

Abstract
The disulfide bond is an important post-translational modification to form and maintain the native structure and biological functions of proteins. Characterization of disulfide bond linkages is therefore of essential interest in the structural elucidation of proteins. Top-down mass spectrometry (MS) of disulfide-bonded proteins has been hindered by relatively low sequence coverage due to disulfide cross-linking. In this study, we employed top-down ESI-MS with Fourier-transform ion cyclotron resonance (FT-ICR) MS with electron capture dissociation (ECD) and collisionally activated dissociation (CAD) to study the fragmentation of supercharged proteins with multiple intramolecular disulfide bonds. With charge enhancement upon the addition of sulfolane to the analyte solution, improved protein fragmentation and disulfide bond cleavage efficiency was observed for proteins including bovine β-lactoglobulin, soybean trypsin inhibitor, human proinsulin, and chicken lysozyme. Both the number and relative abundances of product ions representing disulfide cleavage increase with increasing charge states for the proteins studied. Our studies suggest supercharging ESI-MS is a promising tool to aid in the top-down MS analysis of disulfide-bonded proteins, providing potentially useful information for the determination of disulfide bond linkages.
Keywords: Supercharging, ESI-MS, protein, disulfide bond, top-down MS, Fourier-transform ion cyclotron resonance MS
1. Introduction
The formation of disulfide bonds is an important post-translational modification (PTM) to stabilize the folded structures of proteins [1–3], which is often required for protein activity and biological function. It is estimated that disulfide bonds are present in 15% of the human proteome and are associated with a growing list of protein folding related biological processes and diseases [4, 5], such as protein globular structure formation [1, 6], protein non-covalent interactions, and amyloid fibril formation [7–9]. Therefore, the determination of disulfide bonds is an important aspect for the complete structural elucidation of proteins. However, disulfide bond analysis by mass spectrometry (MS) approaches is challenging, especially for proteins composed of many S-S linkages.
In a conventional “bottom-up” MS approach, disulfide bonds are reductively cleaved, alkylated, and the protein is enzymatically digested prior to identification by MS and liquid chromatography-tandem MS (LC-MS/MS), and therefore information on disulfide bond connectivity can be lost. Alternatively, proteins can be digested under non-reducing or partially reducing conditions to generate disulfide-linked peptides for LC-MS/MS measurements [10–12]. Although the latter approach has been applied to peptides and proteins with simple disulfide connections, it has several noticeable drawbacks. Protein sequence coverage can be less than complete, and therefore could result in loss of disulfide linkage information. This becomes more significant for larger proteins with large numbers of disulfide bonds. The experimental conditions also need to be carefully controlled in order to avoid disulfide bond interchange.
Top-down mass spectrometry has become an increasingly important tool for protein characterization. Compared to bottom-up approaches, top-down MS has several distinct advantages. More simplified sample preparation procedures can be employed. The presence of post-translational modifications can be obtained by an intact protein mass spectrum and the modification sites can be further determined by MS/MS with very high sequence coverage. In the past decade, top-down MS has been successfully applied to a wide range of applications including protein PTM characterization [13, 14] and determination of protein-protein and protein-ligand non-covalent interactions [15, 16]. Top-down MS for protein structure and PTM characterization is not lacking in challenges currently, and these include speed and sensitivity, fragmentation efficiency to achieve complete primary sequence coverage, and efficient software for data interpretation. However, the potential of top-down MS to either compete with and/or complement current bottom-up MS strategies is encouraging.
Several groups previously have used this approach to study disulfide-linked peptides and proteins. McLafferty and coworkers applied top-down MS with electron capture dissociation (ECD), and reported preferential cleavage of disulfide bonds over backbone amide bonds in disulfide-linked peptides [17, 18]. Similar preferential cleavage of disulfide bonds was also reported for deprotonated peptides by collisionally activated dissociation (CAD) fragmentation in negative ion MS mode [19–21]. However, the ECD and CAD efficiency of disulfide bond cleavage in proteins is relatively low. To improve disulfide bond cleavage via tandem MS, various approaches have been implemented including metal cationization [22–24], ultraviolet photodissociation [25], reactive electrospary-assisted laser desoption/ionization [26], electrolytic reduction [12], plasma induced oxidation [27], and other electron based fragmentation methods such as ETD [28] and EDD with moderate success [29].
Recently, several groups extended top-down MS to proteins with more complex disulfide bond connections. Chen et al studied chicken lysozyme, a protein with four intramolecular disulfide bonds, by CAD using an orbitrap mass spectrometer and reported product ions with multiple disulfide bond cleavages [30]. Ge and coworkers studied human salivary α-amylase, a 56 kDa protein with five disulfide bonds, by ECD and CAD and were able to map the disulfide bond connections through a combination of top-down MS and limited digestion [31]. However, thus far, low fragmentation efficiency of disulfide bonds is still a major limiting factor in the top-down MS analysis of proteins.
Previously, our group and others have demonstrated the use of reagents such as sulfolane and m-nitrobenzyl alcohol (mNBA) to significantly increase the charge states of proteins and protein complexes in ESI-MS experiments [32–35]. These supercharging reagents can increase the protein charge state [36, 37], and therefore can be used for the MS analysis of a wide range of proteins and other biomolecules. We have also shown that supercharging can be applied to protein-ligand complex systems to increase the fragmentation efficiency for top-down MS studies and generate more product ions retaining protein-ligand interactions that provide improved sequencing and non-covalent protein-ligand interaction mapping [34]. In this study, we extended supercharging to top-down MS analysis with ECD and CAD of disulfide-bonded proteins. For several proteins with multiple disulfide-bonded networks, we consistently observed improved cleavage of disulfide bonds and more extensive protein backbone fragmentation for the higher charged proteins generated by supercharging compared to the lower charged proteins.
2. Experimental
2.1. Samples and sample preparation
Bovine β-lactoglobulin, soybean trypsin inhibitor, chicken lysozyme, and human proinsulin were purchased from Sigma-Aldrich (St. Louis, MO). Chemicals were purchased from Aldrich (St. Louis, MO) unless otherwise noted. All protein samples were desalted with 10 mM ammonium acetate using centrifugal filter devices (10,000 molecular weight cutoff, Microcon and Amicon Ultra; Millipore Corporation, Billerica, MA) before analysis. All solutions were prepared in Milli-Q water (Millipore Corporation, Billerica, MA). Glass nanoelectrospray emitters were purchased from Proxeon/Thermo Scientific (West Palm Beach, FL).
2.2. Top-down FT-ICR mass spectrometry
Top-down MS of proteins was performed on an ultrahigh resolution 15-Tesla Bruker SolariX hybrid Qq-FTICR mass spectrometer. Proteins (0.5–5 μM) were prepared in denaturing solution conditions with an acetonitrile (ACN): H2O: formic acid (FA) ratio of 49.95: 49.95: 0.1. Supercharging reagent sulfolane was added to a final concentration of 150 mM. The protein solutions were nanoelectrosprayed at flow rates of 20–50 nL/min. MS experiments were performed in the broadband mode from m/z 600–3000 with the following settings: capillary voltage 1000–1200 V; source accumulation time 0.5 sec; ion accumulation time 1 sec; ion cooling time 0.05 sec; time of flight 1 ms. Precursor ions of single charge states were isolated by a quadrupole Q1 with a selection window size of 10–20 m/z. MS/MS experiments were performed with the following settings: ECD, pulse length 0.01 sec; bias 1 V; ECD lens 15 V; CAD, collision energy 8–15 V adjusted for each charge state according to the normalized energy level: CS1 × CE 1 = CS2 × CE 2, where CS denotes the charge state and CE denotes collision energy. 50–100 scans were averaged for each MS experiment. 400 scans were averaged for each ECD experiment in the charge state dependent experiments and 200 scans for each CAD experiment.
2.3. Data analysis
MS/MS data were processed with DataAnalyis and Biotools (Bruker Daltonics). Briefly, monoisotopic masses ([M+H]+) were extracted by DataAnalysis software using a modified Thrash algorithm (SNAP ver 2.0) with the following settings: quality factor threshold 0.5; S/N threshold 2; maximum charge state, ≤ protein precursor charge state. Product ions were assigned using Biotools based on protein sequences determined by accurate mass measurements. Mass accuracy of 15 ppm was used for ECD/CAD product ion assignments. The assigned ions were manually confirmed to ensure the quality of assignments.
Product ion intensities were also extracted from DataAnalysis software for relative abundance of disulfide bond cleavage calculation. Quantification of disulfide bond cleavage in the MS/MS experiments was calculated as:
where Id denotes the ion abundance of product ions with disulfide cleavage, Itotal the total ion abundance.
3. Results and discussion
3.1. Supercharging and top-down MS of β-lactoglobulin
β-Lactoglobulin is an 18 kDa protein present as a major milk component in many mammalian species. Figure 1 shows the high resolution ESI-MS mass spectra of β-lactoglobulin collected under both conventional and supercharging conditions. The addition of sulfolane to 150 mM did not significantly decrease the signal intensity for the proteins studied. However, adding sulfolane significantly increased both the average and maximum charge states of β-lactoglobulin; the most abundant charge state was increased to 15+ with sulfolane, compared to 12+ in the solution without sulfolane. The maximum observed charge state observed with the addition of sulfolane was 18+, compared to 15+ without sulfolane. The measured mass of β-lactoglobulin, 18276.45 Da for the most abundant isotopic mass, compared to the protein’s sequence (P02754, SwissProt/UniProt) corresponds to the presence of two intramolecular disulfide bonds.
Figure 1.

ESI mass spectra of β-lactoglobulin (bovine) (0.5 μM) (a) with 150 mM sulfolane in ACN: H2O: FA (50:50:0.1) (inset: high resolution profile of 11+ charged molecule) and (b) protein in ACN: H2O: FA (50:50:0.1).
To compare the fragmentation of different charge states of β-lactoglobulin, the two most abundant charge states under both supercharging (14+, 15+) and conventional ESI conditions (12+, 13+), respectively, were selected for ECD fragmentation (Figure S1). For all four charge states, the product ions observed were predominantly cleaved from the N-terminal part of the sequence and these ions formed a nearly continuous sequence ladder, from the N-terminus to c65 (Figure 2). Close comparison of the N-terminal product ions from the different charge states indicated more complete sequence coverage was observed for the higher charged precursors (15+, 14+ generated by supercharging). For example, product ions between c18 - c22, c37 - c39, and c57 - c61 were absent for the 12+ charge state, whereas these products were detected for the supercharged 15+ and 14+ precursors, thus demonstrating the improved ECD fragmentation efficiency of the supercharge-generated higher charged protein.
Figure 2.

ECD product ion map of β-lactoglobulin (bovine) for charge states (a) 15+ and (b) 14+ under supercharging conditions, and (c) 13+ and (d) 12+ under non-supercharging conditions.
The lack of product ions detected from c65 to the C-terminus for the 12+ charged precursors suggested this part of the protein sequence is likely enclosed by disulfide link(s). Product ions between Cys66 and Cys160 were completely absent in the 12+ charge state protein. Increasing the precursor charge state resulted in the observation of more product ions in this region, from five (c120, c124, c152, z11·, z22·) in charge state 13+, to eleven (c69, c76, c82, c84, z4·, z6·, z7·, z8·, z11·, z22·, z43·) in charge state 14+, and fourteen (c114, c120, c124, z4·, z6·, z7·, z8·, z11·, z17·, z22·, z26·, z32·, z39·, z43·) in charge state 15+.
ECD disulfide bond cleavage primarily occurred through hemolytic scission of S-S bonds; however there are also a substantial proportion of products corresponding to the C-S bond dissociation that resulted in asymmetric partition of sulfur upon cleavage. Among these product ions, c114 corresponded to the cleavage of two disulfide bonds (Cys66-Cys120, Cys106-Cys119) that was only observed in the charge state 15+ from supercharging. Figure 3 shows representative ECD product ions from disulfide cleavage. Product ion signal-to-noise ratio was significantly increased upon supercharging (i.e., higher charging), which resulted in the identification of more disulfide cleaved ions. The total relative abundance of product ions with disulfide cleavages increase from 0.9% at 13+, to 1.8% at 14+ and 3.3% at 15+, a nearly four fold increase upon supercharging (Figure S2).
Figure 3.

Representative ECD product ions of β-lactoglobulin (bovine) with disulfide bond cleavage. The insets indicate the bond cleavage at the disulfide connection.
Despite the general increase in the number of product ions with disulfide bond cleavages under supercharged conditions, it was also noted that a number of ions were only observed in specific charge states. For example, c69, c76, c82, c84 were only observed for charge state 14+, while these ions were not detected in charge state 15+. This suggests that distinct fragmentation pathways may exist for specific charge states that may be associated with specific protein conformations and charge distributions along the protein chain.
Another interesting type of ion containing potentially cleaved disulfide bonds were large z· ions that contain two disulfide bonds (Figure S3). For example, the sequence of product ions z130·, z139·, z152·, z153·, and z158· include disulfide bonds Cys66-Cys120 and Cys106-Cys119; successive mass increases of 1 Da were observed for these ions that potentially correspond to the addition of hydrogen radicals formed in the ECD process due to the electron affinity for the positively charged protein ions. The attachment of hydrogen radicals to the disulfide bond has been proposed to be an underlying driving force for the preferential cleavage of disulfide bonds in the ECD experiment reported by Zubarev et al [17]. Therefore the presence of these hydrogen radical ions could suggest the cleavage of disulfide bonds in the protein; however, because the protein backbone covered by the disulfide loop was not simultaneously cleaved, these potentially cleaved disulfide moieties remained in the same molecule and could not be further identified from their mass. This suggests also that protein backbone and disulfide bond cleavages are probably independent processes, in contrast to the reported concurrent disulfide and backbone cleavages in ETD experiments for smaller peptides containing disulfide bonds [38]. A recent study by Ganisl and Breuker also suggested ECD did not concurrently cleave disulfide and backbone N-Cα bonds for proteins containing intramolecular disulfide bonds [39]. This may explain the fact that observed ions with disulfide cleavages represents a relatively small proportion of product ions in larger proteins.
The fragmentation efficiency of β-lactoglobulin by CAD was evaluated for six charge states under both conventional and supercharging conditions (Figure 4). The overall fragmentation efficiency increased significantly with increasing charge state, as indicated by the depletion of the precursor ions and the increase of product ions as the charge state increased. For the 12+ charge state, a total of 32 sequence ions were observed, among which one corresponded to the cleavage of a disulfide bond (b134). The number of product ions continuously increased to 40/3 (total ions/disulfide cleavage ions) for charge state 13+, 42/3 for 14+, 47/4 for 15+, 49/5 for 16+, and 64/9 for charge state 17+. For the lower charge states, the predominant product ions were large y ions (e.g., y140, y139, y138, y135). As the charge state increased, the abundance of the large fragment ions decreased and the number of smaller product ions increased, which resulted in a notable increase in the sequence coverage of the protein (e.g., y131, y129, y128, y127, y125, y115, y109, y108, y107, y106, y105, etc) (Figures 5 and S4). Unlike ECD fragmentation, CAD preferentially cleaves the protein sequence at specific residues including proline (e.g., y113, y115, y125, etc) and aspartic acid (e.g., y109, y129, y134, etc). Therefore, product ions had larger variation in their intensity compared with ECD product ions. The improved fragmentation efficiency under supercharging conditions allowed more sequence ions to be obtained for more confident protein sequencing. Among product ions with disulfide bond cleavage, b134 was a common ion observed for all 6 precursor charge states probed, and its abundance increases with increasing charge state from 12+ to 17+ (Figure S5). Formation of this product ion involved concurrent cleavage on the C-terminal side of Glu134 and hemolytic cleavage of the Cys66-Cys160 bond. Cleavage on the C-terminal side of acidic residues (Glu, Asp) is a preferred CAD fragmentation pathway. Therefore the intensity increase of this ion is attributed to the more efficient cleavage of the Cys66-Cys160 bond under supercharging conditions. Both the b134 ion and its complimentary y28 ion were measured with high mass accuracy, 0.7 and 1.5 ppm respectively, which allowed confident identification of these disulfide cleaved ions (Figure S6). In addition, the observed mass of 14969.70 Da matched uniquely with b134. Overall the abundance of product ions with disulfide cleavages increased significantly from below 2% (12+ charge state) to above 25% (17+ charge state) (Figure S7). (Table 1 lists observed product ions with disulfide bond cleavages.)
Figure 4.

CAD mass spectra of β-lactoglobulin (with 150 mM sulfolane and 50:50:0.1 ACN: H2O: FA) for (a) charge state 17+, (b) charge state 16+, and (c) charge state 15+, and β-lactoglobulin without sulfolane for (d) charge state 14+, (e) charge state 13+, and (f) charge state 12+ with similar laboratory-frame collision energy.
Figure 5.

CAD product ion map of β-lactoglobulin (bovine) (see Figure 4 for solution conditions).
Table 1.
β-Lactoglobulin (bovine) CAD/ECD product ions with disulfide bond cleavages.
| Ion Assignment | Observed Mass (Da) | Theoretical Mass (Da) | Mass Error (Da) | Mass Error (ppm) | Note |
|---|---|---|---|---|---|
| b69 | 7596.894 | 7596.924 | −0.030 | −3.9 | Cys66+H; one disulfide cleavage |
| b111 | 12471.391 | 12471.473 | −0.083 | −6.6 | Cys66+SH; Cys106+SH; two disulfide cleavage |
| b133 | 14829.601 | 14829.607 | −0.007 | −0.5 | Cys66−H; one disulfide cleavage |
| b134 | 14961.686 | 14961.673 | 0.013 | 0.8 | Cys66+H; one disulfide cleavage |
| b*134 | 14976.677 | 14976.618 | 0.059 | 3.9 | Cys66+SH; one disulfide cleavage |
| b∘138 | 15493.926 | 15493.920 | 0.006 | 0.4 | Cys66+SH; one disulfide cleavage |
| b∘∘138 | 15475.945 | 15475.885 | 0.059 | 3.8 | Cys66+SH; one disulfide cleavage |
| y4 | 498.217 | 498.213 | 0.007 | 8.1 | Cys160−H; one disulfide cleavage |
| y5 | 660.276 | 660.271 | 0.012 | 7.3 | Cys160+S; one disulfide cleavage |
| y6 | 789.319 | 789.314 | 0.008 | 6.6 | Cys160+S; one disulfide cleavage |
| y21 | 2476.256 | 2476.235 | 0.037 | 8.5 | Cys160−H; one disulfide cleavage |
| y22 | 2571.371 | 2571.350 | 0.051 | 8.2 | Cys160−SH; one disulfide cleavage |
| y23 | 2749.395 | 2749.422 | −0.053 | −9.7 | Cys160+S; one disulfide cleavage |
| y25 | 2949.497 | 2949.526 | −0.040 | −9.8 | Cys160+SH; one disulfide cleavage |
| y*25 | 2931.475 | 2931.492 | −0.017 | −5.8 | Cys160+S; one disulfide cleavage |
| y28 | 3305.749 | 3305.729 | 0.021 | 6.2 | Cys160−H; one disulfide cleavage |
| c69 | 7614.951 | 7614.958 | −0.006 | −0.8 | Cys66+H, one disulfide bond cleavage |
| c76 | 8398.440 | 8398.443 | −0.003 | −0.3 | Cys66+H, one disulfide bond cleavage |
| c82 | 9053.842 | 9053.849 | −0.006 | −0.7 | Cys66+H, one disulfide bond cleavage |
| c84 | 9295.026 | 9295.028 | −0.002 | −0.2 | Cys66+H, one disulfide bond cleavage |
| c114 | 12712.642 | 12712.726 | −0.084 | −6.6 | Cys66−SH, Cys106−SH, two disulfide bond cleavage |
| c124 | 13912.071 | 13912.200 | −0.129 | −9.3 | Cys66+SH, one disulfide bond cleavage |
| z6 | 741.287 | 741.287 | 0.0001 | 0.1 | Cys160−H, one disulfide cleavage |
| z7 | 854.371 | 854.371 | −0.0004 | −0.5 | Cys160−H, one disulfide cleavage |
| z8 | 982.429 | 982.430 | −0.0005 | −0.5 | Cys160+H, one disulfide cleavage |
| z•11 | 1296.591 | 1296.589 | 0.002 | 1.6 | Cys160−H, one disulfide cleavage, mass off |
| z17 | 2047.010 | 2046.994 | 0.016 | 8.0 | Cys160+SH, one disulfide bond cleavage |
| z•22 | 2622.319 | 2622.299 | 0.020 | 7.5 | Cys160−SH, one disulfide cleavage |
| z31 | 3602.850 | 3602.874 | −0.023 | −6.5 | Cys160−H, one disulfide cleavage |
| z•26 | 3017.541 | 3017.570 | −0.029 | −9.6 | Cys160−H, one disulfide cleavage |
| z29 | 3418.785 | 3418.752 | 0.082 | 9.5 | Cys160+H, one disulfide cleavage |
| z39 | 4546.339 | 4546.298 | 0.050 | 9.0 | Cys160+H, one disulfide cleavage |
denotes ammonium neutral loss;
denote one and two water loss ions, respectively.
3.2. Supercharging and MS/MS of trypsin inhibitor
Top-down MS was also performed for soybean trypsin inhibitor, a 20 kDa protein with two disulfide bonds, under both supercharging and conventional ESI conditions. The addition of sulfolane resulted in a significantly higher overall charge state distribution (from 13+ to 20+), compared to those under conventional solution conditions (from 10+ to 17+) (Figure S8). High resolution and accurate mass measurements revealed that two forms of the protein were present in the sample: the mature full chain form of trypsin inhibitor (P01070_ITRA_SOYBN, D[25-205]L) and a truncated form (P01070, D[25-204]S) with the loss of C-terminal Leu. The full chain form only accounted for ~ 15% of the total protein abundance (Figure 6). The major form (D[25-204]S) was selected for top-down MS. For the 14+ precursor, CAD yielded little fragmentation and the precursor ion largely remained intact. As the precursor charge state increased, the fragmentation efficiency increased as indicated by the decrease of the precursor ion intensities and increase of observed product ions (Figure 7). A total of 17 sequence ions (8 b ions, 9 y ions) were observed for charge state 15+, with no disulfide cleaved ions (Figures S9 and S10). For the 16+ precursor, the number of product ions increased to 34 (20 b ions, 14 y ions), with one disulfide cleaved ion (b84). 52 ions (31 b ions, 21 y ions) were observed for charge state 17+, and among them were two disulfide cleaved products (b75, b84). For the 18+ charge state, a total of 4 product ions with disulfide cleavage were observed (b75, b78, b82, b84), and 56 total sequence ions (32 b ions, 24 y ions).
Figure 6.

High resolution mass spectrum of trypsin inhibitor (soybean) showing the 14+ charge profile. The predominant protein form corresponds to the full chain D[25-205]L with the loss of C-terminal Leu.
Figure 7.

(a–c) CAD mass spectra of trypsin inhibitor (soybean; 50:50:0.1 ACN: H2O: FA) with 150 mM sulfolane and (d–e) without sulfolane.
Disulfide cleavage occurred exclusively at the Cys39-Cys86 bond, while no cleavage was observed for the Cys136-Cys145 bond. This may be due to the relative short sequence between the Cys136-Cys145 bond, and therefore lower probability of concurrent cleavages of both the disulfide bond and the protein backbone enclosed by the disulfide loop. For the same reason, relatively fewer product ions with disulfide bond cleavage were observed for the Cys39-Cys86 bond that encompasses 48 residues, compared to the Cys66-Cys160 bond in β-lactoglobulin that includes 95 residues. The intrinsic structural properties of the proteins may also be an important factor in determining the cleavages of the disulfide bonds and the protein backbone enclosed by the disulfide link.
ECD of trypsin inhibitor showed a similar trend, i.e., improved disulfide cleavage for higher precursor charge. Consistent with the previous report by Ganisl and Breuker [39], activation pre- or post-ECD was not effective for cleaving disulfide bonds for large proteins such as trypsin inhibitor. ECD of the 15+ charge state produced 16 c ions and 24 z· ions with one disulfide bond cleavage (z102·). Supercharge-generated 16+ and 17+ precursors produced 18 c ions/26 z· ions and 28 c ions/27 z· ions, respectively, with two disulfide cleaved ions (c62, z102·). Charge state 18+ produced the same disulfide cleaved ions, although the number of backbone fragment ions decreased slightly (18 c ions, 20 z· ions), probably due to the lower precursor ion abundance. Two product ions observed with disulfide cleavages were located within the sequence covered by the Cys39-Cys86 disulfide bond, and no disulfide cleavage was observed for Cys136-Cys145 bond (Figure S11). Surprisingly, no fragment ions were observed for the N-terminal region of the protein, despite this region not enclosed by a disulfide bond. This suggests the N-terminal region could be “protected” by conformational effects. Similar conformational effect has also been observed previously in ECD experiments of native proteins [40, 41] and protein complexes [42–45].
3.3. Supercharging top-down MS of proinsulin and lysozyme
Additional supercharging top-down MS experiments were performed on two proteins with multiple intrachain disulfide bonds: human proinsulin, a 9.3 kDa protein with three disulfide bonds, and 14 kDa chicken lysozyme with four disulfide bonds. The most abundant charge state of proinsulin was increased from 7+ to 9+ after adding sulfolane to the solution (Figure S12). For CAD of the 7+ charged precursor, little protein fragmentation was observed, with 85% of the precursor remaining intact; no product ions were observed corresponding to the cleavage of any of the three disulfide bonds. CAD of the 8+ produced significantly more extensive fragmentation. Because the structure of proinsulin is tightly enclosed by three overlapping disulfide bonds covering 92% of the protein backbone (79 of 86 total residues), a significant proportion of the product ions correspond to internal cleavage ions and only one product ion containing a disulfide bond cleavage (y72) was observed. CAD of the 9+ charge state generated by supercharging marked a major improvement for disulfide bond cleavage and yielded 6 b/y ions with one or two disulfide bond cleavages (Table 2).
Table 2.
CAD product ions containing disulfide bond cleavages observed for 9+ charge human proinsulin (with sulfolane).
| Ion | Observed Mass (Da) | Theoretical Mass (Da) | Mass Error(Da) | Mass Error(ppm) | Note |
|---|---|---|---|---|---|
| b7 | 808.416 | 808.410 | 0.006 | 7.212 | Cys7−SH; one disulfide cleavage |
| b67 | 7208.669 | 7208.653 | 0.016 | 2.236 | Cys7+SH;Cys19-1.0078; two disulfide cleavage; low S/N, mixed with other ions |
| b68 | 7307.726 | 7307.722 | 0.005 | 0.617 | Cys7+SH;Cys19-1.0078; two disulfide cleavage |
| b74 | 7923.867 | 7923.925 | −0.059 | −7.427 | Cys19+H, Cys71−H; or Cys19−H, Cys71+H; two disulfide cleavage |
| b75 | 8034.962 | 8034.994 | −0.032 | −4.021 | Cys19−H; Cys71−H; two disulfide cleavage |
| y*72 | 7865.753 | 7865.763 | −0.010 | −1.294 | Cys72+SH; one disulifide cleavage |
Similar to proinsulin, lysozyme is tightly enclosed by four overlapping intramolecular disulfide bonds that cover 95% of the protein sequence. As a result, ECD provided limited fragmentation of the molecule because of the low efficiency for concurrent cleavages of disulfide bonds and protein backbone. CAD experiments with four different charge states from supercharging (12+ and 13+) and conventional ESI conditions (10+ and 11+) exhibited similar fragmentation patterns as observed for the other disulfide-bonded proteins (Figure S13). For lower charge states, the protein precursor ions largely remained intact and very few fragment ions were formed for the 10+ and 11+ molecules, whereas the supercharged 12+ and 13+ molecules showed increased fragmentation efficiency and product ion yield. Accurate mass measurements also revealed product ions containing disulfide cleavages not observed at lower charge states (Figure S14), which could be attributed to improved signal-to-noise ratio and distinct fragmentation channels associated with proton distribution at supercharged states.
3.4. Effect of charge state on disulfide protein fragmentation
The effect of precursor ion charge state on protein and peptide fragmentation has been observed previously with various activation techniques including CAD [46–50], ECD [51], and ETD [52]. The charge state effect is especially prominent for electron based fragmentation techniques such as ECD [51] and ETD [52], with more sequence ions being produced for higher charge states.
The mechanism of disulfide bond cleavage by ECD and CAD is still not thoroughly understood. It is widely accepted that the high electron affinity of disulfide bonds is a primary driving force for the preferential cleavage of disulfide bonds by ECD and other electron based fragmentation methods, as well as in negative ion MS [17]. Electrons are either directly captured at the disulfide bond or through intramolecular transfer in the form of nascent hydrogen radicals (Scheme 1), although experimental and theoretical studies currently provide more evidence to support the latter scenario [17, 53]. Ganisl and Breuker suggest that electron capture occurs at protonated sites, resulting in backbone cleavage into c/z• fragments, followed by disulfide bond cleavage via secondary radical reactions, provided that the disulfide bond is in close proximity to a radical site from the primary backbone cleavage [39]. Our results are consistent with the Ganisl and Breuker report [39], as the higher charge states from supercharging provide more accessibility of nascent hydrogen radicals, which are conducive to more efficient disulfide bond cleavage.
Scheme 1.
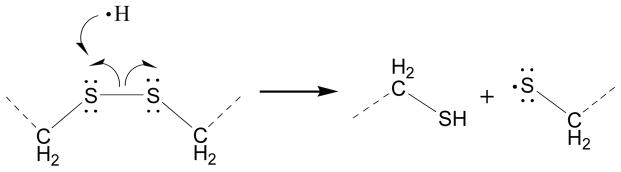
ECD cleavage of disulfide bond involving transfer and attachment of nascent hydrogen radical to the disulfide bond.
The mechanism of disulfide bond cleavage by CAD fragmentation is more elusive and relatively less understood, partly because of the low proton affinity of the disulfide bond and higher energy barrier for breaking disulfide bonds compared to amide bonds [54]. There are several studies supporting more facile disulfide bond cleavage at lower charge states or non-mobile proton conditions [30, 54]. A CAD top-down MS study of lysozyme reported richer fragmentation spectra for protein ions with relatively low charge states (e.g., 9+), and the report hypothesized that precursor ions without any or fewer mobile protons yielded more abundant fragments corresponding to the cleavages of one or more disulfide bonds [30]. Our study demonstrates more efficient disulfide cleavage for higher charged molecules. Compared to the salt bridge mechanism proposed previously [54], our results showing more facile disulfide bond cleavage for higher charge states suggests the participation of protons in the cleavage of the disulfide bond (Scheme 2).
Scheme 2.

Proposed mechanism for CAD cleavage of disulfide bond involving participation of a mobile proton.
3.5. Mapping of disulfide bond connections by top-down MS
Top-down MS of intact proteins with disulfide bonds can provide direct evidence regarding the location of disulfide bonds. CAD fragmentation of soybean trypsin inhibitor represents an exemplary case in this regard. The data obtained from multiple charge states showed a highly reproducible pattern in their product ion distribution. Several continuous sequence ion series from backbone cleavage (b1-b25, b86-b120, b150-b180) suggest the regions lacking product ions are due to the presence of disulfide bonds (Cys39-Cys86, Cys136-Cys145) (Figure S9). Increasing charge states generated more product ions in the region linked by the long-range disulfide bond (Cys39-Cys86). However, the relatively short region covered by the disulfide bond Cys136-Cys145 showed no new product ions at higher charge states.
Proteins with multiple overlapping disulfide bonds present more elusive and challenging cases for the determination of disulfide bond patterns. ECD fragmentation of β–lactoglobulin showed nearly consecutive cleavages on the N-terminal side of the sequence to c65, which provides clear evidence that the N-terminal part of the protein is disulfide free. However, the very sparse ion yield between c65 and the C-terminus could indicate different possibilities. This relatively long sequence could be covered by either overlapping disulfide bonds or tightly spaced disulfide bonds. Our data suggested the first possibility could be more likely the case, since very few ions were observed in the sequence between Cys106 and Cys119, suggesting the presence of a disulfide bond within this sequence. Overall, however, our study also suggests that disulfide bond connectivity studies could be enhanced by a strategy that combines supercharging to generate higher charged precursors for more effective ECD/ETD/CAD (with gas phase cleavage of some S-S bonds) and partial solution phase disulfide bond reduction [31].”
4. Conclusions
Supercharging agents such as sulfolane can promote significant increase in the ESI charging of proteins with intramolecular disulfide bonds. This increased charging allows improved fragmentation efficiency for ECD and CAD of disulfide-bonded proteins. The numbers and relative abundances of product ions representing disulfide cleavages increase with increasing precursor ion charge state, and this yields better overall sequence coverage for protein top-down MS experiments. However, mapping disulfide linkages remain challenging, especially for proteins larger than 20 kDa with complex disulfide bonding patterns. Disulfide bond cleavage by ECD or CAD is still a relatively low yield process, even from the higher charged proteins produced from supercharging ESI. Potentially, supercharging ESI-MS represents one of several tools that can be useful for disulfide mapping of these structurally challenging biomolecules.
Supplementary Material
Acknowledgments
Support from the US National Institutes of Health (R01GM103479, S10RR028893) and the US Department of Energy (UCLA Institute of Genomics and Proteomics; DE-FC03-02ER63421) are acknowledged.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at X.
References
- 1.Betz SF. Disulfide bonds and the stability of globular-proteins. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feige MJ, Buchner J. The Role of Disulfide Bonds in Protein Folding and Stability. In: Buchner J, Moroder L, editors. RSC Biomolecular Sciences. Royal Soc Chemistry, Thomas Graham House; Science Park, Cambridge Cb4 4wf, Cambs, Uk: 2009. pp. 179–191. [Google Scholar]
- 3.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Disulfide bonds and protein folding. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 4.Thomas PJ, Qu BH, Pedersen PL. Defective protein-folding as a basis of human-disease. Tr Biochem Sci. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- 5.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Ann Rev Biochem. 2006:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 6.Thornton JM. Disulphide bridges in globular proteins. J Mol Biol. 151(1981):261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Gong H, Sun Y, Yan J, Cheng B, Zhang X, Huang J, Yu M, Guo Y, Zheng L, Huang K. Dissecting the role of disulfide bonds on the amyloid formation of insulin. Biochem Biophys Res Commun. 2012;423:373–378. doi: 10.1016/j.bbrc.2012.05.133. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Yan J, Zhang X, Huang K. Disulfide bonds in amyloidogenesis diseases related proteins. Proteins. 2013;81:1862–1873. doi: 10.1002/prot.24338. [DOI] [PubMed] [Google Scholar]
- 9.Mossuto MF, Bolognesi B, Guixer B, Dhulesia A, Agostini F, Kumita JR, Tartaglia GG, Dumoulin M, Dobson CM, Salvatella X. Disulfide bonds reduce the toxicity of the amyloid fibrils formed by an extracellular protein. Angew Chem Int Ed. 2011;123:7186–7189. doi: 10.1002/anie.201100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foley SF, Sun Y, Zheng TS, Wen D. Picomole-level mapping of protein disulfides by mass spectrometry following partial reduction and alkylation. Anal Biochem. 2008;377:95–104. doi: 10.1016/j.ab.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 11.Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Dewald HD, Chen H. Online Mass Spectrometric Analysis of Proteins/Peptides Following Electrolytic Cleavage of Disulfide Bonds. J Proteome Res. 2011;10:1293–1304. doi: 10.1021/pr101053q. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Dong X, Hacker TA, Ge Y. Deciphering Modifications in Swine Cardiac Troponin I by Top-Down High-Resolution Tandem Mass Spectrometry. J Am Soc Mass Spectrom. 2010;21:940–948. doi: 10.1016/j.jasms.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Zhang H, Ayaz-Guner S, Chen YC, Dong X, Xu Q, Ge Y. Phosphorylation, but Not Alternative Splicing or Proteolytic Degradation, Is Conserved in Human and Mouse Cardiac Troponin T. Biochemistry. 2011;50:6081–6092. doi: 10.1021/bi2006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Y, Zhang J, Yin S, Loo JA. Top-down ESI-ECD-FT-ICR mass spectrometry localizes noncovalent protein-ligand binding sites. J Am Chem Soc. 2006;128:14432–14433. doi: 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- 16.Yin S, Loo JA. Elucidating the Site of Protein-ATP Binding by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom. 2010;21:899–907. doi: 10.1016/j.jasms.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J Am Chem Soc. 1999;121:2857–2862. [Google Scholar]
- 18.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park J-H, Begley TP, McLafferty FW. Top Down Characterization of Larger Proteins (45 kDa) by Electron Capture Dissociation Mass Spectrometry. J Am Chem Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 19.Chrisman PA, McLuckey SA. Dissociations of Disulfide-Linked Gaseous Polypeptide/Protein Anions: Ion Chemistry with Implications for Protein Identification and Characterization. J Proteome Res. 2002;1:549–557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 20.Bilusich D, Maselli VM, Brinkworth CS, Samguina T, Lebedev AT, Bowie JH. Direct identification of intramolecular disulfide links in peptides using negative ion electrospray mass spectra of underivatised peptides. A joint experimental and theoretical study. Rapid Commun Mass Spectrom. 2005;19:3063–3074. doi: 10.1002/rcm.2149. [DOI] [PubMed] [Google Scholar]
- 21.Zhang MX, Kaltashov IA. Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection. Anal Chem. 2006;78:4820–4829. doi: 10.1021/ac060132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lioe H, Duan M, O’Hair RAJ. Can metal ions be used as gas-phase disulfide bond cleavage reagents? A survey of coinage metal complexes of model peptides containing an intermolecular disulfide bond. Rapid Commun Mass Spectrom. 2007;21:2727–2733. doi: 10.1002/rcm.3122. [DOI] [PubMed] [Google Scholar]
- 23.Kim HI, Beauchamp JL. Identifying the presence of a disulfide linkage in peptides by the selective elimination of hydrogen disulfide from collisionally activated alkali and alkaline earth metal complexes. J Am Chem Soc. 2008;130:1245–1257. doi: 10.1021/ja075698w. [DOI] [PubMed] [Google Scholar]
- 24.Mentinova M, McLuckey SA. Cleavage of multiple disulfide bonds in insulin via gold cationization and collision-induced dissociation. Int J Mass Spectrom. 2011;308:133–136. doi: 10.1016/j.ijms.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fung YME, Kjeldsen F, Silivra OA, Chan TWD, Zubarev RA. Facile disulfide bond cleavage in gaseous peptide and protein cations by ultraviolet photodissociation at 157 nm. Angew Chem Int Ed. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 26.Peng IX, Ogorzalek Loo RR, Shiea J, Loo JA. Reactive-electrospray-assisted laser desorption/ionization for characterization of peptides and proteins. Anal Chem. 2008;80:6995–7003. doi: 10.1021/ac800870c. [DOI] [PubMed] [Google Scholar]
- 27.Xia Y, Cooks RG. Plasma Induced Oxidative Cleavage of Disulfide Bonds in Polypeptides during Nanoelectrospray Ionization. Anal Chem. 2011;82:2856–2864. doi: 10.1021/ac9028328. [DOI] [PubMed] [Google Scholar]
- 28.Gunawardena HP, Gorenstein L, Erickson DE, Xia Y, McLuckey SA. Electron transfer dissociation of multiply protonated and fixed charge disulfide linked polypeptides. Int J Mass Spectrom. 2007;265:130–138. [Google Scholar]
- 29.Håkansson K, Kalli A. Preferential cleavage of SS and CS bonds in electron detachment dissociation and infrared multiphoton dissociation of disulfide-linked peptide anions. Int J Mass Spectrom. 2007;263 [Google Scholar]
- 30.Chen J, Shiyanov P, Zhang L, Schlager JJ, Green-Church KB. Top-Down Characterization of a Native Highly Intralinked Protein: Concurrent Cleavages of Disulfide and Protein Backbone Bonds. Anal Chem. 2010;82:6079–6089. doi: 10.1021/ac1006766. [DOI] [PubMed] [Google Scholar]
- 31.Peng Y, Chen X, Sato T, Rankin SA, Tsuji RF, Ge Y. Purification and High-Resolution Top-Down Mass Spectrometric Characterization of Human Salivary alpha-Amylase. Anal Chem. 2012;84:3339–3346. doi: 10.1021/ac300083y. [DOI] [PubMed] [Google Scholar]
- 32.Lomeli SH, Peng IX, Yin S, Ogorzalek Loo RR, Loo JA. New Reagents for Increasing ESI Multiple Charging of Proteins and Protein Complexes. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomeli SH, Yin S, Ogorzalek Loo RR, Loo JA. Increasing: Charge While Preserving Noncovalent Protein Complexes for ESI-MS. J Am Soc Mass Spectrom. 2009;20:593–596. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin S, Loo JA. Top-down mass spectrometry of supercharged native protein-ligand complexes. Int J Mass Spectrom. 2011;300:118–122. doi: 10.1016/j.ijms.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iavarone AT, Williams ER. Supercharging in electrospray ionization: effects on signal and charge. Int J Mass Spectrom. 2002;219:63–72. [Google Scholar]
- 36.Iavarone AT, Williams ER. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J Am Chem Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogorzalek Loo RR, Lakshmanan R, Loo JA. What Protein Charging (and Supercharging) Reveal about the Mechanism of Electrospray Ionization. J Am Soc Mass Spectrom. 2014 doi: 10.1007/s13361-014-0965-1. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole SR, Ma X, Zhang X, Xia Y. Electron Transfer Dissociation (ETD) of Peptides Containing Intrachain Disulfide Bonds. J Am Soc Mass Spectrom. 2012;23:310–320. doi: 10.1007/s13361-011-0300-z. [DOI] [PubMed] [Google Scholar]
- 39.Ganisl B, Breuker K. Does electron capture dissociation cleave protein disulfide bonds? Chemistry Open. 2012;1:260–268. doi: 10.1002/open.201200038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breuker K, McLafferty FW. Native electron capture dissociation for the structural characterization of noncovalent interactions in native cytochrome c. Angew Chem Int Ed. 2003;42:4900–4904. doi: 10.1002/anie.200351705. [DOI] [PubMed] [Google Scholar]
- 41.Breuker K, McLafferty FW. The thermal unfolding of native cytochrome c in the transition from solution to gas phase probed by native electron capture dissociation. Angew Chem Int Ed. 2005;44:4911–4914. doi: 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Wolff JJ, Van Orden SL, Loo JA. Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem. 2014;86:317–320. doi: 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Wongkongkathep P, Van Orden SL, Ogorzalek Loo RR, LJA Revealing Ligand Binding Sites and Quantifying Subunit Variants of Non-Covalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J Am Soc Mass Spectrom. 2014 doi: 10.1007/s13361-014-0928-6. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Native Electrospray and Electron-Capture Dissociation in FTICR Mass Spectrometry Provide Top-Down Sequencing of a Protein Component in an Intact Protein Assembly. J Am Soc Mass Spectrom. 2010;21:1966–1968. doi: 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chanthamontri C, Liu J, McLuckey SA. Charge state dependent fragmentation of gaseous α-synuclein cations via ion trap and beam-type collisional activation. Int J Mass Spectrom. 2009;283:9–16. doi: 10.1016/j.ijms.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engel BJ, Pan P, Reid GE, Wells JM, McLuckey SA. Charge state dependent fragmentation of gaseous protein ions in a quadrupole ion trap: bovine ferri-, ferro-, and apo-cytochrome c. Int J Mass Spectrom. 2002;219:171–187. [Google Scholar]
- 48.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Charge-State-Dependent Sequence Analysis of Protonated Ubiquitin Ions via Ion Trap Tandem Mass Spectrometry. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 49.Newton KA, Chrisman PA, Reid GE, Wells JM, McLuckey SA. Gaseous apomyoblobin ion dissociation in a quadrupole ion trap: [M+2H]2+-[M+21H]21+ Int J Mass Spectrom. 2001;212:359–376. [Google Scholar]
- 50.Watson DJ, McLuckey SA. Charge state dependent ion trap collision-induced dissociation of reduced bovine and porcine trypsin cations. Int J Mass Spectrom. 2006;255:53–64. [Google Scholar]
- 51.Iavarone AT, Paech K, Williams ER. Effects of Charge State and Cationizing Agent on the Electron Capture Dissociation of a Peptide. Anal Chem. 2004;76:2231–2238. doi: 10.1021/ac035431p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Uggerud E. Electron capture dissociation of the disulfide bond - a quantum chemical model study. Int J Mass Spectrom. 2004;234:45–50. [Google Scholar]
- 54.Lioe H, O’Hair RA. A novel salt bridge mechanism highlights the need for nonmobile proton conditions to promote disulfide bond cleavage in protonated peptides under low-energy collisional activation. J Am Soc Mass Spectrom. 2007;18:1109–1123. doi: 10.1016/j.jasms.2007.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.