

Abstract
All viruses require host cell factors to replicate. A large number of host factors have been identified that participate at numerous points of the human immunodeficiency virus 1 (HIV-1) life cycle. Recent evidence supports a role for components of the trans-Golgi network (TGN) in mediating early steps in the HIV-1 life cycle. The Conserved Oligomeric Golgi (COG) complex is a heteroctamer complex that functions in coat protein complex I (COPI)-mediated intra-Golgi retrograde trafficking and plays an important role in the maintenance of Golgi structure and integrity as well as glycosylation enzyme homeostasis. The targeted silencing of components of lobe B of the COG complex, namely COG5, COG6, COG7 and COG8, inhibited HIV-1 replication. This inhibition of HIV-1 replication preceded late reverse transcription (RT) but did not affect viral fusion. Silencing of the COG interacting protein the t-SNARE syntaxin 5, showed a similar defect in late RT product formation, strengthening the role of the TGN in HIV replication.
Keywords: human immunodeficiency virus, Conserved oligomeric Golgi complex, trans-golgi network, HIV-dependency factors
1. Introduction
Retroviruses, like all viruses, rely on host cell factors for successful infection, replication and progeny virion release (Goff, 2007; Goff, 2008). A wide variety of host proteins have been shown to play critical roles at practically every step in the HIV-1 life cycle from viral entry, uncoating, reverse transcription of the viral genome, nuclear translocation, integration, and transcription to mRNA export, translation, viral packaging and budding. Functional genomic screens have greatly expanded the range of host cell factors associated with productive HIV-1 infection of target cells (Brass et al., 2008; Bushman et al., 2009; Konig et al., 2008; Yeung et al., 2009; Zhou et al., 2008). Among the novel associations identified, there was an overrepresentation of host proteins involved in intracellular trafficking (Bushman et al., 2009). This enrichment in host factors connected to intracellular transport pathways were presumably related to the translation and post-translational modification of HIV-1 proteins required for the proper assembly and maturation of infectious progeny virions. In fact, previous studies implicated the RAB9 GTPase, as well as the RAB9-associated protein PIKfyve, (necessary for late-endosome-to-trans-Golgi vesicular transport) and RAB11A (needed for trans-Golgi-to-plasma-membrane transport) as required for progeny virus production (Murray et al., 2005). However, characterization of several components of the trans-Golgi network (TGN) that were identified through siRNA-based genomic screens supported a role for these factors in early events in the HIV-1 life cycle (Brass et al., 2008). Specifically, the targeted silencing of components of the Golgi-associated retrograde protein/vesicular protein sorting fifty-three (GARP/VFT) complex was shown to inhibit HIV-1 replication prior to gag translation (Brass et al., 2008). In Saccharomyces cerevisiae, the organism in which these transport pathways are the most extensively characterized, the GARP/VFT complex is composed of the vacuolar protein sorting (Vps) proteins, Vps51, Vps52, Vps53, and Vps54 (Bonifacino and Hierro, 2011). The GARP/VFT complex is recruited to the TGN through interactions with the Rab-family member, Ypt6 (yeast protein transport-6, known as RAB6 in mammals) and interacts with the SNAREs (Soluble N-ethylmaleimide sensitive fusion protein (NSF) attachment protein receptors): Tlg1 (Syntaxin 6 (STX6)), Tlg2 (STX16), Vti1 (VT1A), or Snc1/2 (no known mammalian homologs) (Bonifacino and Hierro, 2011). The siRNA-mediated silencing of either RAB6 or VPS53 led to a significant inhibition in HIV-1 replication (Brass et al., 2008). This inhibition occurred very early in the HIV-1 life cycle preceding viral reverse transcription.
In addition to components of the GARP/VFT complex, members of an additional multiprotein tethering factor, the conserved oligomeric golgi (COG) complex, were identified in a RNAi-based functional genomic screen to identify host factors whose silencing impairs the replication of HIV-1 (Brass et al., 2008). Biochemical and structural analyses have shown that the COG complex is made up of eight proteins (COG1-8) that are divided into two subcomplexes or lobes (Lees et al., 2010; Miller and Ungar, 2012; Ungar et al., 2002; Willett et al., 2013a; Willett et al., 2013b). Lobe A is composed of COG1, -2, -3 and -4 while lobe B is composed of COG5, -6, -7 and -8 with the two lobes connected through interactions between COGs 1 and 8. The COG complex interacts with a variety of proteins to facilitate the organization and proper targeting of vesicles during intra-Golgi retrograde trafficking, including members of the Rab-family GTPase (eg. RAB1A, -1B, RAB2A, RAB6A and RAB6B), SNAREs (eg. STX5, STX6, and STX16), and tethering factors (eg. golgin-84, SEC6, TMF) (Willett et al., 2013b). The COG complex coordinates retrograde vesicle transport within the Golgi and is required for maintaining the distribution of glycosylation enzymes. Mutations in components of the COG complex lead to impaired retrograde transport from both early and late endosomes (Miller and Ungar, 2012; Sztul and Lupashin, 2009).
While three of the four components of the COG complex lobe A, specifically COG2, COG3 and COG4, were identified as HIV dependency factors (HDFs) in a high content functional genomic screen (Brass et al., 2008), the components of lobe B were not identified in the same study. This could be a result of technical limitations in the screen such as incomplete silencing of these factors due to poor transduction efficiencies or inefficient siRNA sequences. Therefore, we set out to examine if COG lobe B components were needed for HIV-1 replication. The silencing of COG5, COG6, COG7 or COG8 impaired HIV-1 replication, as measured by intracellular p24 staining. These results suggest that the impairment in HIV-1 replication precedes gag translation. Consistent with the phenotype observed with the silencing of RAB6 and VPS53, the silencing of all four COG components results in lower levels of proviral integration and late RT product formation. This suggests that COGs involvement in HIV-1 replication precedes even the reverse transcription of the incoming viral genome. Pseudotyping with the vesicular stomatitis virus (VSV) glycoprotein was unaffected, demonstrating that the inhibition of viral replication was specific for the HIV-1 envelope coated viral particles. Unlike the silencing of RAB6 and VPS53 that resulted in impaired fusion, the silencing of these COG factors had no significant effect on viral fusion. These results support an important role for the COG complex in HIV-1 replication.
2. Methods and Materials
2.1 Cell culture
The following cell lines were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: H9 cells from Dr. Robert Gallo (Mann et al., 1989), P4R5 MAGI from Dr. Nathaniel Landau (Charneau et al., 1994), and HeLa-CD4 (HeLa-T4+) from Dr. Richard Axel (Maddon et al., 1986). Lenti-X 293T cells were from Clontech. All cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 1.6mM L-glutamine, 100 U/ml of penicillin G, 100ug/ml streptomycin, 6mM HEPES and 50uM β-mercaptoethanol. H9 cells were grown in RPMI-1640, with 10% FBS and 100 U/ml of penicillin G, 100ug/ml streptomycin, 6mM HEPES and 50 μM β-mercaptoethanol.
2.2 siRNA sequences and shRNA cloning and expression
The short interfering RNAs and short hairpin RNAs used in this study are listed in Supplemental table 1. siRNAs were transfected into the P4R5 Maji or HeLa-CD4 cells using Oligofectamine™ (Life Technologies). SiRNA sequences are listed in Supplemental Table 1. For the shRNA, oligonucleotides targeting CD4, COG5, COG6, COG7, COG8, STX5 and a control scrambled shRNA were synthesized as 19-nt inverse repeats separated by a 9-nt loop sequence and cloned into the U6 promoter–expressing lentiviral vectorpLL3.7 as described by Rubinson and colleagues (Rubinson et al., 2003). The lentiviral particle were generated by transducing lentiX-293 T cells with the packaging plasmid psPAX2 and the Vesicular stomatitis virus envelope glycoprotein (VSVg) expressing plasmid pCMV-VSV-G (envelope) using Lipofectamine™ 2000 (Invitrogen). Viral supernatants were harvested 48 hours after transfection.
2.3. Viral propagation
The following HIV-1 strains were obtained through the NIH AIDS Reagent Program: the lab adapted HIV-1 strain HIV-1 IIIB (HTLV-IIIB/H9) from Dr. Robert Gallo(Popovic et al., 1984a; Popovic et al., 1984b; Ratner et al., 1985), and the molecular clones NL4-3 from Dr. Malcolm Martin (Adachi et al., 1986), LAI.2 from Dr. Keith Peden (Peden et al., 1991), and 89.6 from Dr. Ronald G. Collman (Collman et al., 1992). HIV-1 IIIB was propagated in the T cell line H9 in RPMI media supplemented with 10% FBS. The virus containing supernatant was harvested by centrifugation (1,500×g for 10 min). The molecular clones Nl4-3, LAI.2, and 89.6 were transfected into lenti-X 293T cells, the virus containing supernatant harvested 48h later. HIV envelope pseudotyped virus were generated by cotransfecting lenti-X 293T cells (Clontech) with pIIIenv3-1 (HIV envelope glyocoprotein), pUltrahot, and psPAX2 with Lipofectamine™ 2000 (Life Technologies) and collecting the viral supernatant after 48 hours. Vesicular stomatitis virus envelope glycoprotein (VSVg) pseudotyped virus were similarly produced using pCMV-VSVg, pUltrahot, and psPAX2.
2.4. HIV infection
The shRNA or siRNA treated cells were infected 72h later with HIV-1 IIIb, NL4-3, LAI.2 and 89.6. The cells were incubated for 48h at 37°C in 5% CO2 before they were harvested and HIV-1 p24 levels were analyzed by intracellular staining for HIV-1 p24 and flow cytometry.
2.5. Flow Cytometry
To assess CD4 receptor, or CCR5 or CXCR4 coreceptor levels, P4-R5 MAGI cells were transfected with siRNAs targeting the COG complex components, CD4 or scramble control siRNAs. After 48h, the cells were harvested with EDTA, washed and then stained with either the PE conjugated mouse monoclonal anti-human CD4, clone M-T477 (BD Pharmingen), FITC conjugated rat monoclonal anti-Human CD195, clone HEK/1/85a (Biolegend), or rabbit polyclonal antibody to CXCR4 (Abcam) and FITC conjugated goat anti-rabbit secondary antibody (Invitrogen) and analyzed by flow cytometry on the Accuri C6 (BD Biosciences). To determine levels of HIV infection in HeLa-CD4 cells, the siRNA or shRNA-treated cells were infected with HIV-1, harvested 48h later, fixed and permeabilized (Caltag fix and perm solutions, BD Biosciences), incubated with mouse anti-HIV-1 p24-PE antibody (KC57-Phycoerythrin (PE), Beckman Coulter) or a mouse isotype matched PE control antibody and analyzed by flow cytometry.
2.6. HIV integration assay
The shRNA treated HeLa-CD4 were harvested 24h post HIV-1 IIIB infection. Using the HIRT protocol (Hirt, 1967), unintegrated viral DNA and mitochondrial DNA were separated from the chromosomal DNA by the preferential precipitation of chromosomal DNA in the presence of SDS and NaCl. The SDS-NaCI precipitated chromosomal faction was dissolved in water, phenol:chloroform extracted and ethanol precipitated. The purified chromosomal DNA was used to examine viral integration by quantitative Alu-PCR. First, the integrated HIV sequence was amplified using primers designed to anneal to conserved site on the Alu repeat element (Alu1: 5′-TCCCAGCTACTGGGGAGGCTGAGG-3′, Alu2: 5′-GCCTCCCAAAGTGCTGGGATTACAG-3′) in conjunction with an HIV LTR primer extended at its 5′ end with a lambda phage-specific heel sequence (L-Fw: 5′-atgccacgtaagcgaaactGAAGCTGCAGAATGGGATAGAG-3′). Secondly, nested PCR using a heel-specific primer (Fw: 5′- ATGCCACGTAAGCGAAACT-3′) and a HIV LTR primer (Rev: 5′-CCGGTCTACATAGTCTCTAAAGGG-3′) was performed on the product of the Alu-PCR to quantify the level of viral integration. The levels of these products were assessed by qRT-PCR (SYBR green PCR master mix, Applied Biosystems) on the ABI 7900 HT fast real time PCR system. Beta-globin primers were used as control primers (B-glo+: 5′-GAAGAGCCAAGGACAGGTAC-3′, B-glo-: 5′-AAGCAATAGATGGCTCTGCC-3′).
2.7. Late Reverse Transcriptase (RT) assay
The shRNA treated Hela T4 cells were harvested 7h post HIV-1 IIIB infection. Using the same HIRT protocol described in section 2.6 (Hirt, 1967), unintegrated viral DNA and mitochondrial DNA were separated from the chromosomal DNA and purified from the supernatant by ethanol precipitation. Once purified, the extrachromosomal DNA was analyzed for late RT products using primers that amplify the region of the HIV cDNA between the left LTR sequence and the 5′ end of the gag gene (MH531: 5′-TGTGTGCCCGTCTGTTGTGT-3′, MH532: 5′-GAGTCCTGCGTCGAGAGAGC-3′) (Yoder and Fishel, 2008). Primers amplifying part of the mitochondrial genome were used as an internal control (Mit+: 5′-GACGTTAGGTCAAGGTGTAG-3′, Mit-: 5′-CAACTAAGCACTCTACTCTC-3′) (Butler et al., 2001).
2.8 Viral Fusion Assay
The P4R5 MAGI cells were transduced with the appropriate siRNAs and 48h later the cells were incubated with HIV-1 virus containing the chimeric protein beta-lactamase-Vpr (BlaM-Vpr) at 37°C for 3 hours (Cavrois et al., 2011). The cells were washed with CO2-independent DMEM (GIBCO) and loaded with CCF2-AM dye according to the manufacturer’s protocols (Life Technologies). After 2 hours of incubation at room temperature, the cells were washed with PBS, harvested with trypsin, and fixed for flow cytometry analysis.
2.9 Western blot
SiRNA-treated cells were harvested and lysed using RIPA buffer (Thermo scientific) supplemented with Protease Inhibitor cocktail (Sigma). Protein concentrations were determined by Pierce BCA Protein Assay (Thermo scientific). Equal amounts of protein (15ug per lane) were boiled with 4X LDS sample loading buffer and 10X DTT, loaded on a NuPAGE Novex 4–12% Bis-Tris Protein gel and transferred to a nitrocellulose membrane using Bio-Rad Wet/Tank Blotting Systems. The membrane was blocked in I-block™ protein-based blocking reagent (Applied Biosystems) and then incubated with primary antibodies to COG3 (kindly provided by Dr. Vladimir Lupashin, University of Arkansas), COG4 (kindly provided by Dr. Lupashin), or β-tubulin (Invitrogen). The membrane was incubated with horseradish peroxidase (HRP) conjugated anti-mouse (Invitrogen) or rabbit (Santa Cruz Biotechnology) secondary antibodies and the blot was developed with Pierce ECL western blotting reagent (Thermo Scientific) and viewed by FluorChem™ gel viewer system. Quantitation of the appropriate bands was performed using the Image J software. All values were normalized to β-tubulin and made relative to the control siRNA treated sample.
3. Results
3.1 Targeted silencing of components of the COG complex Lobe B inhibits HIV-1 replication
Lobe B of the COG complex is a heterotetramer composed of COG5, COG6, COG7, and COG8which is linked to the COG complex Lobe A through interactions between COG1 and COG8 (Figure 1 A). High content functional genomic screening has linked components of lobe A of the COG complex with HIV-1 replication (Brass et al., 2008). To determine whether components of -lobe B are required for HIV-1 replication, siRNAs targeting each of the 4 lobe B components (COG5, COG6, COG7, or COG8) were transfected into P4R5 HeLa cells that were infected with HIV-1 IIIb 48 hours later. The targeted silencing of lobe B factors decreased the corresponding target gene expression level by 70 to 90% compared to control siRNA treated cells (Figure 1B). Cells treated with siRNAs against all four COG lobe B components significantly inhibited HIV-1 replication compared to control siRNA treated cells as measured by intracellular staining for HIV-1 gag (p24) expression (Figure 1C–D). The stable silencing of the four lobe B components using lentiviral delivered short hairpin (sh)RNAs showed a similar inhibition of HIV-1 replication (Supplemental Figure 1). The COG-specific shRNAs targeted regions distinct from those recognized by the siRNAs. This provides additional evidence that it is the targeting of the COG lobe B components that is resulting in the inhibition of HIV-1 replication and not a non-specific off-target effect of the siRNAs.
Figure 1.
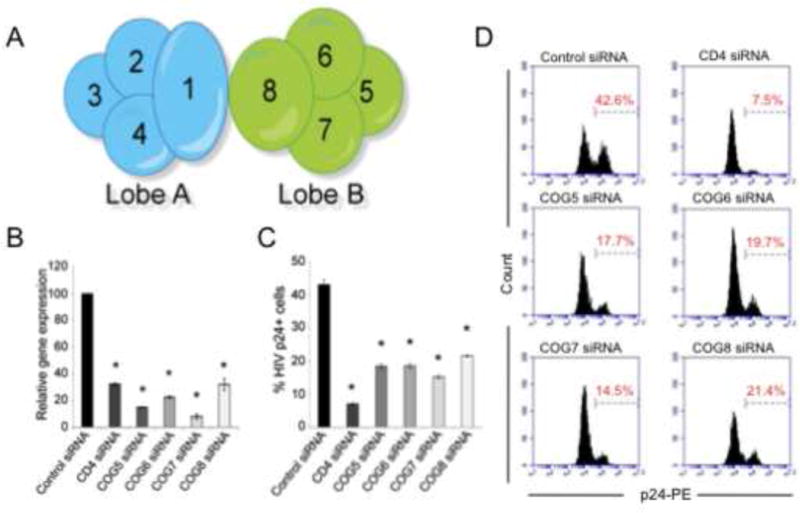
RNAi-mediated silencing of components of the COG complex, COG5, -6, -7 or -8 impairs HIV-1 replication. (A) Schematic representation of the components of the COG complex that are divided into two lobes (lobe A and B) linked through interactions with COGs 1 and 8. (B) P4R5 MAJI cells were transduced with siRNAs targeting COG5, -6, -7, or -8, CD4 or a non-specific control sequence (control siRNA). The silencing of the cognate target genes was measured by quantitative real time PCR (N=3, *p<0.002, two-tailed t-test) relative to expression levels in the control siRNA treated cells. All values were normalized to GAPDH expression. (B and C) The silencing of COG5, -6, -7 or -8 significantly inhibited HIV-1 gag expression compared to the control siRNA-treated cells. The siRNA treated cells were infected with HIV-1 IIIB and the intracellular level of HIV-1 gag (p24) was measured 48 hours later. Shown are the averaged inhibition of p24 expression (±Standard deviation (S.D.)) normalized to the Control siRNA treated cells (C) (N=3, *p<0.002, two-tailed t-test) and representative histograms (D).
To ensure that the inhibition of HIV-1 replication upon the silencing of components of the COG complex lobe B was not limited to the lab-adapted HIV-1 IIIB, the siRNA-treated cells were challenged with the T cell tropic molecular clone HIV-1 NL4-3 or LAI.2 or the dual-tropic HIV-1 clone 89.6. Consistent with the results seen with the HIV-1 IIIB, the silencing of COG5, COG6, COG7, or COG8 significantly impaired the replication of all three HIV clones (Figure 2A–C). Therefore, the inhibition of HIV-1 replication in cells silenced for components of lobe B of the COG complex is not limited to HIV-1 IIIB infection but impairs the replication of additional viral strains.
Figure 2.
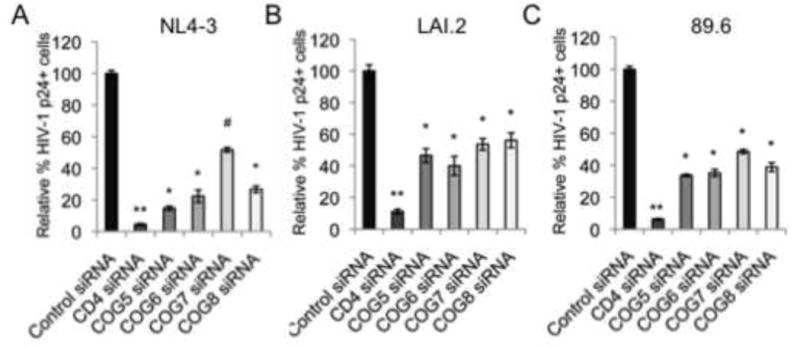
COG5, -6, -7 or -8 silencing impaired replication of multiple strains of HIV-1. The siRNA-mediated silencing of COG5, -6, -7 or -8 impaired the replication of HIV-1 NL4-3 (A), HIV-1 LAI.2 (B) and HIV-1 89.6 (C) as measured by intracellular staining for HIV-1 p24 levels and flow cytometry. Shown are the averaged % inhibition of p24 expression (± S.D.) normalized to control siRNA treated cells (N=3, all p<0.01, two-tailed t-test).
Previous studies have shown that the silencing of a component of the COG complex can alter the level of other COG proteins. For example, Laufman and colleagues showed that silencing of COG8 in HeLa cells resulted in a decrease in the steady state levels of COG1 (~80% decrease) and, to a lesser degree, COG6 (~30% decrease) (Laufman et al., 2013). In addition, the silencing of COG8 had no significant effect on COG5 and COG7 expression and a modest effect on COG2 and COG3 expression (~15–20% decrease compared to the ~80% decrease in COG1) with no effect on COG4 expression (Laufman et al., 2013). Although this study showed that COG8 silencing disrupted the Golgi localization of the COG lobe B, the components of COG lobe A did not show any evidence of mislocalization. The depletion of COG6 resulted in decreases in the lobe B components (~50% for COG5 and COG8 and ~60% for COG7), as well as decreases in the steady state level of COG lobe A components (ranging from 50% decrease in COG1 to an ~30% decrease in COG2) (Laufman et al., 2013). Given that three components of the COG complex lobe A, (COG2, COG3, and COG4) were identified as HDFs through an RNAi-based screen for host genes required for HIV-1 replication, we wanted to ensure that silencing COG lobe B components did not lead to a decrease in COG lobe A components which, in turn, could lead to the inhibition of HIV-1 replication. To that end, immunoblot analysis was performed to examine the levels of COG3 and COG4 proteins in the siRNA-treated cells (Supplemental Figure 2). As expected, the direct silencing of COG3 and COG4 by siRNAs significantly decreased the levels of their respective proteins within the cell. However, the treatment of the P4R5 MAJI cells with siRNAs targeting COG5, COG6, COG7 or COG8 showed no decrease in COG3 expression (even a 20% increase in the COG7 and COG8 siRNA-treated cells). Similarly, there was no significant effect on COG4 expression in the COG5 siRNA treated cells and only a modest decrease (10%) for COG6, COG7 or COG8 siRNA treated cells. Based on these results, the treatment of cells with siRNAs targeting lobe B components doesn’t appear to lead to a significant decrease in the steady state levels of lobe A components. It appears that the inhibition of HIV-1 replication cannot merely be attributed to indirect consequences of lobe A regulation.
3.2. COGs5-8 are required for steps in the HIV-1 life cycle that precede reverse transcription of the viral genome
The reduction in intracellular staining of HIV-1 p24 indicates that the silencing of lobe B components inhibit one of the viral life cycle steps preceding gag translation. To further delineate the impact that decreased COG5-8 expression has on HIV-1 replication, the siRNA treated and HIV-1 IIIB infected cells were analyzed for the levels of different forms of the HIV-1 genome. To determine if the targeted silencing impairs viral integration, COG5-8 siRNA treated cells were infected with HIV-1 IIIB, genomic DNA purified and quantitative real time PCR (qRT-PCR) was performed using a nested PCR strategy (Butler et al., 2001). This involves a first round of PCR using a primer specific to Alu elements – a family of repetitive elements found dispersed throughout the human genome – and a primer specific to HIV-1. In the second round of PCR, a pair of HIV-1 specific primers is used whose product is contained within the region of the HIV-1 genome amplified in the first round of PCR. The silencing of each of the components showed approximately 40% decrease in the amounts of integrated proviral DNA compared to the control siRNA treated cells (Figure 3A). The silencing of the HIV-1 co-receptor CD4 showed a significant inhibition of HIV-1 integration surpassing that seen with the silencing of the COG complex (~60%). The inhibition of HIV-1 integration strongly correlated with the level of inhibition of HIV-1 replication measured by intracellular staining for HIV-1 p24 levels and flow cytometric analysis (Figure 1C–D).
Figure 3.
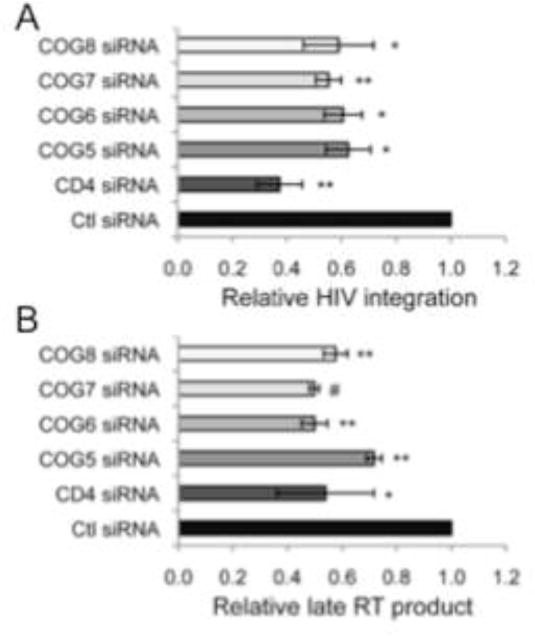
Silencing of COG5, -6, -7 or -8 inhibits HIV-1 replication prior to HIV-1 integration. A) COG5, -6, -7 or -8 depletion blocks HIV prior to viral integration. P4R5 HeLa cells transduced with the indicated shRNA were infected with HIV-1 IIIB and the level of the integrated HIV-1 genome was assessed using a nested PCR strategy. Relative integration indicates percent normalized to control siRNA treated cells (±S.D.) (N=3, *p<0.05, **p<0.01, two-tailed t-test). All values were normalized to β-globin. B) Silencing COG5, -6, -7 or -8 resulted in decreased levels of late RT product formation following HIV-1 IIIB infection. P4R5 HeLa cells treated as indicated were infected with HIV-1 IIIB and 8h later the non-chromosomal DNA was segregated from the chromosomal DNA using a selective salt precipitation. The level HIV-1 late RT product was measured using HIV-1 late RT-specific primers. Percent late RT product indicates percent versus control siRNA treated cells (±S.D.) (N=3, *p<0.05, **p<0.01, #p<0.001, two-tailed t-test). All values were normalized to mitochondrial DNA
Upon release of the viral core into newly infected target cells, the incoming HIV genomic RNA is rapidly reverse transcribed to yield linear viral cDNAs. The amount of this linearized viral cDNA can be measured by partitioning the viral cDNA from the host chromosomal DNA using selective precipitation of the chromosomal and non-chromosomal DNA fractions composed of mitochondrial DNA and viral cDNA (Hirt, 1967). The amount of viral cDNA can be measured in the non-chromosomal DNA fraction using an HIV-1-specific primer pair. Similar to the impairment in integrated viral genome, silencing of COG5-8 decreased the amount of HIV-1 late RT product compared to control siRNA treated cells (Figure 3B). The decrease in late RT product seen in the COG5-8 silenced cells was similar to that found in CD4 siRNA treated cells. Taken together, these results suggest that the components of the COG complex lobe B are required for productive HIV-1 infection and play a role in early events in the viral life cycle.
3.3 Silencing of the COG complex has no effect on surface expression of the HIV-1 receptor CD4, nor the co-receptors CXCR4 or CCR5
The early identification of components of the COG complex was facilitated by screening for CHO cells defective in LDL (low density lipoprotein) receptor activity (Chatterton et al., 1999; Krieger et al., 1981; Podos et al., 1994). This led to the identification of the ldlB and ldlC genes later shown to encode the COG components COG1 and COG2 (Chatterton et al., 1999; Miller and Ungar, 2012; Podos et al., 1994). HIV-1 infection is dependent on the viral envelope glycoprotein (gp120) recognizing of the CD4 cell surface receptor and either the CXCR4 or CCR5 chemokine receptor (HIV-1 co-receptor) on HIV-1 susceptible cells (Barre-Sinoussi et al., 2013). To determine if modulating the levels of COG lobe B components has an effect on the expression levels of the HIV-1 receptor or co-receptors, siRNA treated cells were stained for CD4, CXCR4 or CCR5 and analyzed by flow cytometry. As expected, the CD4 siRNA effectively silenced CD4 surface expression (Figure 4A–B). However, the silencing of lobe B components had no significant effect on the cell surface expression of CD4 as assessed by the percentage of CD4 expressing cells in the culture (Figure 4A) or the mean fluorescence intensity (MFI). Although no significant difference was seen in the percentage of cells expressing the CXCR4 co-receptor, there was a statistically significant but modest decrease in the CXCR4 MFI in the COG5 siRNA treated cells and a slight increase in COG7 and COG8 silenced cells (Figure 4C and D). No difference was seen in the percentage of cells expressing CCR5 on the surface of the COG lobe B silenced cells (Figure 4 E). However, a statistically significant but modest (~20%) decrease in CCR5 MFI was seen in the cells in which COG8 was silenced (Figure 4F). Since the silencing of the COG lobe B components showed, at best, a modest decrease in the surface expression of CD4, CXCR4 or CCR5, it is highly unlikely that these modest changes would be sufficient to account for the level of inhibition of HIV-1 replication seen in Figure 1 and 2. Therefore, the inhibition of HIV-1 infectivity seen upon silencing of the components of COG5-8 does not appear to be a result of altered CD4, CXCR4 or CCR5 expression levels on the HIV-1 susceptible cells.
Figure 4.
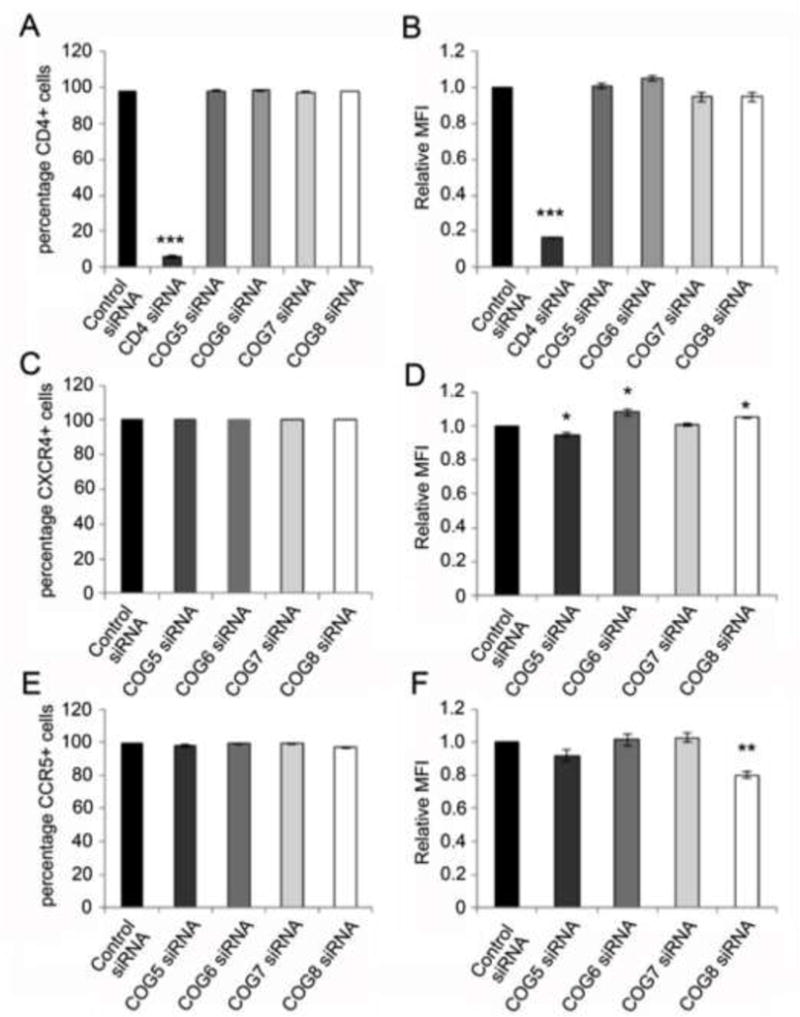
COG5, -6, -7 or -8 silencing had no or a modest effect on CD4, CXCR4 or CCR5 expression levels. The knockdown of the lobe B components had no effect on CD4 expression as measured by the percentage of CD4 expressing cells (A) or the mean fluorescence intensity (MFI) of CD4 expression (B). Although the silencing of COG5-8 had no effect on the percentage of cells staining positive for surface expression of CXCR4 (C), there was a modest decrease in the MFI of CXCR4 stained cells upon COG5 silencing and a modest increase in CXCR4 MFI in the COG8 siRNA treated cells (D). In addition, the percentage of cells staining positively for CCR5 (E) was not changed upon silencing of lobe B components but there was a modest, but statistically significant, decrease in the MFI of CCR5 in the COG8 silenced cells (F). Graphs represents the average of three experiments (± S.D.) (*p<0.05, **p<0.01, two-tailed t-test).
3.4 The defect in viral replication in COG5-8 silenced cells is dependent on the HIV-1 envelope
To determine if COG5-8 silencing causes a block in viral entry, the silenced cells were infected with a recombinant HIV-1 strain expressing the fluorescent protein mcherry (pUltrahot) and pseudotyped with either the vesicular stomatitis virus G (VSVg) envelope glycoprotein or the HIV-1 envelope (pIIIenv3-1). The VSVg envelope pseudotyped virus was only modestly inhibited by the silencing of COG5 and COG6 (~20%) with no significant effect on COG7 and COG8 silenced cells (Figure 5). On the other hand, viral particles packaged with the HIV-1 envelope were greatly inhibited by all 4 lobe B components (40–60% inhibition, Figure 5). Although there is a modest, yet statistically significant, impairment of HIV-1 replication in the VSVg-pseudotyped virus in the COG5 or COG6 silenced cells, the inhibition of replication by the HIV-1 envelope coated virus was significantly stronger than their VSVg coated counterparts (p=3.8×10−5 for COG5 and p=0.003 for COG6). These results suggest that, although there may be some generalized inhibition of viral replication in the COG5 or COG6 depleted cells, the most significant effect of the silencing the COG complex lobe B components is specific to the HIV envelope. The transfection of the COG silenced cells with the proviral cDNA had no negative effect on the expression of dsRed, indicating that silencing of lobe B factors has no effect on transcription and translation from the viral genome (data not shown).
Figure 5.
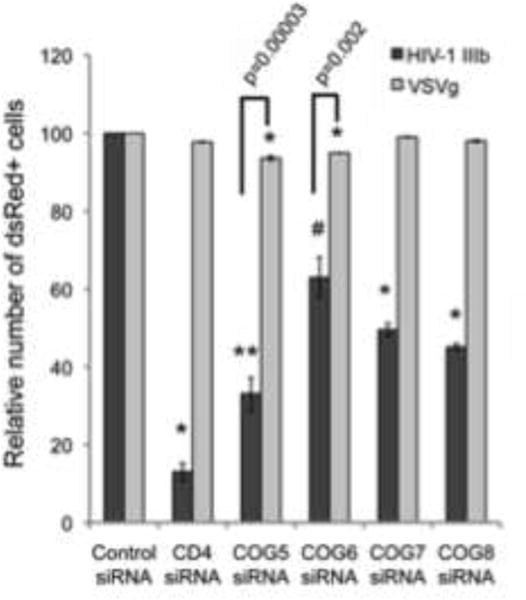
COG5, -6, -7 or -8 depletion inhibits wild type-enveloped HIV-1. The siRNA-treated cells were infected with recombinant HIV-1-dsRED bearing the VSVg envelope glycoprotein (light gray box) or HIV-1 IIIb envelope glycoprotein (dark gray box). Infection was monitored by flow cytometry for the dsRED reporter gene 48h post infection. Percent infection is relative to control siRNA-treated cells (± S.D.) (N=3, *p=0.001, **p<0.01, #p<0.05, two-tailed t-test)
3.5 Silencing of lobe B factors has no effect on viral fusion
In the early stages of the HIV-1 lifecycle, enveloped viruses fuse with the plasma membrane or a vesicle membrane to enter into cells (Miyauchi et al., 2009). To investigate whether the impairment in HIV-1 replication by lobe B factor silencing is before or after viral fusion, a fusion assay utilizing β-lactamase-Vpr chimeric proteins was performed (Cavrois et al., 2002; Cavrois et al., 2011). The siRNA treated cells were incubated in the presence of the Blam-Vpr chimeric and 3h later the cells were loaded with CCF2-AM dye. Viral fusion activity was quantitated by measuring the fluorescence changes upon the cleavage of the CCF2-AM dye by β-lactamase obtained from the fusion of the BlaM-Vpr containing HIV-1 virions with the target cell. As expected, the silencing of CD4 in the target cells decreased the relative number of β-lactamase positive cells. However, the COG5-8 depleted cells showed no significant difference in the percentage of β-lactamase positive cells compared to the control siRNA treatment, indicating that silencing of lobe B components do not impair viral fusion (Figure 6).
Figure 6.

Silencing of COG5, -6, -7 or -8 had no effect on HIV-1 viral fusion. Viral fusion was measured by infecting COG5, -6, -7 or -8 or CD4 or control siRNA treated cells with recombinant viral particles containing the HIV-1 vpr-β-lactamase fusion protein. After 3h of infection, the cells were loaded with CCF2-AM dye and the change in fluorescence was measured by flow cytometry. Representative dot plots are shown in (A). Although CD4 silencing significantly impaired viral fusion, the COG5, -6, -7 or -8 siRNA-treated cells had no significant change in fusion levels compared to Control siRNA treated cells. B) Relative fusion levels in the COG silenced cells compared to CD4 siRNA and Control siRNA treated cells (±S.D.) (N=4, p<0.05, two-tailed t-test).
3.6 Silencing of the COG interacting SNARE STX5 inhibits HIV-1 replication
Members of the COG complex interact both physically and functionally with all the known classes of proteins required for the maintenance of intra-Golgi trafficking, including SNAREs (v-SNAREs and t-SNAREs), SNARE-interacting proteins, Rabs, coiled-coil tethers, vesicular coats and molecular motors (eg. myosin heavy chain 14 (MYH14) and myosin light chain 12 (MYL12)) (Willett et al., 2013b). Willett and colleagues recently showed that both COG6 and COG8 interact with STX5, a member of the t-SNARE (target-SNAP receptor) family of proteins (Willett et al., 2013a). Interestingly, RNAi-based functional genomic screening identified STX5 as a potential HIV-dependency factor (Brass et al., 2008). Silencing STX5 impaired HIV-1 infection when measured by intracellular staining for HIV-1 gag (p24) expression and production of progeny virion. We sought to validate this earlier association and determine if the silencing of STX5 impacts HIV-1 replication in a manner similar to that seen with silencing of COG5-8.
HeLa-CD4 cells were transduced with lentiviral vectors expressing two separate shRNAs targeting STX5, the HIV receptor CD4, or a non-specific control shRNA. The transduced cells demonstrated a 70–90% decrease in mRNA from the appropriate target gene relative to the control shRNA-expressing cells (Figure 7A). The silencing of STX5 led to a significant decrease in the level of HIV-1 infection in the STX5 shRNA compared to the control shRNA treated cells as measure by intracellular staining for HIV-1 gag (p24) expression and flow cytometric analysis (Figure 7B). In fact, both STX5 shRNAs tested showed over 70% inhibition of HIV-1 replication. This confirms our earlier findings from the high content genomic screen (Brass et al., 2008). Consistent with the finding for the silencing of COG5-8, the silencing of STX5 led to decreased levels of both integrated HIV-1 provirus (Figure 7C) and late RT product formation (Figure 7D) compared to the control shRNA treated cells. These results support the findings that targeting components of the retrograde transport pathway inhibits HIV-1 replication. It is noteworthy that our earlier screen implicated additional factors shown to directly interact with the COG complex as potential HIV-dependency factors including RAB1A, RAB2A, RAB6A, GS27 (GOSR2), and SLY1 (Brass et al., 2008; Willett et al., 2013b).
Figure 7.
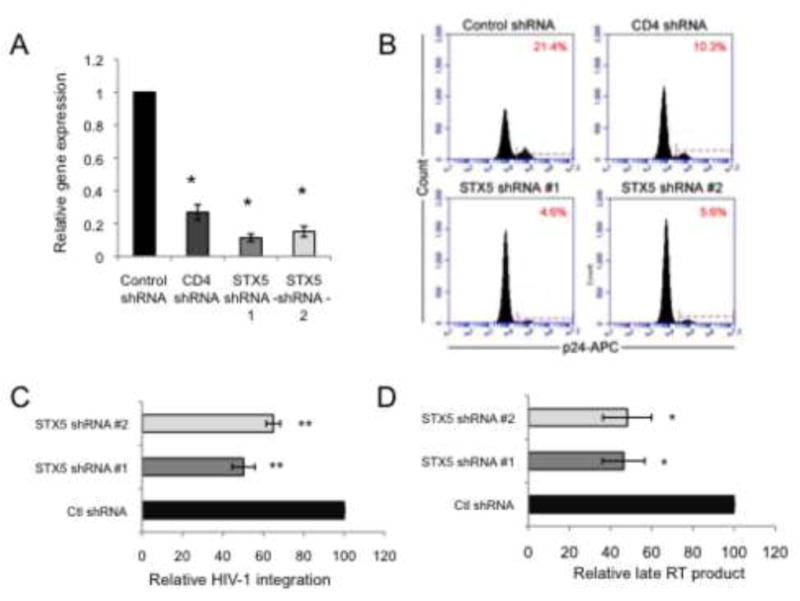
Silencing of STX5 inhibited HIV-1 replication prior to HIV-1 integration. A) RNAi-mediated silencing of STX5 inhibited STX5 expression as measured by qRT-PCR compared to control shRNA treated cells (±S.D.) (N=3, p<0.007, two-tailed t-test). B) Silencing of STX5 inhibited HIV-1 replication as measured by reduced intracellular HIV-1 p24 levels and analysis by flow cytometry. C) STX5 depletion blocks HIV prior to viral integration. The targeted silencing of STX5 significantly impaired viral integration as measured by an Alu-LTR nested PCR strategy. Percent integration indicates percent versus Control shRNA treated cells (±S.D.) (N=3, p<0.03, two-tailed t-test). All values were normalized to β-globin. D) Late RT product formation was significantly inhibited upon silencing of STX5. Percent late RT formation relative to Control shRNA-treated cells (±S.D.) (N=3, p<0.03, two-tailed t-test). All late RT product values are normalized to mitochondrial DNA.
4. Discussion
HIV replication requires a wide variety of cellular factors, HIV dependency factors (HDFs), for effective viral infection and replication. This includes HDFs that participate in virtually every step in the viral lifecycle from the recognition of target cells and entry, through reverse transcription and integration of the viral genome into the host’s genetic material, to progeny virion production, release and maturation (Friedrich et al., 2011; Goff, 2004; Goff, 2007). The secretory system in eukaryotic cells is a series of organized membrane enclosed compartments that includes the endoplasmic reticulum (ER), Golgi cisternae, the TGN, various secretory vesicles and the plasma membrane (Bonifacino and Hierro, 2011; Bonifacino and Rojas, 2006). Proteins and membrane components move through this system in a dynamic and highly regulated manner. Central to these membrane trafficking pathways is the Golgi apparatus that serves as a hub for both anterograde and retrograde trafficking and is essential for the processing and sorting of glycoproteins and glycolipids to their proper location within the cell (Shorter and Warren, 2002). The role that these vesicular trafficking pathways play in HIV-1 progeny virion production has been well documented (Balasubramaniam and Freed, 2011; Garrus et al., 2001; Roeth and Collins, 2006). HIV-1 particle assembly is directed to the plasma membrane by the myristolation of the viral Gag protein which is then processed by HIV-1 protease into the matrix, capsid, nucleocapsid and p6 proteins. In macrophages, budding of assembled viral particles occurs through their integration into multivesicular bodies (MVB) through the interaction of a monoubiquininated HIV-1 p6 protein to TSG101 and AIP1/ALIX (Fujii et al., 2007; Fujii et al., 2009; Garrus et al., 2001; Sette et al., 2010; Strack et al., 2003; Usami et al., 2009). In T cells and tissue culture cell lines, viral egress occurs through both MVBs and late-endosome-trans-Golgi trafficking to the plasma membrane. This latter pathway is dependent on host factors including RAB9p40, TIP47, PIKfyve, and RAB11A (Murray et al., 2005; Ono et al., 2004; Ono and Freed, 2004; Strack et al., 2003; Strack et al., 2002). However, a recent study has called into question the role that TIP-47 plays in HIV-1 replication (Checkley et al., 2013). The silencing of these factors has been shown to impair the production of progeny virions. In contrast, we have recently shown that the silencing of components of the GARP/VFT tethering complex (VPS53) and its associated small GTPase RAB6 impaired viral entry in an HIV-1 envelope-dependent manner (Brass et al., 2008). This suggests that tethering complex components are required not just for the production of progeny virions, but also for the proper infection and replication of HIV-1. In this study, we have shown that the silencing of components of another vesicle tethering complex, the COG complex, have an early defect in HIV-1 replication. Specifically, the targeted silencing of components of lobe B of the COG complex, COGs 5–8, showed a defect in HIV-1 infection that occurred after viral fusion but prior to reverse transcription of the viral genome. Mutation or depletion (e.g. RNAi-mediated silencing (Oka et al., 2005)) of the COG complex subunits leads to disruption of retrograde trafficking of endogenous and exogenous factors, improper glycosylation of glycolipids and glycoproteins, and altered Golgi homeostasis and architecture (Laufman et al., 2011; Mallard et al., 1998; Mallard et al., 2002; Smith et al., 2009; Spelbrink and Nothwehr, 1999; Sun et al., 2007; Ungar et al., 2002; VanRheenen et al., 1999; Whyte and Munro, 2001; Wuestehube et al., 1996; Zolov and Lupashin, 2005).
The first suggestion that the COG complex was required for HIV-1 replication came from an RNAi-based functional genomic screen looking for factors whose silencing inhibits HIV-1 replication. Through that screen, 3 of the 4 COG complex lobe A proteins, COG2, -3 and -4, were identified as HDFs. Although the components of the lobe B of the COG complex where not identified in this screen, given the close association of the two lobes of the COG complex, we sought to determine if the components of the COG complex lobe B were required for HIV-1 replication. The targeted silencing of components of lobe B of the COG complex led to an early block in the HIV life cycle preceding reverse transcription and formation of the cDNA version of the HIV-1 genome. However, viral fusion was not affected by the silencing of COGs 5–8. This is in contrast to our earlier studies on the GARP/VFT tethering complex whose silencing was found to impair fusion (Brass et al., 2008). This could be a result of different techniques used to analyze viral fusion since our initial RNAi screen used a cell-based fusion assay while the current study uses recombinant viral particles that contained a chimeric Blam-Vpr fusion protein (Brass, et al 2008). Alternatively, it is possible that these different tethering complexes play distinct roles in the viral life cycle, a hypothesis that would require more investigation.
To ensure that the silencing of the COG lobe B components were not exerting their effect on HIV-1 through alterations in expression of components of Lobe A of the COG complex, we examined the steady state levels of COG3 and COG4 in cells in which the COG lobe B components were silenced. While COG3 levels were unaffected, a modest decrease in COG4 levels was found in the COG6, -7 and -8 silenced cells. Laufman et al (2013) found that the knockdown of COG6 and -8 resulted in the mislocalization of the COG lobe B components from the Golgi to the cytosol but had no effect on the localization of the COG lobe A components (Laufman et al., 2013). However, the integrity and proper localization of the entire COG complex is required for vesicular tethering and SNARE complex assembly. Therefore, although the silencing of the COG lobe B components failed to appreciably alter the levels of the COG lobe A components, their silencing could impair protein localization and lobe A and B’s interaction, disrupting the functionality of the whole COG complex. Further supporting a role for the COG complex in HIV-1 replication, the RNAi-mediated knock down of the t-SNARE STX5 shown to interact with COG6 and 8 led to a similar early defect in HIV replication (Figure 7) (Willett et al., 2013a; Willett et al., 2013b).
The COG complex has been shown to play important roles in maintaining the organization of the Golgi apparatus and in the distribution of glycosylation enzymes (Oka et al., 2004; Zolov and Lupashin, 2005). As a result, deficiencies in COG components leads to the mislocalization of the glycosylation enzymes, thereby affecting the functionality of a variety of proteins and lipids that are not proper glycosylated. The TGN plays an important role in determining the lipid composition of the secretory pathway components through the transport and recycling of membranes between the plasma membrane, secretory vesicles, TGN and ER (VanRheenen et al., 1998; VanRheenen et al., 1999). The Golgi apparatus serves as the major site for sphingolipid synthesis (Futerman and Riezman, 2005). As such, the Golgi apparatus is essential for the maintenance of membrane homeostasis by serving as a buffer between the glycerolipid-rich ER and the sterol/sphigolipid-rich plasma membrane (Shorter and Warren, 2002). Alterations to the composition of the different cellular membranes will have a drastic impact on their functionality. Interestingly, there is growing evidence that lipid rafts are the preferential sites for HIV-1 virion assembly and infection (Brugger et al., 2006; Jolly and Sattentau, 2005; Kerviel et al., 2012; Leung et al., 2008; Manes et al., 2003; Nguyen and Hildreth, 2000; Ono and Freed, 2001). A reduction in the abundance of lipid rafts has been shown to impair virion production and, subsequently, reduces the infectivity of the released virions (Brugger et al., 2006; Brugger et al., 2007; Kerviel et al., 2012; Zheng et al., 2003; Zheng et al., 2001). Spessott and collegues identified a relationship between the COG complex and sphingomyelin synthesis (Spessott, et al, 2010). Since sphingomyelin plays an important role as a structural component of membranes, an intriguing proposition is that the COG complex regulates the lipid composition and abundance of lipid rafts, and that decreasing the amount of lipid rafts may decrease the susceptibility of the cells to HIV-1 infection. However, the lack of impairment in viral fusion seen upon silencing of the COG lobe B components would suggest that any potential alteration in membrane composition was insufficient to prevent viral infection.
Recently, Gallo and Hope (2012) showed an impairment in HIV-1 replication in cells in which proteins that play a role in cytoskeletal structure and function are silenced(Gallo and Hope, 2012). Following up on two of these proteins, DNAL1 and MAP4, they showed that the silencing of these factors resulted in a post-fusion/pre-reverse transcription block in HIV-1 replication which was similar to that which was seen upon the silencing of components of lobe B of the COG complex. Interestingly, earlier work by Chabin-Brion and colleagues (2001) had shown that the Golgi apparatus served as a microtubule organizing center(Chabin-Brion et al., 2001). It has also been shown that the targeted silencing of COG components led to morphological changes to the Golgi apparatus, including fragmentation of the Golgi (Zolov and Lupashin, 2005). Taken together, these results suggest that the silencing of COG complex components could alter the organization of the Golgi apparatus that, in turn, could disrupt microtubule organization negatively impacting early events in the HIV-1 life cycle.
Alternatively, since the silencing of components of the COG complex have been shown to disrupt the intra-Golgi trafficking of endogenous and exogenous proteins, this could potentially alter the localization or functionality of important HDFs that, in turn, would negatively impact viral infection and replication. It is possible that the effect of COG5, -6, -7 and -8 silencing on HIV-1 replication may be due to alteration in the posttranslational modification and function of a yet to be identified HDF. Although these results and those of our earlier study support a role of the TGN and vesicular trafficking in HIV-1 replication, it is clear that additional work is required to determine the full impact of the COG components on the HIV-1 life cycle.
5. Conclusions
There is a growing appreciation for the important role that vesicular trafficking pathways play in the HIV-1 life cycle, particularly, in the formation, budding and maturation of progeny virions. However, there is increasing evidence suggesting that trafficking pathways play a critical in early events in the viral life cycle. Recent high content functional genomic screens have implicated several components of the endosome-to-TGN trafficking pathway in early events in the viral life cycle including our previous study demonstrating the role of the component of the GARP/VFT complex in regulating viral fusion (Brass et al., 2008). In this study, we extend these findings and show that silencing components of the Golgi tethering complex, the COG complex, impaired HIV-1 replication prior to reverse transcription of the incoming viral RNA but appeared to have no significant effect on viral fusion. The negative effect on HIV-1 replication upon silencing of the lobe B components of the COG complex was found to be dependent on the HIV-1 envelope. A full understanding of the early events in the HIV-1 life cycle that precede the integration of the viral genome into that of the hosts is important for the development of strategies to prevent viral transmission. The identification of components of retrograde vesicular trafficking pathways as key players in early events in the HIV-1 life cycle may not only shed light on HIV biology but may point the way for the development of novel classes of antiviral agents.
Supplementary Material
Supplemental Figure 1. ShRNA-mediated silencing of the components of the COG complex lobe B inhibited HIV-1 IIIB infection of target cells.
Supplemental figure 2. The silencing of COG5, -6, -7, -8 had no significant effect on COG3 or COG4 protein levels. Immunoblot analysis of COG siRNA treated cells using COG3 or COG4 antibodies. The siRNA treatment is indicated in the figure. There was no significant decrease in COG3 and COG4 protein levels in the COG5, -6, -7 or, -8 treated cells indicating that the inhibition of HIV-1 replication seen in cells silenced for these proteins is not due to decreased levels of components of lobe A.
Highlights.
Endosomal trafficking pathways play an important role in HIV-1 replication.
Silencing components of the COG complex impairs early events in the viral life cycle.
The COG-mediated impairment in HIV-1 infection is dependent on the HIV-1 envelope.
Acknowledgments
We would like to thank H. Cukier (University of Miami) for critically reading this manuscript and Dr. V. Lupashin (University of Arkansas) for providing antisera against COG3 and COG4. This research was supported by grant 1R33AI088601 from the National Institutes of Health (NIH) to DMD. We acknowledge support from the Miami Center for AIDS Research (CFAR) at the University of Miami Miller School of Medicine funded by grant (P30AI073961) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
he authors declare they have no competing interests.
Author contributions
L, MD-N and DMD designed and performed the experiments and performed the analysis. SL and DMD drafted the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Sicen Liu, Email: s.liu8@med.miami.edu.
Monika Dominska-Ngowe, Email: mdominska@gmail.com.
Derek Michael Dykxhoorn, Email: DDykxhoorn@med.maimi.edu.
Literature cited
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam M, Freed EO. New insights into HIV assembly and trafficking. Physiology (Bethesda) 2011;26(4):236–51. doi: 10.1152/physiol.00051.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barre-Sinoussi F, Ross AL, Delfraissy JF. Past, present and future: 30 years of HIV research. Nat Rev Microbiol. 2013;11(12):877–83. doi: 10.1038/nrmicro3132. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hierro A. Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 2011;21(3):159–67. doi: 10.1016/j.tcb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7(8):568–79. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A. 2006;103(8):2641–6. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B, Krautkramer E, Tibroni N, Munte CE, Rauch S, Leibrecht I, Glass B, Breuer S, Geyer M, Krausslich HG, Kalbitzer HR, Wieland FT, Fackler OT. Human immunodeficiency virus type 1 Nef protein modulates the lipid composition of virions and host cell membrane microdomains. Retrovirology. 2007;4:70. doi: 10.1186/1742-4690-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD, Malani N, Fernandes J, D’Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, Konig R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5(5):e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7(5):631–4. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20(11):1151–4. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Cavrois M, Neidleman J, Galloway N, Derdeyn CA, Hunter E, Greene WC. Measuring HIV fusion mediated by envelopes from primary viral isolates. Methods. 2011;53(1):34–8. doi: 10.1016/j.ymeth.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabin-Brion K, Marceiller J, Perez F, Settegrana C, Drechou A, Durand G, Pous C. The Golgi complex is a microtubule-organizing organelle. Mol Biol Cell. 2001;12(7):2047–60. doi: 10.1091/mbc.12.7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneau P, Mirambeau G, Roux P, Paulous S, Buc H, Clavel F. HIV-1 reverse transcription. A termination step at the center of the genome. J Mol Biol. 1994;241(5):651–62. doi: 10.1006/jmbi.1994.1542. [DOI] [PubMed] [Google Scholar]
- Chatterton JE, Hirsch D, Schwartz JJ, Bickel PE, Rosenberg RD, Lodish HF, Krieger M. Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc Natl Acad Sci U S A. 1999;96(3):915–20. doi: 10.1073/pnas.96.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley MA, Luttge BG, Mercredi PY, Kyere SK, Donlan J, Murakami T, Summers MF, Cocklin S, Freed EO. Reevaluation of the requirement for TIP47 in human immunodeficiency virus type 1 envelope glycoprotein incorporation. J Virol. 2013;87(6):3561–70. doi: 10.1128/JVI.03299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, Srinivasan A. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66(12):7517–21. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich BM, Dziuba N, Li G, Endsley MA, Murray JL, Ferguson MR. Host factors mediating HIV-1 replication. Virus Res. 2011;161(2):101–14. doi: 10.1016/j.virusres.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol. 2007;5(12):912–6. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Functional role of Alix in HIV-1 replication. Virology. 2009;391(2):284–92. doi: 10.1016/j.virol.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Riezman H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005;15(6):312–8. doi: 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Gallo DE, Hope TJ. Knockdown of MAP4 and DNAL1 produces a post-fusion and pre-nuclear translocation impairment in HIV-1 replication. Virology. 2012;422(1):13–21. doi: 10.1016/j.virol.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Goff SP. Retrovirus restriction factors. Mol Cell. 2004;16(6):849–59. doi: 10.1016/j.molcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Goff SP. Host factors exploited by retroviruses. Nat Rev Microbiol. 2007;5(4):253–63. doi: 10.1038/nrmicro1541. [DOI] [PubMed] [Google Scholar]
- Goff SP. Knockdown screens to knockout HIV-1. Cell. 2008;135(3):417–20. doi: 10.1016/j.cell.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–9. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J Virol. 2005;79(18):12088–94. doi: 10.1128/JVI.79.18.12088-12094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerviel A, Thomas A, Chaloin L, Favard C, Muriaux D. Virus assembly and plasma membrane domains: Which came first? Virus Res. 2012 doi: 10.1016/j.virusres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger M, Brown MS, Goldstein JL. Isolation of Chinese hamster cell mutants defective in the receptor-mediated endocytosis of low density lipoprotein. J Mol Biol. 1981;150(2):167–84. doi: 10.1016/0022-2836(81)90447-2. [DOI] [PubMed] [Google Scholar]
- Laufman O, Freeze HH, Hong W, Lev S. Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes. Traffic. 2013;14(10):1065–77. doi: 10.1111/tra.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufman O, Hong W, Lev S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J Cell Biol. 2011;194(3):459–72. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Yip CK, Walz T, Hughson FM. Molecular organization of the COG vesicle tethering complex. Nat Struct Mol Biol. 2010;17(11):1292–7. doi: 10.1038/nsmb.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K, Kim JO, Ganesh L, Kabat J, Schwartz O, Nabel GJ. HIV-1 assembly: viral glycoproteins segregate quantally to lipid rafts that associate individually with HIV-1 capsids and virions. Cell Host Microbe. 2008;3(5):285–92. doi: 10.1016/j.chom.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47(3):333–48. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- Mallard F, Antony C, Tenza D, Salamero J, Goud B, Johannes L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J Cell Biol. 1998;143(4):973–90. doi: 10.1083/jcb.143.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallard F, Tang BL, Galli T, Tenza D, Saint-Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol. 2002;156(4):653–64. doi: 10.1083/jcb.200110081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes S, del Real G, Martinez AC. Pathogens: raft hijackers. Nat Rev Immunol. 2003;3(7):557–68. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- Mann DL, O’Brien SJ, Gilbert DA, Reid Y, Popovic M, Read-Connole E, Gallo RC, Gazdar AF. Origin of the HIV-susceptible human CD4+ cell line H9. AIDS Res Hum Retroviruses. 1989;5(3):253–5. doi: 10.1089/aid.1989.5.253. [DOI] [PubMed] [Google Scholar]
- Miller VJ, Ungar D. Re’COG’nition at the Golgi. Traffic. 2012;13(7):891–7. doi: 10.1111/j.1600-0854.2012.01338.x. [DOI] [PubMed] [Google Scholar]
- Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137(3):433–44. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JL, Mavrakis M, McDonald NJ, Yilla M, Sheng J, Bellini WJ, Zhao L, Le Doux JM, Shaw MW, Luo CC, Lippincott-Schwartz J, Sanchez A, Rubin DH, Hodge TW. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J Virol. 2005;79(18):11742–51. doi: 10.1128/JVI.79.18.11742-11751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74(7):3264–72. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15(5):2423–35. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Vasile E, Penman M, Novina CD, Dykxhoorn DM, Ungar D, Hughson FM, Krieger M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5- and COG7-deficient mammalian cells. J Biol Chem. 2005;280(38):32736–45. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101(41):14889–94. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98(24):13925–30. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol. 2004;78(3):1552–63. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden K, Emerman M, Montagnier L. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology. 1991;185(2):661–72. doi: 10.1016/0042-6822(91)90537-l. [DOI] [PubMed] [Google Scholar]
- Podos SD, Reddy P, Ashkenas J, Krieger M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J Cell Biol. 1994;127(3):679–91. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic M, Read-Connole E, Gallo RC. T4 positive human neoplastic cell lines susceptible to and permissive for HTLV-III. Lancet. 1984a;2(8417–8418):1472–3. doi: 10.1016/s0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]
- Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984b;224(4648):497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature. 1985;313(6000):277–84. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- Roeth JF, Collins KL. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev. 2006;70(2):548–63. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33(3):401–6. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Sette P, Jadwin JA, Dussupt V, Bello NF, Bouamr F. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J Virol. 2010;84(16):8181–92. doi: 10.1128/JVI.00634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Warren G. Golgi architecture and inheritance. Annu Rev Cell Dev Biol. 2002;18:379–420. doi: 10.1146/annurev.cellbio.18.030602.133733. [DOI] [PubMed] [Google Scholar]
- Smith RD, Willett R, Kudlyk T, Pokrovskaya I, Paton AW, Paton JC, Lupashin VV. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic. 2009;10(10):1502–17. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink RG, Nothwehr SF. The yeast GRD20 gene is required for protein sorting in the trans-Golgi network/endosomal system and for polarization of the actin cytoskeleton. Mol Biol Cell. 1999;10(12):4263–81. doi: 10.1091/mbc.10.12.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–99. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Strack B, Calistri A, Gottlinger HG. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J Virol. 2002;76(11):5472–9. doi: 10.1128/JVI.76.11.5472-5479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Shestakova A, Hunt L, Sehgal S, Lupashin V, Storrie B. Rab6 regulates both ZW10/RINT-1 and conserved oligomeric Golgi complex-dependent Golgi trafficking and homeostasis. Mol Biol Cell. 2007;18(10):4129–42. doi: 10.1091/mbc.E07-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztul E, Lupashin V. Role of vesicle tethering factors in the ER-Golgi membrane traffic. FEBS Lett. 2009;583(23):3770–83. doi: 10.1016/j.febslet.2009.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar D, Oka T, Brittle EE, Vasile E, Lupashin VV, Chatterton JE, Heuser JE, Krieger M, Waters MG. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157(3):405–15. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami Y, Popov S, Popova E, Inoue M, Weissenhorn W, H GG. The ESCRT pathway and HIV-1 budding. Biochem Soc Trans. 2009;37(Pt 1):181–4. doi: 10.1042/BST0370181. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Lupashin VV, Barlowe C, Waters MG. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J Cell Biol. 1998;141(5):1107–19. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen SM, Cao X, Sapperstein SK, Chiang EC, Lupashin VV, Barlowe C, Waters MG. Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J Cell Biol. 1999;147(4):729–42. doi: 10.1083/jcb.147.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JR, Munro S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev Cell. 2001;1(4):527–37. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V. COG complexes form spatial landmarks for distinct SNARE complexes. Nat Commun. 2013a;4:1553. doi: 10.1038/ncomms2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Ungar D, Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem Cell Biol. 2013b;140(3):271–83. doi: 10.1007/s00418-013-1117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuestehube LJ, Duden R, Eun A, Hamamoto S, Korn P, Ram R, Schekman R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics. 1996;142(2):393–406. doi: 10.1093/genetics/142.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung ML, Houzet L, Yedavalli VS, Jeang KT. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem. 2009;284(29):19463–73. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder KE, Fishel R. Real-time quantitative PCR and fast QPCR have similar sensitivity and accuracy with HIV cDNA late reverse transcripts and 2-LTR circles. J Virol Methods. 2008;153(2):253–6. doi: 10.1016/j.jviromet.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Plemenitas A, Fielding CJ, Peterlin BM. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A. 2003;100(14):8460–5. doi: 10.1073/pnas.1437453100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Plemenitas A, Linnemann T, Fackler OT, Peterlin BM. Nef increases infectivity of HIV via lipid rafts. Curr Biol. 2001;11(11):875–9. doi: 10.1016/s0960-9822(01)00237-8. [DOI] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4(5):495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol. 2005;168(5):747–59. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. ShRNA-mediated silencing of the components of the COG complex lobe B inhibited HIV-1 IIIB infection of target cells.
Supplemental figure 2. The silencing of COG5, -6, -7, -8 had no significant effect on COG3 or COG4 protein levels. Immunoblot analysis of COG siRNA treated cells using COG3 or COG4 antibodies. The siRNA treatment is indicated in the figure. There was no significant decrease in COG3 and COG4 protein levels in the COG5, -6, -7 or, -8 treated cells indicating that the inhibition of HIV-1 replication seen in cells silenced for these proteins is not due to decreased levels of components of lobe A.