

Abstract
Recent advances in genetics have spurred rapid progress towards the systematic identification of genes involved in complex diseases. Still, the detailed understanding of the molecular and physiological mechanisms through which these genes affect disease phenotypes remains a major challenge. Here, we identify the asthma disease module, i.e. the local neighborhood of the interactome whose perturbation is associated with asthma, and validate it for functional and pathophysiological relevance, using both computational and experimental approaches. We find that the asthma disease module is enriched with modest GWAS P-values against the background of random variation, and with differentially expressed genes from normal and asthmatic fibroblast cells treated with an asthma-specific drug. The asthma module also contains immune response mechanisms that are shared with other immune-related disease modules. Further, using diverse omics (genomics, gene-expression, drug response) data, we identify the GAB1 signaling pathway as an important novel modulator in asthma. The wiring diagram of the uncovered asthma module suggests a relatively close link between GAB1 and glucocorticoids (GCs), which we experimentally validate, observing an increase in the level of GAB1 after GC treatment in BEAS-2B bronchial epithelial cells. The siRNA knockdown of GAB1 in the BEAS-2B cell line resulted in a decrease in the NFkB level, suggesting a novel regulatory path of the pro-inflammatory factor NFkB by GAB1 in asthma.
Introduction
There is increasing evidence that disease genes in both monogenic and complex diseases are not distributed randomly on the molecular interaction network (interactome), but they rather work together in similar biological modules or pathways (1–3). Moreover, gene products (e.g. proteins) linked to the same phenotype have a strong tendency to interact with each other (2,4,5) and to cluster in the same network neighborhood (6). This suggests the existence of a disease module, a connected sub-network that can be mechanistically linked to a particular disease phenotype (1–3,6–8). The accurate identification of such disease modules could help uncover the molecular mechanisms of disease causation, identify new disease genes and pathways and aid rational drug target identification. Currently, the accurate identification of disease modules is hampered by the incompleteness of the available cellular network maps and disease genes lists. There is an increasing sense, however, that recent advances in interactome mapping and disease gene identification have begun to offer sufficient network coverage and accuracy to enable identification of disease modules for some well-studied complex diseases. The goal of this paper is to demonstrate the maturity and the value of such an approach, through its application to the study of asthma, a major complex disease with an estimated economic burden of $56 billion in the USA for the year 2007 (9).
Despite the identification of many susceptibility alleles and genes by GWAS and other technologies (10), our knowledge of the underlying etiologic mechanisms responsible for asthma remains limited. Given the many genes and environmental factors linked to the disease, traditional single gene or single pathway-based approaches have shown limited utility. In the past few years, several attempts have been made to integrate the topological properties of protein interaction networks with different types of ‘omics’ data to discover novel genes and pathways (11–13). These approaches rely on the local impact hypothesis, assuming that if a few disease components are identified, other disease-related components are likely found in their network vicinity (14,15). Therefore, each disease can be linked to a well-defined local neighborhood of the interactome, called the disease module. Yet, the existence of such a single neighborhood remains a hypothesis that needs to be tested.
Our goal, here, is to determine whether a whole network-based approach can enhance our understanding of the local network neighborhood of a disease using asthma as an example (Fig. 1A). We start by compiling a list of physical and experimentally documented interactions in human cells from the literature, as well as a set of known and well-established disease genes. These seed genes allow us to pinpoint the position of the putative disease module within the interactome (Stage I). Next, we apply a graph theoretic procedure to identify the localization of potential asthma genes that may belong to this disease module (Stage II). In Stage III the obtained disease module is validated for functional and pathophysiological relevance using several asthma-specific biological datasets. We further explore the overlap and the difference between the asthma module and other immune-related disease modules. Finally, in Stage IV we explore the pathways within the module that contain promising therapeutic targets. In particular, we confirm the relevance of the newly identified GAB1 pathway using in vitro experiments.
Figure 1.

Overview of the disease module approach for asthma. (A) The stages of mapping out and validating the asthma disease module. (I) Construction of the interactome and compilation of the consensus disease gene list (seed genes). (II) Mapping the seed genes onto the interactome and identification of the disease module, the sub-network that contains disease-associated components, via DIAMOnD. (III) Bioinformatics validation of the asthma disease module using gene expression data, gene ontology, pathway information and comorbidity analysis. (IV) Detailed biological analysis and in vitro confirmation of novel asthma-related pathways with potential therapeutic relevance. (B) Subgraph of the full interactome showing the connections among the asthma seed genes. The largest cluster (proto-module) contains 37 seed genes, the rest is scattered in smaller fragments. The colors of the nodes indicate the source of each seed gene; the links are colored according to the source of the interaction. (C) Size distribution of the largest connected component expected for 129 randomly distributed genes. The observed proto-module for asthma is highly significant (z-score = 10.7, empirical P-value <10−6).
Results
Disease module identification
Asthma seed genes in the human interactome
Using five complementary data sources, we compiled a list of 144 asthma-associated seed genes (Supplementary Material, Table S1), 129 (89.5%) of which are represented within our human Interactome. Though obviously incomplete and prone to false positives, this list reflects our current knowledge of the molecular underpinning of asthma.
If both the interactome and the seed gene list were complete and accurate, according to the disease module hypothesis the seed genes should form one (or a few) connected subgraph(s), defining the disease module(s). Consistent with this hypothesis, we observe a cluster of 37 highly interconnected asthma seed genes, which we call the proto-module (Fig. 1B). In order to test whether such a proto-module could have emerged by chance, we measured the largest connected component for 129 genes randomly drawn from the network. The resulting distribution (z-score = 10.6, empirical P-value < 10−6) in Figure 1C shows that on average only 3–4 genes are connected in these random controls, suggesting that the observed proto-module is at the core of an asthma disease module. The proteins within this core reflect several biological processes fundamentally implicated in asthma pathobiology, including TH2-mediated (IL4 and IL13 cytokines), immunoglobulin E- (IgE receptors FCER1A and MS4A2) and eosinophil (CCL11-eotaxin)-mediated allergic inflammation, innate immunity (TLR3, TLR4 and CD14) and vitamin D signaling (VDR). Note that despite the existence of a proto-module with significant size, most asthma seed genes are disconnected from it and scattered throughout the interactome. They form nine small isolated clusters of size two to four, and more than half of the seed proteins (72/129, 56%) do not interact with other seed proteins (Fig. 1B). Three factors likely contribute to this fragmentation:
Interactome incompleteness: current protein–protein interaction maps cover <10% of all potential interactions (16–18). Hence many more of the now isolated proteins could be part of a single disease module, but the missing links have isolated them.
False positives: not all genes in the seed gene list have a mechanistic association with asthma. Hence, their isolation from the giant component is real.
Missing disease genes: not yet identified asthma-related genes could link currently isolated seed genes to the putative asthma module.
Problem (i) is currently being addressed by several high-throughput-mapping projects (19,20). Here we focus on the localization of asthma genes that will help us to refine the asthma disease module.
Expanding the proto-module
Locally dense neighborhoods of a biological network, called ‘topological modules', can be uncovered using community detection algorithms (21,22). We first tested whether these topological modules correspond to potential disease modules by implementing three different widely used module-finding algorithms (23–25) and inspecting the predicted topological modules for enrichment with asthma-related genes. Our failure to find modules with meaningful asthma association (see Supplementary Material, Section VIII and Fig. S2) prompted us to choose a tool that simultaneously exploits the knowledge of the network topology and the network position of the known asthma-associated genes to identify the asthma disease module (26). The DIseAse MOdule Detection (DIAMOnD) algorithm starts from the s seed genes and prioritizes the other proteins of the interactome for their putative asthma relevance (see Fig. 2 and Supplementary Material, Section IX for a description of the algorithm). The key ingredient of DIAMOnD is the selection of proteins that have a significant fraction of their interactions with seed proteins, while ignoring proteins that interact with many seed proteins, mainly as a consequence of their high degree (promiscuity), like the yellow protein at the bottom of Figure 2A. Hence the proteins selected by the algorithm are not enriched in hubs, in line with the finding that disease genes tend to avoid hubs (1). We call the genes encoding proteins added by the algorithm ‘DIAMOnD genes’. The union of the seed and DIAMOnD genes represents our putative ‘asthma disease module’ (Fig. 2B and C). For a detailed characterization of DIAMOnD and a critical comparison with other methods, see Ghiassian et al. (26). Furthermore, to systematically estimate the impact of potential noise in the interaction data, we repeated our analysis on a set of modified networks with varying degrees of artificial noise. The results indicate that our main findings are robust over a wide range of noise (see Supplementary Material, Section X).
Figure 2.

DIAMOnD method for disease module identification and its validation: (A) Schematic network configuration with seed genes (red) and their neighbors (yellow). The P-values next to the yellow genes indicate how significant their respective number of links to seed genes are (see Supplementary Material, Section IX for details). (B) At each iteration step of the DIAMOnD algorithm the gene with the most significant number of connections to seeds and previously added genes is agglomerated into the module. (C) Schematic depiction of the relationship between the disease module predicted by DIAMOnD and the seed proto-module. We call the proteins/genes selected by the DIAMOnD algorithm DIAMOnD genes. The union of the DIAMOnD genes and the seed genes connected to them is the predicted disease module. (D) Validation of the DIAMOnD genes. Column 1 shows the number of DIAMONnD genes found in the different validation datasets, column 2 the corresponding statistical significance. Column 3 identifies the considered datasets (see Supplementary Material Sections III–VII for details): (i) differentially expressed genes compiled from nine sources; (ii) 35 asthma-specific pathways from GeneGO; (iii) MSIgDB pathways; (iv) gene ontology (biological processes); (v): genes associated with comorbid diseases. The values for the DIAMOnD genes are show in orange, the values for seed genes and random expectation in red and green, respectively. In Column 2 we used a sliding-window approach in order to compensate for the dependence of P-values on the underlying set size: At each iteration step i, we consider all DIAMOnD genes in the interval [i−129/2, i + 129/2], thereby obtaining sets of the same size as the seed genes that can be compared to each other. Column 1 shows that DIAMOnD always performs better than random expectation (orange line above green line), and in (i) and (ii) even better than the established seed genes themselves (orange line above red line). (E) Comparison of the number of seed genes contained in the final DIAMOnD module with the number obtained in 105 random simulations. Connected sets that are constructed completely at random contain on average 51.6 ± 4.9 seed genes, which is significantly lower than in the real disease module. (F) Same as E, but choosing the connected genes not completely at random, but only from the immediate neighborhood of the seed genes. The random modules contain 76.0 ± 4.5 seed genes, again significantly lower than in the real disease module. (G) Schematic illustration of the disease module, together with a 3D depiction of the actual asthma module within the interactome, consisting of 441 genes (91 seed genes + 350 DIAMOnD genes). (H) Comparison of the enrichment of different gene sets with low P-value genes from the EVE GWAS meta-analysis dataset. The seed genes (red) have the highest enrichment. When the 33 GWAS genes are removed from the seed genes (yellow), their enrichment becomes comparable with the DIAMOnD genes (orange). The two random controls of genes with protein interactions to seed genes (green) and all interactome genes (blue) have significantly lower fractions of low P-value genes.
Validation of the disease module
The DIAMOnD algorithm prioritizes the proteins in the network based on their topological proximity to seed proteins. Hence, the order in which the DIAMOnD proteins are selected can be interpreted as a network-based ranking criterion. Note, however, that DIAMOnD ranks the entire network and we therefore need an additional stopping criterion to define the boundary of the disease module. For this, as well as for a general bioinformatics validation of the ranked genes, we use five different asthma-specific validation data (see Materials and Methods and Fig. 2D). In all five assessments, we observe greater enrichment for asthma-relevant genes among the DIAMOnD genes than expected by chance. Specifically we observe enrichment for differentially expressed genes in a survey of nine asthma gene expression datasets (Supplementary Material, Table S2, Fig. 2D (i)), for genes with asthma-related GeneGO, MSigDB and GO pathways (Fig. 2D(ii), (iii), (iv)), and in an asthma-related comorbidity analysis (Fig. 2D(v)). The comorbidity analysis demonstrates that the DIAMOnD genes are enriched with genes associated with 85 diseases that show significant comorbidity with asthma (relative risk RR > 1). The top enriched comorbid diseases include immune mechanism disorders (RR = 2.2, enrichment P = 3.5 × 10−4, Fisher's exact test) and disorders of lipoid metabolism lipodystrophy (RR = 2.14, P = 2.6 × 10−4). Across the five validation data presented in Figure 2D we find that approximately the first 350 DIAMOnD genes show the most significant asthma association, with minimal gains observed when further genes are considered. Hence these 350 DIAMOnD genes together with 91 directly connected seed genes represent our putative asthma disease module (441 genes in total) (Supplementary Material, Fig. S3). These 91 seed genes include the 37 genes from the proto-module (Fig. 1B), as well as 54 seed genes that were initially disconnected, thus incorporating 71% of the initial 129 seed genes. To test whether the number of previously disconnected genes that were integrated is higher than expected by chance, we compared it with randomly chosen sets of connected genes: we find that connected sets of the same size that were constructed completely at random on average contain 51.6 ± 4.9 seed genes, which is significantly lower than in the real disease module (Fig. 2E, z-score = 6.9, empirical P < 10−6). Even when the random genes are chosen only from the immediate neighborhood of the seed genes we find that the resulting modules contain only 76.0 ± 4.5 seed genes, which is again significantly lower as observed in the DIAMOnD module (Fig. 2F, z-score = 2.0, empirical P = 0.02). These differences suggest that DIAMOnD successfully identifies the most relevant local neighborhood within the interactome.
GWAS analysis
If the asthma disease module is enriched for asthma-causing genes, then the module should also be enriched for disease-susceptibility variants. To assess the significance of the 350 DIAMOnD genes for asthma-associated genetic variants, we used data collected by two meta-analysis GWAS studies conducted by the North American EVE (n = 15 000 subjects) and European GABRIEL (n = 26 475) asthma consortia (27,28). We computed a single P-value for each gene in the interactome using the LDsnpR method (29). Figure 2G and H shows the respective fraction of seed genes, DIAMonD genes, genes connected to seed genes and the rest of the interactome that have a GWAS P-value below a given threshold φ. The results indicate that the seed list has the highest fraction of genes associated with asthma, followed in order by the DIAMOnD list, genes with protein interactions with seed genes and finally the rest of the interactome. Once we remove four seed genes that were selected based on their high GWAS significance (ORMDL3, IL33, IL1RL1 and IL18R1), the DIAMOnD and the seed genes show comparable disease association P-values (Fig. 2H), implying that the DIAMOnD genes are indeed enriched for disease-susceptibility variants. Recently published associations of the DIAMOnD genes CDK2 (rs2069408, GWAS P-value = 1.0 × 10−10) and GAB1 (rs3805236, GWAS P-value = 7.0 × 10−8) with adult asthma in an Asian ancestry population provide further evidence for an elevated asthma association among the DIAMOnD genes (30) (see Supplementary Material, Table S6 for a list of all DIAMOnD genes with GWAS P-value < 0.05).
Comparison with other methods
In recent years, a variety of methods have been developed to infer disease genes from their connectivity patterns within the interactome (31,32). These tools are based on the observation that genes causing similar diseases tend to link to each other in the interactome (14,15,33–36). For example, a study used GRAIL and DAPPLE together with GWAS data to prioritize genes in inflammatory bowel disease (IBD) (14). Similarly, the clustering in protein interaction networks was used to identify true positive hits in siRNA screens (15). To explore the performance of DIAMOnD, we tested it against four different network-based algorithms (see Materials and Methods): random walk (RW) (33), DaDa (36), PRINCE (35), CIPHER (34) and one sequence-based candidate gene prioritization tool: Endeavor (37). We find that with heterogeneous biological data (GO terms, MSIgDB, etc.) that are less specific to asthma, DIAMOnD performs equally or slightly better than the other algorithms (Fig. 3A). When using asthma-specific datasets (GWAS, differential gene-expression, etc.), however, DIAMOnD performs considerably better than the reference tools (Fig. 3B). We further compared the genes identified by DIAMOnD with the genes from DaDa and RW, the two methods that had the best performance for asthma. We find a large overlap between the predicted DaDa and RW genes, but only moderate overlap with DIAMOnD genes (Fig. 3C). Indeed, 223 (64%) DIAMOnD genes are unique and were not identified by either RW or DaDa. An analysis of the genes that are exclusive to each method uncovered two mechanisms that contribute to DIAMOnD's distinct performance: first, we find that RW and DaDa genes are enriched in hubs, which is not the case for DIAMOnD (Fig. 3D). Indeed, DIAMOnD selects proteins based on the statistical significance of the number of links they have to seed proteins, rather than their raw number. As a consequence, the average degrees <k> in the two gene sets are <k> = 44 (DIAMOnD) and <k> = 57 (RW and DaDa), a highly significant difference (P < 3 × 10−3, Mann–Whitney U test). The absence of hubs among the DIAMOnD genes is consistent with evidence that disease genes generally tend to avoid hubs (1). Second, we find that DIAMOnD selects 55 genes that initially have no connections to seed genes, in contrast to eight and six genes selected by RW and Dada, respectively. This means that while RW and DaDa select mainly immediate neighbors of the seed genes, DIAMOnD successfully identifies genes beyond a simple first order association.
Figure 3.

Comparison of DIAMOnD with other methods. (A) Enrichment of the gene sets obtained by five gene-prioritization methods and DIAMOnD with non-asthma-specific validation data. For each method we use the 350 highest ranked genes. All tested methods show comparable performance. (B) Enrichment according to asthma-specific data. Here, DIAMOnD clearly shows stronger enrichment and outperforms the other methods. (C) Venn diagram of the gene sets of the three methods with the best performance in (A) and (B). While there is a significant overlap, each method also has a considerable number of unique genes. (D) Comparison of the degrees of the 232 unique DIAMOnD genes and the 267 combined unique genes from (C). The genes identified by the other two methods contain many network hubs that are absent in DIAMOnD.
Assessing disease heterogeneity, drug response and inflammatory signature genes
Relation to other immune-related diseases
Asthma has an important immune-related component that should also be represented in its disease module. We therefore expect that the asthma module overlaps with modules of other immune-related diseases. To explore this possibility, we compiled seed genes for 11 other immune-related diseases that show extensive co-morbidity among each other as reported previously (38). We find that 9 out of 11 diseases exhibit a significant proto-module (Supplementary Material, Fig. S4). For each of these diseases we then determined a set of 350 DIAMOnD genes and measured their overlap with the asthma module. The highest overlaps were found for Crohn's disease (202 genes), Hashimoto's disease (182 genes) and multiple sclerosis (180 genes), indicating a strong shared pathogenesis between these diseases. Indeed, as shown in the Venn diagram in Figure 4A, the four disease modules exhibit a common core of 72 genes. Most of the shared genes are part of a few important immunological pathways: T-cell differentiation, immune cell signaling, innate immune response and HLA pathways (Supplementary Material, Fig. S5). This indicates that the key components in regulating the immune response are shared among many chronic immune-related diseases. Yet, besides the overlap, the comparison also reveals genes that are specific to each disease. We find that 65 genes within the asthma module do not appear in any other module. Interestingly, they all interact with each other in a single connected component (Fig. 4B), thus pointing towards a region of the interactome that is highly asthma specific (Supplementary Material, Fig. S6A and B).
Figure 4.
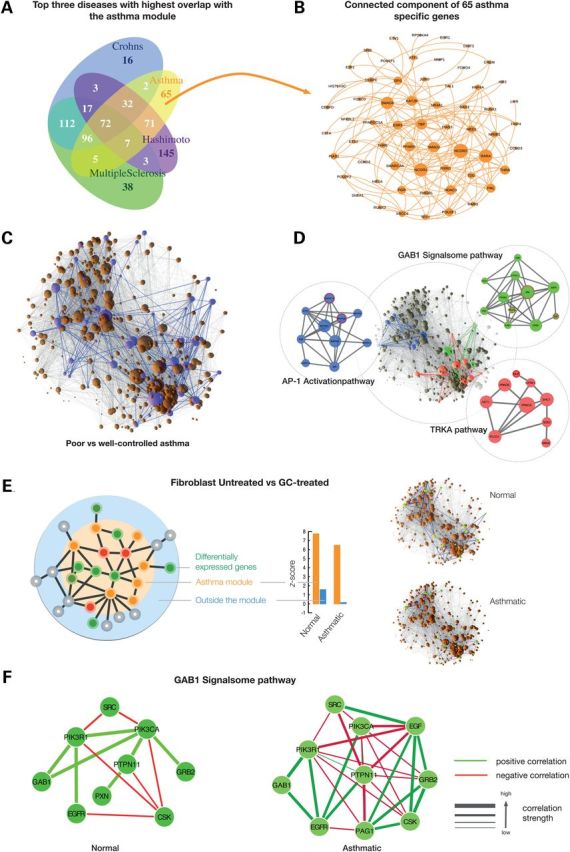
Overlap between Asthma disease module and the 11 other immune-related diseases. (A) Venn diagram of the three immune-related diseases with the highest overlap with the asthma module. Seventy-two genes are common among the four diseases. (B) Connected component in the interactome of the 65 genes unique to the asthma module. (C) The largest connected component of the asthma module with genes highlighted in purple that are differentially expressed between poorly and well-controlled asthma subjects. (D) The three pathways that are fully embedded in the asthma disease module (shown separately on the side). Seed genes are circled in red. (E) Effect of GC treatment in fibroblast cells. We determined the differentially expressed genes from ‘normal-untreated versus normal-treated’ and ‘asthma-untreated versus asthma-treated’ and mapped them to the interactome. The asthma module is significantly enriched with differentially expressed genes. These genes are highly interconnected inside the module (z-score = 7.8 and 6.5 for normal and asthmatic; orange bars), whereas the differentially expressed genes that fall outside the module are scattered and do not form significant clusters (blue bars). (F) Co-expression networks of GAB1 pathway genes in normal and asthmatic fibroblast cell lines.
Asthma severity and disease module
We next explored the relation between the asthma module and two important asthma phenotypes that are central to the classification and management of the disease: (i) asthma severity, defined on the basis of lung function, disability and exacerbation history; and (ii) poor asthma control, a critical harbinger of impending exacerbation characterized by symptom frequency (including interference with sleep and activity limitation) and use of rescue medication. Asthma severity varies widely between patients, with markedly different clinical courses and responses to asthma controller therapies. To understand the asthma module relationships with acute asthma control (AAC), we performed differential expression analyses in a subset of 583 subjects from the Asthma BioRepository for Integrative Genomic Exploration (Asthma BRIDGE) cohort who had genome-wide gene expression data derived from whole blood RNA (see Materials and Methods). AAC was represented by a modified version of the clinically validated Asthma Control Test (mACT) score. We observe that the 63 genes differentially expressed between poor and well-controlled asthmatics are enriched for DIAMOnD genes (P-value = 6 × 10−3, Fisher's exact test). These genes are also significantly connected (z-score = 2.4, empirical P-value = 0.016) within the asthma module (Fig. 4C). Together, these data suggest widespread perturbations of specific asthma disease module components across the spectrum of asthma severity and control, and lend further credence to the biological and clinical importance of the identified disease module in asthma pathogenesis.
Identifying key pathways within the asthma module
A pathway analysis reveals 162 pathways that have at least half of their genes in the asthma disease module (Supplementary Material, Table S7). To single out the most relevant and novel pathways, we integrated the various validation data introduced above, such as asthma-specific gene expression and GWAS meta-analysis signals (see Supplementary Material, Section XI): for each gene in the module, we first compute an integrated score capturing the amount of available evidence for its asthma relevance. We then used the average score of the genes in a pathway to quantify its integrated biological relevance to asthma (Supplementary Material, Table S5). Among the highest ranked pathways are several whose asthma association is well established in immune response, including the IL4, EGF and IGF1 signaling pathways (Table 1). We also find three additional novel pathways that are fully embedded within the asthma disease module (Fig. 4D): the activation of the AP1 family of transcription factors (2 in Table 1), Trka Receptor Signaling (1) and GAB1 signalsome (4). Though not traditionally considered ‘asthma-associated pathways’, the relevance of each of these for asthma pathogenesis has been at least superficially established. Indeed, the promoters of asthma-associated cytokines are enriched for AP-1 DNA-binding sites and inhibition of airway AP-1 expression has been proposed as a therapeutic avenue to control asthma (39). Activation of Trka receptor signaling is involved in airway inflammation and remodeling–cardinal histopathologic features observed in asthma (40). The potential relevance of the GAB1 signalsome is supported by a recent GWAS study identifying the GAB1 locus (GWAS P-value = 1.87 × 10−12) as associated with adult asthma in subjects of Asian ancestry (30). In the disease-connect database, we find that GAB1 is not differentially expressed in any of the other 11 immune-related diseases (disease-connect.org; threshold used: P-value < 10−6). In Gene expression omnibus (GEO), GAB1 is differentially expressed only in multiple sclerosis (adjusted P-value = 1.1 × 10−3, t-test) and Psoriasis (adjusted P-value = 5.9 × 10−6) (Supplementary Material, Table S10). Interestingly, we do not find an expression change of GAB1 in Crohn's disease (Supplementary Material, Table S10), emphasizing the importance of condition- and tissue-specific activation of common genes between the two disease modules.
Table 1.
The top 20 pathways enriched in asthma module
| Pathways prioritized with seed genes | |||||
|---|---|---|---|---|---|
| Pathway name | All genes in asthma module | all pathway genes | coverage | mean score | Genes |
| ACTIVATION OF THE API FAMILY OF TRANSCRIPTION FACTORS | 10 | 10 | 1.00 | 0.72 | MAPK14 MAPK3 MAPK1 MAPK8 MAPK9 MAPK11 MAPK10 FOS ATF2 JUN |
| IL2 PATHWAY | 20 | 22 | 0.91 | 0.61 | IL2RB JAK1 MAPK3 SYK GRB2 MAPK8 STAT5A SOS1 SHC1JAK3 IL2RA LCK ELK1 MAP2K1 CSNK2A1 RAF1 STAT5B HRAS JUN FOS |
| IL6 PATHWAY | 21 | 22 | 0.95 | 0.61 | CEBPB JAK2 JAK1 MAPK3 SRF STAT3 TYK2 SHC1JAK3 IL6ST ELK1 MAP2K1 SOS1 RAF1 PTPN11 HRAS JUN GRB2 FOS CSNK2A1 IL6 |
| EGF PATHWAY | 28 | 31 | 0.90 | 0.61 | JAK1 PIK3R1 PIK3CA SRF PLCG1 JUN GRB2 RAF1 RASA1 STAT5B MAP2K1 EGFR HRAS EGF STAT6 MAPK3 PRKCB PRKCA MAPK8 STAT5A SOS1 SHC1 STAT1 STAT2 ELK1 STAT3 CSNK2A1 FOS |
| EPO PATHWAY | 18 | 19 | 0.95 | 0.61 | JAK2 STAT5B MAPK3 EPOR GRB2 MAPK8 STAT5A SOS1 SHC1 ELK1 MAP2K1 CSNK2A1 HRAS PLCG1 FOS RAF1 JUN PTPN6 |
| TPO PATHWAY | 21 | 24 | 0.88 | 0.60 | CSNK2A1JAK2 PIK3R1 MAPK3 PRKCB PRKCA STAT3 STAT5A STAT5B SHC1 PIK3CA RAF1 MAP2K1 SOS1 HRAS STAT1 FOS GRB2 PLCG1 JUN RASA1 |
| CDMAC PATHWAY | 13 | 16 | 0.81 | 0.59 | MAPK3 MAPK1 PRKCB PRKCA FOS MAP2K1 HRAS MYC RAF1 JUN TNF RELA NFKB1 |
| TRKA PATHWAY | 12 | 12 | 1.00 | 0.59 | NGF PRKCB AKT1 PRKCA NTRK1 GRB2 SHC1 PIK3CA SOS1 PLCG1 HRAS PIK3R1 |
| GLEEVEC PATHWAY | 20 | 23 | 0.87 | 0.58 | JAK2 STAT5B MAPK3 AKT1 GRB2 MAPK8 BAD STAT5A PIK3R1 PIK3CA FOS HRAS MAP2K1 BCR SOS1 RAF1 STAT1 CRKL MYC JUN |
| AKT PHOSPHORYLATES TARGETS IN THE CYTOSOL | 13 | 14 | 0.93 | 0.57 | CDKN1B PDPK1AKT3 CASP9 AKT1 AKT2 GSK3B MDM2 TSC2 GSK3A CDKN1A BAD CHUK |
| IL3 PATHWAY | 13 | 15 | 0.87 | 0.57 | JAK2 STAT5B MAPK3 GRB2 STAT5A SOS1 SHC1 CSF2RB MAP2K1 HRAS FOS RAF1 PTPN6 |
| PDGF PATHWAY | 27 | 32 | 0.84 | 0.57 | JAK1 PIK3R1 PIK3CA PDGFRA SRF PLCG1 JUN GRB2 RASA1 STAT5B HRAS FOS STAT6 PRKCA MAPK3 PRKCB MAP2K1 MAPK8 STAT5A SOS1 SHC1 STAT1 STAT2 ELK1 STAT3 CSNK2A1 RAF1 |
| IGF1 PATHWAY | 19 | 21 | 0.90 | 0.56 | IRS1 SOS1 MAPK3 SRF MAPK8 IGF1R PIK3R1 SHC1 PIK3CA RAF1 ELK1 MAP2K1 CSNK2A1 HRAS PTPN11 FOS GRB2 JUN RASA1 |
| GAB1 SIGNALOSOME | 11 | 11 | 1.00 | 0.56 | GRB2 PXN GAB1 PIK3R1 PIK3CA PAG1 CSK EGFR PTPN11 SRC EGF |
| NGF PATHWAY | 16 | 18 | 0.89 | 0.55 | CSNK2A1 NGF MAPK3 GRB2 MAPK8 PIK3R1 SHC1 PIK3CA ELK1 MAP2K1 SOS1 HRAS PLCG1 JUN RAF1 FOS |
| SIG IL4RECEPTOR IN B LYPHOCYTES | 23 | 27 | 0.85 | 0.55 | JAK1 AKT3 BCL2 BAD PIK3R1JAK3 PIK3CA MAP4K1 GRB2 SOCS1IRS1 AKT1AKT2 GSK3B GSK3A RAF1 MAPK3 IL4R MAPK1 SOS1 SHC1 IRS2 STAT6 |
| IL4 PATHWAY | 9 | 11 | 0.82 | 0.55 | IRS1JAK1 IL4R AKT1 GRB2 IL4 SHC1JAK3 STAT6 |
| INSULIN PATHWAY | 19 | 22 | 0.86 | 0.54 | IRS1 SOS1 MAPK3 MAP2K1 MAPK8 PIK3R1 SHC1 PIK3CA ELK1 SRF CSNK2A1 RAF1 PTPN11 HRAS JUN GRB2 FOS INSR RASA1 |
| GH PATHWAY | 23 | 28 | 0.82 | 0.54 | JAK2 SOS1 MAP2K1 PIK3R1 PIK3CA SRF PLCG1 GRB2 SOCS1 IRS1 HRAS INSR PTPN6 MAPK3 MAPK1 PRKCB PRKCA STAT5A STAT5B SHC1 RPS6KA1 RAF1 GHR |
| RACCYCD PATHWAY | 20 | 26 | 0.77 | 0.52 | MAPK3 CDKN1B CDKN1A CCND1 CHUK AKT1 PIK3R1 PIK3CA RAF1 CDK4 RB1 IKBKB PAK1 CDK2 HRAS E2F1 CDK6 RELA NFKB1 MAPK1 |
| Pathways priortized without seed genes | |||||
| Pathway_name | Only DIAMOnD genes | all pathway genes | coverage | mean score | Genes |
| ACTIVATION OF THE AP1 FAMILY OF TRANSCRIPTION FACTORS | 8 | 10 | 0.80 | 0.59 | MAPK3 MAPK1 MAPK9 MAPK11 MAPK10 FOS ATF2 JUN |
| IL6 PATHWAY | 20 | 22 | 0.91 | 0.58 | CEBPB JAK2 JAK1 MAPK3 SRF STAT3 TYK2 SHC1JAK3 IL6ST ELK1 MAP2K1 SOS1 RAF1 PTPN11 HRAS JUN GRB2 FOS CSNK2A1 |
| EPO PATHWAY | 17 | 19 | 0.89 | 0.57 | JAK2 STAT5B MAPK3 EPOR GRB2 STAT5A SOS1 SHC1 ELK1 MAP2K1 CSNK2A1 HRAS PLCG1 FOS RAF1 JUN PTPN6 |
| AKT PHOSPHORYLATES TARGETS IN THE CYTOSOL | 13 | 14 | 0.93 | 0.57 | CDKN1B PDPK1AKT3 CASP9 AKT1 AKT2 GSK3B MDM2 TSC2 GSK3A CDKN1A BAD CHUK |
| IL3 PATHWAY | 13 | 15 | 0.87 | 0.57 | JAK2 STAT5B MAPK3 GRB2 STAT5A SOS1 SHC1 CSF2RB MAP2K1 HRAS FOS RAF1 PTPN6 |
| CDMAC PATHWAY | 12 | 16 | 0.75 | 0.55 | MAPK3 MAPK1 PRKCB PRKCA FOS MAP2K1 HRAS MYC RAF1 JUN RELA NFKB1 |
| TPO PATHWAY | 19 | 24 | 0.79 | 0.53 | CSNK2A1JAK2 PIK3R1 MAPK3 PRKCB PRKCA STAT3 STAT5A STAT5B SHC1 RAF1 MAP2K1 SOS1 HRAS FOS GRB2 PLCG1 JUN RASA1 |
| GH PATHWAY | 22 | 28 | 0.79 | 0.52 | JAK2 SOS1 MAP2K1 PIK3R1 SRF PLCG1 GRB2 SOCS1 IRS1 HRAS INSR PTPN6 MAPK3 MAPK1 PRKCB PRKCA STAT5A STAT5B SHC1 RPS6KA1 RAF1 GHR |
| IL2 PATHWAY | 17 | 22 | 0.77 | 0.51 | JAK1 MAPK3 GRB2 STAT5A SOS1 SHC1JAK3 IL2RA LCK ELK1 MAP2K1 CSNK2A1 RAF1 STAT5B HRAS JUN FOS |
| TRKA PATHWAY | 10 | 12 | 0.83 | 0.51 | PRKCB AKT1 PRKCA NTRK1 GRB2 SHC1 SOS1 PLCG1 HRAS PIK3R1 |
| IGF1 PATHWAY | 17 | 21 | 0.81 | 0.50 | IRS1 SOS1 MAPK3 SRF IGF1R PIK3R1 SHC1 RAF1 ELK1 MAP2K1 CSNK2A1 HRAS PTPN11 FOS GRB2 JUN RASA1 |
| EGF PATHWAY | 23 | 31 | 0.74 | 0.49 | JAK1 PIK3R1 SRF PLCG1 JUN GRB2 RAF1 RASA1 STAT5B MAP2K1 EGFR HRAS MAPK3 PRKCB PRKCA STAT5A SOS1 SHC1 STAT2 ELK1 STAT3 CSNK2A1 FOS |
| SIGNAL ATTENUATION | 10 | 11 | 0.91 | 0.48 | IRS1 MAPK3 GRB10 GRB2 SOS1 SHC1 DOK1 IRS2 CRK INSR |
| GLEEVEC PATHWAY | 17 | 23 | 0.74 | 0.48 | JAK2 STAT5B MAPK3 AKT1 GRB2 BAD STAT5A PIK3R1 FOS HRAS MAP2K1 BCR SOS1 RAF1 CRKL MYC JUN |
| INSULIN PATHWAY | 17 | 22 | 0.77 | 0.48 | IRS1 SOS1 MAPK3 MAP2K1 PIK3R1 SHC1 ELK1 SRF CSNK2A1 RAF1 PTPN11 HRAS JUN GRB2 FOS INSR RASA1 |
| RACCYCD PATHWAY | 18 | 26 | 0.69 | 0.48 | MAPK3 CDKN1B CDKN1A CCND1 CHUK AKT1 PIK3R1 RAF1 CDK4 RB1 PAK1 CDK2 HRAS E2F1 CDK6 RELA NFKB1 MAPK1 |
| SOS MEDIATED SIGNALLING | 10 | 13 | 0.77 | 0.47 | IRS1 MAPK3 MAPK1 GRB2 SOS1 IRS2 MAP2K1 RAF1 HRAS YWHAB |
| PDGF PATHWAY | 23 | 32 | 0.72 | 0.47 | JAK1 PIK3R1 PDGFRA SRF PLCG1 JUN GRB2 RASA1 STAT5B HRAS FOS PRKCA MAPK3 PRKCB MAP2K1 MAPK8 STAT5A SOS1 SHC1STAT2 ELK1 STAT3 CSNK2A1 RAF1 |
| VDR PATHWAY | 10 | 12 | 0.83 | 0.46 | KAT2B NCOA2 NCOR1 NCOA3 TSC2 EP300 NCOA1 MED1 CREBBP RXRA |
| NGF PATHWAY | 13 | 18 | 0.72 | 0.46 | CSNK2A1 MAPK3 GRB2 PIK3R1 SHC1 ELK1 MAP2K1 SOS1 HRAS PLCG1 JUN RAF1 FOS |
Gene names in bold are ‘seed genes’. Of the 20 pathways, Activation of AP1, TRKA and the GAB1 signalsome pathways are fully embedded in the asthma module.
Identifying new disease mechanisms and predicting drug targets in the asthma module
To experimentally test whether the predicted asthma module offers a perspective for identifying new therapeutic targets, we investigated steroid- and cytokine-induced gene expression in fibroblast cells: if the disease module is relevant to asthma pathogenesis, then treatment of asthma-specific cell lines by corticosteroids, an effective anti-inflammatory therapy for asthma, is expected to lead to gene expression changes localized within the module. Naturally, dampening the inflammation could be achieved by either directly affecting the asthma module, but steroids could also act through other pathways that limit airway inflammation. To experimentally test whether the impact of the steroids is limited to the module, we compared the gene expression profiles between normal untreated fibroblast cells versus normal fibroblasts treated with glucocorticoid (GC) and asthmatic fibroblasts untreated versus asthmatic fibroblast cells treated with GC (Fig. 4E). To check whether the observed enrichment can be attributed to the GCR pathway alone, we re-analyzed the data without GCR pathway genes. We found that the significant enrichment of DIAMOnD genes with differentially expressed genes persists, indicating that the module is indeed influenced by GC response beyond the presence of the GCR pathway (Supplementary Material, Table S8). Overall, three results stand out:
In both normal and asthmatic fibroblast cell lines a statistically significant number of genes responding to GC treatment are located inside the asthma module, but with a different set of genes lighting up in normal and asthmatic subjects. This supports our hypothesis that the module collects genes of potential asthma relevance, where different regions are activated in normal and asthmatics (Fig. 4E). For example, for the normal and asthmatic fibroblast cells we, respectively, find 49 and 44 differentially expressed genes within the asthma module following GC treatment, compared with random expectations of 7.8 and 6.5 (P = 1.67 × 10−9 and 9.02 × 10−9, respectively, Fisher's exact test). The differentially expressed genes of both the normal and the asthmatic state form significant connected components (in both cases z-score = 6.0, empirical P-value < 10−5)
The enrichment of the DIAMOnD genes with differentially expressed genes (38 in normal, and 34 in asthmatic samples, corresponding to P-values 2.6 × 10−8 and 1.3 × 10−7, respectively, Fisher's exact test) is significantly higher than the enrichment of the seed genes (11 differentially expressed genes, P = 0.02). To correct for the different number of genes in the compared gene sets, we also used equal size windows of DIAMOnD genes, the results confirming this conclusion (Supplementary Material, Fig. S8). Further, the mean fold change of seed, asthma module and DIAMOnD genes was significantly higher compared with the background (Supplementary Material, Fig. S9). In summary, these results indicate that the cell's response to GC treatment is heavily localized in the gene set added by the DIAMOnD algorithm, rather than the consensus asthma genes (seeds), thus supporting DIAMOnD's ability to identify genes relevant to therapeutic intervention.
We also observe significant enrichment among genes differentially expressed by GCs within many of the top asthma module pathways, particularly the IGF1 and insulin pathways (Supplementary Material, Table S9). In line with genetic association evidence described earlier, a particularly interesting insight is the emergence of GAB1 as the highest ranked pathway in the response of asthmatic fibroblasts to GC.
Co-expression connection between normal and asthmatic cells for GAB1 pathway genes
Genes in the same biological pathway are more likely to be co-expressed in order to synchronize an array of biochemical reactions (41). We investigated the connectivity pattern of GAB1 pathway genes in our normal and asthmatic fibroblast gene expression data. We observe that in normals only 9 pathway genes are connected with a Pearson correlation r > 0.5 (corresponding P-value <0.05) compared with 11 genes in asthmatic subjects. For example, PAG1 and EGF genes are not significantly correlated with other pathway genes in normal subjects, whereas in asthmatic patients they are negatively correlated with PTPN11, EGFR, PIK3CA and PIK3R1 (Fig. 4F). Also, GAB1, PAG1 and PIKR1 were differentially expressed after the GC treatment in asthmatics (Fig. 4F).
To assess the response of the asthma module to biological perturbations central to the pathogenesis of asthma, we compared the gene expression profiles in fibroblasts from healthy and asthmatic subjects following treatment with either IL13 or IL17. Interleukin-13 has been implicated in the pathogenesis of asthma, and IL-13 blockade has demonstrated efficacy in asthmatics (42). Similarly, Th17-cell-derived IL-17 expression modulates the degree of Th2-mediated inflammation, and has been implicated in the pathogenesis of asthma and other chronic airways diseases (43). Similar to the GC response, the asthma module is enriched for IL13 and IL17 responsive genes compared with the random expectation (Table 2).
Table 2.
Differential expression within the asthma module in normal and asthmatic fibroblast cells under GCs (GC), IL13 and IL17 treatments compared with the random expectation
| Normal | GC Treatment |
IL13 Treatment |
IL17 Treatment |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DE genes in the network: 471 |
p-value | DE genes in the network: 154 |
p-value | DE genes in the network: 96 |
p-value | |||||||
| DE genes | random expectation | FC increase | DE genes | random expectation | FC increase | DE genes | random expectation | FC increase | ||||
| Module genes | 49 | 19.4 | 2.5 | 1.67E-09 | 15 | 6.3 | 2.4 | 0.002 | 10 | 3.9 | 2.5 | 0.0060 |
| DIAMOnD | 38 | 14.2 | 2.7 | 2.58E-08 | 8 | 4.6 | 1.7 | 0.093 | 4 | 2.9 | 1.4 | 0.3264 |
| All seeds | 11 | 5.2 | 2.1 | 0.015 | 7 | 1.7 | 4.1 | 0.002 | 6 | 1.1 | 5.6 | 0.0007 |
| seed in lcc | 11 | 3.4 | 3.2 | 0.001 | 7 | 1.1 | 6.2 | 0.0001 | 6 | 0.7 | 8.6 | 6.93E-05 |
| Asthmatic | DE genes in the network: 419 | p-value | DE genes in the network: 85 | p-value | DE genes in the network: 29 | p-value | ||||||
| DE genes | random expectation | FC increase | DE genes | random expectation | FC increase | DE genes | random expectation | FC increase | ||||
| Module genes | 44 | 17.2 | 2.6 | 9.03E-09 | 12 | 3.5 | 3.4 | 0.0002 | 4 | 1.2 | 3.4 | 0.0297 |
| DIAMOnD | 34 | 12.6 | 2.7 | 1.28E-07 | 7 | 2.6 | 2.7 | 0.0139 | 1 | 0.9 | 1.1 | 0.5877 |
| All seeds | 10 | 4.6 | 2.2 | 0.018 | 5 | 0.9 | 5.3 | 0.0025 | 3 | 0.3 | 9.3 | 0.0039 |
| seed in lcc | 10 | 3.1 | 3.3 | 0.001 | 4 | 0.6 | 6.4 | 0.0034 | 3 | 0.2 | 14.2 | 0.0012 |
The asthma module is significantly enriched with differentially expressed (DE) genes from all the three treatments. In response to treatment, changes in expression of seed genes in the largest connected component (LCC) of the module was significant compared with all seed genes (144).
Interestingly, we find that the seed genes differentially expressed by the three perturbations, (GC, IL13, IL17) are virtually all among the 91 seed genes that reside within the final asthma module. Indeed, only one of the 53 seed genes outside the module was differentially expressed in response to perturbation (P = 0.002, Fisher's exact test). Hence the DIAMOnD algorithm has a unique record in separating pathophysiologically asthma-relevant seed genes from spurious associations. Despite the fact that these 53 genes were considered relevant to asthma in the literature, this marked difference in responsiveness to GC-relevant perturbations suggests that they may be false positives and not part of the asthma disease module.
Replication of GAB1 pathway genes enrichment in the CAMP cohort after dexamethasone treatment
To differentiate the potential role for GAB1 in asthma susceptibility versus drug treatment response, we assessed GAB1 expression in a microarray experiment involving dexamethasone (a glucocorticosteroid)-treated immortalized B-cells derived from 145 childhood asthmatics (44). We find that GAB1 is differentially expressed in dexamethasone-treated cells versus sham controls (FDR-adjusted P-value 8.85 × 10−34). Using a Cox proportional hazards model, higher basal (sham) GAB1 expression was significantly associated with risk of severe asthma exacerbations (hazard ratio 1.46, 95% CI: 1.02–2.11 for each standard deviation of GAB1 expression). Moreover, increased cellular response of GAB1 to dexamethasone resulted in significant protection from severe exacerbations (hazard ratio 0.68, 95% CI: 0.46–0.99 for each standard deviation of GAB1 expression change with dexamethasone). These results support the role of GAB1 in the GC-signaling pathway and as an asthma susceptibility and treatment gene.
Inhibition of NFkB mechanism through GAB1
The topology of the disease module helps us identify potential novel molecular pathways through which GAB1 is involved in asthma, suggesting a relatively proximal link to GCs. Motivated by the local impact hypothesis (45), indicating that the shortest path between two molecules are the most predictive for disease causation, we identified the shortest path within the interactome between the differentially expressed genes in the GAB1 pathway and NR3C1, the GC receptor (Fig. 5A). The local map of the asthma disease module suggests that after the activation by GC, NR3C1 can activate MAPK8 (a seed gene) and several MAPK family members, like MAPK1, MAPK3, MAPK4 (DIAMOnD genes), that could trigger GAB1. In addition to these indirect links, we also note the presence of conserved GC response element (GRE) sequences (46) in the promoter of the GAB1 gene (Fig. 5A, Supplementary Material, Section XIII), suggesting GAB1 as a direct GC (dexamethasone) target. The other member of the family, GAB2, is already known to harbor GREs in neuronal cells (47).
Figure 5.
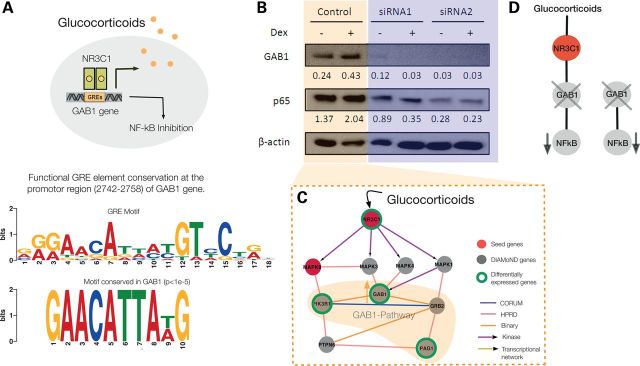
Putative GC mechanism effect via GAB1 in the asthma module. (A) The consensus functional GC response element (GRE) in target genes reads 5′-AGAACAnnnTGTTCT-3′. The probable GRE-binding site conserved in the GAB1 promoter region is 2742–2758 bp. (B) Expression of GAB1 and NFkB (p65) in Beas-2B cells transfected with GAB1-targeted or non-target (NT) control siRNA, as detected by western blot. β-Actin was used as loading control. Band density was quantified using Image J and then normalized to β-actin control. (C) Putative molecular mechanism for the downstream inhibition of NFkB after GC-treatment via the GAB1 signalsome pathway in the asthma module. The activation of NR3C1 via GCs can trigger GAB1 through several MAPK family members. Alternatively, GAB1 may also be a direct GC target as suggested by A. From the 11 genes in the GAB1 signalsome pathway, GAB1, PIK3R1 and PAG1 are differentially expressed both in normal and asthmatic cells. (D) Illustration that GAB1 regulates NFkB regardless of the presence of GCs.
To test these network-based predictions we reasoned that if GC is affecting the level of GAB1, then GAB1 expression should increase with dexamethasone treatment. To test this hypothesis, we performed the in vivo experiment in BEAS-2B bronchial epithelial cells (10 nm, 2 h). As shown in Figure 5B and C, GAB1 levels increase in the presence of dexamethasone, establishing the fact that GAB1 is influenced by the GC treatment.
NFkB controls a variety of processes in asthma, including inflammation. Consequently, regulation of NFkB has been a continued focus of study. GAB1 was shown to promote pro-inflammatory cytokine production by enhancing NFkB activation in macrophages (48). Recently, Vaughan et al. stressed the role of NFkB as a downstream target of GAB1 (49), and it has been postulated that GAB-mediated PI3-K activation has different consequences, depending on the cell type studied (50). However, little is known at the molecular level about the role of GAB1 in asthma. To test whether GAB1 regulates NFkB in BEAS-2B cells, we performed siRNA knockdown of GAB1. Interestingly, we observed decreased expression of NFkB upon GAB1 silencing with or without dexamethasone (Fig. 5D). This observation suggests a new regulatory pathway of the pro-inflammatory factor NFkB by GAB1 in human bronchial epithelial cells.
In summary, our experimental validation indicates that GAB1 positively regulates NFkB, offering a novel mechanistic insight into the control of NFkB and inflammation. Most importantly, this finding offers direct experimental evidence of the utility of the disease module based approach to uncover pertinent novel disease mechanisms.
Discussion
In this paper our goal was to extract the predictive value of the interactome by identifying the localized gene neighborhood associated with asthma. The starting point of our analysis was a list of seed genes with established asthma association. As asthma involves multiple genetic as well as environmental factors, its causal mechanisms are expected to be highly complex. Any list of seed genes must therefore remain incomplete and preliminary. Yet, despite this limitation, we find that the known seed genes already carry enough information to build a robust and predictive disease module.
To date, only a few studies have applied network-based tools to investigate asthma. A study using a protein interaction network suggested hub nodes with high betweenness centrality, such as BRCA1, as potential candidate genes (51). Lu et al. (52), on the other hand, demonstrated that nodes with high connectivity (hubs and superhubs) show little expression variations in asthma, which is consistent with the observation that disease genes tend to be peripheral in the interactome (1). The disease module detection algorithm used in this study avoids hubs by design. Within the identified disease module, we find an inflammatory response signature that is shared with other immune-related diseases, such as Crohn's disease, multiple sclerosis, rheumatoid arthritis and others (Supplementary Material, Section SXIV) (53). This finding is in agreement with recent insights suggesting that similar pathogenetic pathways operate in these inflammatory diseases (54). At the same time, a considerable part of the discovered asthma module is unique, suggesting the existence of a specific region within the interactome that can be used to investigate the functional molecular circuits active only in asthma. In particular, the localization in the interactome allows us to identify and explore high impact pathways that are perturbed in the disease. We find that the GAB1 signalsome is enriched for genes responsive to GC-, IL13 and IL17 treatment. GAB1 variants were associated with asthma susceptibility in a recently published asthma GWAS study conducted in a Japanese population (30). Marginal associations with GAB1 variants in populations of European (GWAS P = 0.006) and Latino (GWAS P = 0.0004), but not of African ancestry have been observed (lowest P-value in the meta-analysis was 0.07) (27). Moreover, strong association of GAB1 expression with risk of severe asthma exacerbations and cellular response of GAB1 to dexamethasone indicates GAB1 being both an asthma gene and a treatment response gene. Abnormal steroid response is a cardinal feature of asthma as exemplified by the identification of NR3C1 as one of our seed genes and by the use of exogenous steroids as the primary drug for asthma treatment. It also suggests that genes for asthma steroid response may also be genes for asthma. Linking the GAB1 signalsome to the GC pathway is a novel finding that emphasizes the importance of abnormal steroid response to asthma development but also suggests possibilities for novel treatments.
The ability to identify specific disease neighborhoods in the interactome opens a series of opportunities to understand the complexity of heterogeneous immune-related diseases. It helps us narrow the vast search space of the full interactome and focus our search for potential disease mechanisms and disease heterogeneity in a well-localized network neighborhood. In this work, we chose to utilize the wide range of available biological information to identify the candidate genes carrying the most significant asthma-associated signals. The cutoff for the selection of the candidate genes is not exact, but the threshold of ∼350 DIAMOnD genes is supported by diverse data. In our view this approach more accurately reflects the diverse biological mechanisms underlying asthma compared with relying solely on the network topology. Moreover, we have focused on a global interactome that does not take tissue-specific interactions into account (Supplementary Material, Section XV and Fig. S10). We expect, however, that incorporating such context specificity can enhance the predictive power of disease modules. The demonstrated success of the approach presented provides a road map for the integrated understanding of the interplay between a disease and the underlying cellular network that can easily be extended to such a scenario.
Materials and Methods
Compiling the interactome and seed genes
The human interactome
We assembled all known physical interactions potentially present in a human cell from appropriate sources. As described in Supplementary Material, Section I, our network contains information from regulatory networks (protein–DNA interaction), high-throughput protein–protein interactions, literature curated protein–protein interactions, metabolic enzyme-coupled interactions, protein complexes and kinase–substrate interactions. In total, the resulting interactome consists of M = 101,032 links between N = 11,643 proteins (Supplementary Material, Fig. S1C).
Disease genes
The seed genes represent a consensus list of genes collected based on their known association with asthma-related phenotypes (26), asthma-related pathology (43), OMIM (19), Gene to MeSH relationship (36) and GWAS data (33) (see Supplementary Material, Table S1 and Section II).
Biological validation datasets
Gene expression data: we selected nine expression data of direct asthma relevance from the Gene Expression database (55). The selection criteria and the details of individual datasets are available in Supplementary Material, Section III.
Asthma-specific pathways: we selected 35 asthma-specific pathways using the GeneGO Meta-Core software (see Supplementary Material, Section IV and Table S2).
General pathways: for broad pathway annotations we used MSIgDB (56) (Supplementary Material, Section V).
Gene Ontologies: to elucidate the biological processes associated with the individual seed genes we used the Gene ontology (GO) biological process category (57) (detailed in Supplementary Material, Section VI).
Comorbidity analysis: we used a large set of Medicare patient medical history data (58) to identify a total of 85 diseases for which asthma patients are at a significantly increased risk (Supplementary Material, Section VII, Table S4). Genes associated with these 85 diseases were considered as validation dataset.
Disease Module Detection Method
The Disease Module Detection (DIAMOnD) algorithm is a topological method to identify the network neighborhood of the seed genes. As documented in Figure 1B and C, seed genes form larger clusters than expected by chance. A hypothesis that can be formulated from this observation is that potential disease genes interact with most of the known disease genes (Supplementary Material, Fig. S1E). For example, IL8 forms 14 connections, of which 4 are known asthma seed genes. Yet, some genes are promiscuous, i.e. they have a large number of interactions to start with. For instance, BRCA1 has three links to asthma seed genes, but 239 links to non-seed genes. For such promiscuous genes, several links to asthma seed genes are not as strong an indication of belonging to the disease cluster than for less connected genes (Fig. 2A). To correct for such biases, we use an algorithm that considers the significance of the number of connections to seed genes as a criterion to prioritize the genes in the network for their asthma relevance. Further details on the method can be found in the Supplementary Material, Section IX.
Childhood Asthma Management Program (CAMP) GCs data
One hundred and forty-five CAMP participants who were randomized to the budesonide (a glucocorticosteroid) treatment arm constituted the cohort in which the longitudinal response to GC therapy was assessed. We focused on severe exacerbations, defined as an emergency room visit or overnight hospitalization due to asthma. Thirty-two of the tested CAMP subjects had at least one exacerbation during the 4-year follow-up. The gene expression analysis was done on immortalized B-cells directly derived from the clinical subjects as described previously (44). We tested the association of GAB1 expression with the time to first severe exacerbations, using a Cox proportional hazards model. All research involving data collected from the CAMP Genetics Ancillary Study was conducted at the Channing Laboratory of the Brigham and Women's Hospital according to the appropriate CAMP policies and regulations for human-subjects protection.
The Asthma BioRepository Genomic Exploration
The Asthma BRIDGE database is an open-access repository of immortalized cell lines from 1435 subjects participating in genetic studies of asthma, with an accompanying database consisting of phenotype data and genome-wide genetic (SNP), genomic (RNA expression) and epigenetic (CpG DNA methylation) data. We performed a gene-set enrichment analysis (GSEA) of the ranked associations using the GSEA2 software (http://www.broadinstitute.org/), assessing whether each signature is significantly enriched for genes in the other. We observed significant enrichment (FDR < 0.25) of 288 MSigDB gene sets among genes whose increased expression was associated with below-median mACT and two gene sets associated with above-median mACT.
Identifying the most relevant asthma pathways within the module
For all pathways in the KEGG, Biocarta and Reactome databases in MSIgDB we compute the enrichment with genes from the asthma module (Fisher's exact test), as well as a mean score that considers all the different datasets used for the biological validation (Supplementary Material, Section XI and XII). For the SNP to gene annotation in the GWAS data, we applied the LDsnpR method that is based on both the physical position of a SNP and its pairwise LD with other SNPs (29). Since the status of an individual gene in differentially expressed gene sets, GWAS signals and other biological datasets directly reflects its relevance to asthma, the mean score of all genes in a pathway is a sensible choice to quantify its overall relevance.
Comparison with other network-based methodologies
For a comparison of the performance of DIAMOnD to existing candidate gene identification algorithms we implemented five methods, four of which being network-based as well:
We choose a method based on a random walk (RW) starting from the seed genes (33), which was found to perform best among several available network-based gene prioritization tools.
DaDa (Degree-aware algorithms for Network-based Disease Gene Prioritization), a set of methods that implement various adjustments for the skewed degree distribution of protein interaction networks (36).
PRINCE (PRIortization and Complex Elucidation) (35), a method that does not only use local information, but considers global network properties as well.
CIPHER (Correlating protein Interaction network and PHEnotype network to pRedict disease genes), which used a regression model to evaluate the functional relatedness of genes (34).
As a final comparison, we tested DIAMOnD against the Endeavour algorithm that uses a basic machine learning approach to rank candidate genes (37). Endeavour is one of the most widely accepted sequence-based candidate gene prioritization tools.
For the general evaluation of these methods we used the same seed genes and interactome, as well as the same validation data as introduced above: (i) GeneGo pathways, (ii) MSIgDB pathways, (iii) Gene ontologies and (iv) comorbidity analysis. For the asthma-specific validation we used (v) gene expression data from the literature (Supplementary Material, Section III), (vi, vii) gene expression data from fibroblast cells under GC treatment and (viii) asthma GWAS data. Supplementary Material, Figure S3A exemplarily shows the number of ‘hits’ of the different methods for the generic gene expression data (compare also with Fig. 2D(i)): for each gene in their respective ranked gene lists we determine, whether it was found to be differentially expressed. While the different methods show comparable performance, DIAMOnD yields the highest number hits. At iteration step 350, corresponding to the total number of DIAMOnD genes we use in the final asthma module, the DIAMOnD genes include 117 differentially expressed genes, the next best method, RW, 111. Note that generally it is difficult to define strict benchmark sets of true positives and true negatives for the disease association of genes. For a comparative analysis of the different algorithms, however, their absolute values are not necessary and we can therefore simply use the genes contained in the respective validation data (i)–(v) as true positives, all others as true negatives.
Steroid and cytokines induced gene expression in fibroblast cells
Bronchial fibroblasts cultured from 10 healthy and 5 fatal asthmatic individuals were treated with fluticasone propionate (20 nm) (Supplementary Material, Tables S11 and S12), IL13 (10 ng/ml) (Supplementary Material, Tables S13 and S14) and IL17 (10 ng/ml) (Supplementary Material, Tables S15 and S16). RNA isolated from the cells was subject to microarray analyses. Raw intensities from all samples were merged, normalized and log2 transformed. We performed statistical t-test for differential mRNA expression between treated and untreated controls and asthmatics fibroblast cells separately. Both controls and asthmatic fibroblast cells (45 000 cells per well) were treated with 20 nm GCs, IL13 (10 ng/ml) and IL17 (10 ng/ml). Differentially expressed mRNAs were defined as having a fold change >1.5 and a raw P-value <0.05 (paired t-test comparisons) between the treated and untreated groups.
GAB1 siRNA knockdown study
Depletion of GAB1 by RNAi transfection
To determine the regulation of GAB1 on the NFkb pathway, we performed knockdown of GAB1 in Beas-2B cells, a human bronchial epithelial cell line using two individual siRNAs as we have done previously (59). Briefly, 40% confluent Beas-2B cells were transfected with 100 pmol/well of GAB1-specific siRNAs or a non-targeting siRNA control (Life Technologies) using lipofectamine 2000 (Life Technologies). Forty-eight hours post transfection; Beas-2B cells were treated with either sham or dexamethasone (10 nm) for 2 h before cells were being collected for western blot. Knockdown efficiency of GAB1 siRNA was confirmed by western blot.
Immunological methods
Western blotting was performed as previously described (60). Antibodies used were β-actin (Sigma, #A5441), GAB1 (Cell Signaling, #3232), NFkB (Cell Signaling, #8242). Secondary antibodies were horseradish peroxidase-linked anti-mouse or anti-rabbit IgG (GE Healthcare). Signals were detected using an enhanced chemiluminescence kit (Perkin Elmer Life Sciences, Inc.) and Imaging auto-developing system (Syngene Inc.). Band densities were quantified by Image J software.
Supplementary Material
Funding
This research received funding from Janssen R&D and was supported in part by National Institutes of Health (NIH) grants P50-HG004233-CEGS, MapGen grant (1U01HL108630-01) and 5P01-HL083069-5, U01 HL065899, P01 HL105339, R01HL111759 and -RC HL10154301.
Supplementary Material
Acknowledgements
We thank Pitchumani Sivakumar, Mike Pratta, Matthew Loza and Anton Bittner from Janssen R&D for their help in data collection and Gabriele Musella for figure design.
Conflicts of Interest statement. A.-L.B. is a consultant to Janssen R&D and has ownership interests in Netsci Consutling. A.-L.B. and J.M. are co-founders and shareholders of DZZOM.
References
- 1.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabasi A.L. (2007) The human disease network. Proc. Natl Acad. Sci. USA., 104, 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oti M., Snel B., Huynen M.A., Brunner H.G. (2006) Predicting disease genes using protein-protein interactions. J. Med. Genet., 43, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gandhi T.K., Zhong J., Mathivanan S., Karthick L., Chandrika K.N., Mohan S.S., Sharma S., Pinkert S., Nagaraju S., Periaswamy B., et al. (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet., 38, 285–293. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J., Zhang C., Lamb J., Edwards S., Sieberts S.K., et al. (2008) Variations in DNA elucidate molecular networks that cause disease. Nature, 452, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ideker T., Sharan R. (2008) Protein networks in disease. Genome. Res., 18, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabasi A.L., Gulbahce N., Loscalzo J. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet., 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J., Li Y. (2006) Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics, 22, 2800–2805. [DOI] [PubMed] [Google Scholar]
- 8.Agusti A., Sobradillo P., Celli B. (2011) Addressing the complexity of chronic obstructive pulmonary disease: from phenotypes and biomarkers to scale-free networks, systems biology, and P4 medicine. Am. J. Respir. Crit. Care Med., 183, 1129–1137. [DOI] [PubMed] [Google Scholar]
- 9.Barnett S.B., Nurmagambetov T.A. (2011) Costs of asthma in the United States: 2002–2007. J. Allergy. Clin. Immunol.,127, 145–152. [DOI] [PubMed] [Google Scholar]
- 10.Akhabir L., Sandford A.J. (2011) Genome-wide association studies for discovery of genes involved in asthma. Respirology, 16, 396–406. [DOI] [PubMed] [Google Scholar]
- 11.Baranzini S.E., Galwey N.W., Wang J., Khankhanian P., Lindberg R., Pelletier D., Wu W., Uitdehaag B.M., Kappos L., Polman C.H., et al. (2009) Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet., 18, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., Cotsapas C., Daly M.J. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet., 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pers T.H., Dworzynski P., Thomas C.E., Lage K., Brunak S. (2013) MetaRanker 2.0: a web server for prioritization of genetic variation data. Nucleic Acids Res., 41, W104–W108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu Z., Argmann C., Wong K.K., Mitnaul L.J., Edwards S., Sach I.C., Zhu J., Schadt E.E. (2009) Integrating siRNA and protein-protein interaction data to identify an expanded insulin signaling network. Genome Res., 19, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkatesan K., Rual J.F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K.I., et al. (2009) An empirical framework for binary interactome mapping. Nat. Meth., 6, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart G.T., Ramani A.K., Marcotte E.M. (2006) How complete are current yeast and human protein-interaction networks? Genome Biol., 7, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidal M., Cusick M.E., Barabasi A.L. (2011) Interactome networks and human disease. Cell, 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walhout A.J., Vidal M. (2001) Protein interaction maps for model organisms. Nat. Rev. Mol. Cell Biol., 2, 55–62. [DOI] [PubMed] [Google Scholar]
- 20.Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Seraphin B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotech., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- 21.Girvan M., Newman M.E. (2002) Community structure in social and biological networks. Proc. Natl Acad. Sci. USA., 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L. (2002) Hierarchical organization of modularity in metabolic networks. Science, 297, 1551–1555. [DOI] [PubMed] [Google Scholar]
- 23.Vincent D., Blondel J.-L.G., Lambiotte Renaud, Lefebvre Etienne. (2008) Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp., 10, P10008. [Google Scholar]
- 24.Ahn Y.Y., Bagrow J.P., Lehmann S. (2010) Link communities reveal multiscale complexity in networks. Nature, 466, 761–764. [DOI] [PubMed] [Google Scholar]
- 25.Enright A.J., Van Dongen S., Ouzounis C.A. (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res., 30, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghiassian S.D., Menche J., Barabási A.L. (2015) A DiseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human Interactome. PLoS Comp. Biol. (in press).
- 27.Torgerson D.G., Ampleford E.J., Chiu G.Y., Gauderman W.J., Gignoux C.R., Graves P.E., Himes B.E., Levin A.M., Mathias R.A., Hancock D.B., et al. (2011) Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nature Gen., 43, 887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moffatt M.F., Gut I.G., Demenais F., Strachan D.P., Bouzigon E., Heath S., von Mutius E., Farrall M., Lathrop M., Cookson W.O. (2010) A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med., 363, 1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christoforou A., Dondrup M., Mattingsdal M., Mattheisen M., Giddaluru S., Nothen M.M., Rietschel M., Cichon S., Djurovic S., Andreassen O.A., et al. (2012) Linkage-disequilibrium-based binning affects the interpretation of GWASs. Am. J. Hum. Genet., 90, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirota T., Takahashi A., Kubo M., Tsunoda T., Tomita K., Doi S., Fujita K., Miyatake A., Enomoto T., Miyagawa T., et al. (2011) Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nature Gen., 43, 893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreau Y., Tranchevent L.C. (2012) Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nature Rev. Gen., 13, 523–536. [DOI] [PubMed] [Google Scholar]
- 32.Hutz J.E., Kraja A.T., McLeod H.L., Province M.A. (2008) CANDID: a flexible method for prioritizing candidate genes for complex human traits. Genet Epidemiol., 32, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohler S., Bauer S., Horn D., Robinson P.N. (2008) Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet., 82, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X., Jiang R., Zhang M.Q., Li S. (2008) Network-based global inference of human disease genes. Mol. Sys. Bio., 4, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanunu O., Magger O., Ruppin E., Shlomi T., Sharan R. (2010) Associating genes and protein complexes with disease via network propagation. PLoS Comp. Biol., 6, e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erten S., Bebek G., Ewing R.M., Koyuturk M. (2011) DADA: degree-aware algorithms for network-based disease gene prioritization. BioData Min., 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aerts S., Lambrechts D., Maity S., Van Loo P., Coessens B., De Smet F., Tranchevent L.C., De Moor B., Marynen P., Hassan B., Carmeliet P., Moreau Y. (2006) Gene prioritization through genomic data fusion. Nature Biotech., 24, 537–544. [DOI] [PubMed] [Google Scholar]
- 38.Zhernakova A., van Diemen C. C., Wijmenga C. (2009) Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nature Rev. Gen., 10, 43–55. [DOI] [PubMed] [Google Scholar]
- 39.Desmet C., Gosset P., Henry E., Garze V., Faisca P., Vos N., Jaspar F., Melotte D., Lambrecht B., Desmecht D., et al. (2005) Treatment of experimental asthma by decoy-mediated local inhibition of activator protein-1. Am. J. Respir. Crit. Care Med., 172, 671–678. [DOI] [PubMed] [Google Scholar]
- 40.Freund-Michel V., Frossard N. (2008) The nerve growth factor and its receptors in airway inflammatory diseases. Pharmacol. Ther., 117, 52–76. [DOI] [PubMed] [Google Scholar]
- 41.Kumari S., Nie J., Chen H.S., Ma H., Stewart R., Li X., Lu M.Z., Taylor W.M., Wei H. (2012) Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS ONE, 7, e50411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corren J., Lemanske R.F., Hanania N.A., Korenblat P.E., Parsey M.V., Arron J.R., Harris J.M., Scheerens H., Wu L.C., Su Z., et al. (2011) Lebrikizumab treatment in adults with asthma. N. Engl. J. Med., 365, 1088–1098. [DOI] [PubMed] [Google Scholar]
- 43.Warner J.A., Davies D.E., Andrews A.L. (2011) Effects of Il-13 and Il-17 on fibroblasts derived from the proximal and distal airways. Am. J. Respir. Crit. Care Med., 183, A3598. [Google Scholar]
- 44.Tantisira K.G., Lasky-Su J., Harada M., Murphy A., Litonjua A.A., Himes B.E., Lange C., Lazarus R., Sylvia J., Klanderman B., et al. (2011) Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N. Engl. J. Med., 365, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gulbahce N., Yan H., Dricot A., Padi M., Byrdsong D., Franchi R., Lee D.S., Rozenblatt-Rosen O., Mar J.C., Calderwood M.A., et al. (2012) Viral perturbations of host networks reflect disease etiology. PLoS Comp. Biolo., 8, e1002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alheim K., Corness J., Samuelsson M.K., Bladh L.G., Murata T., Nilsson T., Okret S. (2003) Identification of a functional glucocorticoid response element in the promoter of the cyclin-dependent kinase inhibitor p57Kip2. J. Mol. Endocrinol., 30, 359–368. [DOI] [PubMed] [Google Scholar]
- 47.Polman J.A., Welten J.E., Bosch D.S., de Jonge R.T., Balog J., van der Maarel S.M., de Kloet E.R., Datson N.A. (2012) A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci., 13, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Y., An H., Yao M., Hou J., Yu Y., Feng G., Cao X. (2010) Scaffolding adaptor protein Gab1 is required for TLR3/4- and RIG-I-mediated production of proinflammatory cytokines and type I IFN in macrophages. J. Immun., 184, 6447–6456. [DOI] [PubMed] [Google Scholar]
- 49.Vaughan T.Y., Verma S., Bunting K.D. (2011) Grb2-associated binding (Gab) proteins in hematopoietic and immune cell biology. Am. J. Blood Res., 1, 130–134. [PMC free article] [PubMed] [Google Scholar]
- 50.Sarmay G., Angyal A., Kertesz A., Maus M., Medgyesi D. (2006) The multiple function of Grb2 associated binder (Gab) adaptor/scaffolding protein in immune cell signaling. Immunol. Lett., 104, 76–82. [DOI] [PubMed] [Google Scholar]
- 51.Hwang S., Son S.W., Kim S.C., Kim Y.J., Jeong H., Lee D. (2008) A protein interaction network associated with asthma. J. Theor. Biol., 252, 722–731. [DOI] [PubMed] [Google Scholar]
- 52.Lu X., Jain V.V., Finn P.W., Perkins D.L. (2007) Hubs in biological interaction networks exhibit low changes in expression in experimental asthma. Mol. Syst. Biol., 3, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang I.M., Zhang B., Yang X., Zhu J., Stepaniants S., Zhang C., Meng Q., Peters M., He Y., Ni C., et al. (2012) Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol., 8, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kidd B.A., Peters L.A., Schadt E.E., Dudley J.T. (2014) Unifying immunology with informatics and multiscale biology. Nat. Immunol., 15, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novershtern N., Itzhaki Z., Manor O., Friedman N., Kaminski N. (2008) A functional and regulatory map of asthma. Am. J. Respir. Cell Mol. Biol., 38, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdottir H., Tamayo P., Mesirov J.P. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Gent., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hidalgo C.A., Blumm N., Barabasi A.L., Christakis N.A. (2009) A dynamic network approach for the study of human phenotypes. PLoS Comput. Biol., 5, e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou X., Qiu W., Sathirapongsasuti J. F., Cho M. H., Mancini J. D., Lao T., Thibault D. M., Litonjua A. A., Bakke P. S., Gulsvik A., et al. (2013) Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics, 101, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou X., Munger K. (2010) Clld7, a candidate tumor suppressor on chromosome 13q14, regulates pathways of DNA damage/repair and apoptosis. Cancer Res., 70, 9434–9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.