

Background: Cellulosomal cohesin-dockerin interactions show intraspecies promiscuity but interspecies specificity.
Results: A combination of computations and experiments reveals single cohesin residue mutations with dramatic effects not only on binding affinity but also on specificity and promiscuity.
Conclusion: Natural interspecies specificity barriers in the cohesin-dockerin interaction are easily overcome by single mutations, indicating considerable plasticity.
Significance: This study sheds light on the malleability and evolvability of a high affinity interaction.
Keywords: carbohydrate-binding protein, cellulosome, Clostridium cellulolyticum, computer modeling, protein complex, substrate specificity, Clostridium thermocellum, cohesin, dockerin, iELISA
Abstract
Interactions between cohesin and dockerin modules play a crucial role in the assembly of multienzyme cellulosome complexes. Although intraspecies cohesin and dockerin modules bind in general with high affinity but indiscriminately, cross-species binding is rare. Here, we combined ELISA-based experiments with Rosetta-based computational design to evaluate the contribution of distinct residues at the Clostridium thermocellum cohesin-dockerin interface to binding affinity, specificity, and promiscuity. We found that single mutations can show distinct and significant effects on binding affinity and specificity. In particular, mutations at cohesin position Asn37 show dramatic variability in their effect on dockerin binding affinity and specificity: the N37A mutant binds promiscuously both to cognate (C. thermocellum) as well as to non-cognate Clostridium cellulolyticum dockerin. N37L in turn switches binding specificity: compared with the wild-type C. thermocellum cohesin, this mutant shows significantly increased preference for C. cellulolyticum dockerin combined with strongly reduced binding to its cognate C. thermocellum dockerin. The observation that a single mutation can overcome the naturally observed specificity barrier provides insights into the evolutionary dynamics of this system that allows rapid modulation of binding specificity within a high affinity background.
Introduction
What determines binding specificity, and how easily can this specificity be changed during evolution? These are fundamental questions regarding molecular interactions in biological systems. Nature manipulates binding specificity and affinity using different strategies (1) ranging from subtle changes at the level of residue point mutations in distinct regions of the interface (2) to more dramatic changes such as loop insertion or deletion (3, 4). Protein binding interfaces consist in general of independent patches of networks of interacting residues (5). Such patches can perform different tasks in the interaction (6).
Anaerobic, cellulose-degrading bacteria produce a sophisticated multiprotein complex called cellulosome that is optimized for efficient degradation of cellulose (7). The different components of the cellulosome are held together by interactions between two distinct modules: cohesin modules that occur in repeats on the scaffoldin protein and dockerin modules that are attached to hydrolases or to additional scaffoldins (Fig. 1A). The cohesin-dockerin interaction (see Fig. 1B) is characterized by very high affinity (8, 9). The small ∼70-residue dockerin module contains two F-hand motif repeats (10, 11) that allow binding of its cohesin partner in two symmetric orientations (see Fig. 1, C and D) (12, 13). The cohesin module folds into a β sandwich and binds to dockerin using one of its β sheets; binding specificity is achieved by the loops at the periphery of this sheet (12–16). Cohesin-dockerin interactions can be classified into three general types according to their sequence similarity (7). In C. thermocellum and C. cellulolyticum, enzyme-associated dockerins are in general grouped as type I, whereas dockerins that participate in attachment of scaffoldins to the cell belong to the type II family (represented in Fig. 1A as CohI-DocI and CohII-DocII, respectively). Within a given species, dockerin and cohesins may interchangeably bind (within the same family type), thereby increasing cellulosomal heterogeneity (notably its enzyme content). In contrast to this binding promiscuity for different cohesin modules of the same species, interactions between dockerin and cohesin modules from different species have not been often observed (Ref.17; for an exception, see e.g. Sakka et al. (18)).
FIGURE 1.

Overview of the cellulosome and the cohesin-dockerin complex in C. thermocellum. A, scheme of the cellulosome organization. The primary scaffoldin (yellow) contains nine type I cohesin modules (CohI), a single cellulose-specific CBM, and a type II dockerin module (XDocII). Each type I cohesin binds a type I dockerin module (DocI). This module occurs in a variety of cellulases and other carbohydrate-active enzymes (shades of blue and green). The primary scaffoldin (yellow) is attached to one of several cell-anchoring scaffolds via interaction between type II dockerin (XDocII) and type II cohesin (CohII). The anchoring scaffoldins (pale orange) contain type II cohesin modules and a surface layer homology (SLH) module that is connected to the cell surface. B, structure of the type I cohesin-dockerin interface (cohesin, red; dockerin, blue; Protein Data Bank code 1OHZ (12)). The hydrophobic patch at the interface is shown in spheres; the residues involved in the hydrogen bond network are depicted as sticks (atoms in this and subsequent figures are colored in Corey-Pauling-Koltun). C and D, sequence conservation at the cohesin-dockerin interface. The hydrophobic patch at the cohesin-dockerin interface is conserved across species (C). In contrast, the network of hydrogen bonds is conserved within C. thermocellum (Ct) but replaced by hydrophobic residues in C. cellulolyticum (Cc) cohesin-dockerin interfaces (D). Multiple sequence alignments are shown for different cohesin repeats (red) and dockerins from different enzymes from C. thermocellum and C. cellulolyticum (blue). The dual binding mode is shown in cartoon representation. The first dockerin segment is colored dark blue and the second is colored light blue; calcium ions are shown as green spheres.
The cohesin-dockerin interaction is an ideal model system for the assessment of minimal changes needed for modulation of binding promiscuity and specificity. How many mutations are needed and where are they located? Rational manipulation of the cohesin-dockerin interaction is a precondition toward more elaborate designs of multimolecular cellulosomes of specific architecture and composition in the future (19–21).
Previous studies identified specificity-determining residues in the dockerin module based on sequence conservation within and across different species (8, 22). In a gene-swapping experiment, Nakar et al. (23) were able to switch the binding specificity of a cohesin from C. cellulolyticum to dockerin of C. thermocellum by the replacement of only three residues. In their large scale assessment of correlated mutations, Halperin et al. (24) harnessed the ample information about intraspecies binding promiscuity and interspecies binding specificity in the cohesin-dockerin interaction to identify interface residues of this interaction in a precise manner.
Here we used a structure-based approach to determine which residues contribute to the high affinity observed in this interaction and which in turn are important for binding specificity. We applied a combination of computational design and binding experiments to identify crucial features in the C. thermocellum cohesin that determine the structural and physical bases for binding affinity and specificity in the type I cohesin-dockerin interface. We characterized residues at the cohesin-dockerin interface by computational modeling using the Rosetta molecular modeling suite (25) and validated these predictions using indirect ELISA (iELISA)4 specifically developed by us to measure effects on binding of high affinity interactions (26, 27). Evaluation of the effect of these mutations on binding by a number of additional computational as well as experimental approaches allowed us to provide a robust picture of the main determinants of binding affinity and specificity of this interaction. Our study identified two types of hot spot residues in C. thermocellum cohesin: affinity hot spots such as Leu83 in the conserved hydrophobic patch of the cohesin-dockerin interface contribute significantly to binding affinity, whereas the specificity hot spot Asn37 in the hydrogen bond network in the C. thermocellum cohesin-dockerin interface plays a crucial role in determining binding specificity.
Experimental Procedures
Computational Protocols
Cohesin and Dockerin Modules and Structures
Throughout this study, we refer to the C. thermocellum complex between the second cohesin module in scaffoldin A (residues 183–322) and the dockerin connected to endo-1,4-β-xylanase Y (Xyn10B; residues 733–788) (Protein Data Bank code 1OHZ (12)) and to the C. cellulolyticum complex between the first cohesin module on scaffoldin C (residues 277–427) and the dockerin connected to endoglucanase A (Cel5A; residues 410–472, A16S/L17T mutant) (Protein Data Bank code 2VN6 (16)). Residue numbering refers to these structures. In this study, we report modeling studies based on the dockerin orientation that positions recognition residues Ser45/Thr46 and Ala47/Phe48 for C. thermocellum and C. cellulolyticum, respectively, at the interface. Structures of the inverse, symmetrical binding mode show a very similar interface (C. thermocellum, Protein Data Bank code 2CCL (13); C. cellulolyticum, Protein Data Bank code 2VN5 (16)) and provide similar results (data not shown).
Correction of an Incorrectly Fitted Side Chain at Cohesin Position Asn37 in the Crystal Structure of the C. thermocellum Cohesin-Dockerin Complex
Electron density maps do not allow distinction between the two different possible planar side chain orientations. The positions of asparagine side chain Oδ1 and Nδ2 atoms (as well as glutamine side chain atoms Oϵ1 and Nϵ2) are therefore usually defined based on optimization of the surrounding polar environment and the optimal satisfaction of hydrogen bonds. The NQ-Flipper protocol (28) suggests that Asn37 in structure 1OHZ is misfitted. Therefore, we used in all computations a starting structure of the C. thermocellum cohesin-dockerin interaction in which the side chain of Asn37 had been flipped for optimal positioning of its side chain hydrogen bond donor and acceptor as also suggested previously (29).
Computational Alanine Scanning and Interface Design
The structure-based computational analysis of the cohesin-dockerin interaction was performed using the Rosetta modeling framework in which structure optimization and sequence design are performed using a stochastic search based on Monte Carlo with minimization and an energy function dominated by tight and clash-free packing, burial of hydrophobic residues, and satisfying hydrogen bonds (25, 30–32). We used Rosetta version 2.3.0 throughout this study, i.e. Rosetta revision 12795 and Rosetta database revision 21964, unless mentioned otherwise. Rosetta is available free of charge to academic users.
We applied the Rosetta interface mode for alanine scanning to identify interface residue hot spots as well as for the design of sequences with modified affinity (33). The contribution of different interface residues to binding was evaluated by mutation to alanine and measuring the effect on binding: ΔΔGbind = ΔGbind mutant − ΔGbind WT where ΔGbind = Gcomplex − Gfree protein A − Gfree protein B. A threshold of ΔΔGbind ≥1.0 kcal/mol was used to select putative hot spot residues (as in e.g. Refs. 31 and 34). The ΔΔG predictions were calculated by introducing minimal changes in the structure as we would not expect major backbone changes (the evaluated residues and their neighbors are restricted: they lie within the β-sheet of the cohesin or within the helix or calcium binding loop of the dockerin). The starting structure was first optimized by minimizing the complex structure. Upon mutation, neighboring side chain atoms were minimized, but side chains were not repacked and therefore not allowed to rearrange significantly.
The command line consists of:

where <INPUT_PDB> can be an experimentally solved structure or a model obtained by docking (see below), -interface invokes the interface mode, the mutations to model (e.g. N37D and N37A/D39A) are specified in the file defined by -mutlist, -output_structure writes the structures modeled during the simulation into Protein Data Bank format files, and the results specifying ΔG and energy values of the wild type and mutants are written to the output file specified by -intout. Torsion angles (both backbone and side chain) and rigid body orientation were minimized prior to the modeling of the mutation.
We compared this protocol (Rosetta 2.3) with a parameterized protocol for modeling effects of point mutations (termed here Rosetta 3.0* and described in detail in Kellogg et al. (34) as protocol16). For these calculations, we used Rosetta revision 34507 and Rosetta database version 40221. The main differences between these two protocols is that the latter allows the repacking of all interface side chain residues together with optimized weights of the scoring function and a combination of soft repulsive repacking followed by standard, hard repulsive minimization of both backbone and side chain atoms under constraints that tether the structure to the starting conformation.
Protocol16 consists of the following command line:

The resfile supplied allows repacking of all residues, including the input side chain conformation, choosing from a rotamer library with extended χ1 and χ2 angle sampling. As an example, the resfile for mutation N37A would contain the following:

In short, this protocol first repacks the input structure and then performs the mutation of interest. Starting both from the wild-type and the mutated structure, 50 independent runs of minimization of all torsion and rigid body degrees of freedom are performed for each. ΔΔG is then calculated as the difference in energy between the minimal energy conformation of the mutant and the wild type (among the 50 simulations of each).
In addition, we compared the Rosetta-based protocols with the following other approaches: 1) FoldX (35) version 3.0 Beta3 run locally using default values; 2) EPPI-2 Orbit interface energy function EPPI-2 (36) for which two different implementations of EPPI-2 were used: EPPI-2 values were transformed to optimize correlation to a large set of 404 experimental ΔΔGbind values (Orbit1) or a restricted set of 53 designed mutations (Orbit2) (corresponding to Fig. 3, C and F, in Ref. 36, respectively); 3) Hunter (37); and 4) Concoord/PBSA (38).
FIGURE 3.

The network of hydrogen bonds in the cohesin-dockerin interface of C. thermocellum contains cohesin residues responsible for both affinity (Asp39 and Glu131) and specificity (Asn37). A, detailed structural view of the polar network of hydrogen bonds that is conserved in C. thermocellum (Ct) cohesin-dockerin interactions. Hydrogen bonds (green dotted lines) mediated by interface residues (sticks; colored in Corey-Pauling-Koltun) and water molecules (cyan) are shown (see Fig. 1D for conservation of hydrogen bond network). B, schematic view of the network highlighting the central cohesin residues involved in the network, Asp39, Asn37, and Glu131. These residues contact the known specificity-determining C. thermocellum dockerin residues Ser45 and Thr46. C, iELISA measurements of the effect on binding of C. thermocellum cohesin mutants at positions Asn37, Asp39, and Glu131 to C. thermocellum dockerin. Removal of charge in E131A and even more so in D39A impairs binding to C. thermocellum dockerin. In contrast, N37A does not significantly influence binding but rather dampens the effect observed for E131A (and to a lesser extent also for D39A) as manifested by the corresponding double mutants N37A/E131A and N37A/D39A. D, iELISA measurements of the effect of binding of C. thermocellum cohesin mutants N37D and D39N to C. thermocellum dockerin. Changes in charge and in the directionality of the network of hydrogen bonds impair the interaction significantly. Cc, C. cellulolyticum. See Fig. 2 legend for more details about the iELISA measurements.
Modeling of Non-cognate Cohesin-Dockerin Interactions Using RosettaDock
A model of the non-cognate interaction was generated using as partners the C. thermocellum cohesin structure from Protein Data Bank structure 1OHZ (12) where the residue Asn37 was truncated to alanine (N37A) and the C. cellulolyticum dockerin structure from Protein Data Bank codes 2VN5 and 2VN6 (16).
We used RosettaDock (with off-rotamer minimization of the side chain conformations (30, 39, 40)) to model the structure of non-cognate cohesin-dockerin pairs based on the monomer structures taken from the cognate C. thermocellum and C. cellulolyticum complex structures (see above). We considered two possible orientations for modeling the complex between C. thermocellum cohesin mutant N37A and the C. cellulolyticum dockerin since either the first (Ala16-Leu17) or second (Ala47-Phe48) dockerin recognition motif can interact with the cohesin module (Protein Data Bank codes 2VN5 and 2VN6, respectively (16)) because of the dual mode of cohesin-dockerin binding. In our calculations, the second orientation predicted better ΔG values (−20.5 versus −17.0 Rosetta energy units) and was therefore chosen for subsequent analysis.
Docking was performed as described previously (40). The docking command line consists of the following:

-dock_pert 3 8 8 will start the refinement protocol from a structure that has been slightly perturbed to sample the local energy landscape: random moves according to Gaussian distributions with 3-Å (8-Å) standard deviation (S.D.) are performed on translations along the axis that connects the centers of mass (and the two perpendicular axes), and according to a Gaussian distribution with 8° S.D. on rotations around the three Euler angles (30), -dock_mcm indicates the docking protocol that involves Monte Carlo sampling with minimization of rigid body orientation, -dock_rtmin allows sampling of off-rotamer side chain conformations, -unbound_rot adds for each position the side chain conformation encountered in the free (i.e. starting) monomer conformation to the rotamer library and assigns it minimal energy to bias toward this conformation, -ex1 and -ex2aro allow the inclusion of additional rotamers (±S.D. for χ1 for all amino acids and for χ2 of aromatic residues), -dock_score_norepack will just rescore the starting structure rather than repacking and minimizing it, -nstruct 1000 will generate 1000 independent models, and -scorefile XXXX.score indicates the name of the score output file.
In a preceding step, the free monomers were prepacked to remove any internal clashes. In this step, the same sampling of side chain conformations was allowed. The command line consists of the following:
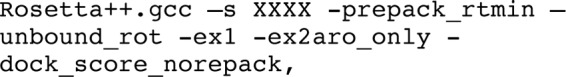
where -prepack_rtmin allows sampling of off-rotamer side chain conformations, and the other parameters are as described above.
Experimental Protocols
Expression Plasmids
Plasmid cassettes of the fusion proteins xylanase-fused dockerin (XynDoc) and carbohydrate-binding module-borne cohesin (CBM-Coh) were produced as described earlier (29, 41, 42). We used the cohesin constructs coh2-CBM of C. thermocellum scaffoldin CipA (the second cohesin module and following cellulose-binding module) and miniCipC of C. cellulolyticum scaffoldin C (CBM, hydrophilic domain, and the first cohesin module) and the dockerin constructs C. thermocellum XynDocS (C. thermocellum Cel48S dockerin) and C. cellulolyticum XynDocA (C. cellulolyticum Cel5A dockerin). Mutations of cohesin residues were performed by site-directed mutagenesis using the QuikChange kit (Stratagene). The mutations were verified by DNA sequencing. Mutant cohesin fragments were restricted with BamHI and XhoI enzymes and ligated into pET28a, which was also digested with the same enzymes. The final construct was verified by sequencing.
Expression and Purification of Proteins
Wild-type and mutant CBM-Coh constructs were expressed and purified by affinity chromatography on a cellulose resin as described by Barak et al. (41). XynDocs were expressed and purified as described previously (41) with the following changes. The proteins were expressed in BL21 in LB medium supplemented with 50 μg/ml kanamycin. The culture was grown at 37 °C until it reached an A600 of ∼0.6–0.8 and then induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 37 °C. The dockerins were purified on a nickel-nitrilotriacetic acid column as described, without heat treatment. Purity of the CBM-Coh and XynDocs proteins was estimated by analytical gel filtration using a Superdex 200 column. Protein concentration was determined by absorption.
iELISA
The relative binding of the cohesin mutants was estimated using the iELISA-based method described previously in detail (26) and briefly below. Different concentrations (1 pm–1 μm) of cohesin mutants were incubated with 100 pm wild-type Xyn-dockerin for 1 h at 37 °C in binding buffer (TBS, 10 mm CaCl2, 0.05% Tween 20, 2% BSA). Next, 100 μl of the mixture was transferred to the 96-well MaxiSorp (Nunc A/S, Roskilde, Denmark) plate coated with wild-type cohesin for incubation for 15–30 min at 37 °C. Preformed complexes were then washed with washing buffer (TBS, 10 mm CaCl2, 0.05% Tween 20) followed by 1-h incubation with rabbit-anti Xyn, washing, and another 1-h incubation with HRP-conjugated goat anti-rabbit antibodies.
The relative binding of the cohesin mutants to C. cellulolyticum dockerin was measured as in previous studies except for a few modifications. 300 pm C. cellulolyticum dockerin was incubated with different concentrations (1 pm–1 μm) of cohesin mutants, and plates coated with the C. thermocellum cohesin N37A mutant were used. This allows presentation of differences in the binding affinity of C. cellulolyticum dockerin to WT C. cellulolyticum, WT C. thermocellum, and mutant C. thermocellum cohesin in one plot.
The obtained binding data were analyzed using GraphPad Prism (version 5.00 for Windows; GraphPad Software, San Diego, CA). Wild-type cohesin binding was used to normalize the experimental scale, and all mutants were standardized according to the wild type. The results were fitted to a sigmoidal dose-response curve (43), and changes in free energy of binding (ΔΔGbind_exp) were calculated relative to the wild type according to the following equation:
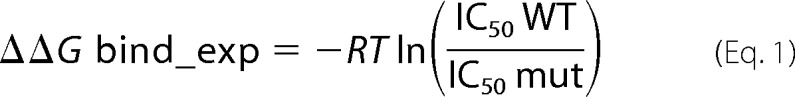 |
Standard errors of the mean of logarithmic values of IC50 did not exceed the half-log value (as suggested by Ref. 43) with the exception of measures for the L83S mutant (see Table 1).
TABLE 1.
Effect of C. thermocellum cohesin mutations on type I cohesin-dockerin binding affinity
Mutations in both the hydrophobic and hydrophilic interface patches (Figs. 2 and 3) can significantly reduce binding. Predicted ΔΔG values were calculated based on the Protein Data Bank structure 1OHZ (12) using the Rosetta interface protocol (33). Predicted hot spots (mutations to alanine with ΔΔG >1.0 kcal/mol) within the interface patches were experimentally validated by iELISA. Fig. 4 summarizes the predicted effect on binding of mutations as calculated by additional computational protocols.
| Mutation | Calculated ΔΔG Rosetta 2.3a | Experimental ΔΔG | Experimental IC50b |
|---|---|---|---|
| REU | kcal/mol | nm | |
| WT | 0 | 1.1b | |
| Hydrophobic patch | |||
| L83A | 2.2 | 3.0 | 123c |
| L83S | 2.4 | 3.4 | 257c,d |
| Polar patch | |||
| N37A | 2.2 | 0 | 1 |
| N37D | 2.1 | 2.5 | 70 |
| N37L | 1.9 | >4e | >103 |
| D39A | 3.0 | >4 | >103c |
| D39N | 0.3 | >4 | >103f |
| E131A | 1.3 | 1.8 | 20 |
| N37A/E120A | 2.2 | 1.2 | 7c |
| N37A/D39A | 5.2 | >4 | >103 |
| N37A/E131A | 3.5 | 1.0 | 6 |
| N37A/D39A/E131A | 6.4 | >4 | >103 |
| Additional positions | |||
| V41A | 0.0 | ||
| V41Y | 0.3 | 0.7 | 4f |
| D70A | 0.1 | ||
| D70S | 0.2 | −0.1 | 1f |
| Y74A | 1.8 | −0.1g | |
| V81S | 0.1 | 1.9 | 30f |
| A85L | 0.5 | −0.1 | 1f |
| E86A | 1.6 | ||
| E86S | 1.6 | 0.4 g | |
| E120A | 0.0 | ||
| D39S/Y74A | 4.4 | 1.1g | |
| D39S/Y74A/E86S | 6 | 2.8g | |
| D39N/D70S/V81S | 0.5 | >3 | >200f |
a In Rosetta energy units (REU); optimized to correlate with kcal/mol.
b IC50 values in this study are normalized relative to the binding of wild-type C. thermocellum cohesin to C. thermocellum dockerin (i.e. 1.1, first row).
c Reported in Ref. 26.
d The standard error of the mean of logarithmic values of IC50 exceeds the half-log value for this measure.
e Very weak binding could not be fitted and was therefore assigned ΔΔG values of >4 kcal/mol corresponding to the largest measured concentration.
f Measured also previously by enzyme-linked interaction assay (29).
g Measured by isothermal titration calorimetry (65).
Protein Cellulose Microarray
Protein cellulose microarray experiments were performed as described previously (17). In short, WT C. thermocellum, WT C. cellulolyticum, and mutant N37A C. thermocellum cohesin modules were printed in rows of 2-fold diluted concentration each. The slides were incubated with either C. thermocellum XynDocS or C. cellulolyticum XynDocA and visualized using anti-xylanase antibody labeled with Cy5.
Results
Computational Alanine Scanning Identifies Interface Residue Hot Spots in Two Distinct Patches at the C. thermocellum Cohesin-Dockerin Interface
The structure of the C. thermocellum cohesin-dockerin interface contains a hydrophobic patch that is conserved in both C. thermocellum and C. cellulolyticum cohesin-dockerin interactions (Figs. 1C and 2A) as well as an extensive network of hydrogen bonds at the center of the interface (Fig. 1D and Fig. 3A). The C. thermocellum dockerin residues Ser45 and Thr46 located in the conserved binding motif of the second F-hand constitute a central part of this network (Fig. 3B). In the corresponding C. cellulolyticum dockerin, this binding motif is replaced by hydrophobic residues Ala47 and Phe48 (Fig. 1D and Ref. 16).
FIGURE 2.
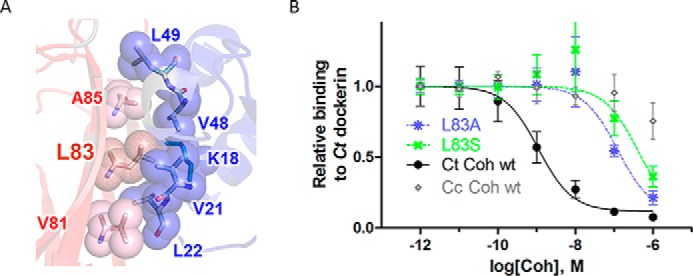
The hydrophobic patch contributes strongly to cohesin-dockerin binding affinity. A, detailed structural view of the hydrophobic interface patch. Residues involved in the cohesin (red)-dockerin (blue) interaction are shown in spheres and labeled (numbering according to Protein Data Bank code 1OHZ (12)) (see also Fig. 1C for sequence conservation of the hydrophobic patch). B, measurement of effect of C. thermocellum (Ct) cohesin mutations L83A (blue) and L83S (green) on binding to C. thermocellum dockerin using iELISA. Various concentrations of cohesin (x axis) were incubated with a constant concentration of dockerin in solution (0.1 nm), and the amount of unbound dockerin was detected on plates coated with wild-type cohesin (y axis; see “Experimental Procedures”). Binding wild-type C. thermocellum and C. cellulolyticum (Cc) cohesins to C. thermocellum dockerin represents positive (black) and negative (i.e. non-cognate, nonspecific; gray) binding controls, respectively. Results represent the average value over four independent experiments (the S.E. is shown as error bars).
To locate interface residues that play a critical role in binding, we first identified putative interface hot spot residues in the C. thermocellum cohesin-dockerin interface with computational alanine scanning using Rosetta (version 2.3). Binding hot spot residues (calculated ΔΔGbind ≥1.0 kcal/mol) are located in 1) the hydrophobic patch (cohesin Leu83 and dockerin Leu22), 2) the network of hydrogen bonds (cohesin Asn37, Asp39, and Glu131 and dockerin Ser45 and Thr46), and 3) other regions of the interface (e.g. cohesin Tyr74 and a conserved intermolecular salt bridge between cohesin Glu86 and dockerin Arg53). Table 1 summarizes predicted and experimental ΔΔG values evaluated in this as well as previous studies.
The Hydrophobic Patch Plays an Important Role in C. thermocellum Cohesin-Dockerin Binding
The hydrophobic patch is centered around the conserved C. thermocellum cohesin residue Leu83 (Figs. 1C and 2A). The strong conservation of this hydrophobic and exposed residue suggests an important functional role in the interaction with dockerin partners. To investigate the contribution of this residue to binding affinity, we replaced Leu83 with a smaller residue that creates a void in the hydrophobic interface patch and is therefore predicted to significantly affect binding (mutations L83A and L83S; see Table 1). We used iELISA (26, 27) to measure the effect of these mutations on the affinity of the C. thermocellum cohesin-dockerin interaction (see “Experimental Procedures”). The IC50 of the wild type C. thermocellum cohesin-dockerin interaction was 1 nm, whereas the IC50 values of the L83A and L83S mutants were 123 and 257 nm, respectively, corresponding to a decrease in binding free energy ΔΔG of 3.0 and 3.4 kcal/mol, respectively (Fig. 2B and Table 1). Both mutations indeed significantly impair binding. In compliance with this result, the corresponding residue in C. cellulolyticum cohesin, Leu87, was shown previously to significantly decrease binding upon mutation to alanine (16). These experimental data validate the crucial role of Leu83 in the interaction of the C. thermocellum cohesin with its cognate dockerin and highlight the importance of the hydrophobic patch at this interface.
Contributions of the Polar Patch to Cohesin-Dockerin Binding Affinity
The network of hydrogen bonds at the interface of the C. thermocellum cohesin-dockerin complex is centered on the specificity-determining dockerin residues Ser45 and Thr46 and on cohesin residues Asn37, Asp39, and Glu131 (Figs. 1D and 3, A and B). Our structure-based analysis and energy calculations suggest that mutations to alanine at these positions will reduce binding affinity significantly (ΔΔG > 1.0 kcal/mol) by disrupting the network of hydrogen bonds and by modifying the electrostatic attraction between the overall negatively charged cohesin and positively charged dockerin modules (Table 1).
Using iELISA experiments to measure the binding of the cohesin single mutants N37A, D39A, and E131A to wild-type C. thermocellum dockerin, we confirmed that D39A and E131A mutations indeed significantly impair binding to the C. thermocellum dockerin (ΔΔG >4.0 kcal/mol for D39A and 1.8 kcal/mol for E131A, respectively). Surprisingly, we observed no such effect for N37A (Fig. 3C and Table 1). In addition, the N37A mutation was found to reduce the effect of the E131 mutant in the polar patch: the double mutant N37A/E131A (ΔΔG = 1.0 kcal/mol) had less impact on binding than the single mutant E131A (ΔΔG = 1.8 kcal/mol) (Fig. 3C and Table 1).
We next investigated two additional mutations at the polar patch, N37D and D39N. These mutations involve only minor changes of one side chain atom but result in a change of the overall charge and affect the network of hydrogen bonds (Fig. 3B). Our calculations predict that N37D will be detrimental to binding, whereas D39N will not affect binding affinity (Table 1). iELISA experiments confirmed the predicted detrimental effect of the N37D mutant on binding (ΔΔG = 2.5 kcal/mol). Contrary to our predictions, however, the iELISA experiment showed an even stronger reduction of binding for the D39N mutant (ΔΔG > 4 kcal/mol) (Fig. 3D and Table 1). Together, these two mutations, N37D and D39N, highlight the sensitivity of this interface to both addition and removal of negative charges.
Structure-based ΔΔG Prediction Protocols Partially Succeed in the Identification of Interface Hot Spots at the Cohesin-Dockerin Interface
For the mutations tested by iELISA experiments for their effect on binding in this study, we found an overall good agreement of predicted and experimentally measured effects except for the mutations N37A and D39N (Table 1). Why then does our model fail to accurately capture the effect of these two mutations? Are other protocols more successful? We recalculated predicted effects on binding using a range of other published and available approaches, namely FoldX (35), Hunter (37), two different versions of the Orbit function optimized for interface prediction EPPI-2 (36) (termed here Orbit1 and Orbit2), and CC/PBSA (38) (see “Experimental Procedures”). In addition, we repeated the prediction with a more recent Rosetta protocol calibrated specifically for the prediction of effect of mutations on protein stability (termed here Rosetta 3.0*; protocol16 in Kellogg et al. (34)). The results of these different approaches are summarized in Fig. 4. In concordance with our results described above, all approaches identify the strong effect on binding upon Leu83 mutation to serine or alanine. However, for mutations of polar residues Asp39 and Asn37, the agreement among the protocols on the effect on binding is lower, and none can correctly describe the whole set. Hunter does not predict a significant effect on binding for any of these mutations (including N37A). The lack of destabilization by N37A is missed by all other methods but Rosetta 3.0*. The latter predicts that the energy contributed by the hydrogen bonds formed by N37A is small due to non-optimal geometry (compared e.g. with the hydrogen bonds formed by Asp39) and therefore cannot compensate for the solvation penalty of burying Asn37. Therefore, mutation to alanine at this position will not significantly affect binding affinity. The strong effect of D39N is also missed by most of the other approaches (including CC/PBSA that is aimed at accurately modeling electrostatic effects) except for Orbit1. This protocol, however, predicts a similar effect for all mutations, and slight modification of the threshold used to define a hot spot (1 kcal/mol) will strongly affect the results.
FIGURE 4.
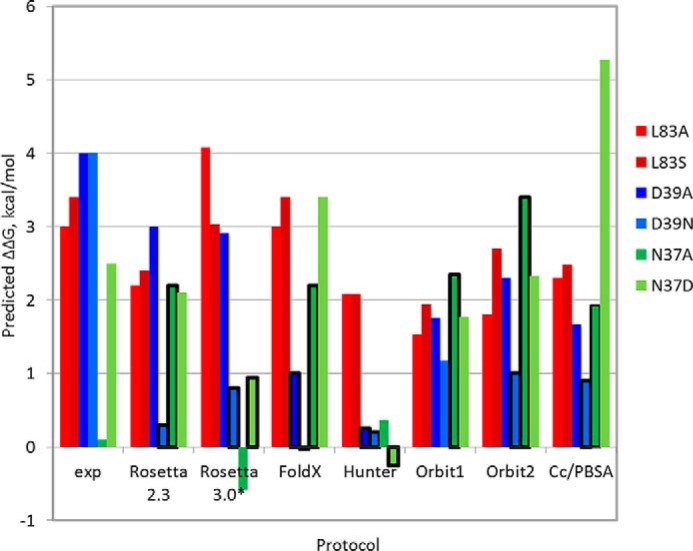
Comparison of predicted and experimental ΔΔG values indicates that none of a range of different computational approaches can accurately predict all tested mutations at C. thermocellum cohesin positions Leu83, Asn37, and Asp39. The plot shows experimental and predicted ΔΔG values (y axis) of the different mutations (colored red for Leu83, blue for Asp39, and green for Asn37). The x axis plots results from the experiment (exp) as well as from a range of different approaches, including different versions of Rosetta alanine scanning (Rosetta2.3 used in this study and a newly calibrated protocol Rosetta 3.0* (34)), FoldX (35), Hunter (37), two versions of the Orbit interface energy function EPPI-2 (36), and CC/PBSA (38). Incorrect predictions are boxed in bold black. All protocols accurately predict the effects of mutations of Leu83 but perform less well on polar mutations. See “Experimental Procedures” for more details about the different protocols.
The N37A Mutation of the C. thermocellum Cohesin Establishes a New Interaction with the Non-cognate C. cellulolyticum Dockerin
We reconfirmed that the C. thermocellum cohesin mutation N37A does not affect binding to its cognate dockerin by independent evaluation of the binding ability of both wild-type C. thermocellum cohesin and C. thermocellum cohesin N37A to C. thermocellum and C. cellulolyticum dockerin using a cellulose-binding microarray assay (17). Surprisingly, the assay revealed that the N37A mutant also binds to non-cognate C. cellulolyticum dockerin unlike the wild-type C. thermocellum cohesin (Fig. 5A). Thus, the cross-species binding barrier observed for wild-type C. thermocellum and C. cellulolyticum cohesin-dockerin pairs is partially overcome in the C. thermocellum cohesin mutation N37A because it binds promiscuously to both C. thermocellum and C. cellulolyticum dockerins.
FIGURE 5.
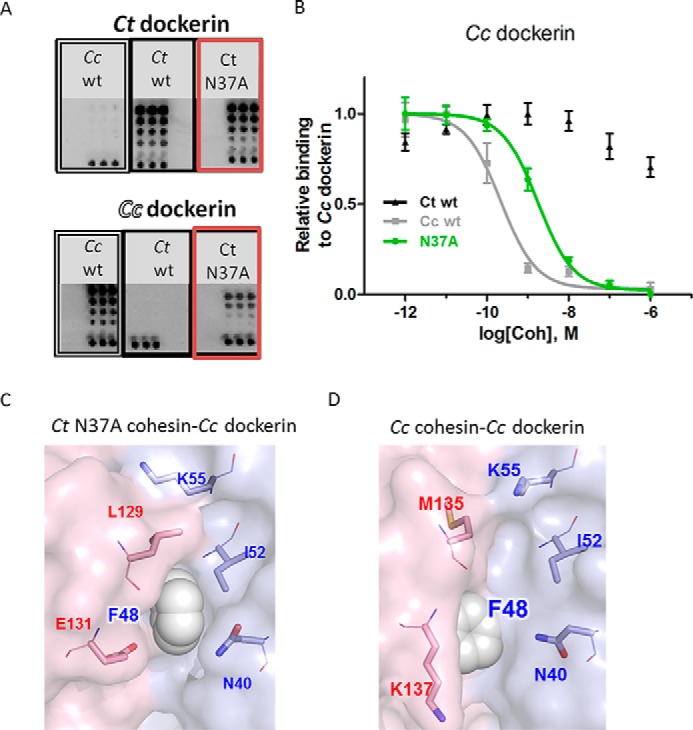
Binding promiscuity of the C. thermocellum cohesin N37A mutant. The N37A mutation allows promiscuous binding of the C. thermocellum (Ct) cohesin both to its cognate C. thermocellum dockerin and non-cognate C. cellulolyticum dockerin. A and B, experimental validation of non-cognate binding. A, a protein cellulose microarray assay reveals that although as expected C. thermocellum dockerin binds only to C. thermocellum cohesins (upper panel) and C. cellulolyticum (Cc) dockerin binds only to C. cellulolyticum cohesins (lower panel) the promiscuous C. thermocellum cohesin N37A mutant binds both to its cognate C. thermocellum dockerin and to the non-cognate C. cellulolyticum dockerin (red boxes). Each row of dots represents a 2-fold dilution; the bottom row contains a CBM-Xyn location marker. B, confirmation of cellulose binding microarray results by iELISA. The C. thermocellum N37A cohesin mutant can indeed bind to both its cognate (C. thermocellum) and non-cognate (C. cellulolyticum) dockerins. C. thermocellum N37A cohesin mutant-covered plates were used in this assay. C and D, structural models of the non-cognate and cognate interactions with C. cellulolyticum dockerin. C, model of the non-cognate interaction of C. cellulolyticum dockerin with the C. thermocellum N37A cohesin mutant (generated using RosettaDock; see “Experimental Procedures”). D, C. cellulolyticum dockerin interacting with its cognate wild-type C. cellulolyticum cohesin (Protein Data Bank code 2VN6 (16)). While in the C. cellulolyticum cohesin-dockerin interaction the C. cellulolyticum dockerin motif residue Phe48 points toward the cohesin (D), the model of non-cognate interaction suggests that Phe48 points into a hydrophobic pocket formed by C. cellulolyticum dockerin residue Ile52 and C. thermocellum cohesin residue Leu129, displacing dockerin residue Lys55 to the solvent (C). Phe48 is shown as spheres, and residues predicted to interact with Phe48 in the non-cognate interaction are depicted as sticks and labeled and colored in red and blue for cohesin and dockerin, respectively. See Fig. 2 legend for more details about the iELISA assay in B.
We further confirmed this non-cognate binding by the corresponding iELISA assay. We compared the affinity of C. cellulolyticum dockerin to the promiscuous C. thermocellum cohesin mutant N37A with its affinity to both its cognate (C. cellulolyticum) and non-cognate (C. thermocellum) wild-type cohesin modules (Fig. 5B). The results indicate that in strong contrast to wild-type C. thermocellum cohesin, which does not bind the C. cellulolyticum dockerin, C. thermocellum N37A mutant cohesin indeed binds to C. cellulolyticum dockerin. Still, C. thermocellum N37A mutant cohesin bound with lower affinity to C. cellulolyticum dockerin than the cognate wild-type C. cellulolyticum cohesin (ΔΔG = +1.3 kcal/mol; Fig. 5B and Table 2). We conclude from these two experiments that mutation of a single amino acid residue in the C. thermocellum cohesin creates a promiscuous cohesin module that can overcome the specificity barrier and bind also to C. cellulolyticum dockerin.
TABLE 2.
Effect of C. thermocellum cohesin mutations on binding to non-cognate C. cellulolyticum dockerins
Single mutations show dramatic and varied effects on both binding affinity and specificity.
| Cohesin | Calculated ΔΔGa Rosetta2.3 | Experimental ΔΔG | Experimental IC50 |
|---|---|---|---|
| REU | kcal/mol | nm | |
| C. cellulolyticum WT | 0.2b | ||
| C. thermocellum WT | 0 | >5 | >4500 |
| C. thermocellum N37A | 0 | 1.3 | 8 |
| C. thermocellum D39A | 1.7 | >5 | >4500 |
| C. thermocellum N37L | −1.1 | 1.7 | 17 |
| C. thermocellum N37A/D39A | 1.7 | 1.5 | 12 |
a ΔΔG values were calculated based on a structural model of the interaction between C. thermocellum cohesin N37A and C. cellulolyticum dockerin generated by docking (see “Experimental Procedures”). REU, Rosetta energy units.
b IC50 values in this study are normalized relative to the binding of wild-type C. cellulolyticum cohesin to C. cellulolyticum dockerin (i.e. 0.2, first row).
We note that in this iELISA experiment, to measure the amount of non-bound dockerin, the preformed complexes were incubated with C. thermocellum N37A cohesin-coated plates, rather than with C. cellulolyticum wild-type cohesin. This optimized our ability to distinguish between the affinities of the three different C. thermocellum cohesin molecules to C. cellulolyticum dockerin (see “Experimental Procedures” and “Discussion”).
A structural model of the C. thermocellum cohesin N37A mutant bound to its non-cognate C. cellulolyticum dockerin (Fig. 5C) suggests how the N37A C. thermocellum cohesin mutant could accommodate this hydrophobic motif region in C. cellulolyticum dockerin. In the cognate C. cellulolyticum interaction, dockerin specificity motif residue Phe48 points into the interface to interact with the side chains of Ala129 and Lys137 and the backbone of Met135 of C. cellulolyticum cohesin (Fig. 1D). Conversely, in the non-cognate interaction, Phe48 points in a different direction toward a newly created hydrophobic patch formed by C. thermocellum cohesin residue Leu129 and C. cellulolyticum dockerin residues Ile52 and Asn40 (Fig. 5C). In the C. cellulolyticum cohesin-dockerin interaction, Ile52 is exposed and only partly covered by dockerin residue Lys55 (Fig. 5D and Ref. 16), whereas in the model of interaction between the C. thermocellum cohesin N37A mutant and the C. cellulolyticum dockerin, Lys55 moves away to the solvent to make room for C. thermocellum cohesin Leu129 (Fig. 5C).
The C. thermocellum Cohesin N37L Mutation: Switching Binding Specificity
If a single mutation overcomes the species barrier and allows cohesin to bind to non-cognate dockerin, how difficult would it be to keep this new interaction and abolish the original cognate interaction, i.e. to create a switch in binding specificity? We designed two mutants, single mutant N37L and double mutant N37A/D39A, based on the solved structures of C. thermocellum and C. cellulolyticum cognate cohesin-dockerin complexes (Protein Data Bank codes 1OHZ (12) and 2VN6 (16), respectively). Although the large and polar asparagine side chain at position 37 is suited for its polar environment in the C. thermocellum cognate complex, it would not fit the corresponding hydrophobic environment in the C. cellulolyticum complex, which could be the reason for its failure to bind to non-cognate C. cellulolyticum dockerin. The mutant N37L in turn is predicted to prefer the hydrophobic non-cognate C. cellulolyticum dockerin over the polar cognate C. thermocellum dockerin (Fig. 6A). Experimental validation of this prediction with the iELISA assay confirmed that indeed the N37L C. thermocellum cohesin mutant barely binds to the cognate C. thermocellum dockerin anymore (ΔΔG > 4 kcal/mol compared with WT C. thermocellum cohesin; Fig. 6B and Table 1). Instead, it now binds to the non-cognate C. cellulolyticum dockerin (ΔΔG = 1.7 kcal/mol compared with WT C. cellulolyticum cohesin) with slightly lower affinity than that of N37A (ΔΔG = 1.3 kcal/mol; Fig. 6C and Table 2).
FIGURE 6.

Toward a new binding specificity to C. cellulolyticum dockerin: C. thermocellum cohesin single mutant N37L (A–C) and double mutant N37A/D39A (D–F). Structural models of C. cellulolyticum (Cc) dockerin bound to C. thermocellum (Ct) cohesin mutants N37L (A) and N37A/D39A (D) and binding assays to C. thermocellum (B and E) and C. cellulolyticum (C and F) dockerin are shown. Both the N37L and N37A/D39A mutants exhibit reduced binding to the cognate C. thermocellum cohesin (B and E) and increased binding to non-cognate C. cellulolyticum cohesin (C and F). In contrast, the promiscuous C. thermocellum cohesin mutant N37A binds to both C. thermocellum and C. cellulolyticum dockerin, whereas the C. thermocellum cohesin mutant D39A does not bind to any of the dockerins. Binding to C. cellulolyticum dockerin was measured using a C. thermocellum cohesin N37A coating. See Fig. 2 legend for more details about the iELISA measurements.
The double mutant N37A/D39A shows a similar binding pattern to C. cellulolyticum dockerin as N37L: impaired binding to its cognate C. thermocellum dockerin (ΔΔG > 4 kcal/mol; Table 1) and new binding to its non-cognate C. cellulolyticum dockerin (ΔΔG = 1.5 kcal/mol; Fig. 6, D–F, and Table 2). In this case, a mutation that abolishes the original binding (D39A) was combined with a mutation that creates new, non-cognate binding (N37A), resulting in a mutant that lost its strong original binding ability but gained binding ability to a new, non-cognate partner. Note, however, that for both the N37L and the N37A/D39A mutants that change their binding specificity from C. thermocellum dockerin to C. cellulolyticum dockerin, preferences of the dockerins to cohesin have not changed (Fig. 6B): C. cellulolyticum dockerin still shows preferential binding to its cognate C. cellulolyticum cohesin rather than the non-cognate C. thermocellum cohesin mutants.
Discussion
In this study, we used a structure-based approach to study binding specificity and promiscuity in the cohesin-dockerin interaction. Our main finding is that the balance between specificity and promiscuity can be controlled by a single residue. We discuss this easy disruption of specificity in light of other studies on protein binding specificity and affinity and its evolution and identify similarities and differences both at functional and structural levels. Finally, we relate to challenges and successes in current, state-of-the-art modeling protocols and experimental assays that are aimed at the accurate characterization of binding affinity and specificity.
Binding Affinity, Specificity, and Promiscuity Is Easily Modulated by Single Mutations even in High Affinity Interactions
The cohesin-dockerin interaction presents a special case of a high affinity interaction whose binding promiscuity and specificity can strongly be affected by one single mutation. Our computational results, confirmed by two independent experimental assays, demonstrate the crucial and central role that residue Asn37 in the C. thermocellum cohesin can play in binding specificity. Although wild-type C. thermocellum cohesin binds only to C. thermocellum dockerin, mutation N37A abolishes this specificity and extends to non-cognate C. cellulolyticum dockerins, and mutation N37L completes a specificity switch toward C. cellulolyticum dockerin. Residue Asn37 can thus be termed a binding specificity hot spot. Interestingly, mutation N37A also has stabilizing effects on D39A and E131A mutations in the polar patch of the interface (Fig. 3C and Table 1), indicating that specificity comes at a price of increased sensitivity to mutations (i.e. the interaction with dockerin is less sensitive to mutations for the promiscuous mutant N37A than for the specific wild-type cohesin).
The new non-cognate binding can be explained in light of the distinct features of C. thermocellum and C. cellulolyticum dockerins: the polar binding motif of C. thermocellum dockerin in the second F-hand contains Ser45 and Thr46, whereas in C. cellulolyticum dockerin, they are replaced by Ala47 and Phe48 at corresponding positions (Fig. 1D). The accommodation of the Phe48 side chain into an existing hydrophobic patch (Fig. 5C) without the need of further evolutionary changes of the interface highlights the preorganized plasticity of interfaces, similar to what has been observed in other experiments of evolution of new binding specificities (2).
Rather than affecting binding affinity on its own, the N37A mutation attenuates the effect on binding affinity of additional mutations. Consequently, rather than an interface binding hot spot, this residue functions as an interface specificity hot spot. We have observed a similar behavior of a cohesin residue in the type III cohesin-dockerin interaction that upon mutation increased binding and attenuated effects by other positions at the interface.5 Thus, mutations that do not impair binding but rather extend the range of binding partners to dockerins from different species (i.e. binding to non-cognate C. cellulolyticum dockerin) can readily appear during evolution.
The dramatic effect on binding specificity of one single mutation as reported for the cohesin-dockerin interaction studied here contrasts with the robustness that has been reported for other systems such as the colicin-immunity protein interaction (6). Reducing or switching specificity in the colicin-immunity protein interaction involves a number of mutations that were found only in an extended in vitro evolution experiment (2) but not identified by computation (44, 45). In contrast, the cohesin-dockerin interaction shows a much simpler picture in which major effects on specificity may be achieved by a single residue as was demonstrated previously on the dockerin module (8) and is shown here on the cohesin module.
It therefore seems that functional rather than biophysical constraints have shaped the differential plasticity of these two high affinity associations. The interaction between cohesin and dockerin plays a very different biological role compared with the interaction between colicin and its immunity protein. The binding of the immunity protein to the bacterial endonuclease is crucial as failure to do so will result in active nuclease molecules within the cell and lead to bacterial death. Therefore, this interaction has evolved to be robust to strong effects on binding by single mutations. Consequently, crossing the strain-specific barriers would be difficult as this would involve mutations that reduce binding affinity at the price of promiscuity.
In contrast, we speculate that the cohesin-dockerin interaction within a species might have evolved toward promiscuity to facilitate extensive diversity in the hydrolase enzyme composition of cellulosome subpopulations (because the cohesin module repeats can each bind to a range of different dockerin modules, each connected to a distinct hydrolase). This platform for diversity can be fine-tuned on demand toward maximal degradation efficiency of specific cellulosic substrates. Expression of specific hydrolases is regulated at the transcriptional level by specific σ factors, which are released from carbohydrate sensor proteins upon their binding to specific plant cell wall polysaccharide substrates (46–48). Because of the promiscuous binding between dockerin modules of the different hydrolases to the different repeats of cohesin modules on the scaffoldin, this malleable platform can be fine-tuned based on the specifically induced hydrolases, resulting in cellulosomes with optimized composition for degradation of the specific substrate(s).
Binding Affinity and Specificity Are Determined by Two Distinct Regions
Despite the fundamental differences between the cohesin-dockerin and the colicin-immunity protein interaction described above, their underlying interface architecture shows similar organization. In-depth mutational analysis of the colicin-immunity protein interaction has suggested a so-called “dual recognition mechanism” (6, 49) that involves two distinct patches at the interface: one conserved patch is responsible for binding affinity (i.e. two conserved central tyrosine residues), and the second patch governs binding specificity (i.e. the network of hydrogen bonds) (Note that this dual recognition mechanism should not be confused with the “dual binding mode” of the cohesin-dockerin interaction that allows binding of dockerin in two opposite orientations described in the Introduction (13, 16)). The interface of the C. thermocellum cohesin-dockerin interaction is also composed of a conserved hydrophobic patch and a specific polar patch. We have shown here that the hydrophobic patch plays an important role in binding affinity because the mutation of a central position, L83A, significantly impairs binding (Fig. 2). Despite its hydrophobic and exposed nature, the high degree of cross-species sequence conservation of this residue (Fig. 1C) indicates a more general role of this patch for cohesin-dockerin binding affinity. In contrast, binding specificity is obtained by the polar patch that is conserved only within C. thermocellum and replaced by a hydrophobic patch in C. cellulolyticum. This patch in C. thermocellum contains at least one residue, Asn37, that upon mutation to alanine can overcome the species barrier and create a promiscuous cohesin that binds both its cognate (C. thermocellum) and non-cognate (C. cellulolyticum) dockerin (Fig. 6).
Polar Interactions across the Interface and Charges Play a Major Role in the C. thermocellum Cohesin-Dockerin Interaction but Are Challenging to Model
The large number of charged residues at the C. thermocellum cohesin-dockerin interface suggests that this interaction is governed by electrostatics. The detrimental effect on binding of the C. thermocellum cohesin D39N mutation (first reported in Ref. 29 and reconfirmed in this study by an iELISA assay; Fig. 3D) suggests that this is due to removal of a charge. The importance of this charge at position 39 could not be foreseen by molecular modeling attempts (see Fig. 4) even after including a Coulomb electrostatic term in the Rosetta scoring function, the use of a generalized Born-based treatment of electrostatic solvation energy implemented into the Rosetta modeling suite (50), and a detailed calculation of the pKa of Asp39 using the APBS modeling suite (51).
Molecular dynamics simulations of the wild-type cohesin and the D39N mutant suggested that removal of the charge at position Asp39 leads to a significant destabilization of a nearby region, which is not observed in the wild type (52). However, these simulations originated from the crystal structure solved by Carvalho et al. (12) (Protein Data Bank code 1OHZ) in which the critical Asn37 side chain has been incorrectly fitted. In the wild-type simulation, Asn37 is indeed flipped at the very beginning of the simulation, whereas in the simulation of the D39N mutant, it is not. Consequently, the pronounced effect observed in the Xu et al. (52) study likely reflects a non-equilibrated starting conformation.
The ability to identify interface residue hot spots is the basis for more complex redesign of interfaces and binding specificity (53, 54). Considerable effort has therefore been put into the development of computational approaches to model the effect of point mutations on protein stability and binding affinity (33, 34, 36, 55, 56). Despite good overall accuracy, these approaches are still challenged by complex polar effects at the interface as also demonstrated in the present study. Nevertheless, together with structural analysis, they enabled us to pinpoint the crucial interactions in the interface that contribute to binding in different ways.
Outlook
In addition to our ability to address fundamental questions about specificity and promiscuity of the cohesin-dockerin interaction, our study bears biotechnological implications for the fabrication of artificial designer cellulosome complexes (19–21, 57–64). Specific cohesin-dockerin pairs could allow the generation of cellulosome complexes of predetermined content and spatial architecture. This study provides initial support for the feasibility of such an application. Future steps will involve the generation of additional specific cohesin-dockerin pairs.
Acknowledgments
We thank Oz Sharabi and Julia Shifman for evaluating the effect on binding of different mutants with the Orbit energy function. We also thank Tim Whitehead for reading the manuscript and for helpful suggestions.
This work was supported, in whole or in part, by the Israel Science Foundation (ISF) funded by the Israel Academy of Science and Humanities (Grants 306/06 and 319/11 to O. S.-F., 1349/13 to E. A. B., and 24/11 to R. L. by The Sidney E. Frank Foundation through the ISF); the United States-Israel Binational Science Foundation, Jerusalem, Israel (Grants 2009418 to O. S.-F. and 2013284 to E. A. B.); the European Research Council (Starting Grant 310873 to O. S.-F.); the European Union, Area NMP.2013.1.1-2: (Selfassembly of naturally occurring nanosystems: CellulosomePlus Project 604530 to E. A. B.), and a European Research Area-Industrial Biology Consortium (EIB.12.022, acronym FiberFuel, to E. A. B.).
M. Slutzki, I. Noach, and E. A. Bayer, unpublished results.
- iELISA
- indirect ELISA
- Xyn
- xylanase
- Doc
- dockerin
- CBM
- carbohydrate-binding module
- Coh
- cohesin
- CC/PBSA
- Concoord/PBSA.
References
- 1. Schreiber G., Keating A. E. (2011) Protein binding specificity versus promiscuity. Curr. Opin. Struct. Biol. 21, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levin K. B., Dym O., Albeck S., Magdassi S., Keeble A. H., Kleanthous C., Tawfik D. S. (2009) Following evolutionary paths to protein-protein interactions with high affinity and selectivity. Nat. Struct. Mol. Biol. 16, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 3. Akiva E., Itzhaki Z., Margalit H. (2008) Built-in loops allow versatility in domain-domain interactions: lessons from self-interacting domains. Proc. Natl. Acad. Sci. U.S.A. 105, 13292–13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaneko T., Huang H., Zhao B., Li L., Liu H., Voss C. K., Wu C., Schiller M. R., Li S. S. (2010) Loops govern SH2 domain specificity by controlling access to binding pockets. Sci. Signal. 3, ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reichmann D., Rahat O., Albeck S., Meged R., Dym O., Schreiber G. (2005) The modular architecture of protein-protein binding interfaces. Proc. Natl. Acad. Sci. U.S.A. 102, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kleanthous C., Walker D. (2001) Immunity proteins: enzyme inhibitors that avoid the active site. Trends Biochem. Sci. 26, 624–631 [DOI] [PubMed] [Google Scholar]
- 7. Bayer E. A., Belaich J. P., Shoham Y., Lamed R. (2004) The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 58, 521–554 [DOI] [PubMed] [Google Scholar]
- 8. Mechaly A., Fierobe H. P., Belaich A., Belaich J. P., Lamed R., Shoham Y., Bayer E. A. (2001) Cohesin-dockerin interaction in cellulosome assembly: a single hydroxyl group of a dockerin domain distinguishes between nonrecognition and high affinity recognition. J. Biol. Chem. 276, 9883–9888 [DOI] [PubMed] [Google Scholar]
- 9. Sakka K., Kishino Y., Sugihara Y., Jindou S., Sakka M., Inagaki M., Kimura T. (2009) Unusual binding properties of the dockerin module of Clostridium thermocellum endoglucanase CelJ (Cel9D-Cel44A). FEMS Microbiol. Lett. 300, 249–255 [DOI] [PubMed] [Google Scholar]
- 10. Lytle B. L., Volkman B. F., Westler W. M., Wu J. H. (2000) Secondary structure and calcium-induced folding of the Clostridium thermocellum dockerin domain determined by NMR spectroscopy. Arch. Biochem. Biophys. 379, 237–244 [DOI] [PubMed] [Google Scholar]
- 11. Pagès S., Bélaïch A., Bélaïch J. P., Morag E., Lamed R., Shoham Y., Bayer E. A. (1997) Species-specificity of the cohesin-dockerin interaction between Clostridium thermocellum and Clostridium cellulolyticum: prediction of specificity determinants of the dockerin domain. Proteins 29, 517–527 [PubMed] [Google Scholar]
- 12. Carvalho A. L., Dias F. M., Prates J. A., Nagy T., Gilbert H. J., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M. (2003) Cellulosome assembly revealed by the crystal structure of the cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 100, 13809–13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carvalho A. L., Dias F. M., Nagy T., Prates J. A., Proctor M. R., Smith N., Bayer E. A., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M., Gilbert H. J. (2007) Evidence for a dual binding mode of dockerin modules to cohesins. Proc. Natl. Acad. Sci. U.S.A. 104, 3089–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adams J. J., Gregg K., Bayer E. A., Boraston A. B., Smith S. P. (2008) Structural basis of Clostridium perfringens toxin complex formation. Proc. Natl. Acad. Sci. U.S.A. 105, 12194–12199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams J. J., Pal G., Jia Z., Smith S. P. (2006) Mechanism of bacterial cell-surface attachment revealed by the structure of cellulosomal type II cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 103, 305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pinheiro B. A., Proctor M. R., Martinez-Fleites C., Prates J. A., Money V. A., Davies G. J., Bayer E. A., Fontesm C. M., Fierobe H. P., Gilbert H. J. (2008) The Clostridium cellulolyticum dockerin displays a dual binding mode for its cohesin partner. J. Biol. Chem. 283, 18422–18430 [DOI] [PubMed] [Google Scholar]
- 17. Haimovitz R., Barak Y., Morag E., Voronov-Goldman M., Shoham Y., Lamed R., Bayer E. A. (2008) Cohesin-dockerin microarray: diverse specificities between two complementary families of interacting protein modules. Proteomics 8, 968–979 [DOI] [PubMed] [Google Scholar]
- 18. Sakka K., Sugihara Y., Jindou S., Sakka M., Inagaki M., Sakka K., Kimura T. (2011) Analysis of cohesin-dockerin interactions using mutant dockerin proteins. FEMS Microbiol. Lett. 314, 75–80 [DOI] [PubMed] [Google Scholar]
- 19. Moraïs S., Barak Y., Caspi J., Hadar Y., Lamed R., Shoham Y., Wilson D. B., Bayer E. A. (2010) Contribution of a xylan-binding module to the degradation of a complex cellulosic substrate by designer cellulosomes. Appl. Environ. Microbiol. 76, 3787–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moraïs S., Barak Y., Hadar Y., Wilson D. B., Shoham Y., Lamed R., Bayer E. A. (2011) Assembly of xylanases into designer cellulosomes promotes efficient hydrolysis of the xylan component of a natural recalcitrant cellulosic substrate. MBio 2, e00233–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moraïs S., Morag E., Barak Y., Goldman D., Hadar Y., Lamed R., Shoham Y., Wilson D. B., Bayer E. A. (2012) Deconstruction of lignocellulose into soluble sugars by native and designer cellulosomes. MBio 3, e00508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weinstein J. Y., Slutzki M., Karpol A., Barak Y., Gul O., Lamed R., Bayer E. A., Fried D. B. (2015) Insights into a type III cohesin-dockerin recognition interface from the cellulose-degrading bacterium Ruminococcus flavefaciens. J. Mol. Recognit. 28, 148–154 [DOI] [PubMed] [Google Scholar]
- 23. Nakar D., Handelsman T., Shoham Y., Fierobe H. P., Belaich J. P., Morag E., Lamed R., Bayer E. A. (2004) Pinpoint mapping of recognition residues on the cohesin surface by progressive homologue swapping. J. Biol. Chem. 279, 42881–42888 [DOI] [PubMed] [Google Scholar]
- 24. Halperin I., Wolfson H., Nussinov R. (2006) Correlated mutations: advances and limitations. A study on fusion proteins and on the cohesin-dockerin families. Proteins 63, 832–845 [DOI] [PubMed] [Google Scholar]
- 25. Das R., Baker D. (2008) Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 77, 363–382 [DOI] [PubMed] [Google Scholar]
- 26. Slutzki M., Barak Y., Reshef D., Schueler-Furman O., Lamed R., Bayer E. A. (2012) Indirect ELISA-based approach for comparative measurement of high-affinity cohesin-dockerin interactions. J. Mol. Recognit. 25, 616–622 [DOI] [PubMed] [Google Scholar]
- 27. Slutzki M., Barak Y., Reshef D., Schueler-Furman O., Lamed R., Bayer E. A. (2012) Measurements of relative binding of cohesin and dockerin mutants using an advanced ELISA technique for high-affinity interactions. Methods Enzymol. 510, 417–428 [DOI] [PubMed] [Google Scholar]
- 28. Weichenberger C. X., Sippl M. J. (2006) NQ-Flipper: validation and correction of asparagine/glutamine amide rotamers in protein crystal structures. Bioinformatics 22, 1397–1398 [DOI] [PubMed] [Google Scholar]
- 29. Handelsman T., Barak Y., Nakar D., Mechaly A., Lamed R., Shoham Y., Bayer E. A. (2004) Cohesin-dockerin interaction in cellulosome assembly: a single Asp-to-Asn mutation disrupts high-affinity cohesin-dockerin binding. FEBS Lett. 572, 195–200 [DOI] [PubMed] [Google Scholar]
- 30. Gray J. J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B., Rohl C. A., Baker D. (2003) Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 331, 281–299 [DOI] [PubMed] [Google Scholar]
- 31. Kortemme T., Kim D. E., Baker D. (2004) Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, pl2. [DOI] [PubMed] [Google Scholar]
- 32. Kuhlman B., Baker D. (2000) Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kortemme T., Baker D. (2002) A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. U.S.A. 99, 14116–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kellogg E. H., Leaver-Fay A., Baker D. (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharabi O., Dekel A., Shifman J. M. (2011) Triathlon for energy functions: Who is the winner for design of protein-protein interactions? Proteins 79, 1487–1498 [DOI] [PubMed] [Google Scholar]
- 37. Potapov V., Cohen M., Inbar Y., Schreiber G. (2010) Protein structure modelling and evaluation based on a 4-distance description of side-chain interactions. BMC Bioinformatics 11, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benedix A., Becker C. M., de Groot B. L., Caflisch A., Böckmann R. A. (2009) Predicting free energy changes using structural ensembles. Nat. Methods 6, 3–4 [DOI] [PubMed] [Google Scholar]
- 39. Schueler-Furman O., Wang C., Baker D. (2005) Progress in protein-protein docking: atomic resolution predictions in the CAPRI experiment using RosettaDock with an improved treatment of side-chain flexibility. Proteins 60, 187–194 [DOI] [PubMed] [Google Scholar]
- 40. Wang C., Schueler-Furman O., Baker D. (2005) Improved side-chain modeling for protein-protein docking. Protein Sci. 14, 1328–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barak Y., Handelsman T., Nakar D., Mechaly A., Lamed R., Shoham Y., Bayer E. A. (2005) Matching fusion protein systems for affinity analysis of two interacting families of proteins: the cohesin-dockerin interaction. J. Mol. Recognit. 18, 491–501 [DOI] [PubMed] [Google Scholar]
- 42. Lapidot A., Mechaly A., Shoham Y. (1996) Overexpression and single-step purification of a thermostable xylanase from Bacillus stearothermophilus T-6. J. Biotechnol. 51, 259–264 [DOI] [PubMed] [Google Scholar]
- 43. Motulsky H., Christopoulos A. (2004) Fitting Models to Biological Data Using Linear and Nonlinear Regression: a Practical Guide to Curve Fitting, pp. 256–295, Oxford University Press, Oxford [Google Scholar]
- 44. Joachimiak L. A., Kortemme T., Stoddard B. L., Baker D. (2006) Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J. Mol. Biol. 361, 195–208 [DOI] [PubMed] [Google Scholar]
- 45. Kortemme T., Joachimiak L. A., Bullock A. N., Schuler A. D., Stoddard B. L., Baker D. (2004) Computational redesign of protein-protein interaction specificity. Nat. Struct. Mol. Biol. 11, 371–379 [DOI] [PubMed] [Google Scholar]
- 46. Bahari L., Gilad Y., Borovok I., Kahel-Raifer H., Dassa B., Nataf Y., Shoham Y., Lamed R., Bayer E. A. (2011) Glycoside hydrolases as components of putative carbohydrate biosensor proteins in Clostridium thermocellum. J. Ind. Microbiol. Biotechnol. 38, 825–832 [DOI] [PubMed] [Google Scholar]
- 47. Kahel-Raifer H., Jindou S., Bahari L., Nataf Y., Shoham Y., Bayer E. A., Borovok I., Lamed R. (2010) The unique set of putative membrane-associated anti-σ factors in Clostridium thermocellum suggests a novel extracellular carbohydrate-sensing mechanism involved in gene regulation. FEMS Microbiol. Lett. 308, 84–93 [DOI] [PubMed] [Google Scholar]
- 48. Nataf Y., Bahari L., Kahel-Raifer H., Borovok I., Lamed R., Bayer E. A., Sonenshein A. L., Shoham Y. (2010) Clostridium thermocellum cellulosomal genes are regulated by extracytoplasmic polysaccharides via alternative σ factors. Proc. Natl. Acad. Sci. U.S.A. 107, 18646–18651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li W., Hamill S. J., Hemmings A. M., Moore G. R., James R., Kleanthous C. (1998) Dual recognition and the role of specificity-determining residues in colicin E9 DNase-immunity protein interactions. Biochemistry 37, 11771–11779 [DOI] [PubMed] [Google Scholar]
- 50. Onufriev A., Bashford D., Case D. A. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394 [DOI] [PubMed] [Google Scholar]
- 51. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu J., Crowley M. F., Smith J. C. (2009) Building a foundation for structure-based cellulosome design for cellulosic ethanol: Insight into cohesin-dockerin complexation from computer simulation. Protein Sci. 18, 949–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mandell D. J., Kortemme T. (2009) Computer-aided design of functional protein interactions. Nat. Chem. Biol. 5, 797–807 [DOI] [PubMed] [Google Scholar]
- 54. Schreiber G., Fleishman S. J. (2013) Computational design of protein-protein interactions. Curr. Opin. Struct. Biol. 23, 903–910 [DOI] [PubMed] [Google Scholar]
- 55. Guerois R., Nielsen J. E., Serrano L. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 [DOI] [PubMed] [Google Scholar]
- 56. Potapov V., Cohen M., Schreiber G. (2009) Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng. Des. Sel. 22, 553–560 [DOI] [PubMed] [Google Scholar]
- 57. Caspi J., Barak Y., Haimovitz R., Irwin D., Lamed R., Wilson D. B., Bayer E. A. (2009) Effect of linker length and dockerin position on conversion of a Thermobifida fusca endoglucanase to the cellulosomal mode. Appl. Environ. Microbiol. 75, 7335–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caspi J., Irwin D., Lamed R., Li Y., Fierobe H. P., Wilson D. B., Bayer E. A. (2008) Conversion of Thermobifida fusca free exoglucanases into cellulosomal components: comparative impact on cellulose-degrading activity. J. Biotechnol. 135, 351–357 [DOI] [PubMed] [Google Scholar]
- 59. Fierobe H. P., Mingardon F., Mechaly A., Bélaïch A., Rincon M. T., Pagès S., Lamed R., Tardif C., Bélaïch J. P., Bayer E. A. (2005) Action of designer cellulosomes on homogeneous versus complex substrates: controlled incorporation of three distinct enzymes into a defined trifunctional scaffoldin. J. Biol. Chem. 280, 16325–16334 [DOI] [PubMed] [Google Scholar]
- 60. Mingardon F., Chanal A., López-Contreras A. M., Dray C., Bayer E. A., Fierobe H. P. (2007) Incorporation of fungal cellulases in bacterial minicellulosomes yields viable, synergistically acting cellulolytic complexes. Appl. Environ. Microbiol. 73, 3822–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mingardon F., Chanal A., Tardif C., Bayer E. A., Fierobe H. P. (2007) Exploration of new geometries in cellulosome-like chimeras. Appl. Environ. Microbiol. 73, 7138–7149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vazana Y., Moraïs S., Barak Y., Lamed R., Bayer E. A. (2010) Interplay between Clostridium thermocellum family 48 and family 9 cellulases in cellulosomal versus noncellulosomal states. Appl. Environ. Microbiol. 76, 3236–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moraïs S., Shterzer N., Grinberg I. R., Mathiesen G., Eijsink V. G., Axelsson L., Lamed R., Bayer E. A., Mizrahi I. (2013) Establishment of a simple Lactobacillus plantarum cell consortium for cellulase-xylanase synergistic interactions. Appl. Environ. Microbiol. 79, 5242–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vazana Y., Barak Y., Unger T., Peleg Y., Shamshoum M., Ben-Yehezkel T., Mazor Y., Shapiro E., Lamed R., Bayer E. A. (2013) A synthetic biology approach for evaluating the functional contribution of designer cellulosome components to deconstruction of cellulosic substrates. Biotechnol. Biofuels 6, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miras I., Schaeffer F., Béguin P., Alzari P. M. (2002) Mapping by site-directed mutagenesis of the region responsible for cohesin-dockerin interaction on the surface of the seventh cohesin domain of Clostridium thermocellum CipA. Biochemistry 41, 2115–2119 [DOI] [PubMed] [Google Scholar]