.
To obtain a refined description of our data set, we performed a cluster analysis in order to identify typical quantitative patterns of RAR subunit time-dependent occupancy level; for this we only considered a subset of 3174 regions displaying a highly significant occupancy variation between any time point relative to the untreated condition. This subset is referred to as dynamic binding regions hereafter. The various clusters (Figure 2D) can be distinguished on the basis of initial occupancy level, with clusters B, D and F being more occupied on average than the others (Supplementary Figure S3 panel A) and on the basis of their occupancy pattern over time. Interestingly, while most regions (clusters A–D) are characterized by a maximum occupancy after 2 h followed by a decreased occupancy at later time points, a subset of regions exhibits a different profile characterized by the maintenance of their occupancy between 2 h and 24 h (cluster E to F) or even a maximum occupancy at 24 or 48 h after treatment (Figure 2D, cluster G to I).
The observed differences between these RAR/RXR binding patterns likely mirror different molecular mechanisms. Indeed, RAR/RXR binding process could be modulated by intrinsic (receptor-related) parameters like concentration, cellular localization, interaction with partners, or extrinsic (DNA target-related) parameters like HRE motif type, chromatin accessibility, transcription factors binding sites composition.
To start to decipher which of these mechanisms could be at work, we first investigated whether molecular events intrinsically affecting RAR/RXR binding capabilities may play a role in the observed binding dynamics. Several recent studies highlight the effect of RA-induced MAP kinase-dependent phosphorylation of RARA and RARG paralogs, on their ability to interact with target sequences (50–53). To test whether such mechanisms may be responsible for the massive and quick occupancy increase observed in most identified binding regions, independently of other molecular features, we analyzed RAR binding in presence and absence of RA and p38MAPK inhibitor SB203580. Results of ChIP-qPCR analysis for the binding regions located upstream (open bars) and downstream (dark blue bars) of Cyp26a1 locus (Figure 2E) are presented in Figure 2F and show that p38 inhibitor blocks the RA-induced early increase in RAR binding regions occupancy. Similar observations were made on other identified RAR/RXR targeted regions (Supplementary Figure S4). Interestingly, downstream binding site occupancy is also affected by MAP kinase inhibition but remains weakly occupied. This observation suggests that while MAP kinase dependent activation of RAR/RXR binding maybe a general phenomenon, occupancy level could be limited by other parameters. Furthermore, the strong difference in the observed kinetics enables us to safely hypothesize that occupancy profiles characterized by delayed maximum occupancy (cluster G to I) are most probably linked to other molecular mechanisms.
To identify these mechanisms, we tested two non-mutually exclusive hypotheses. The first one was based on the known induction of Rarb2 paralog expression (54–56) (Supplementary Figures S10 and S11) and postulated that late RAR/RXR recruitment mirrored an increase in concentration of this specific paralog. We therefore tested whether late recruitment on specific binding sites could be detected in Rarb2 knockout F9 cells (57). Result of ChIP-qPCR occupancy analysis of binding regions located upstream and downstream of Cyp26a1 locus which respectively exhibit early and late maximum occupancy, is shown in Figure 2G. Notably, we observed a similar recruitment pattern in wild-type and Rarb−/− F9 cells on both sites indicating that late recruitment is not linked to RA-induced expression of Rarb2 mRNA. However, this does not preclude that such mechanism may be at work in others late bound loci.
The second hypothesis postulated that late recruitment might be linked to chromatin remodeling events resulting in the unmasking of RAR/RXR binding sites. To assess loci specific dynamic changes of chromatin environment, we used the so-called FAIRE technique (Formaldehyde-Assisted Isolation of Regulatory Elements) coupled with qPCR analysis (42) to quantitatively detect nucleosome-depleted DNA regions and active regulatory elements from total chromatin. As previously, we analyzed RAR/RXR bound regions located upstream and downstream of the Cyp26a1 locus (Figure 2E). Interestingly, while FAIRE signal associated with the upstream binding site is only slightly altered upon RA stimulation, the downstream site associated FAIRE signal is increased in a time-dependent manner upon RA treatment (Figure 2H), indicating an increased accessibility of chromatin that correlates with the increased RAR/RXR occupancy at this specific genomic location. Of note, FAIRE signal in vehicle treated F9 cells differs between the two regions and represents 9% and 0.4% of the input DNA for upstream and downstream sites, respectively, indicating a difference in accessibility of these sites in untreated cells that is consistent with the observed difference in RAR/RXR basal occupancy level.
While it is difficult to extend these results to all identified binding sites, they confirm the idea that rapid occupancy increase is likely associated with the previously demonstrated role of RA-induced MAP kinase activation on RAR/RXR DNA binding through RARA and RARG phosphorylation, and that delayed occupancy increase appears linked to modulation of chromatin accessibility. Notably, in addition to its effect on RARA and RARG ability to interact with DNA, RA-induced p38MAPK was shown to activate the downstream kinase MSK1 that catalyzes the phosphorylation of histones H3 on serine 10 (H3S10), thereby making RAREs more accessible to RAR/RXR binding (58). The lack of variation of the FAIRE signal associated with the upstream binding site indicates that this mechanism may not contribute significantly to the observed early occupancy increase at the analyzed loci and/or in our cellular model.
Motif Analyses of RAR/RXR binding sites reveals a wide variety of response elements
In addition to chromatin accessibility, the binding site sequence variability is another extrinsic parameter that may modulate RAR occupancy. We therefore implemented a careful characterization of putative transcription factor binding motifs present in RAR/RXR bound genomic loci. To explore this issue, we first searched for enriched motifs using the de novo motif discovery algorithms composing the MEME-ChIP suite (59). As expected, we identified several motifs reminiscent of the canonical nuclear receptor core binding motif RGKTSA with different spacing and relative orientation. Notably, most predicted motifs exhibit a clear enrichment at the center of the identified regions (Supplementary Table S2 and Supplementary Figure S6 panel A). The DREME algorithm (60) highlighted two short motifs (Figure 3A, middle and low panel) that are closely related to the Position Weight Matrix (PWM) of the core motif previously proposed by Balmer and Blomhoff (15) (Figure 3A top panel). As a second more directed attempt, we choose to use the latter to scan our sequence data set for all possible relative orientations (Direct Repeat, Inverted Repeat and Everted Repeat) and for spacers ranging from 0 to 9 nucleotides. The same motifs were used to scan a control data set of randomly generated sequences of same length and G+C composition. Motif hits in both sequence data sets were used to compute Receiver Operating Characteristic (ROC) curves (Figure 3B). The corresponding area under the curve (AUC) for all tested motifs is shown in Figure 3C. In this context, the AUC can be interpreted as the probability for a bound region to have a better PWM alignment score than a non-occupied region.
Figure 3.
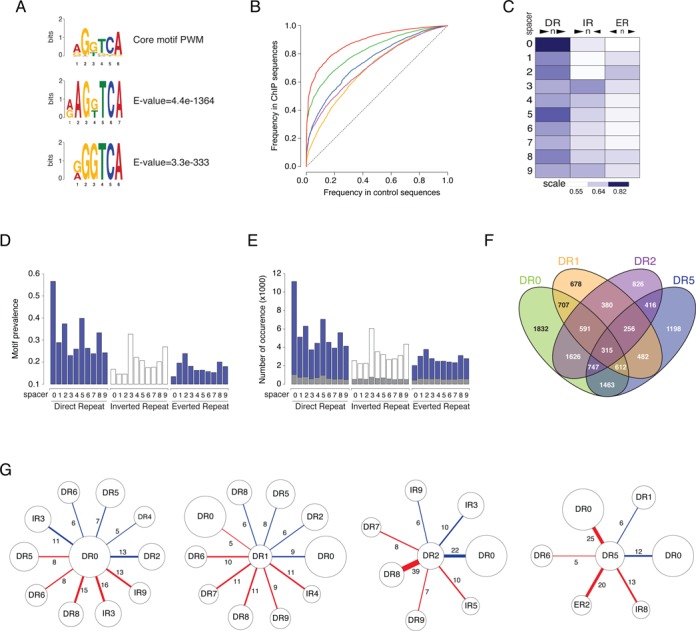
Analysis of Hormone Response Elements (HRE) in RAR/RXR target regions. (A) Position weight matrix (PWM) of core half-site motif. Matrices predicted by Balmer and Blomhoff (upper panel) and by the de novo motif-discovery algorithm DREME (middle and lower panel). (B) ROC curves. Enrichment in the various types of HRE was assessed by comparing the frequency of discovery of the consensus in the RAR/RXR bound regions (y-axis) and in a set of randomly generated sequences (same size, same GC content, x-axis). Results show compound motif (DR0–1–2–5) (red curve) and DR0 (green curve), DR1 (yellow curve), DR2 (purple curve) and DR5 (blue curve) motifs enrichment. (C) Areas under the curve measuring enrichment of RAR/RXR binding region in Direct Repeat (DR), Inverted Repeat (IR) and Everted Repeat (ER) with spacer from 0 to 9 nucleotides are shown as heat map. (D) Prevalence of the different motifs (DR-IR-ER with spacer 0–9) in the RAR/RXR binding region set. (E) Number of occurrences per motif in the RAR/RXR binding region set. Grey bars represent the number of predicted isolated motifs in each category. (F) The number of identified regions encompassing at least one DR0, DR1, DR2 or DR5 motif. Results are shown as four-way Venn diagrams representing the number of region in each subcategory. In D-F panels, motifs are predicted based on their alignment score with the corresponding PWM. Score threshold was defined as the score giving 10% of positive matches in control sequences. (G) Motif association analysis. Results show the percentage of predicted HRE (DR0, DR1, DR2 or DR5) that share one of the half core with another predicted motif. Blue link represent the percentage of embedded motifs and red link represent the percentage of motifs sharing the central half core.
As expected, the previously identified non-canonical motif DR0 (green curve), and the canonical motifs DR5 (blue curve), DR2 (purple curve) and DR1 (yellow curve) were found to be the most significantly enriched (Figure 3B and C and Supplementary Figure S5). Additionally, other motifs showed up with by order of importance DR8, IR3 and, to a lesser extent, all other possible combinations of two core motifs (Figure 3C). With an AUC of 0.89, the compound DR0–1–2–5 motif (Figure 3B, red curve) was thus found to be a specific marker of RAR/RXR binding.
In order to predict true HRE with an acceptable trade-off between sensitivity and false-positive rate, we made use of the computed curves to fix a motif-specific threshold for PWM alignment scores that resulted in 10% prevalence for the considered motif in the control sequence data set. Motif-specific prevalence and occurrence number at the computed threshold are shown in Figure 3D and E respectively. With this setting, 87.5% of the identified binding regions encompass at least one DR0, DR1, DR2, or DR5 motif and 74.3% if we exclude the non-canonical DR0 motif. Noticeably, as shown in Figure 3F, 7,595 regions encompass more than one motif type (DR0, DR1, DR2, or DR5). In addition to motif diversity, the total number of occurrences for DR0, DR1, DR2 and DR5 greatly exceeds the number of predicted regions (Figure 3E) thereby confirming that multiple repetitions of the RARE-associated motifs are obviously present in single binding regions. The distribution of the number of motifs per region further substantiates this observation (Supplementary Figure S6 panel B).
These observations led us to further investigate the exact organization of multiple motifs found in RAR/RXR binding regions. We first considered the number of isolated occurrences of all tested motifs. As HREs are often composed of a repetition of the same hexameric DNA core half-site, we defined isolated HREs as motifs exhibiting no other core half site within 9 nucleotides of their 5′ or 3′ end. The results shown in Figure 3E indicate that the proportion of such isolated HREs (gray bars) is low and that most predicted HREs exhibit additional core half site in the vicinity thereby forming composite HREs encompassing more than two repetition of the hexameric DNA core half-site. Such composite HREs could be seen as a single functional motif or as two overlapping functional units. We therefore asked whether specific HREs associations are more likely to occur by computing the frequency of association for each couple of motifs. We distinguished between associated motifs that share either their 5′ or 3′ core half site (red link), referred to as embedded motifs, and motifs in which 3′ half core of the first motif constitute the 5′ half core of the second one (blue link) referred to as overlapping motifs (Supplementary Figure S6 panel C). The results obtained are shown in Figure 3G and in Supplementary Figure S6 panel D. DR0 and DR1 motifs appear to be promiscuous elements exhibiting little preference for the associated motif. By contrast, DR2 and DR5 are often found associated with DR0. The overlapping of DR2 and DR0 motifs lead to the formation of a DR8 motif recently characterized as a functional RARE (25). Interestingly, DR0 as well as ER2 are often found embedded with DR5, indicating that the spacer sequence of DR5 is frequently involved in the formation of these composite elements.
Collectively, these results show that while a single copy of DR0, DR1, DR2 and DR5 motifs are sufficient to recruit RAR/RXR in vitro, in vivo targeted regions often encompass more than one bona fide RARE. The presence of RARE undoubtedly remains the hallmark of RAR/RXR targeted regions and the lack of detection of RARE-associated motifs in a small proportion of targeted regions suggests that our experiment occasionally captures physical interactions between distant genomic regions. We may however not rule out that this observation may account for the existence of degenerated RARE motifs, associated with a low PWM alignment score.
In addition, RAREs are frequently more complex than canonical direct repeats and often encompass more than two repetitions of the half core motif. Whether these so-called composite motifs confer specific binding properties remains to be established. It is however, worth noticing that while some predicted HREs are not characterized as functional response elements and may be coincidental by-products of the local accumulation of hexameric core half-sites, others are known as bona fide response elements for various nuclear receptors (61). It is therefore tempting to propose that a majority of the identified RAR/RXR targeted regions may serve as a DNA hub allowing the interaction with multiple nuclear receptors through the recognition of composite motifs or distinct but closely localized HREs.
RAR/RXR binding sites coincide with loci targeted by self-renewal associated transcription factors
The motif enrichment analysis of our data set by algorithms composing the MEME-ChIP suite also highlighted several binding motifs for transcription factors, including NRs, known to be associated with self-renewal/pluripotency (i.e. POU5F1, SOX2, KLF4, TCFCP2L1, STAT3, ZFX, ESRRB, NR5A2) or differentiation (i.e. SOX17, NR2F1, NR2F2) (Supplementary Table S2), thereby agreeing with the previous observation made by Mahony and colleagues in RA-treated mES cells on a smaller set of RAR binding regions (22). To confirm the prediction obtained by motif enrichment analysis, we comparatively analyze the binding profiles of these factors with RAR/RXR binding region data set.
Using published data sets listing binding regions of transcription factors or cofactors known to play an important role in pluripotency and/or self-renewal (e.g. ESRRB, NR5A2, SOX2, NANOG, POU5f1, TBX3, ZFX, TCF3, PRDM14, SMAD1, STAT3, E2F1, TCFCP2L1, EP300, SUZ12), reprogramming and stem cell maintenance (e.g. KLF4, MYC, MYCN) or differentiation (e.g. SOX17) of mES or F9 EC cells (7,26–27,40), we computed the correlation coefficient between regions bound by RAR/RXR heterodimers and these transcription factors (Figure 4A) and represented the distance distribution of the annotated TF binding sites relative to the center of RAR/RXR bound regions (Figure 4B–D and Supplementary Figure S7).
Figure 4.
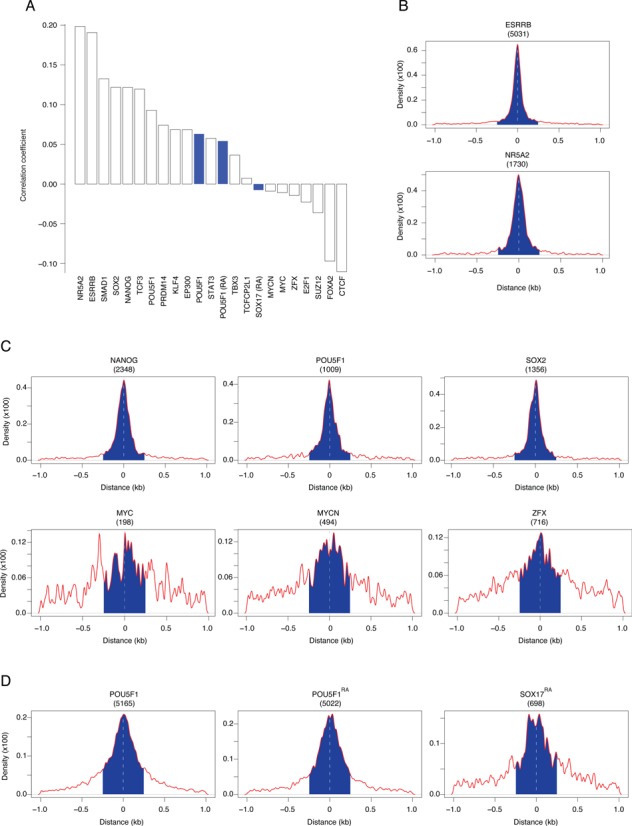
RAR/RXR targeted region coincide with pluripotency- and differentiation-associated transcription factors binding sites. (A) Correlation between RAR/RXR and the indicated factors binding sites. Data sets used for the correlation study were obtained in mouse ES cells (open bars) or mouse F9 EC cells (blue bars). (B) Distance distribution of the orphan nuclear receptors ESRRB (upper panel) and NR5A2 (lower panel) binding regions identified in mouse ES cells that overlap with RAR/RXR binding regions identified in F9 EC cells relative to RAR/RXR region center. (C) Distance distribution of the pluripotency-associated transcription factors binding regions identified in mouse ES cells that overlap with RAR/RXR binding regions identified in F9 EC cells relative to RAR/RXR region center. (D) Distance distribution of POU5F1 and SOX17 binding regions that overlap with RAR/RXR binding regions identified in F9 EC cells relative to RAR/RXR region center. The number between brackets represents the absolute number of RAR/RXR binding regions (peak summit ± 250 bp, blue area) that overlap with the indicated transcription factor.
In agreement with the concept of nuclear receptor binding hub, ESRRB and NR5A2 binding sites exhibit the highest correlation with RAR/RXR binding regions (Figure 4A). More than one third of all RAR/RXR binding sites were also identified as ESRRB binding sites in proliferating mouse ES cells (Figure 4B, upper panel). Similarly, more than 10% of RAR/RXR were also annotated as NR5A2 binding regions (Figure 4B, lower panel). Unfortunately, the ChIP-seq approach identifies binding regions rather than actual binding motifs due to its limited spatial resolution, but the analysis of the relative positions of ESRRB, NR5A2 and RAR/RXR peak summits (Figure 4B) suggests that these receptors recognize either composite motifs or distinct motifs localized in close proximity.
Confirming the results of our motif enrichment analysis, the pluripotency/self-renewal regulators SOX2, NANOG, POU5F1, as well as KLF4, SMAD1, STAT3, TCF3, EP300, PRDM14 binding sites also exhibit positive correlation with RAR/RXR binding regions. By contrast ZFX, MYC, MYCN, E2F1, SUZ12, FOXA2 and CTCF binding sites repertoire exhibit a negative correlation when compared to all RAR/RXR identified regions indicating that DNA regions interacting with these factors are poorly associated with RAR/RXR. As observed for ESRRB and NR5A2, the distance distribution of positively correlated transcription factors in overlapping regions shows that they likely recognize DNA motifs localized in the immediate vicinity of RAR/RXR binding sites (Figure 4C and Supplementary Figure S7 panel A). By contrast, negatively correlated transcription factors exhibit a more uniform distribution indicating that, in overlapping regions, their binding sites are poorly associated with RAR/RXR.
Most interestingly, a recent study determined POU5F1 and SOX17 binding site repertoires in differentiating F9 EC cells and demonstrated that POU5F1 binding sites are redistributed during the differentiation process by forming alternative heterodimers with either SOX2 or SOX17 (40,62). The correlation analysis of POU5F1 (in untreated and RA-treated F9 EC cells) binding sites with all annotated RAR/RXR regions confirmed that POU5F1 co-localizes with RAR/RXR binding regions despite its spatial redistribution in RA-treated cells relative to untreated ones. By contrast, SOX17 binding regions appears slightly negatively correlated with RAR/RXR binding data set. As observed previously, POU5F1 binding sites overlap with a large fraction of RAR/RXR binding regions and the distance distribution in these regions shows that its binding sites are indeed closely associated with RAR/RXR binding. By contrast, SOX17 co-localizes with RAR/RXR in few instances and exhibits a slightly more uniform distribution suggesting that its binding occurs in a subset of RAR/RXR binding regions and that binding motifs are likely more distant.
The absolute intersection between RAR/RXR regions and the positively correlated TFs (Figure 4B–D and Supplementary Figure S7 panel A) shows a large number (typically >1000) of co-occupied regions. The fraction of co-occupied RAR/RXR regions should however be interpreted with caution, as it is strongly biased by each TF detection sensitivity. We noted though that higher quality peaks tend to be better associated with our RAR/RXR regions (Supplementary Figure S7 panel B).
Interestingly, the two groups of factors that could be defined on the basis of their positive or negative correlation with RAR/RXR, are strongly reminiscent of the previously identified groups of transcription factors known to co-localize on pluripotent cell-specific enhanceosomes (26). The observed correlation of RAR/RXR binding sites with SOX2, POU5F1 and NANOG and their relative position in overlapping regions suggest that retinoic acid signaling pathway interacts directly with the pluripotency core circuitry (63). Similarly, the positive correlation (Figure 4A) and the positional enrichment (Supplementary Figure S7) observed for SMAD1, STAT3 and TCF3 interacting regions suggest the existence of crosstalks between the retinoic acid pathway and the bone morphogenetic proteins (BMPs), the leukemia inhibitory factor (LIF) and the non-canonical Wnt pathway respectively. Notably, these three pathways are also known to play a crucial role in pluripotency maintenance (64–66).
Collectively, these observations show that RAR/RXR heterodimers share numerous targets with pluripotency-associated factors and is likely associated with previously identified pluripotent cell-specific enhanceosomes (26). Moreover, we have shown above that RAR/RXR binding displays multiple time-dependent profiles (Figure 2D). Rather than estimating the correlation between RAR/RXR and other factors occupancy globally, we next compared transcription factors sites enrichment in relation with RAR/RXR occupancy profiles.
RAR/RXR binding dynamics is associated with binding site-specific features
We next investigated whether the RAR/RXR occupancy profiles identified by clustering (Figure 2D) were specifically associated with molecular features such as the presence of RARE motifs or transcription factor binding sites experimentally identified in F9 and ES cells. To this end, we performed Fisher's exact tests to detect features whose prevalence varied significantly in one cluster compared to all other RAR/RXR binding regions. The results are presented in Figure 5A and Supplementary Figure S8, where red (resp. green) bars indicate features that are significantly enriched (resp. depleted) in the cluster. First we observe that each binding pattern is characterized by a unique combination of molecular features. For instance, the regions assigned to clusters exhibiting high average occupancy in untreated F9 cells are characterized by their association with ESRRB and POU5F1 binding events (clusters B, D and F in Figure 2D and Supplementary Figure S3 panel A). Also, all clusters exhibiting a maximum occupancy in the early phase of the RA-induced PrE differentiation process, namely clusters A–D, are associated with a higher prevalence of DR0 type motif. Among these four clusters, subtle binding pattern variations appear coupled with specific core pluripotency factor binding sites layouts and are independent or negatively associated with PrE differentiation-associated transcription factor like SOX17. Cluster A, which exhibits only a transient occupancy at 2 h after RA stimulation, is only associated with DR0 enrichment while clusters B, C and D are also associated with NR5A2 and SMAD1 binding but differ in their association with SOX2, NANOG, POU5F1, TCF3 and PRDM14 binding events and the presence of DR2 motif.
Figure 5.
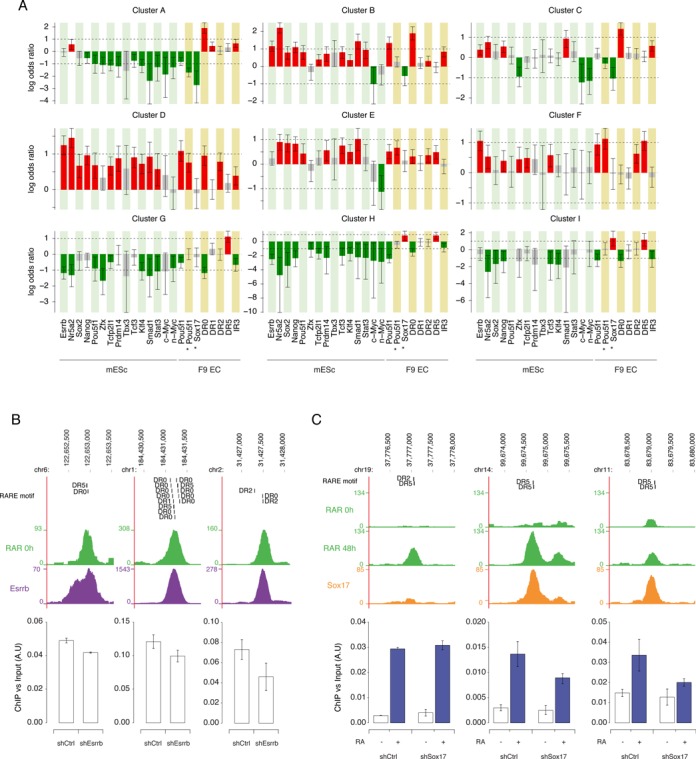
RAR/RXR binding dynamics and region-associated features. (A) Each bar represents the log odds ratio from Fisher's exact test between a cluster assignment and the presence of a feature within 0.5kb of the RAR/RXR peak summit. A red (resp. green) bar indicates that a feature is significantly more frequent (resp. less frequent) in the cluster than in other RAR/RXR binding regions. Error bars indicate 95% confidence intervals. (B) Effect of Esrrb KD on RAR binding intensity. Upper panels show a screenshot (http://genome.ucsc.edu) of RAR and ESRRB binding signal on the genomic loci analyzed in wild-type F9 cells and ES cells respectively. Predicted RARE motifs present under the peak are indicated. Lower panels show ChIP-qPCR quantification of RAR binding in Control and Esrrb KD F9 cells. RAR binding intensity is expressed relative to input amount. The data shown represent mean ± SD of replicate experiments. (C) Effect of Sox17 KD on RAR binding intensity. Upper panels show a screenshot (http://genome.ucsc.edu) of RAR and SOX17 binding signal on the genomic loci analyzed in untreated and RA treated wild-type F9 cells. Predicted RARE motifs present under the peak are indicated. Lower panels show ChIP-qPCR quantification of RAR binding in untreated and RA treated Control and Sox17 KD F9 cells. RAR binding intensity is expressed relative to input amount. The data shown represent mean ± SD of replicate experiments.
By contrast with early-occupied genomic loci, binding regions that reach their maximal occupancy level at a later time point (24 and/or 48 h, clusters G–I), are independent or even depleted in pluripotency/self renewal factors binding events. They also exhibit a significant association with DR5 motif while being depleted in DR0 motif, indicating that RAR/RXR binding shifts from DR0-enriched regions in non-differentiated cells to DR5-enriched regions binding in differentiated cells. Most interestingly, clusters H and I are also characterized by the binding of the differentiation-associated factor, SOX17. The lack of strongly discriminating associated binding events and the independence of these two clusters with respect to several other tested transcription factors are likely reflecting the lack of data on RA-modulated transcription factors binding repertoire.
Clusters E and F have overall smaller absolute log odds ratio and display characteristics of both early and late occupied regions. Together, these observations suggest that clusters E and F could be made of regions peaking between 2 and 24 h and could thus be imperfectly resolved due to the lack of observation in this interval.
Of note, the binding sites of some transcription factors exhibit limited co-localization with RAR/RXR (i.e. MYC, MYCN, ZFX) and are negatively associated with a specific binding pattern (Figure 5, cluster C). By contrast, the sites of other transcription factors such as KLF4 or TCFCP2L1 (Supplementary Figure S7) globally coincide with RAR/RXR binding sites but do not associate with any specific occupancy pattern.
To confirm that these observed statistical associations may be of functional significance, we tested whether the inhibition of Esrrb or Sox17 expression altered the binding of RAR/RXR on specific targets. We therefore generated stable F9 clones expressing control or target-specific shRNAs, the latter exhibiting reduced levels of expression of the targeted mRNA compared to wild-type condition (Supplementary Figure S9). We then measured RAR binding levels in untreated cells (for Esrrb and Sox17) and at 48 h after induction (for Sox17 only). The results we obtained are shown in Figure 5B for Esrrb and 5C for Sox17.
As we previously noticed (Figure 5A and Supplementary Figure S3A), clusters with enriched Esrrb binding in ES cells display a stronger RAR binding in untreated F9 cells, suggesting a cooperative binding mechanism. We selected three DR0-containing regions bound both by RAR in untreated F9 cells and Esrrb in untreated ES cells (Figure 5B, upper panel). We indeed observed a replicable decreased binding in the Esrrb knocked down (KD) F9 cells in all three regions (Figure 5B). The moderate effect size is consistent with the difference observed between Esrrb-dependent (clusters B, D and F) and Esrrb-independent clusters (Supplementary Figure S3A), the partial inhibition of Esrrb in KD cells and the putative redundant effect of Nr5a2.
As mentioned earlier, Sox17 is a transcription factor induced upon RA treatment of F9 cells and involved in the primitive endoderm differentiation process. Consistently, we observed the association of Sox17 with late RAR/RXR bound regions (clusters H and I). We therefore measured the induction of RAR binding at 48 h after treatment in control and Sox17 KD cells. We selected three DR5-containing regions exhibiting their maximum occupancy after 48 h, two of them displaying a bona fide Sox17 peak. The third selected region is the previously mentioned RAR/RXR region downstream of Cyp26a1 locus (Figure 2E), which lacks Sox17 binding in RA treated F9 cells, and for which we showed that the increased RAR/RXR binding coincides with an increased chromatin accessibility (Figure 2H). In this region, the knock down of Sox17 did not alter RAR binding levels (Figure 5C, left panel), while the other two regions displayed a substantially decreased induction, consistent with the partial inhibition of Sox17 expression in KD cells (Figure 5C, central and right panel). These results confirm the existence of a cooperative DNA binding mechanism between RAR/RXR and Sox17, and show that late RAR/RXR binding during PrE differentiation can be accounted for by both Sox17-dependent and Sox17-independent processes.
Overall we found that RAR/RXR binding dynamics can be classified in a handful of typical time-dependent profiles exhibiting specific molecular signatures involving different RARE motifs and experimentally identified TF binding signals in ES and F9 cells. Most importantly, we highlighted that these profiles and their associated signatures are consistent with the known function of these TF in pluripotency maintenance or PrE differentiation processes, suggesting a tight dependence of RAR/RXR binding repertoire with the cell differentiation status.
A natural follow-up to these observations was to ask whether the occupancy profile of RAR/RXR regions could be associated to specific transcription profiles in neighboring genes.
RAR/RXR mediated transcriptional regulation in differentiating F9 EC cells
RA exerts its biological effect primarily through gene transcription modulation (67). Transcriptomic analyses were therefore performed in various biological systems using microarray-based approaches and several data sets are already available in public databases (68). To obtain a more detailed and accurate transcriptomic profile of RA-treated F9 cells, we implemented a time course analysis of gene expression 0, 6, 12, 24 and 48 h after RA stimulation using high-throughput RNA sequencing (RNA-seq). We could unambiguously detect 23598 of the 36381 annotated loci in Ensembl mouse genome (v63, jun 2011), estimating at 65% the percentage of expressed genes at any time of the differentiation process.
To identify the genes that are significantly modulated during RA-induced PrE differentiation process, we performed a differential gene expression analysis between vehicle treated cells and RA-treated cells using DESeq (45). Using a (Benjamini-Hochberg) adjusted P-value cutoff of 0.05, we detected a total of 3971 genes exhibiting significant differential expression at one or more of the RA treatment time points (Figure 6A and Supplementary Table S3). The differential expression of several genes was validated by high-throughput RT-qPCR (Supplementary Figure S10).
Figure 6.
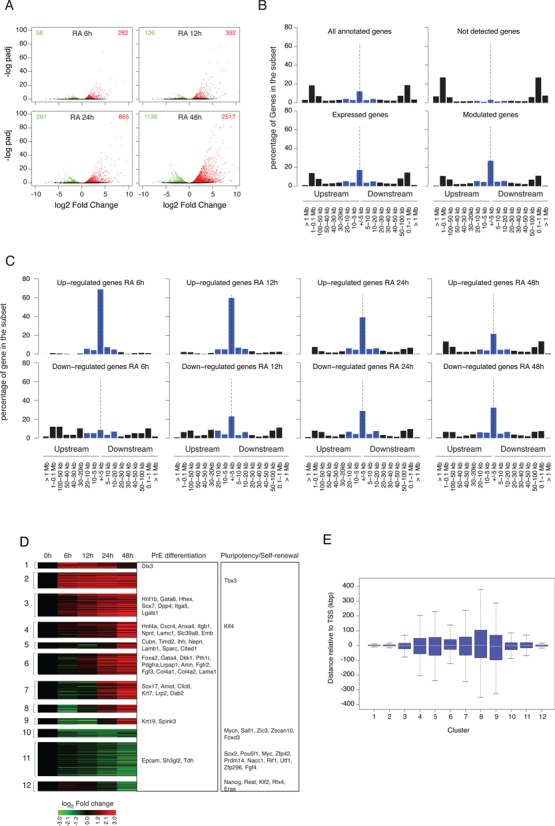
Gene expression profiles during RA-induced PrE differentiation of mouse F9 EC cells. (A) Identification of differentially expressed genes at different time points after RA stimulation relative to their untreated expression level. Results are shown as a volcano plot representing Benjamini-Hochberg adjusted P-value versus log2 fold change on the y- and x-axes, respectively. Up- and down-regulated genes are shown in red and green, respectively. (B) Proportion of genes in the indicated set that exhibit an RAR/RXR target regions within the indicated distance range from their closest annotated TSS. Blue bars correspond to the percentage of genes exhibiting an RAR/RXR target region at less than 20 kb upstream or downstream of their annotated TSS. (C) Same as (B) panel for gene up- or down-regulated at the indicated time point after RA stimulation. (D) Clustering of differentially expressed gene expression profiles. Gene associated to Primitive Endoderm differentiation or pluripotency maintenance are indicated in the corresponding annotated boxes. (E) Boxplot representing the distance distribution of the closest RAR/RXR target region relative to annotated TSS of differentially expressed genes for the previously defined (D) expression clusters.
We confirmed and largely extended the previously published repertoire of 410 differentially expressed genes (68) during the first 24 h of the RA-induced primitive endoderm differentiation process. A Gene Ontology analysis of DE genes data sets and expression of key genes involved in pluripotency/PrE differentiation are presented in Supplementary Table S3 and Supplementary Figure S11, respectively. To the best of our knowledge, our results show that in vitro differentiation of RA-treated F9 cells recapitulates the known regulatory events implicated in the exit from pluripotency and in the earliest step of primitive endoderm induction in vivo.
Differentially expressed genes are enriched in genes encoding chromatin interacting proteins and transcription factors, thus suggesting that primary RA-dependent transcriptional response modulate targeted TF-associated genetic programs and chromatin remodeling events. Differentially expressed genes list also encompasses genes encoding proteins involved in key signaling pathways associated with pluripotency maintenance and differentiation (e.g. TGFB/BMP, WNT, LIF) (Supplementary Figure S11), indicating that crosstalks between the RA and these pathways occur a multiple levels, from extra-cellular ligands expression to pathway-associated specific transcription factors binding.
To unravel how RA signaling triggers pluripotent EC cell PrE differentiation, we implemented an integrative analysis of RAR/RXR binding pattern and of expression data. We first analyzed the distribution of RAR/RXR binding regions relative to annotated TSS for gene subsets defined by their expression status in RA-treated F9 cells. We found that 47% of the differentially expressed gene set did encompass at least one RAR/RXR binding region within 20 kb of their TSS. This proportion is reduced to 32.5% and 9.6% of the constitutively expressed and silent gene set, respectively (Figure 6B). These observations show that while RAR/RXR enrichment is biased toward modulated genes, the presence of an RAR/RXR binding region in the vicinity of a TSS does not predict accurately RA responsiveness. Indeed, the proportion of DE genes relative to all genes exhibiting an RAR/RXR binding region in a given distance range is maximal (26%) for genes encompassing an RAR/RXR binding region within 2.5 kb of their TSS and rapidly decreases with distance (Supplementary Figure S12, top panel). Although transient and/or undetectable transcriptional rate variations could not be ruled out, this observation suggests either that other regulatory inputs are required to achieve gene regulation or that insulation mechanisms are protecting genes from RAR/RXR-mediated transcriptional regulatory effects.
A simple but widely accepted idea postulates that early response upon transcription factor activation is a good indicator of its direct transcriptional effect. To further assess the association between RAR/RXR binding and RA-induced gene transcriptional modulation, we analyzed all time points of the differentiation kinetic individually and distinguished between up- and down-regulated genes. Results presented in Figure 6C highlight the time dependent decrease of the association between gene up-regulation and presence of an RAR/RXR binding region in the vicinity of the TSS. While at 6 h after RA stimulation, nearly all up-regulated genes (91%) exhibit an RAR/RXR binding region within 20 kb around their TSS, only 40% of the genes up-regulated at 48 h after RA stimulation does. Strikingly, down-regulated genes exhibit an opposite behavior with a significant time-dependent increase of RAR/RXR binding regions associated genes from 29% at 6 h to 55% at 48 h. A statistical analysis of the difference in distance distribution for up- versus down-regulated genes (Supplementary Figure S13), strongly suggests that early-detected down-regulatory events are often independent of RAR/RXR-mediated transcriptional effect.
The time-dependent increase in the number of down-regulated genes associated with RAR/RXR binding may seem paradoxical at first sight, but can be accounted for as follows. The mRNA level of a specific transcript is not a direct measure of the gene's transcriptional activity, but integrates multiple biological processes, namely transcription initiation, elongation, pre-mRNA processing, and mRNA degradation. In particular, a decreased transcription rate can only be observed as a decreased mRNA level after a delay related to the transcript half-life. Therefore, many down-regulations we observe at 48 h may in fact result from earlier RAR-mediated repression events.
Although informative, individual time point expression analysis does not capture the dynamics of the biological response. We therefore performed a clustering analysis of all genes exhibiting a significant differential expression relative to the untreated condition. This analysis identified 12 different classes that mostly recapitulate the main steps leading pluripotent cells to a PrE differentiated state (Figure 6D). Clusters 10–12 encompass down-regulated genes among which most if not all genes that are responsible for pluripotency maintenance (e.g. Sox2, Pou5f1, Nanog, Zfp42). By contrast, clusters 1–9 contain genes associated to the PrE differentiation process, and are strongly reminiscent of the sequence of molecular events identified in vivo (Gata6, Gata4, Sox7, Sox17, Lamb1). Interestingly, the transition of F9 cells from an undifferentiated to a PrE differentiated state is accompanied by a down-regulation of Fgf4 and a parallel raise of its cognate receptor Fgfr2, thereby recapitulating in a cell autonomous manner the in vivo PrE differentiation process.
We next analyzed the distribution of the distance of the closest RAR/RXR binding region relative to the TSS in the various expression classes. Results presented in Figure 6E show significant differences between classes and, in agreement with the previous analysis, highlight the close physical association between RAR/RXR binding regions and TSS of genes exhibiting rapid up-regulation (clusters 1 and 2) and late down-regulation (cluster 12).
Integration of RAR-dependent transcriptional regulation in differentiating F9 EC cells
In embryonic pluripotent cells, pluripotency maintenance and inhibition of differentiation are supported by a group of transcription factors working in a coordinated manner (7,26). To better assess the role of RA-dependent signaling pathway in the exit of pluripotency and triggering of differentiation, we analyzed how RAR/RXR-dependent signaling was integrated with the activity of other key transcription factors to achieve a precise regulation of gene expression. For that purpose, we used the previously described approach to assess occupancy profiles dependency, and tested independence between various cis-regulatory features and expression clusters. Results are shown in Figure 7. As previously described, early up-regulated and down-regulated genes are both characterized by their association with core pluripotency/self renewal factors such as SOX2, NANOG and POU5F1. However, they could be further distinguished by their association with a group of factor encompassing MYC, MYCN and ZFX that exhibit a specific enrichment in the regulatory region of down-regulated genes (26,69), thereby validating our methodology. More subtle expression profile variations are associated with specific combinations of TCF3, SMAD1 and STAT3 binding sites. Notably, these transcription factors are not transcriptionally modulated in RA-treated F9 cells but are known mediators of the genomic action of extracellular signaling pathways (WNT, BMP and LIF, respectively) modulated upon RA stimulation (Supplementary Figure S11 and Supplementary Table S3).
Figure 7.
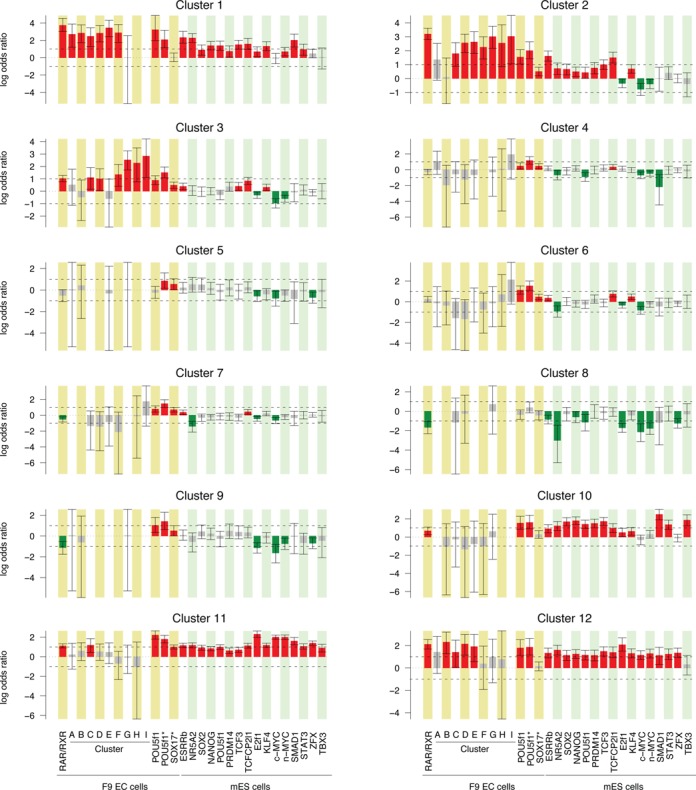
Gene expression profiles and associated regulatory region features. The results are shown as the association score between specific gene expression cluster assignment and presence of the indicated feature within a 40 kb window centered on the annotated TSS. Each bar represents the enrichment of the indicated feature in each cluster with respect to all differentially expressed genes and represents the log odds ratio and 95% confidence interval as computed by Fisher's exact test. The red and green bars correspond to the significantly enriched and depleted features, respectively.
Most interestingly, clusters 1 and 2 and 10–12 are also associated with RAR/RXR binding sites, but could be distinguished by the dynamic occupancy of these sites. Genes assigned to cluster 1 are characterized by an early but transient up-regulation and by their physical association with early bound RAR/RXR binding sites (clusters A–F). In contrast, genes assigned to cluster 2, which are characterized by an early and sustained up-regulation, are associated with RAR/RXR regions exhibiting sustained or induced occupancy over time (clusters C–I) and display weak but significant enrichment in Sox17 binding event. Moreover, genes assigned to cluster 3, which are characterized by a slow or delayed up-regulation, are independent of core pluripotency/self-renewal factor presence but as previously noted for cluster 2, associated to RAR/RXR binding regions exhibiting sustained or induced occupancy over time and to Sox17 bound regions. While the gene expression clusters that are characterized by an early up-regulation (clusters 1–3) or a down-regulation (clusters 10–12) exhibit specific association patterns with cis-regulatory events, gene assigned to clusters characterized by late up-regulation (clusters 4–9) which encompass most PrE differentiation associated genes, are less clearly defined, most likely due to the lack of binding data in differentiating ES and EC cells for most transcription factors except RAR/RXR, POU5F1 and SOX17. However, it is worth noting that these clusters are all (except for cluster 8) characterized by their significant association with POU5F1 and SOX17 binding sites identified in RA-treated F9 cells in agreement with their gene content. This observation is in agreement with the recently demonstrated function of POU5F1 in primitive endoderm differentiation and its relocation following partner exchange (40,70). Furthermore, our analysis highlights the modest but statistically significant enrichment of clusters 3 and 6 in TCFCP2L1 and KLF4 binding, suggesting that these transcription factors which are constitutively expressed or induced in RA-treated EC cells respectively, may be involved in the differentiation process.
Altogether, these results highlight the link between RAR/RXR binding dynamic and gene expression profiles and demonstrate the close functional relation between RAR/RXR and the core pluripotency factors network and to a lesser extent between RAR/RXR and differentiation-associated factors (i.e; Sox17, Pou5f1). Furthermore, they suggest that RA dependent RAR/RXR activation globally counteracts core pluripotency factors action and cooperate with differentiation factors in differentiating F9 cells.
DISCUSSION
Previous contributions demonstrated that RAR/RXR heterodimers are the functional unit transducing retinoid signal during RA-induced PrE differentiation of F9 EC cells (71,72). In this study, we identified and characterized the RAR/RXR heterodimers binding site repertoire throughout the first 48 h of the PrE differentiation process and investigated how binding site occupancy varies over time. We showed that a significant proportion of the more than 13000 identified RAR/RXR heterodimers binding sites exhibit loci specific occupancy profiles that differ in their initial occupancy level and/or time-dependent occupancy pattern. The characterization of these dynamically occupied binding sites shows that occupancy variations are likely resulting from multiple and interlaced molecular mechanisms acting at different time scales and on multiple binding parameters, ultimately resulting in the precise implementation of cell specific genetic program.
Altogether, our observations and published data lead us to propose an integrated model of RAR/RXR binding dynamics during RA-induced PrE differentiation of F9 EC cells (Figure 8). In view of their extensive similarities with ES cells, we propose that this model also applies to RA-induced differentiation of this later cell type.
Figure 8.
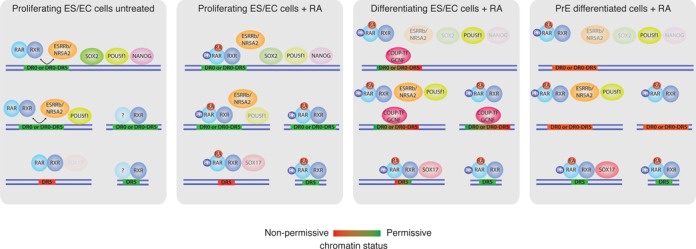
Integrated model of RAR/RXR binding dynamics during RA-induced PrE differentiation of F9 EC cells.
The classical model for the action of nuclear receptors postulates that a major effect of ligand-dependent activation is to convert preexisting DNA-bound NR dimers from an inhibitory to an activating complex. As previously reported in ES cells (22) and by contrast with this model, we show that the most evident consequence of RA stimulation is a massive increase in the number of RAR/RXR bound genomic loci within 2 h of treatment. Hence the widely accepted mechanism of repression by unliganded RAR operates at most on a limited fraction of the retinoid target sites. Furthermore we found that a majority of the binding sites bound upon RA stimulation are initially occupied by non-RAR RXR heterodimers. Indeed, we show that in absence of RA, while most RAR/RXR binding regions identified in the early phase of RA stimulation show only weak or even undetectable level of RAR subunit occupancy, they already exhibit significant binding of RXR subunit indicating that RAR/RXR binding sites are primarily occupied by non-RARs/RXRs dimers and that the RXR dimeric partner is likely ‘exchanged’ upon RA stimulation. Notably, a similar process has already been described for PPARG/RXR binding sites in an in vitro model of adipocyte differentiation (73), suggesting that this phenomenon may be a general feature of RXR heterodimers binding. In this study, the authors found that PPARG/RXR binding region are primarily occupied by non-PPAR/RXR dimers and that PPARG subunit recruitment occurs after several days, following PPARG induction and the increased production of endogenous PPARG agonist. The characteristics of our system significantly differ from this model of adipocyte differentiation as F9 cells constitutively express RARA and RARG paralogs and respond to an exogenous RA stimulation (hence the difference in exchange kinetic). In both systems however, the ligand seems to play a crucial role in inducing the exchange process. Additional binding studies of potential RXR partners are required to further substantiate this exchange hypothesis and identify RXR partner(s) if any. Yet the previously described non-genomic effect mediated by RA-activated RARs may provide a molecular basis for this observation. Indeed, several studies demonstrated that RA stimulation activates p38 MAP kinase in an RAR-dependent manner and triggers a phosphorylation cascade resulting in RARA and RARG phosphorylation and increased interaction with their target sequences (52,58). RA-dependent RAR subunit recruitment is therefore likely reflecting the increased ability of phospho-RAR/RXR heterodimers to interact with DNA. Furthermore, RXR subunit is also a downstream target of MAP kinase (74) and its phosphorylation may favor RAR/RXR heterodimer formation or disrupt non-RAR/RXR dimers.
Why a significant number of sites are nevertheless significantly occupied by RAR/RXR in absence of RA remains unclear but may reveal the presence of a limited pool of DNA-binding-competent RAR/RXR heterodimers. Alternatively, these binding sites may be associated with particular transcription factor binding patterns, chromatin structures and/or epigenetic markings which could participate in creating a favorable environment for RAR/RXR binding through direct physical interactions or other molecular mechanisms. In agreement with this last hypothesis, our data demonstrate that multiple RAR/RXR binding sites in F9 cells coincide with ESRRB and NR5A2 occupied sites in ES cells, and that these sites are associated with a higher RAR/RXR basal occupancy level. The decreased RAR/RXR binding in Esrrb KD F9 cells strongly suggests the existence of such a cooperative binding. The exact mechanism remains to be established on a case-by-case basis to take into account the particular composition and spatial configuration of response elements under each peak.
One longstanding issue in the field of gene expression regulation deals with the identification of the molecular mechanisms that lead to the selective expression of genetic information. In the case of RAR/RXR, the characterization of the various RAREs provides the molecular basis for a specific interaction with DNA but does not explain tissue or cell specific response to RA stimulation. The identification of RAR/RXR binding sites throughout the PrE differentiation process of F9 cells enabled us to address this issue by comparing molecular features associated with genomic loci occupied in undifferentiated versus differentiated cells. Notably, genomic loci exhibiting a maximum occupancy level in the early phase of the RA-induced PrE differentiation process are best characterized by an increased relative prevalence of the DR0 motif often associated with a significant enrichment in pluripotency-associated transcription factors binding sites. By contrast, genomic loci reaching their maximum occupancy in the late phase of the differentiation process are characterized by a decreased prevalence of DR0 motif and an increased relative prevalence of the classical RARE-associated motif DR5, as well as by a depletion in pluripotency-associated transcription factors binding sites and a significant enrichment in the PrE-associated transcription factor SOX17 binding (Figure 5, compare clusters A–D with clusters G–I). While the precise molecular mechanism supporting this observation remains elusive, our data clearly suggest the existence of a cooperative DNA binding mechanism between RAR/RXR and Sox17, as attested by the decreased RAR binding in RA treated Sox17 KD F9 cells (Figure 5C). Nevertheless this mechanism is clearly not the only one at work, as we also showed that the RAR/RXR binding site located downstream of the Cyp26a1 locus displays a late RAR/RXR occupancy profile and is Sox17-independent (Figures 2E–H and 5C left panel).
Furthermore, our integrated cistromic/transcriptomic analysis provides original observations on the transcriptional consequences of RAR/RXR relocation upon RA stimulation of F9 cells. Our results indeed suggest that RAR/RXR activation globally counteracts core pluripotency factors action in pluripotent cells and cooperates with differentiation-associated transcription factors to repress pluripotency and induce PrE differentiation (Figure 7). The elucidation of the molecular mechanisms supporting these observations will require the acquisition of dynamic binding data for pluripotency- and differentiation-associated transcription factors throughout the differentiation process to precisely identify the sequence of events.
More broadly, our study provides important hints for the understanding of the cell or tissue specific response to RA stimulation by strongly suggesting that RAR/RXR binding site selection and consequently RA-induced gene specific expression modulation are closely associated with the binding of master regulators that define the cellular identity.
As hypothesized, our results support the idea that the interaction with DR0-containing regions is favored in undifferentiated pluripotent embryonic cells and that the differentiation process is accompanied by a drastic reorganization of RAR/RXR binding repertoire. They also raise the questions of the role of DR0 motifs in regulatory region of pluripotency-associated genes and of the molecular mechanisms supporting the relocation of RAR/RXR binding sites from DR0- to DR5-enriched regions.
Notably, DR0 motif is the known binding motif for several nuclear receptors that are modulated during RA-induced PrE differentiation, namely the orphan nuclear receptors GCNF and COUP-TFI and II (75). All three receptors are indeed induced upon RA stimulation; most likely through the direct action of RAR/RXR heterodimers as they all exhibit RAR/RXR binding in the vicinity of their respective TSS (Supplementary Figure S11 and Supplementary Table S3). They are also known as important factors in driving pluripotent cells toward differentiation by repressing expression of the pluripotency-associated genes Pou5f1 and Nanog (76–80) through an interaction with DR0-containing regulatory elements. The detailed analysis of Pou5f1 proximal promoter region demonstrated that this region indeed encompasses a composite HRE formed by the overlap of a DR1 and a DR0 motif, and recognized by RAR/RXR, ESRRB (26), NR5A2 (27), COUP-TFs (76) and GCNF (78) (Supplementary Figure S14). The RA-induced increased expression of COUP-TFs and GCNF was shown to displace RAR/RXR heterodimers and to induce the transcriptional repression of Pou5f1 expression. Importantly, active repression was followed by epigenetic silencing through the GCNF-dependent recruitment of the de novo methyltransferases DNMT3A-B and active de novo methylation of Pou5f1 proximal promoter (81,82). Notably, COUP-TF1 was also shown to be able to recruit DNMT3A-B, although in another cell system (83).
The described molecular mechanism supporting Pou5f1 down-regulation and RAR/RXR release from its proximal promoter region provides a seducing explanation for the observed decrease of DR0 prevalence in lately occupied regions. The analysis of the binding site repertoire of GCNF and COUP-TFs orphan receptors in differentiating pluripotent cells will be required to further substantiate this hypothesis. However, our results and previous studies cited above highlight the pivotal role of DR0 motif in nuclear receptor-associated pluripotent cell decision process to differentiate or to maintain undifferentiated pluripotent state. Indeed, all nuclear receptor superfamily members involved in pluripotency maintenance and somatic cell reprogramming, or exit from pluripotent state and differentiation, are all able to interact with either DR0 motif or single monomeric core encompassing a 5′ TCA extension also referred to as SF1 response element (SFRE). Whether the capacity of RAR/RXR heterodimers to participate in somatic cell reprogramming process (9) rely on their ability to interact with DR0 remains to be formally demonstrated but may provide a tempting although partial explanation of this paradoxical observation.
CONCLUSION
The data we collected in this work provide a picture of unprecedented detail of RAR/RXR location throughout primitive endoderm differentiation. Our data confirm the previously observed ligand-induced RAR subunit occupancy increase but highlight the occupancy of RAR/RXR binding sites primarily by non-RARs/RXRs dimers. Our study also demonstrates the redistribution of RAR/RXR binding sites during the RA-induced PrE differentiation process. This observation enables us to propose a coherent scenario that explains the transition between undifferentiated cell types (where RAR/RXR binds predominantly to DR0) and differentiated cell types, where RAR is known to bind to canonical (DR1, DR2, DR5) elements. Altogether, our data show that the selection of RAR/RXR binding sites and the control of their occupancy level integrate several mechanisms and are associated with the expression and binding of master transcription factors that define cell identity.
DATA ACCESS
The data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession No. GSE56893.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank B. Jost, C. Keime and the members of the IGBMC (Strasbourg, France) sequencing platform for ChIP sequencing library preparation and Solexa sequencing; J. Lachuer, F. Barbet and the members of the ProfileXpert sequencing platform (Lyon, France) for RNA-seq libraries sequencing and J. Moore (Fluidigm Corporation, South San Francisco, California 94080, USA) for technical help with DE genes RT-qPCR validation. We also would like to thank I. Masse for establishing RAR and RXR ChIP protocol and M. Paris, B. Boussau, O. Gandrillon, S. Gonin-Giraud, F. Flamant for critical reading of the manuscript and fruitful discussions. This work was performed using the computing facilities of the CC LBBE/PRABI.
Footnotes
Present address: Amandine Chatagnon, Plateforme de Biologie Moléculaire, IBP, CHU Grenoble, 38043 Grenoble, France.
Former address: Amandine Chatagnon, Philippe Veber, Gérard Triqueneaux, and Gérard Benoit, IGFL, Université de Lyon, Université Lyon 1, CNRS, INRA; Ecole Normale Supérieure de Lyon, 69007 Lyon, France.
FUNDING
P.V. was supported by a fellowship of the Fondation pour la Recherche Médicale. Agence Nationale pour la Recherche [ANR-09-SYSC-009–02 to G.B. and F.A.B., ANR-2011-BSV8–023–02 to G.B.]; Institut National du Cancer [PLBIO08–083 to G.B.). Funding for open access charge: Centre National de la Recherche Scientifique.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mark M., Ghyselinck N.B., Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006;46:451–480. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- 2.Sabella J.D., Bern H.A., Kahn R.H. Effects of locally applied vitamin A and estrogen on rat epidermis. Proc. Soc. Exp. Biol. Med. 1951;76:499–503. doi: 10.3181/00379727-76-18537. [DOI] [PubMed] [Google Scholar]
- 3.Wilson E.L., Reich E. Plasminogen activator in chick fibroblasts: induction of synthesis by retinoic acid; synergism with viral transformation and phorbol ester. Cell. 1978;15:385–392. doi: 10.1016/0092-8674(78)90007-7. [DOI] [PubMed] [Google Scholar]
- 4.Wolbach S.B., Howe P.R. Tissue changes following deprivation of fat-soluble a vitamin. J. Exp. Med. 1925;42:753–777. doi: 10.1084/jem.42.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuspa S.H., Harris C.C. Altered differentiation of mouse epidermal cells treated with retinyl acetate in vitro. Exp. Cell Res. 1974;86:95–105. doi: 10.1016/0014-4827(74)90653-3. [DOI] [PubMed] [Google Scholar]
- 6.Mummery C.L., Feyen A., Freund E., Shen S. Characteristics of embryonic stem cell differentiation: a comparison with two embryonal carcinoma cell lines. Cell Differ. Dev. 1990;30:195–206. doi: 10.1016/0922-3371(90)90139-n. [DOI] [PubMed] [Google Scholar]
- 7.Ivanova N., Dobrin R., Lu R., Kotenko I., Levorse J., DeCoste C., Schafer X., Lun Y., Lemischka I.R. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Wang W., Yang J., Liu H., Lu D., Chen X., Zenonos Z., Campos L.S., Rad R., Guo G., Zhang S., et al. Rapid and efficient reprogramming of somatic cells to induced pluripotent stem cells by retinoic acid receptor gamma and liver receptor homolog 1. Proc. Natl. Acad. Sci. U.S.A. 2011;108:18283–18288. doi: 10.1073/pnas.1100893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mader S., Leroy P., Chen J.Y., Chambon P. Multiple parameters control the selectivity of nuclear receptors for their response elements. Selectivity and promiscuity in response element recognition by retinoic acid receptors and retinoid X receptors. J. Biol. Chem. 1993;268:591–600. [PubMed] [Google Scholar]
- 11.Perlmann T., Rangarajan P.N., Umesono K., Evans R.M. Determinants for selective RAR and TR recognition of direct repeat HREs. Genes Dev. 1993;7:1411–1422. doi: 10.1101/gad.7.7b.1411. [DOI] [PubMed] [Google Scholar]
- 12.Umesono K., Evans R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57:1139–1146. doi: 10.1016/0092-8674(89)90051-2. [DOI] [PubMed] [Google Scholar]
- 13.Evans R.M., Mangelsdorf D.J. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangelsdorf D.J., Thummel C., Beato M., Herrlich P., Schutz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balmer J.E., Blomhoff R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J. Steroid Biochem. Mol. Biol. 2005;96:347–354. doi: 10.1016/j.jsbmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Umesono K., Murakami K.K., Thompson C.C., Evans R.M. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang Y.S., Cho J.Y., Cho H.A., Kim H.J., Chang J., Ahn C.M., Kim S.K. 9-cis retinoic acid induces insulin-like growth factor binding protein-3 through DR-8 retinoic acid responsive elements. Cancer Biol. Ther. 2006;5:586–592. doi: 10.4161/cbt.5.6.2658. [DOI] [PubMed] [Google Scholar]
- 18.Fujisawa K., Umesono K., Kikawa Y., Shigematsu Y., Taketo A., Mayumi M., Inuzuka M. Identification of a response element for vitamin D3 and retinoic acid in the promoter region of the human fructose-1,6-bisphosphatase gene. J. Biochem. 2000;127:373–382. doi: 10.1093/oxfordjournals.jbchem.a022618. [DOI] [PubMed] [Google Scholar]
- 19.Carter M.E., Gulick T., Moore D.D., Kelly D.P. A pleiotropic element in the medium-chain acyl coenzyme A dehydrogenase gene promoter mediates transcriptional regulation by multiple nuclear receptor transcription factors and defines novel receptor-DNA binding motifs. Mol. Cell. Biol. 1994;14:4360–4372. doi: 10.1128/mcb.14.7.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee C.H., Wei L.N. Characterization of an inverted repeat with a zero spacer (IR0)-type retinoic acid response element from the mouse nuclear orphan receptor TR2–11 gene. Biochemistry. 1999;38:8820–8825. doi: 10.1021/bi9903547. [DOI] [PubMed] [Google Scholar]
- 21.Hua S., Kittler R., White K.P. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–1271. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahony S., Mazzoni E.O., McCuine S., Young R.A., Wichterle H., Gifford D.K. Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol. 2011;12:R2. doi: 10.1186/gb-2011-12-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martens J.H., Brinkman A.B., Simmer F., Francoijs K.J., Nebbioso A., Ferrara F., Altucci L., Stunnenberg H.G. PML-RARalpha/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell. 2010;17:173–185. doi: 10.1016/j.ccr.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 24.Mendoza-Parra M.A., Walia M., Sankar M., Gronemeyer H. Dissecting the retinoid-induced differentiation of F9 embryonal stem cells by integrative genomics. Mol. Syst. Biol. 2011;7:538. doi: 10.1038/msb.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moutier E., Ye T., Choukrallah M.A., Urban S., Osz J., Chatagnon A., Delacroix L., Langer D., Rochel N., Moras D., et al. Retinoic acid receptors recognize the mouse genome through binding elements with diverse spacing and topology. J. Biol. Chem. 2012;287:26328–26341. doi: 10.1074/jbc.M112.361790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J., et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 27.Heng J.C., Feng B., Han J., Jiang J., Kraus P., Ng J.H., Orlov Y.L., Huss M., Yang L., Lufkin T., et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell. Stem Cell. 2010;6:167–174. doi: 10.1016/j.stem.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Kashyap V., Gudas L.J. Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J. Biol. Chem. 2010;285:14534–14548. doi: 10.1074/jbc.M110.115345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowalczyk A., Bedo J., Conway T., Beresford-Smith B. The poisson margin test for normalization-free significance analysis of NGS data. J. Comput. Biol. 2011;18:391–400. doi: 10.1089/cmb.2010.0272. [DOI] [PubMed] [Google Scholar]
- 33.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Hoon M.J., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 36.Han J., Yuan P., Yang H., Zhang J., Soh B.S., Li P., Lim S.L., Cao S., Tay J., Orlov Y.L., et al. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature. 2010;463:1096–1100. doi: 10.1038/nature08735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J., et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu C., Lu X., Chen E.Z., He Z., Uyunbilig B., Li G., Ma Y., Hui L., Xie B., Gao Y., et al. Genome-wide roles of Foxa2 in directing liver specification. J. Mol. Cell. Biol. 2012;4:420–422. doi: 10.1093/jmcb/mjs037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaji M., Ueda J., Hayashi K., Ohta H., Yabuta Y., Kurimoto K., Nakato R., Yamada Y., Shirahige K., Saitou M. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell. Stem Cell. 2013;12:368–382. doi: 10.1016/j.stem.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Aksoy I., Jauch R., Chen J., Dyla M., Divakar U., Bogu G.K., Teo R., Leng Ng C.K., Herath W., Lili S., et al. Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J. 2013;32:938–953. doi: 10.1038/emboj.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acland A., Agarwala R., Barrett T., Beck J., Benson D.A., Bollin C., Bolton E., Bryant S.H., Canese K., Church D.M., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giresi P.G., Lieb J.D. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S., Pyl P.T., Huber W. HTSeq — A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rambaud J., Triqueneaux G., Masse I., Staels B., Laudet V., Benoit G. Rev-erbalpha2 mRNA encodes a stable protein with a potential role in circadian clock regulation. Mol. Endocrinol. 2009;23:630–639. doi: 10.1210/me.2008-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji X., Li W., Song J., Wei L., Liu X.S. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 2006;34:W551–W554. doi: 10.1093/nar/gkl322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lalevee S., Anno Y.N., Chatagnon A., Samarut E., Poch O., Laudet V., Benoit G., Lecompte O., Rochette-Egly C. Genome-wide in silico identification of new conserved and functional retinoic acid receptor response elements (direct repeats separated by 5 bp) J. Biol. Chem. 2011;286:33322–33334. doi: 10.1074/jbc.M111.263681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margulies E.H., Blanchette M., Haussler D., Green E.D. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bour G., Plassat J.L., Bauer A., Lalevee S., Rochette-Egly C. Vinexin beta interacts with the non-phosphorylated AF-1 domain of retinoid receptor gamma (RARgamma) and represses RARgamma-mediated transcription. J. Biol. Chem. 2005;280:17027–17037. doi: 10.1074/jbc.M501344200. [DOI] [PubMed] [Google Scholar]
- 51.Gaillard E., Bruck N., Brelivet Y., Bour G., Lalevee S., Bauer A., Poch O., Moras D., Rochette-Egly C. Phosphorylation by PKA potentiates retinoic acid receptor alpha activity by means of increasing interaction with and phosphorylation by cyclin H/cdk7. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9548–9553. doi: 10.1073/pnas.0509717103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lalevee S., Bour G., Quinternet M., Samarut E., Kessler P., Vitorino M., Bruck N., Delsuc M.A., Vonesch J.L., Kieffer B., et al. Vinexinß, an atypical ‘sensor’ of retinoic acid receptor gamma signaling: union and sequestration, separation, and phosphorylation. FASEB J. 2010;24:4523–4534. doi: 10.1096/fj.10-160572. [DOI] [PubMed] [Google Scholar]
- 53.Samarut E., Amal I., Markov G.V., Stote R., Dejaegere A., Laudet V., Rochette-Egly C. Evolution of nuclear retinoic acid receptor alpha (RARalpha) phosphorylation sites. Serine gain provides fine-tuned regulation. Mol. Biol. Evol. 2011;28:2125–2137. doi: 10.1093/molbev/msr035. [DOI] [PubMed] [Google Scholar]
- 54.de The H., Marchio A., Tiollais P., Dejean A. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J. 1989;8:429–433. doi: 10.1002/j.1460-2075.1989.tb03394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de The H., Vivanco-Ruiz M.M., Tiollais P., Stunnenberg H., Dejean A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature. 1990;343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- 56.Sucov H.M., Murakami K.K., Evans R.M. Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5392–5396. doi: 10.1073/pnas.87.14.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faria T.N., Mendelsohn C., Chambon P., Gudas L.J. The targeted disruption of both alleles of RARbeta(2) in F9 cells results in the loss of retinoic acid-associated growth arrest. J. Biol. Chem. 1999;274:26783–26788. doi: 10.1074/jbc.274.38.26783. [DOI] [PubMed] [Google Scholar]
- 58.Bruck N., Vitoux D., Ferry C., Duong V., Bauer A., de The H., Rochette-Egly C. A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARalpha to target promoters. EMBO J. 2009;28:34–47. doi: 10.1038/emboj.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machanick P., Bailey T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cotnoir-White D., Laperriere D., Mader S. Evolution of the repertoire of nuclear receptor binding sites in genomes. Mol. Cell. Endocrinol. 2011;334:76–82. doi: 10.1016/j.mce.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 62.Aksoy I., Jauch R., Eras V., Chng W.B., Chen J., Divakar U., Ng C.K., Kolatkar P.R., Stanton L.W. Sox transcription factors require selective interactions with oct4 and specific transactivation functions to mediate reprogramming. Stem Cells. 2013;31:2632–2646. doi: 10.1002/stem.1522. [DOI] [PubMed] [Google Scholar]
- 63.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niwa H., Burdon T., Chambers I., Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ying Q.L., Nichols J., Chambers I., Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 66.Okita K., Yamanaka S. Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr. Stem. Cell. Res. Ther. 2006;1:103–111. doi: 10.2174/157488806775269061. [DOI] [PubMed] [Google Scholar]
- 67.Samarut E., Rochette-Egly C. Nuclear retinoic acid receptors: conductors of the retinoic acid symphony during development. Mol. Cell. Endocrinol. 2012;348:348–360. doi: 10.1016/j.mce.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 68.Eifert C., Sangster-Guity N., Yu L.M., Chittur S.V., Perez A.V., Tine J.A., McCormick P.J. Global gene expression profiles associated with retinoic acid-induced differentiation of embryonal carcinoma cells. Mol. Reprod. Dev. 2006;73:796–824. doi: 10.1002/mrd.20444. [DOI] [PubMed] [Google Scholar]
- 69.Ouyang Z., Zhou Q., Wong W.H. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2009;106:21521–21526. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frum T., Halbisen M.A., Wang C., Amiri H., Robson P., Ralston A. Oct4 cell-autonomously promotes primitive endoderm development in the mouse blastocyst. Dev. Cell. 2013;25:610–622. doi: 10.1016/j.devcel.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiba H., Clifford J., Metzger D., Chambon P. Specific and redundant functions of retinoid X Receptor/Retinoic acid receptor heterodimers in differentiation, proliferation, and apoptosis of F9 embryonal carcinoma cells. J. Cell Biol. 1997;139:735–747. doi: 10.1083/jcb.139.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chiba H., Clifford J., Metzger D., Chambon P. Distinct retinoid X receptor-retinoic acid receptor heterodimers are differentially involved in the control of expression of retinoid target genes in F9 embryonal carcinoma cells. Mol. Cell. Biol. 1997;17:3013–3020. doi: 10.1128/mcb.17.6.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nielsen R., Pedersen T.A., Hagenbeek D., Moulos P., Siersbaek R., Megens E., Denissov S., Borgesen M., Francoijs K.J., Mandrup S., et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Solomon C., White J.H., Kremer R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J. Clin. Invest. 1999;103:1729–1735. doi: 10.1172/JCI6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cooney A.J., Tsai S.Y., O'Malley B.W., Tsai M.J. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol. Cell. Biol. 1992;12:4153–4163. doi: 10.1128/mcb.12.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ben-Shushan E., Sharir H., Pikarsky E., Bergman Y. A dynamic balance between ARP-1/COUP-TFII, EAR-3/COUP-TFI, and retinoic acid receptor:retinoid X receptor heterodimers regulates Oct-3/4 expression in embryonal carcinoma cells. Mol. Cell. Biol. 1995;15:1034–1048. doi: 10.1128/mcb.15.2.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fuhrmann G., Chung A.C., Jackson K.J., Hummelke G., Baniahmad A., Sutter J., Sylvester I., Scholer H.R., Cooney A.J. Mouse germline restriction of Oct4 expression by germ cell nuclear factor. Dev. Cell. 2001;1:377–387. doi: 10.1016/s1534-5807(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 78.Gu P., LeMenuet D., Chung A.C., Mancini M., Wheeler D.A., Cooney A.J. Orphan nuclear receptor GCNF is required for the repression of pluripotency genes during retinoic acid-induced embryonic stem cell differentiation. Mol. Cell. Biol. 2005;25:8507–8519. doi: 10.1128/MCB.25.19.8507-8519.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 79.Sylvester I., Scholer H.R. Regulation of the Oct-4 gene by nuclear receptors. Nucleic Acids Res. 1994;22:901–911. doi: 10.1093/nar/22.6.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhuang Y., Gudas L.J. Overexpression of COUP-TF1 in murine embryonic stem cells reduces retinoic acid-associated growth arrest and increases extraembryonic endoderm gene expression. Differentiation. 2008;76:760–771. doi: 10.1111/j.1432-0436.2007.00258.x. [DOI] [PubMed] [Google Scholar]
- 81.Gu P., Xu X., Le Menuet D., Chung A.C., Cooney A.J. Differential recruitment of methyl CpG-binding domain factors and DNA methyltransferases by the orphan receptor germ cell nuclear factor initiates the repression and silencing of Oct4. Stem Cells. 2011;29:1041–1051. doi: 10.1002/stem.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sato N., Kondo M., Arai K. The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochem. Biophys. Res. Commun. 2006;344:845–851. doi: 10.1016/j.bbrc.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 83.Gallais R., Demay F., Barath P., Finot L., Jurkowska R., Le Guevel R., Gay F., Jeltsch A., Metivier R., Salbert G. Deoxyribonucleic acid methyl transferases 3a and 3b associate with the nuclear orphan receptor COUP-TFI during gene activation. Mol. Endocrinol. 2007;21:2085–2098. doi: 10.1210/me.2006-0490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.