

Abstract
Background
A guanine-rich strand within the promoter of the KRAS gene can fold into an intra-molecular G-quadruplex structure (G4), which has an important role in the regulation of KRAS transcription. We have previously identified indolo[3,2-b]quinolines with a 7-carboxylate group and three alkylamine side chains (IQ3A) as effective G4 stabilizers and promising selective anticancer leads. Herein we investigated the anticancer mechanism of action of these compounds, which we hypothesized due to stabilization of the G4 sequence in the KRAS promoter and subsequent down-regulation of gene expression.
Methodology/Principal Findings
IQ3A compounds showed greater stabilization of G4 compared to duplex DNA structures and reduced KRAS promoter activity in a dual luciferase reporter assay. Moreover, IQ3A compounds showed high anti-proliferative activity in HCT116 and SW620 colon cancer cells (IC50 < 2.69 μM), without eliciting cell death in non-malignant HEK293T human embryonic kidney, and human colon fibroblasts CCD18co. IQ3A compounds significantly reduced KRAS mRNA and protein steady-state levels at IC50 concentrations, and increased p53 protein steady-state levels and cell death by apoptosis in HCT116 cells (mut KRAS, wt p53). Furthermore, KRAS silencing in HCT116 p53 wild-type (p53(+/+)) and null (p53(-/-)) isogenic cell lines induced a higher level of cell death, and a higher IQ3A-induced cell death in HCT116 p53(+/+) compared to HCT116 p53(-/-).
Conclusions
Herein we provide evidence that G4 ligands such as IQ3A compounds can target G4 motifs present in KRAS promoter, down-regulate the expression of the mutant KRAS gene through inhibition of transcription and translation, and induce cell death by apoptosis in colon cancer cell lines. Thus, targeting KRAS at the genomic level with G4 ligands may be a new anticancer therapy strategy for colon cancer.
Introduction
The KRAS gene encodes a G-protein which serves as a molecular switch between the endothelial growth factor receptor and the nucleus, controlling several signaling pathways important for cell growth and survival. KRAS GTPase exists in two states, a GTP-bound active state and a GDP-bound inactive state. Further, KRAS mutations increase KRAS affinity for GTP leading to the constitutive activation of the protein. Importantly, deletion of mutant KRAS allele in colon cancer cell lines dramatically reduces cellular proliferation [1], highlighting the fact that many tumors harboring mutant KRAS are KRAS-dependent. KRAS mutations are mostly prevalent in pancreatic (90–60%), colorectal (30–50%) and lung (20–30%) carcinomas [2]. Due to the high incidence of these cancers worldwide [3] and the increased resistance to conventional chemotherapy [4], the search for new targets has been intensified in past recent years.
The importance of therapeutic modulation of KRAS signaling have been widely recognized and several approaches have been reported in the past, but none has provided an approved new anticancer drug to date [5]. An innovative therapeutic approach being studied is the use of miRNAs, since they play an important role in chemo-sensitization [6]. We have previously demonstrated that miRNA-143, which also reduces KRAS expression, chemosensitizes colon cancer cells to 5-fluorouracil [7], and reduces tumor growth in vivo, with increased apoptosis and reduced proliferation [8].
Continuing our studies that aim to discover novel and selective anticancer drugs, pursued through the development of several chemical systems to be used in diverse therapeutic strategies [9], [10],[11], we report here on an approach to targeting KRAS signaling in cancer cells by directly modulating KRAS expression at the gene level. It has been recently demonstrated that a guanine-rich strand within the promoter of KRAS can fold into an intra-molecular G-quadruplex structure (G4), which has an important role in the regulation of KRAS transcription [12],[13]. G4 arrangements are nucleic acid higher-order structures, formed by sequences containing repetitive guanine(G)-rich tracts [14]. Several studies have provided evidence supporting the existence of G4s in eukaryotic telomeres and oncogene promoters, including those of KRAS, HRAS, HSP90, c-MYC, c-KIT, BCL-2 and VEGF genes, and that small molecules stabilizing G4 structures are able to down-regulate oncogene transcription in tumor cell lines, inhibit telomerase activity and induce cancer cell growth arrest [15],[16],[17]. G4 structures have also been found in RNA sequences, including in the 5′ untranslated region (UTR) of KRAS mRNA, and shown to have translation regulatory functions [18],[19],[20].
Indoloquinolines are natural alkaloids able to target DNA structures, some of which have potential for development into anticancer drugs [21],[22]. Indolo[3,2-b]quinoline derivatives have been shown to be potent G4 ligands, and to inhibit cell proliferation and oncogene (c-MYC) transcription [21],[23],[24]. Moreover, we have recently discovered that indolo[3,2-b]quinolines with a 7-carboxylate group and three alkylamine side chains (1a and 2a in Fig 1) are promising selective anticancer leads [25]. These compounds selectively inhibited (100-fold) the growth of KRAS mutant HCT116 colon cancer cells compared to primary rat hepatocytes, while also decreasing KRAS protein levels. In order to exploit this scaffold towards the discovery of novel and improved anticancer drugs, we have extended the chemical diversity of these indoloquinolines and studied their potential anticancer mechanism of action. Previous structure-activity studies with mono-alkylamine indolo[3,2-b]quinolines have shown that optimal G4 stabilization was induced by compounds with propyl side chains and basic amine groups (pKa ≥ 8) [25]. Thus, compounds 1a-d and 2a-d (Fig 1) were designed, synthesized and evaluated for selective G4 thermal stabilization comparing to duplex DNA, together with inhibition of cancer cell proliferation, induction of apoptosis and down-regulation of KRAS and HSP90 transcription and protein expression. In order to improve the anticancer activity profile and KRAS oncogene down-regulation capacity of our target indoloquinolines, we have used four cell lines with differing KRAS and TP53 genotypes, as well as two positive controls, the anticancer drug 5-fluorouracil (5-FU) and the G4 ligand TMPyP4 (Fig 1).
Fig 1. Structures of studied compounds.

7-carboxylate indolo[3,2-b]quinoline tri-alkylamine derivatives (IQ3A) 1a-d and 2a-d; G4 ligand porphyrin derivative TMPyP4 and 5-fluorouracil (5-FU) anticancer drug.
Results and Discussion
Compounds 1a-d and 2a-d were synthesized in four steps following the procedure previously described [25] with some modifications (S1 Text). Structures of 1a-d and 2a-d were completely elucidated by bidimensional 1H (COSY and NOESY) and 13C heterocorrelation NMR experiments (HMQC and HMBC) and purity (> 95%) confirmed by HPLC-ELSD-MS (S1 Fig).
The capacity of compounds 1a-d, 2a-d and the standard G4 ligand TMPyP4 (Fig 1) [16] to bind and stabilize KRAS [26] and HSP90A [27] G4 DNA structures as well as duplex DNA (T-loop) was evaluated by a Fluorescence Resonance Energy Transfer (FRET) melting assay. The increase in the melting temperatures induced by different concentrations of compounds is presented in Table 1 and S2 Fig. Our results show that tri-alkylamine indolo[3,2-b]quinolines (IQ3A) are potent and selective ligands for the KRAS and HSP90A G4 structures. As previously observed for mono- and di-alkylamine analogues [25] and other polyaromatic-fused G4 ligands [28],[29], compounds with propylamine side chains (1d and 2d) are superior G4 stabilizers (ΔT m between 18 and 23°C at 2 μM of ligand) than compounds with shorter alkylamine side chains (1a-b and 2a-b; ΔT m values between 7 and 17°C at 2 μM ligand concentration). It was observed that the basicity of side chains correlates positively with thermal G4 stabilization of all DNA sequences up to an optimal pKa ~ 8.0–9.0 (Fig 2). Heterocyclic amines at the end of alkyl side chain (1b and 2b) appear to improve binding to G4s and complex stabilization compared to the di-ethylamine group (1a and 2a), which cannot be explained by differences in basicity between terminal groups. Additionally, and in line with what was previously observed for di-alkylamine indoloquinolines [25], this study suggests that N5,N10,COO tri-substitution with heterocyclic amine groups at alkyl side chain termini increase ligand affinity to G4, as 2b,d-G4 DNA complexes have higher melting temperatures than complexes of the correspondent isomers 1b,d. However, this was not observed for the isomeric pair 1a / 2a. Despite the increased G4 stabilization capacity, ligands 2b and 2d were not able to significantly discriminate between the two DNA G4 structures (Table 1).
Table 1. Melting temperatures (ΔTm) of DNA G-quadruplex (KRAS21, HSP90) and duplex (T-Loop) structures stabilized by compounds 1a-d, 2a-d and TMPyP4 at 1 and 2 μM.
| Comp. | ΔTm (°C) (a) | |||||
|---|---|---|---|---|---|---|
| KRAS21 | HSP90A | T-Loop | ||||
| 1 μM | 2 μM | 1 μM | 2 μM | 1 μM | 2 μM | |
| 1a | 8 | 12 | 14 | 20 | <2 | 3 |
| 1b | 5 | 10 | 12 | 16 | <2 | 6 |
| 1c | <2 | <2 | 4 | 7 | <2 | <2 |
| 1d | 9 | 18 | 15 | 21 | <2 | 10 |
| 2a | 4 | 7 | 11 | 15 | <2 | <2 |
| 2b | 10 | 16 | 14 | 19 | 3 | 6 |
| 2d | 13 | 22 | 19 | 25 | 4 | 12 |
| TMPyP4 | 25 | n.d. | n.d. | n.d. | 16 | n.d. |
(a) standard deviation < 0.2°C;
n.d. = not determined.
Fig 2. Basicity of side chains correlates positively with thermal G4 stabilization.
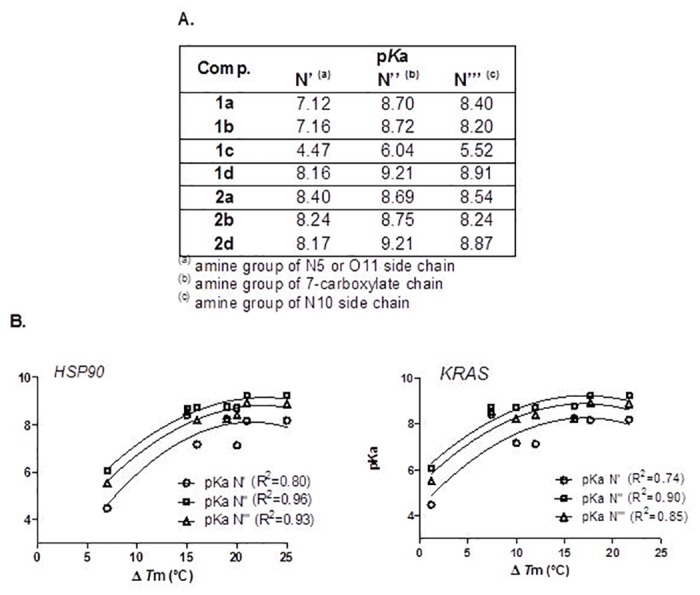
A. Calculated pKa values of side chain amine groups by SPARC (v. 4.6). B. Plots of variation of G4 thermal stabilization by compounds (ΔT m) with basicity (pKa) of side chains groups.
To study the effect of IQ3A compounds on cancer and non-cancer cells, we selected compound 2d showing the best G4 stabilization capacity in vitro and the pair 1a and 2a, which despite showing lower ΔT m values for the respective complexes with G4 DNA, are able to reduce KRAS expression when incubated in HCT116 colorectal cancer cells, as we have previously shown [25].
The short-term effect of compounds at 72 h on cell growth was studied using colorectal carcinoma cell lines with different KRAS and TP53 genotypes: human colorectal carcinoma HCT116 (mut Kras, wild-type (wt) p53), and human metastatic colorectal adenocarcinoma SW620 (mut Kras, mut p53). In parallel, we also used the immortalized human embryonic kidney cell line HEK293T (wt Kras, wt p53) and normal human colon fibroblast cells CCD18co (wt Kras, wt p53). The IC50 and IC65 values in Table 2 show that compounds 2a and 2d display superior anti-proliferative activity compared to 1a and 5-FU, particularly in metastatic SW620 cells, where 2d gave an IC50 value (0.28 μM), almost 20-fold lower than that of the standard anticancer drug 5-FU (IC50 = 5.39 μM). Interestingly, colorectal cancer cells HCT116 and SW620, which express mutant KRAS, were particularly insensitive to the porphyrin derivative TMPyP4, in contrast to non-malignant HEK293T cells expressing wild type KRAS. In addition, IQ3A compounds and 5-FU, were not selective for cancer cells expressing mutant KRAS, since they were equally active against HCT116, SW620 and HEK293T cells. Nevertheless, we observed some selectivity (S.I. > 2.4; Table 2) toward the colon cancer cell line HCT116 compared to normal colon fibroblasts (CCD18co).
Table 2. Antiproliferative activity (IC50 and IC65 in μM) a of compounds 1a, 2a, 2d, TMPyP4 and 5-fluorouracil (5-FU) in malignant (HCT116, SW620) and non-malignant (HEK293T, CCD18co) cell lines.
| Comp. | HCT116 | SW620 | HEK293T | CCD18co | SI (b) | |||
|---|---|---|---|---|---|---|---|---|
| IC50 | IC65 | IC50 | IC65 | IC50 | IC65 | IC50 | ||
| 1a | 1.88 c | 3.42 | 2.69 | 4.12 | 3.23 | 6.06 | 4.63 | 2.46 |
| 2a | 0.99 c | 1.60 | 0.67 | 1.44 | 0.67 | 0.94 | 5.41 | 5.46 |
| 2d | 0.40 | 0.58 | 0.28 | 0.45 | 0.70 | 1.12 | 2.18 | 5.45 |
| TMPyP4 | 12.39 | 14.21 | > 20 | > 20 | 2.97 | 4.11 | 38.17 | 3.08 |
| 5-FU | 2.38 c | 4.13 | 5.39 | 8.32 | 2.66 | 4.13 | >100 | > 42 |
(a) Standard errors ≤ 0.1;
(b) Selective index (SI) = IC50 CCD-18co / IC50 HCT116;
(c) Previously reported IC50 of 4.5(1a),5.7(2a) and 3.79(5-FU) μM using different cell culture conditions.[25]
Subsequently, the Guava ViaCount assay was used to evaluate the effects of IQ3A compounds on cell death induction in cancer (HCT116 and SW620) and non-malignant (HEK293T and CCD18co) cells, compared to 5-FU and TMPyP4 at equitoxic concentrations (IC50 and IC65). Down-regulation of mutant KRAS expression by antisense oligonucleotides in colorectal cancer cells [7],[8],[30] and by a MAZ-binding oligonucleotide decoy in pancreatic cancer cells [31] has been associated with increased apoptosis and cell growth arrest. Also, anticancer drugs, such as 5-FU and G4 stabilizers of telomeric and oncogene promoter sequences such as TMPyP4, are known to induce a generalized cell response leading to cell growth arrest and cell death by apoptosis (DNA damage response) [15],[32], which involves activation of the pro-apoptotic transcription factor p53 among other pathways in the case of 5-FU [34]. Fig 3 shows that our test compounds have distinct effects on cells, depending on compound structure and probably also on the genetic background of an individual cell line. In HCT116 cells, all compounds induced cell death by apoptosis, but the IQ3A compounds showed the most significant apoptotic effect, with a trend of 1a > 2a > 2d, and causing 30–70% cell death at concentrations ranging from 0.58 to 3.42 μM (IC65) (Fig 3B, upper left panel). Conversely, all compounds were unable to induce significant cell death in HEK293T and CCD18co cells (Fig 3A and 3B, lower panels), whereas in SW620 cells only 5-FU was able to induce apoptosis in a dose-dependent manner (Fig 3A and 3B, upper right panels). The effect of TMPyP4 on SW620 was not studied, as this cell line was not sensitive to this compound up to a 20 μM concentration.
Fig 3. Exposure to IQ3A compounds induces cell death, mainly by apoptosis in HCT116 cells.

Cell populations were obtained by Guava ViaCount flow cytometry following 72 h incubation of HCT116 colon cancer, SW620 metastatic colon cancer, CCD18co human colon fibroblasts and HEK293T embryonic kidney cell lines with 5-FU, TMPyP4 and IQ3A at equitoxic (IC50 and IC65) concentrations, or DMSO (vehicle control). Results are expressed as the mean percentage (%) of viable, mid-apoptotic and dead cells ± SEM, of at least three different experiments, for A. IC50, and for B. IC65 compound concentrations. a, p < 0.05; b, p < 0.01 from DMSO (vehicle control); c, p < 0.05; and d, p < 0.01 from 5-FU.
To further validate the effect of the IQ3A compounds on the induction of apoptosis in HCT116 cells, apoptosis was ascertained by evaluating changes in nuclear morphology by Hoechst staining, and also by the Nexin assay, following 72 h of incubation with IQ3A compounds at the IC50 concentrations. CCD18co cells were used in parallel to confirm that IQ3A do not elicit cell death in normal cells. These results showed that IQ3A significantly induce apoptosis in HCT116 cells, with compound 1a producing a marked increase in apoptosis compared to vehicle (Fig 4A and 4B). In addition, we also confirmed that IQ3A compounds do not induce apoptosis in normal colon fibroblasts CCD18co. The p53 protein plays a central and pivotal role in human cancers [33], having a potent tumor suppressive activity via pleiotropic mechanisms. Therefore, the steady-state levels of p53 protein were evaluated by immunoblotting, following 72 h of incubation with IQ3A compounds at the IC50 concentrations, to ascertain their involvement on the mechanism of apoptosis elicited by IQ3A. Our data (Fig 5A) clearly demonstrate that IQ3A increased p53 protein steady state levels in HCT116 by 4–7 fold (p < 0.01) (Fig 5A, left panel), which may be correlated with the higher cell death verified by Guava ViaCount assay. In SW620 cells, no significant increase in p53 steady-state expression was detected, possibly due to the mutant status of p53 (Fig 5A, middle panel). Finally, in HEK293T cells, IQ3A showed no influence on p53 protein expression, in accord with the absence of cell death in the viability assay (Fig 5A, right panel). In addition, we further explored the relevance of p53 in the mechanism of apoptosis elicited by IQ3A, by silencing KRAS in HCT116 p53 wild-type (p53(+/+)) and null (p53(-/-)) isogenic cell lines, and evaluating its impact on cell death, and on IQ3A-induced cell death (Fig 5B). Our results clearly show that KRAS silencing induced a higher level of cell death compared to siRNA control (p < 0.05), and a higher level of IQ3A-induced cell death in HCT116 p53(+/+) compared to HCT116 p53(-/-) (p < 0.05 for 2a and 1a). Further, in siRNA control transfected cells, all IQ3A significantly induced a higher level of cell death in HCT116 p53(+/+) compared to HCT116 p53(-/-) (p < 0.05). However, in p53 mutant SW620 cells, IQ3A compounds did not significantly elicit cell death (Fig 3), nor did they increase p53 protein steady-state expression (Fig 5). In agreement, KRAS and/or HSP90 silencing were unable to induce cell death in this cell line, whereas KRAS silencing induced a dose-dependent effect in the reduction of SW620 cell proliferation (S3 Fig). Collectively, these data further confirm the importance of p53 for IQ3A induction of apoptosis.
Fig 4. Exposure to IQ3A compounds increases apoptosis in HCT116 cells.

A. Nuclear morphology and representative images of HCT116 human colon cancer cells and CCD18co human colon fibroblasts after Hoechst staining, evaluated by fluorescence microscopy after 72 h exposure to equitoxic (IC50) concentrations of 5-FU, TMPyP4 and IQ3A or DMSO (vehicle control) at 400x magnification. White Arrows indicate nuclear fragmentation and chromatin condensation, and B. Apoptotic cell populations obtained by Guava Nexin flow cytometry following 72 h incubation of HCT116 and CCD18co cells with 5-FU, TMPyP4 and IQ3A at equitoxic (IC50) concentrations, or DMSO (vehicle control). Results are expressed as the mean percentage (%) of early (LR quadrant) and late (UR quadrant) apoptotic cells. Results are expressed as mean ± SEM of at least three independent experiments; *p < 0.05 and §p < 0.01 from DMSO (vehicle control); and †p <0.05 and ‡p < 0.01 from 5-FU.
Fig 5. Exposure to IQ3A compounds increases p53 steady-state protein expression in HCT116 cells.

A. p53 protein steady-state expression evaluated by immunoblot relative to DMSO (vehicle control), after 72 h exposure to equitoxic (IC50) concentrations of 5-FU, TMPyP4 and IQ3A. B. Cell populations obtained by Guava ViaCount flow cytometry following 72 h incubation of 5-FU, TMPyP4 and IQ3A at equitoxic (IC50) concentrations, or DMSO (vehicle control), in HCT116 p53 (+/+) and p53 (−/−) isogenic cell lines transfected with 40 nM siRNA KRAS or siRNA control. Results are expressed as mean ± SEM of at least three independent experiments. A. §p < 0.01 from DMSO (vehicle control); and ‡p < 0.01 from 5-FU. B. *p < 0.05 from siRNA control in P53 (+/+) cells; §p < 0.05 from siRNA control in p53 (-/-) cells; and †p <0.05 from siRNA KRAS in p53 (-/-) cells.
Finally, the capacity of IQ3A compounds to repress KRAS expression was evaluated by quantifying KRAS protein in cancer cell lines. Since IQ3As have also shown to be effective stabilizers of G4 sequences in the HSP90 oncogene promoter region (Table 1), the expression of this protein was also evaluated. Fig 6A shows that G4 ligands 1a, 2a, 2d and TMPyP4 down-regulated the expression of mutant KRAS by 35–60% in HCT116 and SW620 cells, with exception of 2a (p < 0.01). In addition, IQ3A also reduced HSP90 protein steady levels, but to a lesser extent compared to KRAS. Therefore, we next investigated the ability of IQ3A compounds to down-regulate KRAS transcription in colon cancer cells. KRAS mRNA steady-state levels were evaluated by RT-PCR after 72 h incubation of cells with the compounds at their IC50 concentrations and compared with the effect of TMPyP4 at equitoxic concentrations. G4 ligands such as TMPyP4 have been shown to bind to G4 structures from the KRAS promoter region and from the 5’-UTR of KRAS mRNA, and also repress both gene transcription and translation [13,19]. Fig 6B shows that 1a, 2a and 2d were able to down-regulate KRAS transcription by ca 40% in HCT116 cells, but not significantly so in SW620 cells. A possible explanation is the time point at which we evaluated KRAS mRNA and protein steady-state levels. After 72 h of IQ3A exposure, there may no longer be significant repression of KRAS promoter activity in the SW620 cell line, whereas protein levels remain reduced as a result of accumulated IQ3A effects. Also, TMPyP4 was unable to reduce KRAS mRNA steady-state levels in the HCT116 cell line, in contrast to a decrease of ca 80% of the protein steady-state levels (Fig 6A and 6B). These results are in agreement with the reported ability of TMPyP4 to preferentially accumulate in the cytoplasm of cells [19], where it can inhibit KRAS mRNA translation.
Fig 6. Exposure to IQ3A compounds decreases HSP90 and KRAS mRNA, protein expression and KRAS transcription.

A. KRAS and HSP90 protein steady-state expression evaluated by immunoblot relative to DMSO (vehicle control), after 72 h exposure to equitoxic (IC50) concentrations of 5-FU, TMPyP4 and IQ3A treatment; B. KRAS mRNA steady-state expression was evaluated by Taqman Real-time RT-PCR using specific Taqman Assays for KRAS and β-Actin for normalization. KRAS mRNA steady-state expression levels were calculated by the ΔΔCt method, using DMSO (vehicle control) for calibration; and C. HEK293T cells were co-transfected with pGL3-basic vector (empty vector control), or with KRAS promoter luciferase reporter construct PGL-Ras0.5, or PGL-Ras2.0, together with pRL-TK. Twenty-four hours later, cells were replated in 96-well plates, at 5.000 cells per well. Subsequently, 24 h after replating, cells were exposed to IC50 equitoxic concentration of test compounds IQ3A, TMPyP4 and vehicle (DMSO); D. HCT116, SW620 and HEK293T cells were co-transfected with pGL3-basic vector (empty vector control), or with KRAS promoter luciferase reporter construct PGL-Ras0.5, together with pRL-TK. Twenty-four hours later, cells were replated in 96-well plates, at 5,000 cells per well and exposed to IC50 equitoxic concentration of test compounds IQ3A, TMPyP4 and vehicle (DMSO). KRAS promoter activity levels were evaluated by Dual-Luciferase assay 72 h (C.) or 24 h (D.) after compound exposure. Results are expressed as the luciferase signal ratio of pGL-Ras2.0 or pGL-Ras0.5 to pGL3-basic vector transfected cells, after normalization with Renilla Luciferase. Results are expressed as mean ± SEM of at least three independent experiments; *p < 0.05 and §p < 0.01 from DMSO (vehicle control); and †p <0.05 and ‡p < 0.01 from 5-FU.
To validate that the mechanism of anti-proliferative activity and apoptotic induction by IQ3A compounds involves repression of KRAS gene expression as a result of the stabilization of G4-forming sequences present in the promoter, the effects of compounds on the KRAS gene promoter were directly evaluated by a luciferase reporter assay. For this purpose, we used two different size promoter constructs containing the G4 region of the KRAS gene promoter, cloned into the pGL3 Basic backbone: pGL-Ras0.5, pGL-Ras2.0, and pGL3 Basic empty (Firefly Luciferase negative control/no promoter), co-transfected together with pRL-TK (transfection efficiency normalization) into HEK293T cells, as a G4 negative control. This construct does not harbor G4 sequences and is insensitive to G4-related effects/regulation. Our data clearly demonstrate that IQ3A compounds, similarly to TMPyP4, were able to significantly reduce KRAS transcription, we suggest by interacting with the G4 region of the KRAS gene promoter, suppressing downstream coding-region expression from 40 to 60% versus DMSO control (p < 0.01) (Fig 6C). Using both plasmids, with 500 and 2000 bp upstream to the transcription start site, we also show that the target region of the IQ3A compounds is within this region, thus coinciding with the polypurine G-rich strand responsible for G-quadruplex structure assembly [12]. Importantly, we were also able to show that IQ3A compounds, similarly to TMPyP4, were able to significantly reduce KRAS promoter activity in HCT116 and SW620 cells (Fig 6D).
Conclusions
We have investigated in this study the ability of a group of 7-carboxylate indolo[3,2-b]quinoline tri-alkylamine derivatives (IQ3A), which are potent stabilizers of DNA G4 structures present in the KRAS promoter, to down-regulate KRAS expression and induce cell death by apoptosis, particularly in KRAS-dependent colon cancer cell lines. The IQ3A compounds markedly showed anti-proliferative activity in an IC50 range lower than 2.69 μM in HCT116 and SW620 cells. Particularly, in HCT116 cells, IQ3A compounds at the IC65 concentration increased cell death up to 4.7 fold (p < 0.01) and 2.4 fold (p < 0.01) in comparison to DMSO or 5-FU, respectively. In addition, in non-malignant cell lines the IQ3A compounds did not cause significant cell death. Further, they markedly reduced KRAS mRNA expression and protein levels in colon cancer cells possibly through direct transcriptional repression of the KRAS promoter. Our data to date shows a correlative relationship between transcriptional repression and binding to the G4 region within this promoter, although further studies are needed in order to demonstrate a direct link between the two. The repression effect was accompanied by increased p53 protein steady-state levels in HCT116, and only a slight reduction of HSP90 levels in the three cell lines tested, indicating that these compounds are not also having an effect on some other possible G4 targets such as in the HSP90 promoter [27]. Rather the IQ3A compounds are selectively targeting the KRAS driver genes in the colon cancer cell lines. Collectively these results show that G4-binding ligands, such as these indoloquinoline derivatives, display potential to be employed as novel anticancer therapeutic agents, especially for the treatment of human cancers driven by KRAS mutations, which are still in large part cancers of high unmet clinical need.
Materials and Methods
Chemicals
7-carboxylate indolo[3,2-b]quinoline tri-alkylamine derivatives 1a-d and 2a-d (IQ3A) were synthesized as described in S1 Text. 5-fluorouracil (5-FU) and TMPyP4 were purchased from Sigma-Aldrich, St Louis, MO, USA.
Oligonucleotides sequences
All oligonucleotides were purchased from Eurofins MWG Synthesis GmbH, Germany. The labeled oligonucleotides used in the FRET assays had attached the donor fluorophore FAM (6-carboxyfluorescein) and the acceptor fluorophore TAMRA (6-carboxytetramethylrhodamine): KRAS21R (5’-FAM-AGGGCGGTGTGGGAAGAGGGA-TAMRA-3’); HSP90A (5’- [FAM]-GGGCCAAAGGGAAGGGGTGGG-[TAMRA]-3’); T-Loop (5’-FAM-TATAGCTATATTTTTTTATAGCTATA-TAMRA-3’). Each oligonucleotide was initially diluted to a storage solution at 100 μM in nuclease free water (not DEPC-treated), purchased from Ambion Applied Biosystems UK.
FRET melting assay
The ability of IQ3A and TMPyP4 to stabilize G-quadruplex and duplex DNA sequences was investigated using a Fluorescence Resonance Energy Transfer (FRET) assay. Stock solution of the oligonucleotides sequences at 20 μM and subsequent dilutions were obtained from storage solutions diluting with K-cacodylate buffer pH = 7.4, containing 60 mM K+ (10 mM potassium cacodylate, 50 mM KCl). The FRET probe sequences were diluted from stock to the correct concentration (0.4 μM) and then annealed by heating to 95°C for 10 min, followed by cooling to RT. Test compounds were prepared as 1 mM in 10% DMSO and 10% 1mM HCl in HPLC-grade water. The rest of the dilutions were performed using FRET buffer. Annealed DNA (50 μL) and compound solution (50 μL) were distributed across 96-well RT-PCR plates (BioRad; MJ Research, Waltham, MA, USA). Relevant controls were also performed to check for interference with the assay. Fluorescence readings were made on a DNA Engine Opticon (MJ Research) with excitation at 450–495 nm and detection at 515–545 nm, taken at intervals of 0.5°C in the range 30–100°C, with a constant temperature being maintained for 30 s prior to each reading to ensure a stable value. Experiments were performed in triplicate. Final analysis of the data was carried out with GraphPad Prism 5.0 software (GraphPad Software, Inc). The advanced curve-fitting function in GraphPad Prism was used for calculation of ΔT m values and associated standard deviations.
Cell culture
SW620 human colorectal adenocarcinoma cells (European Collection of Cell Cultures, ECACC, Porton Down, Wiltshire, UK), HEK293T human embryonic kidney cells (ATCC, LGC Standards, UK) HCT116 human colon carcinoma cells (ECACC), HCT116 p53(+/+) and p53(−/−) isogenic human colon cancer cells (GRCF Cell Center and Biorepository, the Johns Hopkins University, School of Medicine, Baltimore, MD, USA), were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, and 1% antibiotic/antimycotic (Invitrogen, Grand Island, NY, USA), CCD18co human colon fibroblasts (ATCC, CRL-1459) were grown in Minimum Essential Medium (MEM) supplemented with 10% fetal bovine serum, 1% antibiotic/antimycotic, 2 mM GlutaMAX, 0,1 mM Non-essential amino acids (NEAA) (Invitrogen) and 0.57 mM Recombinant Human TNF-α (Peprotech, USA) and maintained at 37°C in a humidified atmosphere of 5% CO2.
Evaluation of cell viability and general cell death
Cell viability was evaluated by the tetrazolium dye (MTS) Short-Term Cytotoxicity Assay. In brief, cells were seeded in 96 well plates at 5,000 cells/well. Twenty-four hours after cell plating, media was removed and replaced with fresh media containing test compounds IQ3A, TMPyP4 and 5-FU, or vehicle control (DMSO). Following 72 h of compound exposure, cell viability was evaluated using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA), using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) as previously described [34] [35]. Cell viability data were expressed as mean ±SD from at least three independent experiments. IC50 and IC65 values were determined using GaphPad Prism v.5.00 (GraphPad Software). In selected experiments, cell viability was also assessed by trypan blue exclusion assay [34], and general cell death by LDH activity from cell culture supernatants [7].
Guava ViaCount assay
ViaCount assay was used with Guava easyCyte 5HT Flow cytometer (Guava Technologies, Inc., Hayward, CA, USA), as previously described [36] to evaluate viable, apoptotic and dead cell populations, on HCT116, SW620, HEK293T and CCD18co cells exposed to test compounds IQ3A, TMPyP4 and 5-FU at IC50 and IC65 concentrations, and a vehicle control (DMSO). The ViaCount Assay distinguishes viable mid apoptotic and dead cells based on differential permeability of two DNA-binding dyes in the GuavaVia Count Reagent. The nuclear dye stains only nucleated cells, while the viability dye brightly stains dying cells. HCT116, SW620, HEK293 T and CCD18co cells were seeded in 24-well plates 50,000 cells/well. Twenty-four hours later, cells were exposed to compounds for 72 h. After treatment, cell culture supernatants were collected and adherent cells were detached with TrypLE (Invitrogen). Next, detached cells were pooled with cell culture supernatants and centrifuged for 5 min (650 g). Supernatants were discarded and the cells were resuspended in 50–500μl phosphate buffered saline (PBS) with 2% FBS. Subsequently, 15 μl of cell suspension were mixed with 135 μl of Guava ViaCount reagent, and incubated for 5 min at room temperature. Sample acquisition and data analysis were performed using the ViaCount software module.
Guava Nexin assay
Nexin assay was used with Guava easyCyte 5HT Flow cytometer (Guava Technologies, Inc., Hayward, CA, USA) to evaluate viable, early and late apoptotic cell populations, on HCT116 and CCD18co cells exposed to either test compounds IQ3A, TMPyP4 and 5-FU at IC50 concentration, or vehicle control (DMSO). The Nexin Assay distinguishes viable, early and late apoptotic cells based on the externalization of phosphatidylserine to the cell surface, where Annexin V can readily bind them. The membrane dye stains with higher intensity early and late apoptotic cells. HCT116 and CCD18co cells were seeded in 24-well plates 50,000 cells/well. Twenty-four hours later, cells were exposed to compounds for 72 h. After treatment, cell culture supernatants were collected and adherent cells were detached with TrypLE (Invitrogen). Next, detached cells were pooled with cell culture supernatants and centrifuged for 5 min (650 g). Supernatants were discarded and the cells were resuspended in 50–500 μl phosphate buffered saline (PBS) with 2% FBS. Subsequently, 50 μl of cell suspension were mixed with 50 μl of Guava Nexin reagent, and incubated for 20 min at room temperature. Sample acquisition and data analysis were performed using the Nexin software module.
Total protein extraction and immunoblotting
HCT116, SW620 and HEK293 T cells were seeded in 35 mm plates at 300,000 cells per well. Test compounds IQ3A, TMPyP4, 5-FU and a vehicle (DMSO) were added to the cells 24 h after plating, at IC50 equitoxic concentration. After 72 h of compound exposure, cells were collected and processed for total protein extraction, as previously described [8]. Briefly, samples were homogenized in ice-cold 1:1 solution of buffer A [10 mM Tris•HCl pH 7.6, 5 mM MgCl2, 1.5 mM KAcO, 2 mM dithiothreitol (DTT), and Halt Protease and Phosphatase inhibitor cocktail, EDTA-free (#78445, Thermo Scientific)] and buffer 2X (10 mM Tris•HCl pH 7.6, 1% Nonidet-P40, and Halt Protease and Phosphatase inhibitor cocktail), by vigorous vortexing and incubated on ice for 30 min. Samples were then sonicated (two cycles of 15 s sonication and 30 s ice incubation, using a compact ultrasonic device with amplitude adjusted to 80% and pulse to 90%; model UP100H, Hielscher Ultrasonics GmbH, Teltow (Germany); 100 W, ultrasonic frequency: 30 kHz) and centrifuged at 10,000 g for 10 min at 4°C. The clear supernatants containing the total protein extracts were transferred to a fresh tube and stored at −80°C. Protein concentrations were determined using the BioRad protein assay kit according to the manufacturer’s instructions. Steady-state levels of KRAS and Hsp90 protein were determined by immunoblot analysis. Briefly, 50–100 μg of total protein extracts were separated by 12% SDS-PAGE. After electrophoretic transfer onto nitrocellulose membranes, immunoblots were blocked with 5% milk solution, and next incubated overnight at 4°C with primary mouse monoclonal antibody reactive to KRAS (#sc-30; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and to Hsp90 (#sc-13119; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Finally, immunoblots were incubated with secondary anti-mouse antibody conjugated with horseradish peroxidase (BioRad) for 3 h at room temperature. The membranes were processed for protein detection using Super Signal substrate (Pierce, Rockford, IL, USA). β-Actin (#A-5441, Sigma—Aldrich) was used as a loading control. Steady-state protein levels were expressed as mean ±SEM from at least three independent experiments.
Total RNA extraction and Taqman Real-time RT-PCR
HCT116, SW620 and HEK293 T cells were seeded in 35 mm plates at 300,000 cells per well. Test compounds IQ3A, TMPyP4, and 5-FU and vehicle (DMSO) were added to the cells 24 h after plating, at IC50 equitoxic concentration. After 72 h of compound exposure, cells were collected and processed for total RNA extraction using TRIZOL reagent (Invitrogen) according to the manufacturer’s instructions. Samples were homogenized in TRIZOL reagent using a motor-driven Bio-vortexer (No1083; Biospec Products, Bartlesfield, OK) and disposable RNAse/DNAse free sterile pestles (Thermo Fisher Scientific, Inc., Chicago, IL). RNA was quantified using a NanoDropH spectrophotometer, and typically showed A260/280 ratios between 1.9 and 2.1. Evaluation of steady-state expression of KRAS mRNA was performed by Taqman Real-time PCR. In brief, RT reactions were performed using 1.5 μg of total RNA, using High Capacity RNA to cDNA kit, according to the manufacturer`s instruction. Next, Real-time PCR reactions were performed using Taqman Universal Master Mix II, no UNG, and primers specific to human KRAS (assay ID Hs00000174_rf, # 4465807) primers and to human β-Actin (ACTB # 401846) for normalization to endogenous control (both from Applied Biosystems Inc). Triplicate reactions were run per sample. Data was collected with 7300 System Sequence Detection Software, version 1.2.3 (Applied Biosystems Inc). The comparative threshold cycle method was used to calculate the amplification factor, where the threshold cycle (Ct) is defined as the cycle number at which the fluorescence passes the fixed threshold intensity level. KRAS expression levels in different samples were calculated on the basis of ΔΔCt method. For each cell line, vehicle control (DMSO) was used as the calibrator. The n-fold change in KRAS expression was obtained using the formula: 2-ΔΔCt.
KRAS and Hsp90 silencing
HCT116 p53 (+/+) and p53 (−/−) cells were seeded in 35 mm plates at 300,000 cells per well. Twenty-four hours later, cells were transfected with 80 nM KRAS Silencer Select Pre-Designed & Validated siRNA (siRNA KRAS) or 80 nM Silencer Negative Control siRNA (siRNA control) (both from Applied Biosystems, Foster City, CA, USA). In parallel SW620 cells were seeded in 35 mm plates at 300,000 cells per well. Twenty four hours later, cells were transfected with 50 or 100 nM siRNA KRAS, siRNA control, siRNA HSP90 (HSP90α/β siRNA (h) (sc-35608 Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and co-transfected with 50 nM siRNA KRAS plus 50 nM siRNA HSP90. Transfections were performed using lipofectamine 3000 (Invitrogen), according to manufacturer’s instructions. Twenty-four h later, cells were replated in 24-well plates at 50,000 cells/well and 72 h later Guava ViaCount, trypan blue dye exllusion, MTS metabolism and LDH release assays.
KRAS promoter activity Luciferase Reporter Assay
HEK293T cells were seeded in 35 mm plates at 300,000 cells per well. Twenty-four h later, cells were transiently co-transfected with pGL3-basic vector (empty vector control), or with KRAS promoter luciferase reporter construct PGL-Ras0.5, or PGL-Ras2.0, together with pRL-TK (Promega, Madison, WI, USA). KRAS promotor luciferase reporters respectively harbor 500 bp and 2000 bp of the human KRAS promotor region, kindly provided by Prof. Kim Nam-Soon. pGL3 Basic empty was used as negative control, and pRL-TK simultaneously for transfection efficiency normalization and as a G4 negative control. This construct does not harbor G4 sequences, therefore being insensitive to G-quadruplex-related effects/regulation. Transfections were performed using Lipofectamine 3000 (Invitrogen), according to the manufacturer's instructions. Twenty-four h after transfection, cells were replated in 96-well plates, at 5,000 cells per well. Subsequently, 24 h after replating, test compounds IQ3A, TMPyP4, and vehicle (DMSO) were added to the cells at IC50 equitoxic concentration. Finally, 72 h after compound incubation, cells were lysed and firefly and renilla luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega). KRAS promoter activity levels were expressed as the luciferase signal ratio of pGL-Ras2.0 or pGL-Ras0.5 to pGL3-basic vector transfected cells, after normalization with Renilla Luciferase. In parallel, HCT116, SW620 and HEK293T cells were transfected with PGL-Ras0.5 together with pRL-TK (Promega, Madison, WI, USA), replated in 96-well plates and simultaneously exposed to test compounds IQ3A, TMPyP4, and vehicle (DMSO) at IC50 equitoxic concentration. Twenty-four h later, cells were lysed and renilla luciferase activities were measured and expressed as above. The results are expressed as the mean ± SEM fold-change compared to DMSO exposure, from three independent experiments.
Supporting Information
(PDF)
(PDF)
SW620 cells were transfected with 50 or 100 nM siRNA KRAS, siRNA HSP90 or siRNA control, and co-transfected with 50 nM siRNA KRAS plus 50 nM siRNA HSP90. Twenty-four h later, cells were replated in 24 well plates at 50,000 cells/well, and 72 h later (96 h of transfection) cells were processed for: A. Evaluation of steady-state expression of KRAS and HSP90 protein by immunoblot; B. Guava ViaCount assay (upper panel) and trypan blue exclusion assay (lower panel); C. LDH release assay (upper panel) and MTS metabolism Assay (lower panel). Results are expressed as mean ± SEM of three independent experiments; §p < 0.01 from siRNA control (50 nM); and ‡p < 0.01 from siRNA control (100 nM).
(TIF)
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Authors from iMed-ULisboa acknowledge Fundação para a Ciência e Tecnologia (FCT), Portugal, for financial support through project grants EXPL/QEQ-MED/0502/2012, HMSP-ICT/0018/2011, and PEst-OE/SAU/UI4013/2014. HB, CMPR and PMB also thank Sociedade Portuguesa de Gastroenterologia and the Korea Human Gene Bank (KHGB), Medical Genomics Research Center, KRIBB, Republic of Korea. JL acknowledges FCT for the Post-Doctoral grant SFRH/BPD/72903/2010 and SAS for PhD grant SFRH/BD/80162/2011. The Neidle laboratory acknowledges support from the Pancreatic Cancer Research Fund and the Medical Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dunn EF, lida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, et al. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene. 2011;30: 561–574. 10.1038/onc.2010.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Krens LL, Baas JM, Gelderblom H, Guchelaar HJ. Therapeutic modulation of k-ras signaling in colorectal cancer. Drug Discovery Today. 2010;15: 502–516. 10.1016/j.drudis.2010.05.012 [DOI] [PubMed] [Google Scholar]
- 3. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136: E359–386. 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 4. Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14 Suppl 1: 35–48. [DOI] [PubMed] [Google Scholar]
- 5. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov. 2014;13: 828–851. 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsujii M. Search for Novel Target Molecules for the Effective Treatment or Prevention of Colorectal Cancer. Digestion. 2012;85: 99–102. 10.1159/000334678 [DOI] [PubMed] [Google Scholar]
- 7. Borralho PM, Kren BT, Castro RE, da Silva IB, Steer CJ, Rodrigues CM. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. Febs J. 2009;276: 6689–6700. 10.1111/j.1742-4658.2009.07383.x [DOI] [PubMed] [Google Scholar]
- 8. Borralho PM, Simoes AES, Gomes SE, Lima RT, Carvalho T, Ferreira DM, et al. miR-143 Overexpression Impairs Growth of Human Colon Carcinoma Xenografts in Mice with Induction of Apoptosis and Inhibition of Proliferation. Plos One. 2011;6(8):e23787 10.1371/journal.pone.0023787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Capucha V, Mendes E, Francisco AP, Perry MJ. Development of triazene prodrugs for ADEPT strategy: New insights into drug delivery system based on carboxypeptidase G2 activation. Bioorg Med Chem Lett. 2012;22: 6903–6908. 10.1016/j.bmcl.2012.09.029 [DOI] [PubMed] [Google Scholar]
- 10. Monteiro AS, Almeida J, Cabral G, Severino P, Videira PA, Sousa A, et al. Synthesis and evaluation of N-acylamino acids derivatives of triazenes. Activation by tyrosinase in human melanoma cell lines. Eur J Med Chem. 2013;70: 1–9. 10.1016/j.ejmech.2013.09.040 [DOI] [PubMed] [Google Scholar]
- 11. Perry MJ, Mendes E, Simplicio AL, Coelho A, Soares RV, Iley J,et al. Dopamine- and tyramine-based derivatives of triazenes: Activation by tyrosinase and implications for prodrug design. Eur J Med Chem. 2009;44: 3228–3234. 10.1016/j.ejmech.2009.03.025 [DOI] [PubMed] [Google Scholar]
- 12. Cogoi S, Xodo LE. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34: 2536–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cogoi S, Paramasivam M, Spolaore B, Xodo LE. Structural polymorphism within a regulatory element of the human KRAS promoter: formation of G4-DNA recognized by nuclear proteins. Nucleic Acids Res. 2008;36: 3765–3780. 10.1093/nar/gkn120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34: 5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neidle S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. Febs J. 2010;277: 1118–1125. 10.1111/j.1742-4658.2009.07463.x [DOI] [PubMed] [Google Scholar]
- 16. Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10: 261–275. 10.1038/nrd3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohnmacht SA, Neidle S. Small-molecule quadruplex-targeted drug discovery. Bioorg Med Chem Lett. 2014;24: 2602–2612. 10.1016/j.bmcl.2014.04.029 [DOI] [PubMed] [Google Scholar]
- 18. Collie GW, Parkinson GN. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem Soc Rev. 2011;40: 5867–5892. 10.1039/c1cs15067g [DOI] [PubMed] [Google Scholar]
- 19. Faudale M, Cogoi S, Xodo LE. Photoactivated cationic alkyl-substituted porphyrin binding to g4-RNA in the 5 '-UTR of KRAS oncogene represses translation. Chem Commun. 2012;48: 874–876. 10.1039/c1cc15850c [DOI] [PubMed] [Google Scholar]
- 20. Bugaut A, Balasubramanian S. 5'-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012;40: 4727–4741. 10.1093/nar/gks068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavrado J, Reszka AP, Moreira R, Neidle S, Paulo A. C-11 diamino cryptolepine derivatives NSC748392, NSC748393 and NSC748394: Anticancer profile and G-quadruplex stabilization. Bioorg Med Chem Lett. 2010;20: 7042–7045. 10.1016/j.bmcl.2010.09.110 [DOI] [PubMed] [Google Scholar]
- 22. Lavrado J, Moreira R, Paulo A. Indoloquinolines as Scaffolds for Drug Discovery. Curr Med Chem. 2010;17: 2348–2370. [DOI] [PubMed] [Google Scholar]
- 23. Caprio V, Guyen B, Opoku-Boahen Y, Mann J, Gowan SM, Kelland LM et al. A novel inhibitor of human telomerase derived from 10H-indolo[3,2-b]quinoline. Bioorg Med Chem Lett. 2000;10: 2063–2066. [DOI] [PubMed] [Google Scholar]
- 24. Ou TM, Lu YJ, Zhang C, Huang ZS, Wang XD, Tan JH, et al. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J Med Chem. 2007;50: 1465–1474. [DOI] [PubMed] [Google Scholar]
- 25. Lavrado J, Borralho PM, Ohnmacht SA, Castro RE, Rodrigues CMP,Moreira R, et al. Synthesis, G-Quadruplex Stabilisation, Docking Studies, and Effect on Cancer Cells of Indolo[3,2-b]quinolines with One, Two, or Three Basic Side Chains. Chemmedchem. 2013;8: 1648–1661. 10.1002/cmdc.201300288 [DOI] [PubMed] [Google Scholar]
- 26. Paramasivam M, Cogoi S, Xodo LE. Primer extension reactions as a tool to uncover folding motifs within complex G-rich sequences: analysis of the human KRAS NHE. Chem Commun (Camb). 2011;47: 4965–4967. 10.1039/c1cc10269a [DOI] [PubMed] [Google Scholar]
- 27. Ohnmacht SA, Micco M, Petrucci V, Todd AK, Reszka AP, Gunaratnam M, et al. Sequences in the HSP90 promoter form G-quadruplex structures with selectivity for disubstituted phenyl bis-oxazole derivatives. Bioorg Med Chem Lett. 2012;22: 5930–5935. 10.1016/j.bmcl.2012.07.065 [DOI] [PubMed] [Google Scholar]
- 28. Moore MJB, Schultes CM, Cuesta J, Cuenca F, Gunaratnam M, Tanious FA, et al. Trisubstituted acridines as G-quadruplex telomere targeting agents. Effects of extensions of the 3,6-and 9-side chains on quadruplex binding, telomerase activity, and cell proliferation. J Med Chem. 2006;49: 582–599. [DOI] [PubMed] [Google Scholar]
- 29. Collie GW, Promontorio R, Hampel SM, Micco M, Neidle S, Parkinson GN. Structural Basis for Telomeric G-Quadruplex Targeting by Naphthalene Diimide Ligands. JACS. 2012;134: 2723–2731. 10.1021/ja2102423 [DOI] [PubMed] [Google Scholar]
- 30. Lebedeva IV, Su ZZ, Emdad L, Kolomeyer A, Sarkar D, Kitada S, et al. Targeting inhibition of K-ras enhances Ad.mda-7-induced growth suppression and apoptosis in mutant K-ras colorectal cancer cells. Oncogene. 2007;26: 733–744. [DOI] [PubMed] [Google Scholar]
- 31. Cogoi S, Zorzet S, Rapozzi V, Geci I, Pedersen EB, Xodo LE. MAZ-binding G4-decoy with locked nucleic acid and twisted intercalating nucleic acid modifications suppresses KRAS in pancreatic cancer cells and delays tumor growth in mice. Nucleic Acids Res. 2013;41: 4049–4064. 10.1093/nar/gkt127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang JS, Chen Y, Ren JS, Zhao CQ, Qu XG. G-Quadruplex binding enantiomers show chiral selective interactions with human telomere. Nucleic Acids Res. 2014;42: 3792–3802. 10.1093/nar/gkt1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1: a001883 10.1101/cshperspect.a001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borralho PM, Moreira da Silva IB, Aranha MM, Albuquerque C, Nobre Leitao C, Steer CJ, et al. Inhibition of Fas expression by RNAi modulates 5-fluorouracil-induced apoptosis in HCT116 cells expressing wild-type p53. Biochim Biophys Acta. 2007;1772: 40–47. [DOI] [PubMed] [Google Scholar]
- 35. Silva TF, Martins LM, Guedes da Silva MF, Fernandes AR, Silva A, Borralho PM, et al. Cobalt complexes bearing scorpionate ligands: synthesis, characterization, cytotoxicity and DNA cleavage. Dalton Trans. 2012;41: 12888–12897. 10.1039/c2dt11577h [DOI] [PubMed] [Google Scholar]
- 36. Mansoor TA, Borralho PM, Dewanjee S, Mulhovo S, Rodrigues CM, Ferreira MJ. Monoterpene bisindole alkaloids, from the African medicinal plant Tabernaemontana elegans, induce apoptosis in HCT116 human colon carcinoma cells. J Ethnopharmacol. 2013;149: 463–470. 10.1016/j.jep.2013.06.051 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
SW620 cells were transfected with 50 or 100 nM siRNA KRAS, siRNA HSP90 or siRNA control, and co-transfected with 50 nM siRNA KRAS plus 50 nM siRNA HSP90. Twenty-four h later, cells were replated in 24 well plates at 50,000 cells/well, and 72 h later (96 h of transfection) cells were processed for: A. Evaluation of steady-state expression of KRAS and HSP90 protein by immunoblot; B. Guava ViaCount assay (upper panel) and trypan blue exclusion assay (lower panel); C. LDH release assay (upper panel) and MTS metabolism Assay (lower panel). Results are expressed as mean ± SEM of three independent experiments; §p < 0.01 from siRNA control (50 nM); and ‡p < 0.01 from siRNA control (100 nM).
(TIF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.