

Abstract
Although physiological steroid levels are often pulsatile (ultradian), the genomic effects of this pulsatility are poorly understood. By utilizing glucocorticoid receptor (GR) signaling as a model system, we uncovered striking spatiotemporal relationships between receptor loading, lifetimes of the DNase I hypersensitivity sites (DHSs), long-range interactions, and gene regulation. We found that hormone-induced DHSs were enriched within ±50 kb of GR-responsive genes and displayed a broad spectrum of lifetimes upon hormone withdrawal. These lifetimes dictate the strength of the DHS interactions with gene targets and contribute to gene regulation from a distance. Our results demonstrate that pulsatile and constant hormone stimulations induce unique, treatment-specific patterns of gene and regulatory element activation. These modes of activation have implications for corticosteroid function in vivo and for steroid therapies in various clinical settings.
The pulsatile nature of many regulatory and activating signals is largely unappreciated, and the genomic effects of transient stimuli on chromatin structure and dynamics as well as on the long-range interactions between promoters and distal enhancers are virtually unknown.
Glucocorticoids, the GR-activating hormones are released from the adrenal glands in a circadian, as well as highly pulsatile (ultradian) fashion—e.g., short (20 min) hourly pulses (Lightman et al. 2002, 2008; Young et al. 2004; Atkinson et al. 2006; Lightman 2006; Droste et al. 2008; Lightman and Conway-Campbell 2010; Conway-Campbell et al. 2011; Walker et al. 2012). This pulsatile hormone release pattern is further influenced by the physiological and the psychological status of the animal. For example, stress response is associated with longer (1–2 h) exposure to high levels of glucocorticoids, and the removal of the stress signal restores the pulsatile hormone release pattern (Droste et al. 2008). Although the continuous supply of glucocorticoids is important for coping with natural stressors, pathological effects of prolonged glucocorticoid elevation, either as a result of chronic stress or long-term exogenous administration of synthetic hormones, are also well documented (Sapolsky et al. 2000; Schacke et al. 2002). Thus, different temporal patterns of hormone exposure are associated with diverse physiological outcomes, but the molecular bases for these diverse effects are not well understood.
We have previously demonstrated that pulsatile hormone stimulation leads to a cyclic GR association with regulatory elements in living cells (MMTV promoter array), whereas continuous hormone stimulation is characterized by a constant engagement of the receptor with these elements (Stavreva et al. 2009). Moreover, the binding of the glucocorticoid receptor coincides with accessible chromatin sites (John et al. 2008) as measured by chromatin sensitivity to DNase I digestion (Wu 1980; Elgin 1988; Gross and Garrard 1988; Sabo et al. 2006). The majority of these hypersensitive sites are preexisting, although a small number of them (∼5%) are created de novo by glucocorticoid action (John et al. 2011). However, the genomic consequences of transient versus prolonged GR association with GR regulatory elements (GREs) for chromatin conformation and for the lifetime of the GR-induced hypersensitivity are largely unknown.
Chromatin conformation determines access to a variety of DNA regulatory elements, shaping transcriptional responses in a tissue- and cell-type-specific manner (Stalder et al. 1980; John et al. 2008; Siersbæk et al. 2011). Here we analyze the hormone-induced GR binding patterns, DNase I hypersensitivity, RNA polymerase II occupancy, and chromatin conformation upon pulsed and constant hormone stimulation on a genomic scale. Addressing the relationship between the temporal changes in chromatin accessibility and gene regulation in response to the naturally occurring pulsatile activating signals is critical for understanding transcription regulation in this dynamic context.
Results
Pulsed hormone treatment induces transient GR binding genome-wide, whereas prolonged stimulation increases GR binding levels
We previously discovered that GR interactions with a tandem mouse mammary tumor virus (MMTV) promoter array of GR regulatory elements (GREs) closely follows hormone level fluctuations in the cellular milieu (Stavreva et al. 2009), as demonstrated by single cell imaging (Fig. 1B) or by chromatin immunoprecipitation (ChIP) (Supplemental Fig. S1A). To study this phenomenon genome-wide, we performed ChIP against GR followed by high-throughput sequencing (ChIP-seq) using mouse mammary adenocarcinoma cells (3134 cell line) subjected to pulsed or continuous hormone stimulation (Fig. 1A). Genome-wide, the GR binding pattern demonstrated that ultradian hormone stimulation results in transient GR binding at endogenous GR regulatory elements (Fig. 1C,F; Supplemental Fig. S1B–E). Although the type of genomic regions where GR bound after 20 or 60 min of hormone stimulation did not differ significantly (Fig. 1D,E), GR interactions with the genome were clearly time-dependent. Constant (60 min) hormone stimulation, reminiscent of the hormone release pattern under stress conditions (Droste et al. 2008), increased the level of GR binding at the GREs (Fig. 1C; Supplemental Fig. S1B–E). It was also associated with the engagement of additional GREs throughout the genome, illustrated by the doubling of the number of the GR sites (Fig. 1G), which could be an integral part of the physiological stress response.
Figure 1.

Divergent GR binding responses to pulsed (ultradian) and continuous hormone stimulation. (A) Schematic representation of pulsed and continuous hormone stimulation. (B) GFP-GR association with the MMTV promoter array in the presence of hormones (yellow arrows) and its rapid redistribution upon hormone withdrawal is the basis for the cyclic GR interactions with regulatory elements in response to naturally occurring hormone level fluctuations. Continuous stimulation with corticosterone (60 min constant) results in a persistent GR enrichment at the array (yellow arrows). (Scale bar) 5 µm. (C) GR enrichment at sites situated upstream and downstream, as well as in the body of a GR regulated gene, Tgm2, (20 min sample and 60 min constant sample). (D,E) Genomic location of the GR peaks at 20 and 60 min of constant hormone stimulation did not differ significantly. (F) An aggregate plot demonstrating that GR peaks diminish upon hormone washout (60′ pulsed condition) while increasing in tag density upon continuous hormone stimulation (60 min constant condition). (G) GR interaction with the GREs throughout the genome is time-dependent with more GR peaks appearing at 60 min continuous hormone stimulation.
Temporal dynamics of the hormone-induced chromatin accessibility
Considering the aforementioned hormone-dependent GR interactions with GREs, as well as the fact that the GR binding coincides with accessible chromatin sites (John et al. 2008), we applied the DNase-seq approach to test whether the transient and pulsed hormone stimulations were associated with divergent patterns of chromatin hypersensitivity.
The examination of the MMTV locus by DNase-seq revealed that the pulsed treatment induces a transient hypersensitive site coinciding with the major GR peak, whereas the constant treatment sustained the hypersensitivity of the site (Fig. 2A,B). Interestingly, an early study demonstrated a transient hypersensitive site at an enhancer located 2.5 kb upstream of the start site of the hormone-responsive rat tyrosine aminotransferase gene (Reik et al. 1991). Another work in a different system (chicken bone marrow cells infected in vitro with a temperature-sensitive avian erythroblastosis virus) described a persistent DHS, maintained over several cell divisions even in the absence of the initial inducing signal (Weintraub et al. 1982). Our initial analyses of the DNase-seq data revealed the existence of transiently induced endogenous DHSs (Fig. 2C; Supplemental Fig. S2A). However, we also discovered persistent DHSs that remained hypersensitive even after hormone withdrawal (Fig. 2D; Supplemental Fig. S2B). Constant treatment sustained the hypersensitivity of both the transient and the persistent sites (Fig. 2C,D; Supplemental Fig. S2A,B). We concluded that the transient GR loading at GREs as a result of the pulsed hormone stimulation is associated with both transient and persistent changes in chromatin hypersensitivity.
Figure 2.

Changes in chromatin accessibility at the MMTV array locus and at endogenous sites in response to pulsed and constant hormone stimulation. (A) High-resolution mapping of the GR binding profile at the MMTV locus demonstrates an excellent concordance between the results obtained by single cell imaging (Fig. 1B) and the GR ChIP-seq method. (B) DNase I digestion reveals the presence of a transient DNase I hypersensitive site (DHS) at the MMTV locus coinciding with the major GR peak (black arrows). Hypersensitivity of this site is contingent upon GR binding, suggesting a direct involvement of the receptor in the DHS establishment and maintenance. (C) Examples of endogenous transient DHSs. (D) Examples of endogenous persistent DHSs.
Hue-saturation-value (HSV) color-space representation of DHS dynamics
To further characterize the dynamics of the entire population of endogenous hypersensitive sites as well as to avoid the arbitrary allotment of DHSs to discrete categories, we developed and implemented a novel computational approach that represents the “shape” of a time profile as a single variable (Fig. 3A–C). Our perception of the “hue” of a color (e.g., red, orange, yellow, green, etc.) describes the relative proportions of the three primary light colors for the human eye: red, green, and blue (Nixon and Auado 2012). In computer graphics, the HSV color system is a simple mathematical transformation used to express any color (i.e., any combination of red, green, and blue) in terms of three parameters, one of which is the hue. Our method for representing genome-wide DHS time profiles is based on this HSV transformation (Fig. 3A). For a given time profile, we ascribed the experimental values at the 0, 20, and 60 min time points to the intensity of a pseudo blue, green, and red light source, respectively. The color resulting from their additive mixture is represented by the HSV color system. The “hue” parameter (Fig. 3B) describes the shape of the profile and projects the entire spectrum of possible DHS time courses onto a continuous angular scale. For instance, a hue of 120° (green) represents a DHS that hypersensitivity increases after a 20-min pulse of hormones and reverses exactly to the pretreatment level after the washout. A hue of 60° (yellow) represents an increase of hypersensitivity after a 20 min pulse that remains unchanged after washout. The “saturation” and “value” parameters convey the amplitude and the baseline of the time profile (Fig. 3C). Applying the HSV method to the population of endogenous hypersensitive sites reveals the kinetic behavior of the entire DHS pool in both pulsed and constant treatment in an unbiased way and free of any threshold (Fig. 3D,E). Each point on these scatter plots represents the time profile of hypersensitivity at a DHS. The horizontal axis (hue) indicates the shape of the time profiles, and the vertical axis (saturation) describes its amplitude (difference between the minimum and the maximum of all three time points as a percentage of the maximum).
Figure 3.

Hue-saturation-value (HSV) color-space interpretation of DHS time profiles. (A) The method presented here for analyzing time profiles is inspired from a color representation used in computer graphics that mimics our natural perception. Any color, i.e., any relative proportion of the three primary light colors (blue, green, and red) can be represented in this so-called HSV color space, yielding three parameters: hue, saturation, and value. This transformation is used here to interpret triplets of experimental data points (e.g., tag density at a DHS site at 0, 20, and 60 min over the course of an experiment) as if each of them were describing the intensity of a primary color. The resulting three parameters have the following interpretation: (B) “Hue” is a circular measure (i.e., an angle) that naturally sorts out as a single continuous variable all the possible shapes of the time profile, regardless of its minimal and maximal values. (C) “Saturation” and “Value” parameters describe the range over which the data points are spanning, independently of the shape of the profile. The “Value” is the maximum of all three time points, and the “Saturation” represents the amplitude of the variations as a fraction of the maximum (0% meaning no variations and 100% meaning the smallest time point is null). Using this representation, strong and highly responsive sites are bright and colorful, whereas dimmer colors mean fewer variations, and grayer colors mean weaker sites. (D,E) DHS time profiles are represented using the HSV method. (F,G) The partial recovery in the 60°–120° range can be mapped directly to a half-life of DHS, which can be further compared with preaccessibility levels (shown in G are only sites having at least a 25 tags increase between 0 and 20 min).
Interestingly, the shape indicator of DHS time profile for the pulsed treatment (Fig. 3D) can be directly mapped, in the 60°–120° region, to a measure of hypersensitivity lifetime: Assuming that a site decays exponentially after the washout and will eventually return to its pretreatment level, its half-life (Fig. 3F) is given by −40 min/log2(2 h/60°), where the shape indicator is noted h (for hue). In addition, in the 60°–120° region, a strong saturation (vertical axis) indicates a low preaccessibility level: de novo sites are necessarily close to 100%, whereas preexisting sites have a lower saturation. Hence, the fact that the HSV scatter plot peaks toward 120° hue at high saturation (Fig. 3D) suggests a correlation between preaccessibility and lifetime of hormone-induced hypersensitivity. This correlation is further evident by representing the DHS half-lives of a site versus its level of preaccessibility (Fig. 3G). Not only can a preaccessible site be expected to return to its basal pretreatment level, but it does so more slowly than a de novo site. Although preaccessible sites display a very broad spectrum of lifetimes, de novo and weakly accessible sites necessarily have a short half-life (<25 min), the corollary being that long-lasting hypersensitivity can only occur at preaccessible sites.
Next, we correlated the DHS kinetic behavior with various genomic features (Supplemental Fig. S3). This revealed distinct DHS subpopulations. For instance, sites showing a transient profile with more or less recovery in the pulsed treatment, and an increasing profile or a profile approaching a plateau in the constant treatment, are mostly highly loaded by GR (Supplemental Fig. S3B), not at promoter (TSSs ± 2.5 kb) (Supplemental Fig. S3E), and slightly enriched in gene distal upstream regions (Supplemental Fig. S3D). All the other complex kinetic behaviors correspond to sites that are either not or are weakly loaded by GR, and at promoters (i.e., reflecting most likely indirect effect). Importantly, the observed segregations hold down to ∼20% saturation (e.g., Supplemental Fig. S3B,C,E), showing that the hue indicator accurately categorizes the shape of the time profile even for very moderate variations, although to lesser precision due to experimental noise.
Representation of the hormone-responsive DNase-seq time courses in HSV color space
To be able to isolate the population of dynamic DHSs, responding solely to the hormone treatment and not to the media replacement (or to a combination of both), we performed mock experiments using hormone-free media. Upon filtering out all nonspecific changes in hypersensitivity, we applied the HSV approach to the remaining hypersensitive sites (Fig. 4). We found that the dominant DHS behaviors were a transient increase of hypersensitivity in response to a pulsed treatment and a continuous increase in response to a constant treatment (Fig. 4A,B; Supplemental Movie S2), indicating that hypersensitivity at many sites followed the kinetic mode of hormone treatment. However, we also observed a continuum spanning from transient (120°) to plateauing (60°) profiles and beyond, demonstrating the existence of a range of DHS behaviors.
Figure 4.
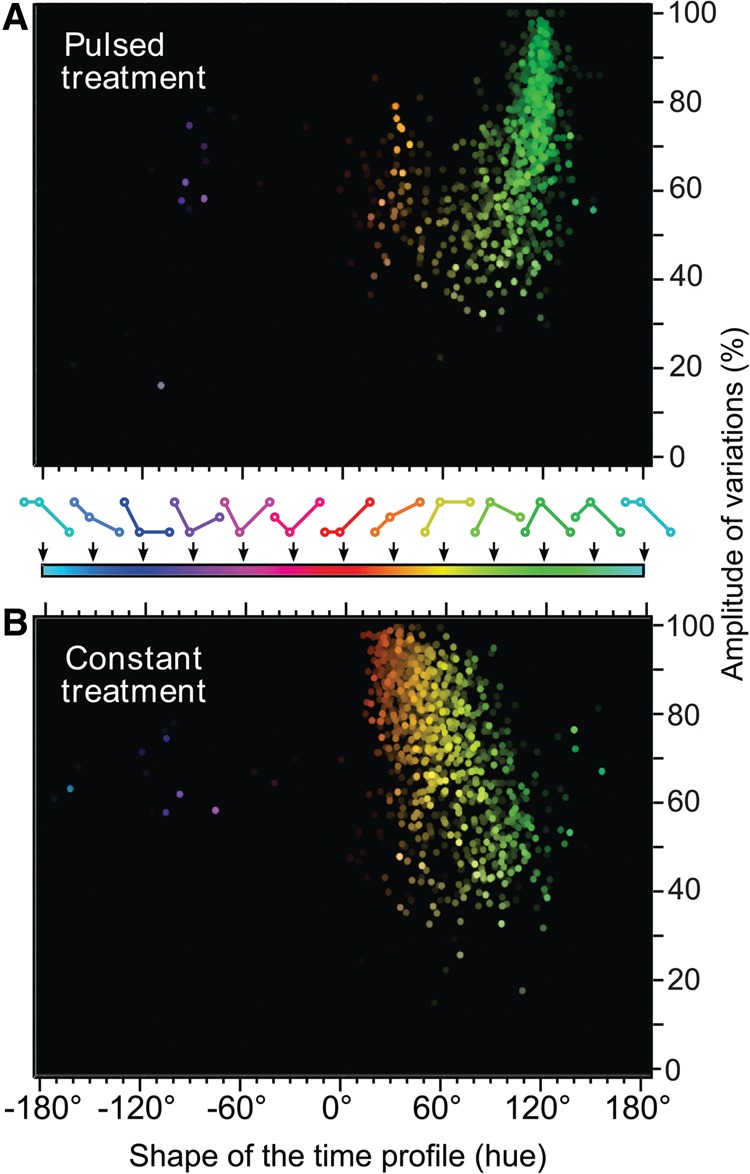
Characterization of the hormone-responsive DHS time profiles. The hue-saturation-value color-space approach is used here in HSV scatter plots to depict both pulsed (A) and constant (B) treatments. Data are filtered for the effects of the mock treatment on DHS dynamics and reveal the kinetic behavior of the remaining DHS pool in an unbiased way and free of any threshold (see Supplemental Movie S2 for 3D versions). Sites displaying highly marked variations (i.e., one or two time points are almost 0) rise to the top of the graph as colorful dots, whereas sites with mild or no variations vanish to the bottom toward gray. Stronger sites, in terms of maximal readout, appear bright at the foreground, and weaker sites naturally disappear on the black background.
We concluded that both the pulsed and constant treatments created unique and diverse landscapes of DHS dynamics (Fig. 4A,B; Supplemental Movie S2). In contrast, GR ChIP-seq time courses, analyzed with the same method, revealed rather uniform GR binding kinetics (Supplemental Fig. S4A,B; Supplemental Movie S1). The majority of the sites were only transiently occupied by GR in response to pulsed hormone stimulation, whereas constant hormone treatment resulted in an increasing GR occupancy, with very few cases of complete saturation upon 20-min treatment (Supplemental Fig. S4A). The shape of the HSV scatter plot in the constant treatment reveals that the accessibility of “de novo” sites (saturation close to 100%, vertical axis) keeps increasing from 20 to 60 min of treatment, whereas preaccessible sites (lower saturation) tend to plateau after 20 min of hormone stimulation. It should be noted that a small fraction of sites retains a trace of GR ChIP-seq signal even after the washout (Fig. 2C,D; Supplemental Fig. S4A). It is difficult to assess whether this is due to actual long-lived binding or rather an incomplete hormone washout. However, this GR binding lifetime is largely uncorrelated with the DNase-seq hypersensitivity lifetime (Supplemental Fig. S5A). Only in very stringent conditions does a modest correlation appear (Supplemental Fig. S5B).
Upon examination of the motifs associated with transient and persistent hypersensitive sites (defined by their shape parameter) (Methods), we found that the top motif for the transient DHSs were GR response element (GRE), JUN (also known as AP1), and RUNX1, which is also considered a half-GRE (Supplemental Fig. S6A). The top motifs for the persistent sites were JUN, an unknown motif, and CTCF (Supplemental Fig. S6B). Both categories of sites shared a JUN motif, but the unknown motif and the CTCF were specific to the persistent sites. Interestingly, the fourth most prevalent motif for the persistent DHSs was a half-GRE (RUNX1), whereas the full GRE motif ranked only twelfth. This suggests that in contrast to the transient sites, where the GR is recruited directly, the GR recruitment at some of the persistent sites might be achieved through interactions with other DNA-binding factors (e.g., tethering). Thus, GR is the key regulator of chromatin accessibility at transient sites, since receptor dissociation upon hormone withdrawal leads to a loss of accessibility. In contrast, receptor dissociation from persistent sites did not lead to reversion of their hypersensitivity to the preinduction levels but remained elevated, suggesting that chromatin accessibility of the persistent sites might be regulated in a more complex combinatorial manner.
To assess the status of transient and persistent DHSs prior to glucocorticoid treatment, we performed additional ChIP-seq experiments for several factors. Our analysis revealed that persistent sites are on average more likely to be bound by JUN, as well as by EP300 (also known as p300), remodeling complexes (SMARCA4, also known as BRG1), and CTCF. This is consistent with the fact that chromatin at persistent DHSs is accessible even prior to hormone treatment and is more permissive to binding (Supplemental Fig. S6C,D).
Next, we sought to determine to what extent the DHS-associated enhancers are active prior to glucocorticoid treatment. We analyzed the genome-wide distribution of EP300, which marks active enhancers (Visel et al. 2009) and discovered that it is enriched at persistent and some preexisting transient DHSs, but is largely absent from the transient de novo sites (Supplemental Figs. S6D, S7A,B,D), indicating a lower or absent enhancer activity at transient sites before stimulation. Upon hormone treatment, transient sites gain EP300, which is consistent with their hormone-dependent activation (Supplemental Fig. S7B). We further investigated EP300 levels at a number of transient sites under pulsed and constant hormone stimulation by manual ChIP and observed strong and consistent EP300 enrichment at these sites in the presence of hormone (Supplemental Fig. S7C). In contrast, EP300 loading profile at persistent DHSs under pulsed and constant hormone stimulation was much more variable (Supplemental Fig. S7E). Although EP300 was enriched at some persistent sites upon hormone stimulation, at other sites it was either unchanged or even strongly inhibited by the hormone treatment (Supplemental Fig. S7E). This finding further corroborates the notion that persistent DHSs are a diverse group, regulated in more a complex manner than transient DHSs.
Representation of Pol II ChIP-seq time courses in HSV color space
We have previously demonstrated cyclic RNA polymerase II [Pol II, also POLR2F] occupancy at the MMTV array in response to pulsatile hormone treatment, whereas continuous hormone stimulation induces continuous Pol II engagement with this locus (Supplemental Fig. S8A,B; Stavreva et al. 2009). We have also shown that a pulsed stimulation is associated with a transient release of nascent RNA from many endogenous GR up-regulated genes (Stavreva et al. 2009; Supplemental Fig. S8C–H). We performed ChIP and ChIP-seq experiments with an antibody recognizing the S5 phosphorylated form of polymerase II. The resulting data confirmed the transient, hormone-dependent association of this activated form of polymerase II with the MMTV locus as well as at endogenous GR-induced genes (Supplemental Fig. S8A–H). It also mirrored the changes observed at nascent (intron-containing) RNA levels (Supplemental Fig. S8C–H), suggesting that the Pol II-S5 signal is a reliable marker for detection of the transient changes in gene activity.
To account for the full range of hormone-dependent changes in transcription activity (as measured by Pol II-S5 occupancy over gene bodies, up to 10 kb downstream from the TSS), we applied the HSV color-space approach (Fig. 3A–C) to the Pol II-S5 ChIP-seq data (Fig. 5). In addition to seeing transient gene activation for pulsed treatment (Stavreva et al. 2009; Supplemental Fig. S8C–H), we observed other gene response profiles and discovered that the two modes of hormone stimulation (pulsed versus constant) were associated with divergent genome-wide transcriptional patterns (Fig. 5E,F; Supplemental Movie S3).
Figure 5.

Diverse Pol II occupancy time profiles over gene bodies. (A–D) Examples of Pol II-S5 ChIP-seq signal over gene bodies in response to pulsed and continuous hormone stimulations. (E,F) HSV scatter plots are used to visualize the variety of time profiles of Pol II-S5 occupancy over the gene body of hormone-responsive genes (see Methods) in the case of both pulsed (E) and constant (F) hormone stimulations (see Supplemental Movie S3). Upper and lower panels in E and F, respectively, represent the distributions of time profiles (hue) in both conditions. The black lines between E and F connect the respective Pol II-S5 time profiles for each individual gene under pulsed and constant treatments, highlighting, in particular, that genes responding to a pulsed treatment with a transient activation (∼90° to 130° hue) display a steadily increasing response in the case of a constant treatment (∼10° to 50° hue) and merge with the broad category of genes showing an activation in both treatments. (G,H) Comparison of the effects of natural and synthetic hormones (corticosterone and dexamethasone, respectively) on GR-mediated transcriptional responses revealed striking ligand-specific effects (see also Supplemental Fig. S8I). (G) Dexamethasone revealed much stronger inhibitory properties than corticosterone (P-value <5 × 10−9, binomial test), whereas the latter (H) was a more potent transcriptional activator (P-value <2 × 10−16, binomial test).
A broad category of genes displayed behaviors that depended on the type of treatment (pulsed or constant). These genes range between transient and persistent time profiles (60°–120°) in response to a pulsed treatment, but have mostly a progressively increasing response (0°–40°) when hormone stimulation is constant. Moreover, the skewed shape of this population of genes on the HSV scatter plot indicates that genes with no basal activity prior to stimulation (amplitude of variations close to 100%, vertical axis) return faster to their pretreatment levels after a hormone pulse (e.g., Orm3, Per1) (Fig. 5A). On the other hand, genes that retain a long memory of the hormone pulse (60°–90°) necessarily have some degree of basal activity prior to hormone treatment (e.g., Atp6v1g1) (Fig. 5A). Another category of responsive genes showed little difference in their reactions to both treatments. These genes, centered around 0° in both cases (e.g., Id1 or Hes1) (Fig. 5A), displayed a continuous or late increasing time profile whether the hormone stimulation was pulsed or constant. Several exceptions from these groups were notable, including a group of repressed genes (e.g., Adamts1 and Zfp36I1) (Fig. 5B,C) as well as a gene responding exclusively upon constant hormone stimulation (Plin4) (Fig. 5D). Interestingly, we observed strong ligand-specific effects when comparing transcriptional responses of the natural hormone (corticosterone) with a synthetic hormone (dexamethasone) upon 60 min of continuous stimulation (Supplemental Fig. S8I). In contrast to the natural hormone, dexamethasone was a stronger repressor of gene transcription (Fig. 5G; Supplemental Fig. S8I), whereas corticosterone activated transcription of a significantly larger number of genes (Fig. 5H; Supplemental Fig. S8I). This observation substantiates the well-known inhibitory (anti-inflammatory and immunosuppressive) effects of dexamethasone for which this drug is widely used in the clinics.
Chromatin dynamics and gene regulation
To gain further insights into the relationship between hormone-mediated gene responses, GR loading, and DHS behavior, we examined the 100-kb window around the TSS of all hormone-responsive genes. It should be noted that the majority of the DHSs associated with gene promoters were preexisting and much less affected by the hormone treatment (John et al. 2008; Supplemental Fig. S3E). Thus, promoter-associated DHSs (TSS ± 2.5 kb) were excluded from the analyses. After evaluating the behavior of the remaining hypersensitive sites, we discovered that they are more likely to be in the vicinity of hormone-responsive genes than near other genes (Fig. 6A) (P-value <10−15, Fisher's exact test). This correlation suggests a possible role for the dynamic hypersensitive sites in gene regulation through long-range interactions.
Figure 6.

Spatiotemporal correlations between lifetimes of the DNase I hypersensitivity sites and transcriptional activity. (A) Dynamic DHSs are enriched within ±50 kb of the TSS of hormone-responsive genes, suggesting a possible role for these hypersensitive sites in gene regulation (promoter DHSs, i.e., TSS ± 2.5 kb was excluded). (B) Real-time qPCR experiments, using primers recognizing the nascent RNA, demonstrate that transcription of the Tsc22d3 gene is regulated in a hormone-dependent manner (see also Pol II ChIP-seq panel in D). Error bars represent the mean ± SEM, n = 4. (C) Pol II-S5 occupancy over the Tsc22d3 gene (lower panel) and the nearby DHSs (upper panel) all have a transient behavior in response to pulsed hormone stimulation (∼120° hue, vertical axis) and are the main hormone responsive elements in this genomic locus. (D) ChIP-seq and DNase-seq tracks displayed for the gray region in C reveal that the Tsc22d3 gene and the DHS cluster are enclosed by CTCF sites. In addition, although all distal DHSs are bound by GR, the promoter of this gene is largely lacking GR, suggesting that its transcriptional response to hormone stimulation depends on the long-range interactions with distal regulatory elements. (E) A modified chromosome conformation capture approach using the most distal DHS (DHS5) as a bait (gray shading) demonstrates a hormone-dependent increase in the interaction frequency of this DHS with the TSS region of the gene (yellow shading) as well as with another GR-bound DHS (purple shading). (F) Interaction frequency of DHS5 with a region close to the TSS (yellow shading) doubles in response to the hormone stimulation and reverts to its original level upon hormone withdrawal. Error bars represent the mean ± SEM, n = 4. (G) A similar trend was observed for the interaction of the DHS5 with the promoter proximal DHS1 (pink shading). Error bars represent the mean ± SEM, n = 4.
To test this possibility, we selected a glucocorticoid-induced gene, Tsc22d3, with a number of unique features (Fig. 6C,D). First, it is located in a region of Chromosome X (Chr X: 136,500,000–137,500,000) without other glucocorticoid-responsive genes in its vicinity; and second, its transcription strictly follows hormone level fluctuations (Fig. 6B,D), although its promoter-associated DHS is not bound by GR upon short (20 min) hormone stimulation and only weakly bound after 60 min (Fig. 6D; Supplemental Fig. S9H, note the change of the scale). This suggests that the hormone-dependent transcriptional activation of Tsc22d3 may rely predominantly on the long-range interactions between the TSS of the gene and distal GR-responsive regulatory elements. Interestingly, several GR peaks, associated with dynamic hypersensitive sites, are found at a distance ∼12–40 kb downstream from the gene [Fig. 6C (upper panel), D]. When considering the topological domain encompassing the Tsc22d3 gene, it is notable from Hi-C data (Supplemental Fig. S9E; Dixon et al. 2012) that the gene and the aforementioned hypersensitive sites exhibit high levels of interactions clustered in the subdomain confined by the closest CTCF binding sites around the gene body (Fig. 6D). To directly test whether the gene interacts with the downstream dynamic hypersensitive sites, we prepared 4C DNA (van de Werken et al. 2012) using two four-cutter restriction enzymes (NlaIII and Csp6I) and a fragment near the Tsc22d3 TSS as the bait. Next, we utilized the amplified 4C material in a number of qPCR reactions using tiled primer pairs covering a large number of NlaIII/Csp6I fragments surrounding the Tsc22d3 gene (for details on the method and a full list of the used primers, see Supplemental Fig. S9A; Supplemental Material; Supplemental Table S1). The resulting profile of the control sample demonstrated a much higher frequency of interaction of the bait with the GR-binding sites downstream from the gene and a much lower interaction frequency with loci upstream of the gene (Supplemental Fig. S9G). This suggests preexisting local conformation favoring these interactions. More importantly, we discovered highly reproducible hormone-dependent changes of the interaction frequency of the bait with the most distal DHS (DHS5) and to a lesser extend with the second most distal DHS (DHS4) (Supplemental Fig. S9I). For the more closely situated hypersensitive sites, significant differences were observed only upon 60 min of hormone treatment (Supplemental Fig. S9G), and it is likely that the closer proximity of the bait to these sites obstructs the detection of small changes. To further verify the finding that the distal DHS (DHS5) and the TSS of the gene were interacting in a hormone-dependent manner, we performed additional experiments using this DHS as the bait (Fig. 6E). We discovered that the GR binding and the local chromatin remodeling at this distal DHS doubled its chances of interacting with the Tsc22d3 promoter region (Fig. 6E,F). A similar but less significant trend was observed for the interaction of the DHS5 with the promoter proximal DHS1 (Fig. 6E,G), and this trend diminished with the increased proximity of the hypersensitive site to the bait. We concluded that transient DHSs contribute to gene regulation through dynamic long-range interactions.
In contrast, when we used the region around a persistent DHS as the bait and applied the aforementioned method (Supplemental Fig. S9A; for a full list of the used primers, see Supplemental Table S2), we found a moderate elevation in the interaction frequency of this DHS and the promoter of the nearby persistent gene Atp6v1g1 (Supplemental Fig. S10E–G, purple and yellow shading in A–E). More importantly, this elevated interaction frequency was sustained even upon hormone withdrawal. We concluded that changes in chromatin accessibility induced by the glucocorticoid treatment could impact the frequency of interaction between a gene and its regulatory elements and lead to functional consequences.
Discussion
Although glucocorticoids are among the most widely prescribed drugs worldwide, GR-mediated gene responses are not fully understood, and the treatments with these drugs are associated with numerous side effects (Sapolsky et al. 2000; Schacke et al. 2002). Release of glucocorticoids from the adrenal glands follows two superimposed temporal patterns: a daily (circadian) pattern and an hourly ultradian pattern (Lightman et al. 2002, 2008; Young et al. 2004; Atkinson et al. 2006; Lightman 2006; Droste et al. 2008; Lightman and Conway-Campbell 2010; Walker et al. 2012). The effects of GR activation are further influenced by the cell-specific chromatin landscape (Stalder et al. 1980; John et al. 2008, 2011; Biddie et al. 2011; Siersbæk et al. 2011; Mandrup and Hager 2012) and by the action of other transcription factors and chromatin modifiers (Biddie et al. 2011; Voss et al. 2011; Miranda et al. 2013b; Morris et al. 2014) genome-wide. This reveals a complex and dynamic picture of the GR-mediated transcription regulation (Stavreva et al. 2012; Miranda et al. 2013a), which is still not fully appreciated.
Here we have compared transient versus constant hormone stimulation, reminiscent of the glucocorticoid release under normal and stress conditions, respectively, and characterized their effects on receptor loading, chromatin dynamics, and gene regulation genome-wide. Our previous work (Stavreva et al. 2009) as well as other studies (Conway-Campbell et al. 2010, 2011) has demonstrated that pulsatile hormone stimulation induces transient GR interaction with regulatory elements. We investigated this phenomenon on the genomic scale and have unexpectedly discovered that transient GR binding induces diverse lifetimes of chromatin accessibility. It can create hypersensitive sites de novo, but it can also influence the degree of hypersensitivity of preaccessible sites. More importantly, upon hormone withdrawal, the hypersensitivity of the de novo sites is rapidly lost, whereas the effects of GR activation at some of the preaccessible sites persist. Thus, the pulsed treatment can induce transient GR activation with marked effects on chromatin structure and accessibility, sometimes persisting longer than the activating signal. In contrast, constant hormone application not only increases the level of GR binding and induced hypersensitivity but also results in the engagement of GR at many GREs that are not bound upon pulsed treatment. This is likely the basis for the divergent transcriptional programs associated with the pulsed and the constant hormone stimulations.
Consistent with earlier studies (John et al. 2011; Grøntved et al. 2013), we demonstrated that the majority of GR binding happens at sites distant from promoters, suggesting that long-range interactions play an important role in GR action. GR binding also coincides with hypersensitive sites (John et al. 2008, 2011; Grøntved et al. 2013), and it was previously demonstrated that the majority of long-range genomic contacts involve hypersensitive sites (Hakim et al. 2011). Considering the dynamics in chromatin accessibility observed here, we hypothesized that it could play a role in gene transcription regulation from a distance.
The interphase nucleus is highly organized into thousands of megabase-long topologically associating domains (TADs) (Dixon et al. 2012; Nora et al. 2012), which are relatively conserved between different cell types. The most specific long-range interactions between promoters and enhancers were found within the boundaries of TADs (Burd et al. 2012), and the loci within a TAD are proposed to be sufficiently dynamic to allow reproducible contacts between a gene and its regulatory elements (Gibcus and Dekker 2012, 2013). Consistent with this idea, we found that the 100-kb regions around GR-responsive genes are preferentially associated with hormone-responsive, dynamic DHSs. Moreover, using a chromosome conformation capture-based technique, we found that the changes in chromatin accessibility at distal enhancers predict changes in the frequency of interactions between the enhancers and their gene targets.
A recent study in human cells generated a high-resolution map of the three-dimensional chromatin interactome (Jin et al. 2013), but it did not detect any changes in the long-range interactions upon 1-h stimulation with TNF (also known as TNF-alpha) and concluded that these interactions are not dynamic. In contrast, we discovered a striking hormone-dependent increase in the frequency of the long-range interactions between a transient hypersensitive site and the TSS region of a GR-responsive gene (Tsc22d3) accompanied by an increase in EP300 levels. When we used a persistent hypersensitive site as the bait, we found that its elevated hypersensitivity upon hormone withdrawal was predictive of an increased interaction frequency of this DHS with the promoter region of a nearby gene, Atp6v1g1. This increased interaction frequency was accompanied by an elevated transcription during the withdrawal period. We concluded that the treatment-induced hypersensitivity influenced the frequency of the long-range contacts in a preset spatial conformation facilitating these contacts. Thus, our findings reveal a previously unsuspected mechanism for gene transcription regulation from a distance. They support a model in which the switch from inaccessible to accessible chromatin at a distal regulatory site plays a major role in gene regulation. Moreover, even a moderate change in chromatin accessibility can result in a change in the strength of long-range interactions at a regulatory site and can impact transcriptional responses of gene targets (Fig. 7).
Figure 7.
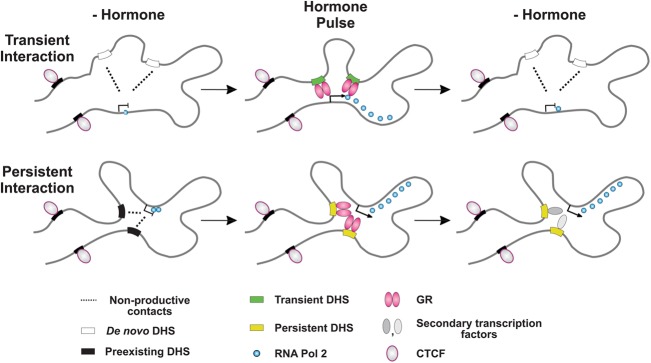
Chromatin accessibility dynamics at distal enhancers impacts the strength of their long-range interactions with gene targets and contributes to gene regulation. (Top) A schematic depicting reversible and hormone-dependent long-range interactions between transient, de novo DHSs (similar to DHS5) (Fig. 6) and the promoter of a gene target. The preexisting organization of the locus favors transient, nonproductive interactions between them, even in the absence of corticosterone (-Hormone). However, GR binding to these DHSs upon hormone addition (Hormone Pulse) facilitates their retention at the TSS resulting in higher interaction frequency. This allows an increased Pol II loading and promotes a hormone-dependent gene transcription. (Bottom) A schematic depicting prolonged interactions of preexisting, but still hormone-responsive, DHSs with a gene target. In contrast to the transient DHSs, the accessibility of the persistent DHSs and the interaction frequency between them and the gene remain elevated even upon hormone withdrawal. We envision the involvement of opportunistic factor(s) binding to these DHSs and sustaining their hypersensitivity as well as gene transcription even after hormone withdrawal.
Finally, our data reveal treatment-specific gene activation patterns. The most striking example is the Plin4 gene, which was specifically induced by the constant treatment (Fig. 5D). Interestingly, perilipin, the product of this gene, has been implicated in the regulation of basal lipolysis (Londos et al. 1995, 1999; Souza et al. 1998), and lower perilipin levels have been associated with leaner body constitution (Martinez-Botas et al. 2000). It is tempting to speculate that increased perilipin levels as a result of constant receptor stimulation, under stress conditions or exogenous administration of synthetic glucocorticoids, could play a role in the process of visceral fat accumulation associated with these conditions (Paredes and Ribeiro 2014).
In summary, we discovered an array of DHS lifetimes with certain DHSs retaining the “memory” of the hormone pulse. This memory correlates with the DHS preaccessibility levels (i.e., de novo sites versus preexisting sites), suggesting a common mechanism at play. In addition, we found previously undocumented dynamic, hormone-induced changes in the long-range interactions between DHSs and a gene promoter. These changes correlate with both DHS memory and gene activity. Thus, transient changes in chromatin accessibility contribute to gene regulation through long-range interactions much more dynamically than previously anticipated. Our data suggest that the interplay between different temporal hormone release patterns and the spatiotemporal chromatin dynamics reflecting transient activation of distal regulatory elements might well provide the basis for the extraordinary range of transcriptional response and physiological outcomes associated with GR signaling (Sapolsky et al. 2000).
Methods
Cell lines and culture conditions
The 3134 murine mammary epithelial cell line originally derived from a subclone of 904.13 (Fragoso et al. 1998). It contains a large tandem array (∼200 copies) of a mouse mammary tumor virus/Harvey viral ras (MMTV/v-Ha-ras) reporter. The 3617 cell line is a derivative of the 3134 cell line expressing a green fluorescent protein (GFP)-tagged version of GR (GFP-GR) from a chromosomal locus under control of the tetracycline-repressible promoter. Both cell lines were described previously (Walker et al. 1999). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen), supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals), 2 mM L-glutamine, and 0.5 mg/mL penicillin-streptomycin in a 37°C incubator with 5% CO2. For experiments, cells were plated in DMEM supplemented with 10% charcoal-dextran treated FBS for 24 h.
For the pulsed treatment, cells were incubated with 600 nM corticosterone for 20 min, the hormone was removed afterward, and cells were incubated in a hormone-free media for an additional 40 min. For the continuous treatment, cells were incubated with 600 nM corticosterone for 60 min. To prevent cell stress, these experiments were performed in a specially adapted incubator, allowing media replacement under conditions of stable CO2 and temperature levels throughout the duration of an experiment. To ensure complete hormone withdrawal during the wash period, cells were washed once with an excess of hormone free media and replaced with fresh media. Control cells were subjected to identical media replacement without hormone. Cells were collected before hormone treatment (0 min), after hormone pulse (20 min), at the end of the wash period (60 min pulse), as well as after continuous stimulation (60 min constant).
Microscopy experiments
To detect the effect of hormone withdrawal on GFP-GR loading at the array, 3617 cells were grown overnight on 22 mm2 coverslips in DMEM medium containing 10% charcoal stripped serum (Hyclone) without tetracycline (to allow the expression of the GFP-GR) and induced for 20 or 60 min with 600 nM corticosterone (purchased from Sigma). For the withdrawal experiments, cells were kept in hormone-free medium for an additional 40 min after the initial 20-min stimulation. All cells were then fixed with 3.5% paraformaldehyde in phosphate-buffered saline (PBS). Cells were further mounted on a glass slide using VECTRASHIELD mounting medium with DAPI (Vector Laboratories, Inc., Cat # H-1200). Cells were examined using Leica 100× 1.3-N.A. oil immersion objective. The images were processed using Metamorph software (Universal Imaging Corp.).
Chromatin immunoprecipitation assay (ChIP)
Chromatin immunoprecipitations were performed as per standard protocols (Upstate). Briefly, cells were treated with either vehicle, 600 nM corticosterone for 20 or 60 min, corticosterone for 20 min followed by 40 min of hormone withdrawal, as well as with 100 nM dexamethasone for 1 h. Cells were cross-linked for 10 min at 37°C in 1% formaldehyde followed by a quenching reaction for 10 min with 150 mM glycine. A single chromatin immunoprecipitation contained 400 μg of sonicated, soluble chromatin and a cocktail of antibodies to the glucocorticoid receptor (7.5 μg PA1-511A antibody [ABR], 15 μg MA1-510 antibody [ABR], and 3 μg sc-1004 [Santa Cruz]), RNA polymerase II phosphoS5 (15 μg ab5131-50 antibody [Abcam]), CTCF (15 μg 07-729 anti-CTCF antibody, [Millipore]), EP300 (10 μg anti-p300 [EP300] antibody, a kind gift from Dr. K. Gardner [NCI]), or SMARCA4 (BRG1) antibody (1 µg anti-BRG1 [SMARCA4] antibody [Epitomics]). The ChIP reaction was scaled 4× for ChIP-seq. DNA isolated from ChIPs was validated and/or confirmed by real-time quantitative PCR amplification using SYBR green mix (BioRad). GR ChIP primers for the promoter region of MMTV (Nuc B) used for amplification: forward TTTCCATACCAAGGAGGGGACAGTG′, and reverse CTTACTTAAGCCTTGGGAACCGCAA. Pol II ChIP primers for the Ras region of the MMTV array: forward CGTGAGATTCGGCAGCATAAA, and reverse GACAGCACACACTTGCAGCTC. Primer sequences used in the EP300 ChIP are listed in Supplemental Table S3.
Preparation of RNA and quantitative real-time PCR (qPCR) analysis
RNA was extracted from cells grown in media with or without corticosterone using the RNeasy Mini Kit (Qiagen). All RNA samples were treated with RNase-free DNase (Qiagen). Following purification, reverse transcription of total RNA was performed using the BioRad cDNA Synthesis Kit via the manufacturer's instructions and analyzed by real-time qPCR using SYBR green (BioRad). Transcription levels of the indicated genes were normalized using the housekeeping gene beta-actin and presented as a fold change of the control. Primer sequences are listed in Supplemental Table S4.
Preparation of DNase I-digested DNA
DNase I digested DNA was prepared for sequencing as previously described with minor modifications (John et al. 2011). Briefly, nuclei from harvested cells were isolated and digested with 60–80 units/mL DNase I (Roche) for 3 min at 37°C. Digested DNA was incubated at 55°C with 10 μg/mL RNase A (Roche) for a few hours to overnight followed by addition of 25 μg/mL Proteinase K (Ambion) and incubation at 55°C for at least 4 h. DNA fragments were purified by phenol/chloroform extraction and ultracentrifugation through a sucrose gradient. After purification, fragments (between 100 and 500 bp in size) were pooled, precipitated, and assembled into libraries for sequencing.
ChIP-seq and DNase-seq data analysis
Sequence reads (36-mer) were generated for ChIP-seq and DNase-seq experiments on the Illumina Genome Analyzer platform, and the tags were uniquely aligned to the mouse reference genome (UCSC mm9 assembly). Regions of enriched tags known as “hotspots” were called and determined significant using algorithms and methods previously described with minor modifications (Siersbæk et al. 2011). Briefly, ChIP-seq and DNase-seq data set tag density values were normalized to 10 million reads to adjust for differences in sequencing depth and to allow for cross data set comparisons, before hotspots were called at 0.5% FDR and thresholding five for the DNase-seq data set and 0% FDR and no thresholding for the GR ChIP data set. ChIP-seq data sets were additionally normalized by subtracting tags found in the corresponding input data. In all data, artifacts from sequencing (small regions of high density tags) were filtered out, including satellites, long interspersed repetitive elements, and short single tandem repeats, after extending these regions on either side to 150 bp. In comparisons of data sets, regions were considered to overlap if at least 2 bp were shared. Changes in chromatin accessibility in the presence or absence of hormone were determined by initially filtering each set of hotspots against hotspots generated in the control. Changes in GR loading at 20 min versus 60 min of hormone treatment were determined by filtering the 20 min set of hotspots against hotspots generated in the 60-min treatment.
Modified chromosome conformation capture assay
The 4C library preparation was performed as described previously (van de Werken et al. 2012) with minor modifications. Briefly, 10 million cultured mouse 3134 epithelial breast carcinoma cells per condition were fixed with 2% formaldehyde for 10 min, after treating the cells with 600 nM corticosterone in either pulsed or constant manner (see Fig. 1A). The reaction was quenched by the addition of glycine (final concentration of 0.125 M). Cells were then washed with cold PBS and lysed (10 mM Tris-HCl, pH 8.0, 10 mM NaCl, 0.2% NP-40, 1× complete protease inhibitors [Roche]) for 1 h at 4°C while rotating. Nuclei were incubated for 1 h at 37°C in 500 μL of restriction buffer (New England Biolabs CutSmart buffer for HindIII) containing 0.3% SDS while shaking (∼900 rpm). The SDS was quenched by adding Triton X-100 to the mix (2% final concentration) and incubating it for 1 h at 37°C while shaking. DNA digestion was performed with 1600 U of HindIII overnight at 37°C shaking (∼900 rpm). After heat inactivation (for 30 min at 65°C), the reaction was diluted to a final volume of 14 mL with ligation buffer containing 100 units T4 DNA Ligase (Roche) and incubated overnight at 16°C. Samples were then treated with 10 μL Proteinase K (10 mg/mL, Ambion) and incubated overnight at 65°C to reverse formaldehyde crosslinking. DNA was then purified by phenol extraction and ethanol precipitation. For circularization, the ligation junctions were digested with Csp6I (Fermentas) overnight at 37°C. After enzyme inactivation and phenol extraction, the DNA was religated in a 14-mL volume (100 units T4 DNA Ligase, Roche). The 4C library DNA was than amplified with the Expand Long template PCR System (Roche). Thermal cycle conditions were DNA denaturing for 2 min at 94°C, followed by 30 cycles of 15 sec at 94°C, 1 min at 60°C, and 3 min at 68°C. The thermal cycles were followed by 7 min at 68°C, and ∞12°C. Primers used to PCR amplify 4C DNA were either the Tsc22d3 promoter proximal site as a bait (5′-GATTTGCAAACAATGGAGCA-3′ and 5′-TAGGCAGATGGAGGACTTCG-3′), the Tsc22d3 distal DHS (DHS5) as a bait (primers: 5′-ttggtccttgctttgatgaac-3′ and 5′-ttattggcaaacctggcagt-3′), or the site upstream of the Atp6v1g1 gene (5′-CCATCCCAAAAGTCCCCCT-3′ and 5′-TCTAGGTGAGGTGGCACACA-3′). Libraries from two independent biological experiments were used. For each library amplification, we combined eight PCR reactions (200 ng template, 50 μL total volume) per condition to assure a good representation of all ligation products in the final bait-amplified 4C library. We conducted from two to four technical replicas of independent library amplifications per biological experiment. Samples were purified using the QIAquick PCR Purification Kit (Qiagen, # 28104). Upon purification, the resulting material was diluted in water and used as a template for qPCR (SYBR green [BioRad] reagent) at a concentration of 3 ng per reaction. We used an array of (over 30) primer pairs tiling a number of NlaIII/Csp6I fragments surrounding the TSS of the Tsc22d3 gene (for the full list of primers see Supplemental Table S1) and the TSS of the Atp6v1g1 gene (for the full list of primers see Supplemental Table S2). Primer pairs were first tested in RT-qPCR reactions using 4C DNA before amplification as template (3–5 ng per reaction), and the measures’ starting quantity values were normalized to the average value for all primer pairs, providing a baseline for the relative abundance of the fragments in the 4C library before amplification. The resulting values were not significantly different from the values obtained when using NlaIII/Csp6I digested genomic DNA as template. Next, these primer pairs were used in RT-qPCR reactions using the bait-amplified 4C libraries. The starting quantity value obtained for each primer pair was first normalized to the previously determined baseline value (measured using the unamplified 4C library pool) and finally renormalized to the average of all normalized starting quantity values to provide means for comparison within and between the different treatment conditions. The RT-qPCR reactions were performed in duplicate. Data were analyzed using the statistical and graphical functions of SigmaPlot 11 (SPSS, Inc.). From the repeated experiments, the mean value was calculated for each sample. The mean values were used in a one-way analysis of variance test. If a significant F-value of P < 0.05 was obtained, a multiple comparisons versus control group (Holm-Sidak method) was conducted.
Time profiles: generation and significance score
To generate the time profiles of both GR ChIP-seq and DNase-seq at a given DHS site, we computed the maximal tag density observed within the peak region for each condition (0, 20, 60 min with pulsed treatment, and 60 min with constant treatment). RefSeq gene annotations were used to generate Pol II occupancy time profiles. Genes that are shorter than twice the fragment length (2 × 150 bp) were excluded from further analysis (mostly microRNAs). For each gene and each condition, we computed the average tag density over the gene body, up to 10 kb downstream from the transcription start site (in order to avoid giving very long genes too much statistical significance over short ones; see below). RefSeq entries corresponding to different forms of the same gene (e.g., alternative transcription start or termination sites, splicing variants) were eliminated as follows: When several entries correspond to overlapping regions on the same strand, only the one giving the highest significance score (see below) is retained for further analysis.
We assessed the significance of the difference between data sets using the edgeR package (Robinson et al. 2010) in R (Furey 2012; Dillies et al. 2013; R Core Team 2014). For each DHS, we calculated the minimal P-values obtained when comparing all the conditions in the regular treatment with one another (Ptreat), then all the conditions in the mock treatment with one another (Pmock), and finally all pairs of similar conditions between the regular and the mock treatments (Ptreat-vs-mock). We only retained DHSs where Ptreat < 0.01, where Ptreat-vs-mock < 0.02, and where Ptreat < Pmock. Applying the same method to Pol II occupancy data over gene bodies, we only retained genes where Ptreat < 0.001, where Ptreat-vs-mock < 0.001, and where Ptreat < Pmock.
HSV transformation
We use the HSV color system to interpret any three-time-point profile (Fig. 3A–C). The HSV color system is a simple mapping of any color expressed as a weighted sum of the three primary light colors blue, green, and red into three other parameters. Denoting b, g, and r, the three data points of a given time profile (0, 20, and 60 min, respectively), hue, saturation, and value are computed as follows:
Whatever the unit of b, g, and r (e.g., tag density), h describes the shape of the time profile (it is an angle measured in degrees, and the time profiles corresponding to −180° and 180° are the same); s describes the extent of the variations over the profile relative to its maximum (it is dimensionless and expressed as a percentage); and v is simply the maximum value over the profile (it has the same unit as the data b, g, and r). Note that in the HSV scatter plots, whereas the numerical value v reported on the axes or on the legend corresponds directly to that obtained from the above equation, the color shown on the figure is modified so that it visually reflects the log scale, i.e., the color is multiplied by log(v)/v. This affects neither the hue nor the saturation of the displayed color.
De novo DNA sequence motif discovery analysis
Analysis of de novo DNA sequence motifs was performed on DHSs using the MEME algorithm (Bailey and Elkan 1994; Bailey and Gribskov 1998). From the hormone-responsive DHSs shown on Figure 4A, two groups of sites were defined: transient site with a shape parameter between 100° and 130°, and persistent site with a shape parameter between 0° and 100°. Each group was analyzed using a width of 150 bp. The minimum and maximum motif size was 8 and 40 bp, respectively, with a maximum of 50 motifs used for the search. Following MEME analysis, motif comparisons and identification of enriched sequences in unknown motifs (MEME E-values < 10–2) were performed using a TOMTOM (Gupta et al. 2007) search against the TRANSFAC database of characterized transcription factor motifs. Matches were considered significant if the majority of sequence nucleotides were shared and P-values were <10–4.
Data access
ChIP-seq and DNase-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE61236.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and National Institute of Diabetes and Digestive and Kidney Diseases. This project was also funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We acknowledge the assistance of Tatiana Karpova, LRBGE Fluorescence Imaging Facility. The work of L.S. was supported by OPVK project CZ.1.07/2.3.00/30.0030.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.184168.114.
References
- Atkinson HC, Wood SA, Kershaw YM, Bate E, Lightman SL. 2006. Diurnal variation in the responsiveness of the hypothalamic-pituitary-adrenal axis of the male rat to noise stress. J Neuroendocrinol 18: 526–533. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14: 48–54. [DOI] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CJ, Ward JM, Crusselle-Davis VJ, Kissling GE, Phadke D, Shah RR, Archer TK. 2012. Analysis of chromatin dynamics during glucocorticoid receptor activation. Mol Cell Biol 32: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway-Campbell BL, Sarabdjitsingh RA, McKenna MA, Pooley JR, Kershaw YM, Meijer OC, de Kloet ER, Lightman SL. 2010. Glucocorticoid ultradian rhythmicity directs cyclical gene pulsing of the clock gene period 1 in rat hippocampus. J Neuroendocrinol 22: 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway-Campbell BL, George CL, Pooley JR, Knight DM, Norman MR, Hager GL, Lightman SL. 2011. The HSP90 molecular chaperone cycle regulates cyclical transcriptional dynamics of the glucocorticoid receptor and its co-regulatory molecules CBP/p300 during ultradian ligand treatment. Mol Endocrinol 25: 944–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, et al. 2013. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14: 671–683. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JM, Linthorst AC. 2008. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology 149: 3244–3253. [DOI] [PubMed] [Google Scholar]
- Elgin SC. 1988. The formation and function of DNase I hypersensitive sites in the process of gene activation. J Biol Chem 263: 19259–19262. [PubMed] [Google Scholar]
- Fragoso G, Pennie WD, John S, Hager GL. 1998. The position and length of the steroid-dependent hypersensitive region in the mouse mammary tumor virus long terminal repeat are invariant despite multiple nucleosome B frames. Mol Cell Biol 18: 3633–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. 2012. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet 13: 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, Dekker J. 2012. The context of gene expression regulation. F1000 Biol Rep 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, Dekker J. 2013. The hierarchy of the 3D genome. Mol Cell 49: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. 2013. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32: 1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Garrard WT. 1988. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 57: 159–197. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. 2007. Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim O, Sung MH, Voss TC, John S, Splinter E, Sabo PJ, Thurman RE, Stamatoyannopoulos JA, de Laat W, Hager GL. 2011. Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res 21: 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. 2013. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503: 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, et al. 2008. Interaction of the glucocorticoid receptor with the global chromatin landscape. Mol Cell 29: 611–624. [DOI] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightman SL. 2006. Patterns of exposure to glucocorticoid receptor ligand. Biochem Soc Trans 34: 1117–1118. [DOI] [PubMed] [Google Scholar]
- Lightman SL, Conway-Campbell BL. 2010. The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat Rev Neurosci 11: 710–718. [DOI] [PubMed] [Google Scholar]
- Lightman SL, Windle RJ, Ma XM, Harbuz MS, Shanks NM, Julian MD, Wood SA, Kershaw YM, Ingram CD. 2002. Hypothalamic-pituitary-adrenal function. Arch Physiol Biochem 110: 90–93. [DOI] [PubMed] [Google Scholar]
- Lightman SL, Wiles CC, Atkinson HC, Henley DE, Russell GM, Leendertz JA, McKenna MA, Spiga F, Wood SA, Conway-Campbell BL. 2008. The significance of glucocorticoid pulsatility. Eur J Pharmacol 583: 255–262. [DOI] [PubMed] [Google Scholar]
- Londos C, Brasaemle DL, Gruia-Gray J, Servetnick DA, Schultz CJ, Levin DM, Kimmel AR. 1995. Perilipin: unique proteins associated with intracellular neutral lipid droplets in adipocytes and steroidogenic cells. Biochem Soc Trans 23: 611–615. [DOI] [PubMed] [Google Scholar]
- Londos C, Brasaemle DL, Schultz CJ, Segrest JP, Kimmel AR. 1999. Perilipins, ADRP, and other proteins that associate with intracellular neutral lipid droplets in animal cells. Semin Cell Dev Biol 10: 51–58. [DOI] [PubMed] [Google Scholar]
- Mandrup S, Hager GL. 2012. Modulation of chromatin access during adipocyte differentiation. Nucleus 3: 12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. 2000. Absence of perilipin results in leanness and reverses obesity in Leprdb/db mice. Nat Genet 26: 474–479. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Morris SA, Hager GL. 2013a. Complex genomic interactions in the dynamic regulation of transcription by the glucocorticoid receptor. Mol Cell Endocrinol 380: 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grøntved L, Schiltz RL, Hager GL. 2013b. Reprogramming of the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 73: 5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, Hager GL. 2014. Overlapping chromatin remodeling systems collaborate genome-wide at dynamic chromatin transitions. Nat Struct Mol Biol 21: 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon MS, Auado AS. 2012. Feature extraction and image processing for computer vision. Academic Press, NY. [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, Ribeiro L. 2014. Cortisol: the villain in Metabolic Syndrome? Rev Assoc Med Bras 60: 84–92. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Reik A, Schutz G, Stewart AF. 1991. Glucocorticoids are required for establishment and maintenance of an alteration in chromatin structure: induction leads to a reversible disruption of nucleosomes over an enhancer. EMBO J 10: 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, et al. 2006. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods 3: 511–518. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21: 55–89. [DOI] [PubMed] [Google Scholar]
- Schacke H, Docke WD, Asadullah K. 2002. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96: 23–43. [DOI] [PubMed] [Google Scholar]
- Siersbæk R, Nielsen R, John S, Sung MH, Baek S, Loft A, Hager GL, Mandrup S. 2011. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J 30: 1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza SC, de Vargas LM, Yamamoto MT, Lien P, Franciosa MD, Moss LG, Greenberg AS. 1998. Overexpression of perilipin A and B blocks the ability of tumor necrosis factor α to increase lipolysis in 3T3-L1 adipocytes. J Biol Chem 273: 24665–24669. [DOI] [PubMed] [Google Scholar]
- Stalder J, Larsen A, Engel JD, Dolan M, Groudine M, Weintraub H. 1980. Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell 20: 451–460. [DOI] [PubMed] [Google Scholar]
- Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. 2009. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol 11: 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavreva DA, Varticovski L, Hager GL. 2012. Complex dynamics of transcription regulation. Biochim Biophys Acta 1819: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Werken HJ, de Vree PJ, Splinter E, Holwerda SJ, Klous P, de Wit E, de Laat W. 2012. 4C technology: protocols and data analysis. Methods Enzymol 513: 89–112. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL. 2011. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146: 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Htun H, Hager GL. 1999. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods 19: 386–393. [DOI] [PubMed] [Google Scholar]
- Walker JJ, Spiga F, Waite E, Zhao Z, Kershaw Y, Terry JR, Lightman SL. 2012. The origin of glucocorticoid hormone oscillations. PLoS Biol 10: e1001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Beug H, Groudine M, Graf T. 1982. Temperature-sensitive changes in the structure of globin chromatin in lines of red cell precursors transformed by ts-AEV. Cell 28: 931–940. [DOI] [PubMed] [Google Scholar]
- Wu C. 1980. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 286: 854–860. [DOI] [PubMed] [Google Scholar]
- Young EA, Abelson J, Lightman SL. 2004. Cortisol pulsatility and its role in stress regulation and health. Front Neuroendocrinol 25: 69–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.