

Abstract
Background
Mechanical compression of cells during mesenchymal condensation triggers cells to undergo odontogenic differentiation during tooth organ formation in the embryo. However, the mechanism by which cell compaction is stabilized over time to ensure correct organ specific cell fate switching remains unknown.
Results
Here, we show that mesenchymal cell compaction induces accumulation of collagen VI in the extracellular matrix (ECM), which physically stabilizes compressed mesenchymal cell shapes and ensures efficient organ-specific cell fate switching during tooth organ development. Mechanical induction of collagen VI deposition is mediated by signaling through the actin-p38MAPK-SP1 pathway, and the ECM scaffold is stabilized by lysyl oxidase (LOX) in the condensing mesenchyme. Moreover, perturbation of synthesis or cross-linking of collagen VI alters the size of the condensation in vivo.
Conclusions
These findings suggest that the odontogenic differentiation process that is induced by cell compaction during mesenchymal condensation is stabilized and sustained through mechanically-regulated production of collagen VI within the mesenchymal ECM.
Keywords: cell compaction, mesenchymal condensation, mechanical, extracellular matrix, actin, p38MAPK, SP1, LOX, tooth development, organogenesis
INTRODUCTION
In the embryo, formation of many organs including tooth, cartilage, bone, muscle, tendon, kidney, and lung, is induced by a mesenchymal condensation process in which previously dispersed connective tissue cells compact into a tightly packed cell mass. This compaction process then triggers expression of genes encoding tissue-specific transcription factors and morphogens that drive organ formation (Hall and Miyake, 2000). Recent studies revealed that the physical compression and rounding of mesenchymal cells that results from this compaction process is the key trigger for cell fate switching that induces tooth organ formation (Mammoto et al., 2011). In the tooth, the condensation process is driven by an opposing gradient of factors that stimulate (fgf8) and repulse (semaphorin 3F) motility; however, it is unclear how this condensation can be sustained over a period of days, which is required for effective tooth formation.
Extracellular matrix (ECM), which is synthesized, deposited and remodeled by epithelial and mesenchymal cells during embryonic development, plays a crucial role in organogenesis by controlling cell fate (e.g., growth versus differentiation, apoptosis or motility) locally and thereby, directing morphogenesis (Ingber and Jamieson, 1985; Rozario and DeSimone, 2010). Mechanical traction forces generated in the actin cytoskeleton contribute significantly to this form of developmental control as they drive various changes in cell shape and polarity that govern cell fate switching as well as directed migration that are crucial for tissue and organ formation (Mammoto et al., 2013). For example, physical interactions between the contractile cytoskeleton and the ECM are required for establishment of local differentials of cell growth that drive epithelial morphogenesis and angiogenesis during embryonic lung formation (Moore et al., 2005). In addition, the actin cytoskeleton acts as a ‘mechanosensor’ in that it orients much of the cells signaling machinery and thereby provides a way for mechanical signals to integrate with intracellular signaling pathways that regulate expression of genes which govern cell fate switching (Mammoto and Ingber, 2009; Huveneers and de Rooij, 2013; Smith et al., 2014). Thus, in the present study we set out to explore how ECM and the actin cytoskeleton contribute to tooth formation triggered by mesenchymal condensation. Our studies revealed that physical compaction of the mesenchymal cells induces formation of a natural ECM scaffold of collagen VI through the actin-p38MAPK-SP1 pathway. We also found that mesenchymal condensation and cell compaction-dependent cell fate switching are stabilized through formation of this natural ECM scaffold, which restrains cells in a compressed form and sustains odontogenic differentiation over a period days.
RESULTS
Mesenchymal condensation in the forming tooth in the mouse embryo occurs when a local thickening of the dental epithelium (DE) called the epithelial placode buds into the underlying mesenchyme on embryonic day 12 (E12) (Fig. 1A). Connective tissue cells that are initially sparsely distributed beneath the placode compact together to form a dense cell mass directly beneath the invaginating DE by day E13-14 (Fig. 1A), with local mesenchymal cell densities increasing by almost 2-fold in these regions (Fig. 1B). This cell compaction process triggers local expression of odontogenic transcription factors, including Pax9 (Fig. 1C), Msx1 and BMP4 (not shown), which then drives odontogenic differentiation and tooth organ development, as previously described (Mammoto, 2011).
Figure 1. Cell compaction-dependent odontogenic induction is reversible.

(A) Hematoxylin and eosin stained (H&E) histological sections showing development of the lower molar tooth in the mouse embryo. (B) Graph showing mesenchymal cell densities measured in non-condensed area (NCA) or condensed area (CA). (C) Fluorescence immunomicrograph showing Pax9 protein in the tooth germ at E14. (D) Phase contrast micrographs showing mesenchymal cells cultured for 16 hr on microfabricated circular FN islands (500 μm diameter) in vitro at low or high plating density (0.2 or 2.5X105 cells/cm2, respectively). (E) Graph showing induction of odontogenic factors (Pax9 and Msx1) in mesenchymal cells cultured under the same conditions as in (D). (F) Graph showing induction of Pax9 in the cells plated and re-plated at various combinations of low (L) and high (H) density. Note that Pax9 induction is cell compaction-dependent and reversible. Dashed lines indicate the epithelial-mesenchymal interface, and tip of white arrows abut on the lower edge of the condensed mesenchyme. Scale bars represent 50 μm for A and C, 100 μm for D; *p < 0.01.
To explore how this induction process is stabilized, we isolated undifferentiated (E10) dental mesenchymal cells and cultured them at a high plating density (2.4 × 105 cells/cm2) on large, circular (500 μm diameter), fibronectin (FN)-coated, adhesive islands created with microcontact printing to artificially produce a level of cell compaction (Fig. 1D) similar to that observed in the regions of mesenchymal condensation in vivo. When mesenchymal cells were cultured under these dense conditions, expression of the odontogenic transcription factors Pax9 and Msx1 increased by about 2.5-fold compared to cells cultured at a low plating density (2 × 104 cells/cm2) on the same FN islands (Fig. 1E) that mimicked the density of cells within non-condensed regions of embryonic mesenchyme (Mammoto et al., 2011). Furthermore, when we harvested the cells that were induced to express Pax9 by culturing at a high density in vitro, and re-plated them at a low density, which promotes cell spreading on the FN islands, the cell compaction-induced expression of Pax9 reversed as detected by qPCR (Fig. 1F). In contrast, high levels of Pax9 expression were maintained when these cells were re-plated at a high density (Fig. 1F) that maintains cell shape in a compressed rounded form (Fig. 1D), and as expected, sparsely plated cells that exhibit low Pax9 expression abruptly increased their expression when re-plated at higher density (Fig. 1F). These findings suggest that cells within the condensed mesenchyme must be physically restrained in a compressed state to maintain organ-specific cell fate switching that drives subsequent tooth development.
Various molecules and cellular mechanisms have been suggested to stabilize the mesenchyme in a condensed form, including local deposition of ECM components, such as FN and hyaluronate (Knudson and Toole, 1985; Kulyk et al. 1989), and alterations in cell-cell or cell-ECM interactions that prevent centrifugal cell movement away from the condensed cell mass (Knudson and Toole, 1985; Frenz et al., 1989a; Frenz et al., 1989b; Kulyk et al., 1989; Oberlender and Tuan, 1994). To explore this mechanism in embryonic tooth development, we carried out transcriptional profiling of various ECM components expressed in dental mesenchyme isolated at different stages of development (E10-E13) to identify ECM components that might be responsible for stabilizing mesenchymal cell compaction. Multiple ECM components, including various collagens, emillin, fibulin, laminin, tenascin C and versican were expressed at higher levels in E13 mesenchyme relative to E10 and E11 samples (Fig. 2A).
Figure 2. Transcriptional profiling of ECM components.

(A) Transcriptional profiling of ECM components expressed in DM at E10, 11 and 13 (all gene-profiling results are normalized to GAPDH levels). Arrows indicate ECMs that are expressed >2-fold higher in the DM at E13 compared with E10. (B) Transcriptional profiling of ECM components expressed in DM cells that are plated on FN islands at low or high plating density for 16 h. Arrows indicate ECMs that are expressed >2-fold higher in the DM, plated at a high density compared with the one plated at a low density. (C) Graph showing induction of collagen VI and TNC in mesenchymal cells cultured under the same conditions as in Fig. 1D.
We hypothesized that ECM molecules that stabilize the condensation process might be specifically induced by the physical process of cell compaction. To test this hypothesis, we cultured E10 mesenchymal cells on FN islands at low or high plating densities and performed transcriptional profiling and q-PCR. Importantly, collagen VI and tenascin C were specifically upregulated in a cell compaction-dependent manner in these in vitro assays (Fig. 2B,C). Moreover, when we screened for expression of ECM components that are induced in the condensing mesenchyme in vivo at E14 using immunohistochemistry, we confirmed that collagen VI, and to a lesser degree tenascin C, specifically accumulate around cells in the condensing mesenchyme at this time (Fig. 3A). Similarly, when the mesenchymal cells were cultured for 16 h at the high plating density to induce compaction, collagen VI expression again increased to a greater degree than tenascin C (Fig. 3B). Thus, the mechanical compaction process not only induces odontogenic transcription factors that drive organ-specific cell fate switching, but it also stimulates accumulation of ECM proteins, such as collagen VI, that might serve to sustain cells in this compact form and thereby, stabilize the differentiation process.
Figure 3. Natural ECM scaffold of collagen VI in condensing mesenchyme.

(A) Fluorescence micrographs showing protein levels of collagen (Col) I, III, IV, VI, tenascin C (TNC) and laminin in the tooth germ at E14. Dashed lines indicate the epithelial-mesenchymal interface, and tip of white arrows abut on the lower edge of the condensed mesenchyme. (B) Fluorescence micrographs showing protein levels of Col VI and TNC in mesenchymal cells cultured for 16 hr on FN islands (500 μm diameter) in vitro at low or high plating density (0.2 or 2.5X105 cells/cm2, respectively). Scale bars represent 50 μm for A and 100 μm for B.
Collagen VI has been reported to assemble into microfibrils that form networks surrounding cells during tissue development (Engvall et al., 1986; Baldock et al., 2003). Deregulation of collagen VI synthesis or assembly also disrupts cellular organization, inhibits ECM fibril formation, and contributes to various congenital disorders, including some muscular dystrophies (Lampe and Bushby, 2005; Maraldi et al., 2009). Given that collagens interact with a number of ECM-modifying molecules such as small leucine-rich proteoglycans (SLRPs) (e.g. biglycan, decorin, lumican, osteoglycan) (Kalamajski and Oldberg, 2010) and the cross-linking enzymes lysyl oxidase (LOX) (Risteli et al., 2009) and transglutaminase (mTG) (Lucero and Kagan, 2006) to assemble into the ECM scaffolds, we carried out transcriptional profiling of these molecules in mesenchymal tissues isolated at different stages of tooth development (E10-E13). The microarray analysis revealed that several molecules, including biglycan, decorin, LOX and lumican were upregulated at E13 relative to E10 in these tissues (Fig. 4A). When we used immunohistochemistry to analyze expression of these molecules in the condensing mesenchyme at E13, we found that only LOX is specifically expressed in the region of cell compaction at this time (Fig. 4B). These data raised the possibility that LOX could play a crucial role in the stabilization of collagen VI-containing ECM scaffolds.
Figure 4. Expression of collagen assembling molecules in the condensing mesenchyme.
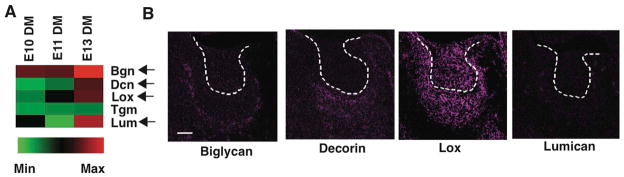
(A) Transcriptional profiling of ECM assembling molecules expressed in DM at E10, 11 and 13 (all gene profiling results are normalized to GAPDH levels). Arrows indicate ECM assembling molecules that are expressed >2-fold higher in the DM at E13 compared with E10. (B) Fluorescence micrographs showing protein levels of biglycan, decorin, LOX, and lumican in the tooth germ at E14. Dashed lines indicate the epithelial-mesenchymal interface. Scale bar, 50 μm.
To further explore this mechanism, we analyzed the mechanical signaling mechanism by which cell rounding influences collagen VI expression. Mechanical signals can produce changes in biochemistry and gene expression by triggering intracellular signaling pathways at the cell membrane, in the cytoskeleton or within the nucleus during development (Mammoto and Ingber, 2010; Mammoto et al., 2012). When we inhibited mechanical signaling at the membrane using blocking antibodies against β1 integrin, pertussis toxin inhibition of G-protein coupled receptors, gadolinium suppression of mechanosensitive ion channel activity, N-cadherin, or a nitric oxide (NO) inhibitor L-NAME, we did not detect any effects on collagen VI production induced by cell compaction in vitro (Fig. 5A). Use of modulators of cytoskeletal mechanotransduction, including the inhibitors of Rho/ROCK signaling C3 and Y27632, Rho activator CNF 1, myosin light chain kinase inhibitor ML7, and Rac/Cdc42 activator CN02 were similarly ineffective (Fig. 5B). However, inhibition of Rac using NSC23766 partially suppressed this response, and disruption of the actin cytoskeleton using cytochalasin D (Cooper, 1987) inhibited it completely (Fig. 5B). While many modulators of intracellular mechanotransduction including inhibitors of PKA (H89), a PKC activator (PMA), PI3 kinase (LY294002), MEK 1/2 (U0126), JNK (SP600125), Src (PP1), and TGF-b1 type I receptor (SB431542) were ineffective, the PKC inhibitor C1 produced partial inhibition and the p38MAPK inhibitor SB203580 suppressed it almost completely (Fig. 5C). Furthermore, because AP1 and SP1 transcription factors play important roles in the transcriptional control of collagen synthesis (Fabbro et al., 1999; Verrecchia et al., 2001; Goto et al., 2006), we also tested specific inhibitors of these factors (SR11302 and mithramycin, respectively), and found that only SP1 inhibition by mithramycin prevents cell compaction-dependent increases in collagen VI production (Fig. 5D).
Figure 5. Mechanotransduction for cell compaction-dependent collagen VI synthesis.

Cells were cultured for 16 h on circular FN islands (500 μm diameter) at low or high plating density (0.2 or 2.4 × 105 cells/cm2, respectively) with or without various inhibitors and activators of mechanical signaling and Col VI mRNA levels were measured. Modulators include (A) anti-Integrin alpha V neutralizing Ab (INTG Ab: 100 μg/ml), pertussis toxin (P-toxin; 25 ng/ml), gadolinium (500 μM), Anti-N-cadherin neutralizing Ab (N-cad Ab: 200 μg/ml) and L-NAME (2 μM); (B) cytochalasin D (CytD: 2.5 μM), C3 (2 μg/ml), Y27632 (10 μM), cytotoxic necrotizing factor (CNF1: 100 ng/ml), ML-7 (10 μM), NSC23766 (50 μM) and CN02 (0.2 U/ml); (C): H89 (10 μM), C1 (10 μM), PMA (100 nM), LY294002 (10 μM), U0126 (10 μM), SB203580 (500 nM), SP600125 (100 nM), PP1 (10 μM) and SB431542 (1 μM); (D): SR11302 (1 μM) and mithramycin (250 nM). *p < 0.01.
To confirm the role of p38MAPK in this signaling pathway, we measured its activity in compacted cells cultured at a high density and found its level to be significantly increased compared to spread cells plated at a low density, and this activity could be suppressed using both the specific p38MAPK inhibitor SB203580 and by disrupting the actin cytoskeleton with cytochalasin D (Fig. 6A). Immunocytochemical staining for F-actin with FITC-phalloidin revealed that mesenchymal cells exhibit well-developed linear stress fibers across their basal surface when spread, whereas actin staining was predominantly cortical in highly compact rounded cells in vitro (Fig. 6B), which was similar to differences in F-actin distribution observed in non-condensed or condensed mesenchyme in vivo, respectively (Fig. 6C). Treatment of cultured mesenchymal cells with cytochalasin D resulted in break up of long stress fibers in spread cells and a more diffuse pattern of F-actin distribution staining in the round cells (Fig. 6B vs. D), suggesting that the densely packed cortical organization of F-actin is required for p38MAPK activation and associated collagen VI production in round cells.
Figure 6. Synthesis of collagen VI is mediated by the mechanosensitive actin-p38MAPK-SP1 signaling pathway.

(A) Immunoblots showing active p38MAPK in mesenchymal cells cultured at low or high plating density with or without CytD (2.5 μM) or SB20358 (500 nM). (B) Fluorescence micrographs showing actin filaments in the mesenchymal cells cultured at low and high plating density in vitro. (C) Fluorescence micrographs showing actin filaments in the mesenchymal cells of non-condensed area (NCA) and condensed area (CA) in E14 tooth germ, respectively. Dashed lines indicate the epithelial-mesenchymal interface. (D) Fluorescence micrographs showing actin filaments in the mesenchymal cells cultured at low and high plating density with or without CytD (2.5 μM) in vitro. (E) Immunoblots showing collagen (Col) VI and GAPDH levels in the mesenchymal cells (Ct or MMK3 transfected), treated with CytD (2.5 μM) or mithramycin (250 nM). Scale bars represent 20 μm for C and 10 μm for B and D.
To explore whether signaling through actin and p38MAPK are linked physiologically, we transiently transfected the mesenchymal cells with the constitutively active MMK3 form of p38MAPK (Brancho et al., 2003) and found that artificially increasing p38MAPK activity increased expression of collagen VI in spread cells plated at low density (Fig. 6E). Moreover, this induction of collagen VI production by the constitutively active form of p38MAPK could be inhibited by treating cells with the SP1 inhibitor mithramycin or with cytochalasin D (Fig. 6E). These findings suggest that cell compaction induces formation of cortical actin structures that stimulate collagen VI production by supporting p38MAPK activation and related downstream stimulation of SP1. However, the finding that cytochalasin D was still able to inhibit collagen VI expression in these cells suggests that the cortical actin cytoskeleton also regulates collagen VI expression through another signaling mechanism as well.
Importantly, when we injected either the SP1 inhibitor mithramycin or the LOX inhibitor β-aminopropionitrile (BAPN) into timed-pregnant CD1 mice at E11-E12, immunohistocytochemical analysis revealed inhibition of mesenchymal condensation, decreased collagen VI levels and reduced Pax9 expression in the condensing mesenchyme of developing molar tooth germs at E14 (Fig. 7A, B). SP1 inhibition by mithramycin appeared to produce greater inhibition of collagen VI accumulation, whereas the collagen VI that remained in BAPN-treated tissues appeared to be less dense, as might be expected from inhibition or reversal of mesenchymal cell compaction. Since the LOX promoter region contains a GC-rich region that binds SP1 (Vindevoghel et al., 1997), mithramycin may influence its expression as well as collagen VI expression, thereby producing a greater impact on mesenchymal condensation. The reduction in collagen VI production induced by BAPN treatment might be due to the finding that less cross-linked collagens are more susceptible to proteolytic degradation (van der Slot-Verhoeven et al., 2005). Taken together, these findings demonstrate that collagen VI accumulation and cross-linking are required to sustain the well-formed condensed mesenchyme that is required for organ-specific fate switching in the developing tooth germ in vivo.
Figure 7. Inhibition of collagen VI synthesis or cross-linking deregulates Pax9 expression and size of mesenchymal condensation.

(A) Fluorescence immunomicrographs showing collagen (Col) VI (upper) and Pax9 (middle) protein expression and distribution in the tooth germs in E14 embryos treated with vehicle (Ct), mithramycin (Mith) (25 μg/kg) or BAPN (1mg/kg) in utero at E12 and E13. Light micrographs of H&E tissue section (bottom) showing tooth germs in E14 embryos treated with mithramycin or BAPN in utero at E12 and E13. Dashed lines indicate the epithelial-mesenchymal interface, and tip of black arrow abuts on the lower edge of the condensed mesenchyme. Scale bar, 50 μm. (B) Graphs showing areas of Col VI and Pax9 positive and condensing areas in Ct, Mith and BAPN treated embryos. *p < 0.05.
DISCUSSION
Embryonic tissue development and organ formation are controlled through complex interplay between chemical and mechanical cues (Mammoto, 2013). In a past study, we demonstrated that epithelium-derived antagonistic motility factors drive mesenchymal condensation, producing physical compaction of cells that induces expression of organ-specific transcription factors through activation of Rho GTPase during tooth development (Mammoto et al., 2011). In the present study, we extended this work and discovered that this same physical compaction process also increases the synthesis of collagen VI through actin-p38MAPK-SP1 signaling in dental mesenchymal cells. In addition, we demonstrated that accumulation and cross-linking of collagen VI by LOX are also required to stabilize cell compaction and organ-specific cell fates in the condensed mesenchyme that are required for tooth formation.
Mechanical forces regulate many aspects of cell behavior, including growth, differentiation, apoptosis and motility through a process of mechanochemical transduction that involves force transmission from ECM across integrins and to the cytoskeleton (Ingber, 1997). The actin cytoskeleton plays a central mechanotransduction role in the condensing mesenchyme of the tooth as chemical disruption of actin cytoskeleton inhibits cell compaction-dependent activation of p38MAPK and collagen VI production. The P38MAPK signaling pathway has been previously shown to allow cells to interpret a wide range of external mechanical signals (e.g., osmotic pressure, strain, deformation) and to respond appropriately by modulating various cell behaviors (Coulthard et al., 2009; Cuadrado and Nebreda, 2010; Whitmarsh, 2010). SP1 is a zinc finger DNA-binding protein that binds a putative GC-rich element (Lemon and Tjian, 2000) and plays a central role in control of transcription of collagen (Goto et al., 2006; Magee et al., 2005) and collagen cross-linking enzymes (Hamalainen et al., 1993). Our data indicate that physical compaction of cells sensed by changes in the actin cytoskeleton activates p38MAPK and stimulates collagen VI transcription by activating SP1. However, the finding that disruption of the actin cytoskeleton still inhibits collagen VI expression in cells expressing constitutively active p38MAPK suggests that there is another unidentified mechanism by which cortical actin contributes to this response. We also found that expression of collagen cross linker, LOX is upregulated in a cell compaction-dependent way to further stabilize the condensation and odontogenic cell fate, as measured by Pax9 induction. Given that LOX promoter contains potential binding sequences for SP1 (Hamalainen et al., 1993), cell compaction-dependent LOX expression also may be regulated by the actin-p38MAPK-SP1 signaling pathway.
Collagen VI is ubiquitously expressed in the ECMs of various organs, including tendons, ligaments, muscles, skin, cornea and cartilage (Keene et al., 1988), and it can be produced by both epithelium and stromal cells (Bruns et al., 1986; Dassah et al., 2014). In this study, we identified collagen VI as a major component of the mechanosensitive ECM scaffold of the condensing mesenchyme in the developing tooth, although it also was expressed by the dental epithelium when mesenchymal condensation reached its maximum. Importantly, collagen VI is synthesized and assembled into microfibrils that form networks that link to neighboring cells (Engvall et al., 1986; Baldock et al., 2003) and basement membrane during embryogenesis, and deregulation of its synthesis leads to disruption of cellular organization and altered ECM mechanics associated with various congenital disorders, including congenital muscular dystrophies (Lampe and Bushby, 2005; Maraldi et al., 2009). We similarly found that inhibition of collagen VI synthesis or cross-linking alters the size of the condensed mesenchyme, further supporting the concept that endogenous ECM scaffolds play crucial roles in the morphogenesis of organ formation. Interestingly, however, significant tooth defects have not been reported in collagen VI null mice (Izu et al., 2011). Thus, there may be some structural redundancy in the ECM components that comprise the scaffold (e.g., TNC is also expressed in the condensing mesenchyme; Fig. 3A). Although the function of collagen VI in the epithelium is not known, it is possible that it could similarly provide a structural supporting role in the tooth germ before mineralization (accumulation of enamel and dentin) occurs.
In conclusion, these studies show that during embryonic tooth formation in the mouse, the physical compaction of cells within the condensing mesenchyme stimulates accumulation and cross-linking of an ECM scaffold rich in collagen VI, which physically stabilizes the cells in a compacted form and sustains organ-specific cell fate switching that is required for subsequent organ development. In the condensing mesenchyme, mechanical cues stimulate collagen VI synthesis through activation of actin-p38MAPK-SP1 signaling in a cell compaction-dependent manner, and perturbation of this pathway alters the size of the condensed mesenchyme. Given that the molecules involved in this mechanism are widely expressed, it is possible that similar interplay between physical and chemical cues might contribute to the control of other organogenetic or regenerative processes.
EXPERIMENTAL PROCEDURES
Materials
Gadolinium, anti N-cadherin neutralizing antibody, L-name, ML7, PMA and BAPN were from Sigma; anti integrin blocking antibody, Anti-collagen I, III, IV and VI, anti-TNC, anti-laminin, anti-biglycan, anti-decorin, anti-lumican, anti-LOX and anti-Pax9 antibodies were from Abcam; C3 and CN02 were from Cytoskeleton Inc; Y27632 and LY294002 were from Calbiochem; pertussis toxin, NSC23766, cytochalasin D, H89, C1, U0126, SB203580, SP600125, PP1, SB431542, SR11302 and mithramycin were from Tocris; and cytotoxic necrotizing factor 1 (CNF 1) was prepared as described previously (Mammoto et al., 2004).
Animal Experiments
All animal studies were reviewed and approved by the Animal Care and Use Committee of Boston Children’s Hospital. For transplacental reagents delivery (treatment in utero), mithramycin or BAPN was injected intravenously through the retro-orbital vein into timed-pregnant dams (E12 and E13) under isoflurane anesthesia (Mammoto et al., 2011). Histological assays were performed as described previously (Mammoto et al., 2011), and morphometric analysis was performed using ImageJ software (National Institutes of Health).
Cell Culture
Using sterile technique, the first pharyngeal arch was dissected from E10 embryos and treated with Dispase II (2.4 U/ml; Roche) and DNase I (QIAGEN) at 37°C for 23 min. After the epithelium and mesenchyme were separated using fine forceps, the presumptive DM was dissected out and physically triturated several times using fire-polished Pasteur pipette before being cultured on FN (Becton Dickinson)-coated glass bottom dishes (MatTek Corporation) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS (Mammoto et al., 2011). We confirmed the purity of the isolated DE for cell culture using GFP-labeled DE cells isolated from keratin (K)-14/GFP transgenic mice from The Jackson Laboratory. The DM cells were passaged by using FN-coated microcarrier beads for the first several passages (Thermo Scientific). DM functional capacity, as measured by its ability to undergo cell compaction-dependent odontogenic (Pax9/Msx1) induction, is preserved in cultured DM cells for at least 7 cell passages, and all studies utilized cells at passage 4. The constitutively active MMK3 plasmid was from Open Biosystems and transient transfection was performed using FuGENE® transfection reagent (Promega).
Cellular and Molecular Biological Methods
qRT-PCR was performed with the QuantiTect SYBR Green RT-PCR kit (QIAGEN) using ABI7300 real-time PCR system (Applied Biosystems); the primers used are shown in Table 1. p38MAP kinase was measured by nonradioactive p38 MAP kinase assay kit (Cell Signaling Technology) according to the instruction manual.
Table 1.
The sequences for primers for q-PCR.
| Forward | Reverse | |
|---|---|---|
| Mouse Collagen VI | CTGCTGCTACAAGCCTGCT | CCCCATAAGGTTTCAGCCTCA |
| Mouse Msx1 | TGCTGCTATGACTTCTTTGCC | GCTTCCTGTGATCGGCCAT |
| Mouse Pax9 | CATTCGGCTTCGCATCGTG | CTCCCGGCAAAATCGAACC |
| Mouse Tenascin C | TGCTGCTATGACTTCTTTGCC | GCTTCCTGTGATCGGCCAT |
Fabrication of Micropatterned Substrates
Our microcontact printing method for producing glass substrates containing micrometer-sized ECM islands has been described previously (Mammoto et al., 2011). In brief, PDMS stamps were cast, cured, and removed from master templates, which were created using photolithographic methods. Stamps were coated with FN (50 mg/ml) for 1 hr, dried with compressed nitrogen. Flat and thin PDMS substrates were prepared on the cover glass and UV oxidized for 5 min (Ellsworth Adhesives), stamped with FN, blocked with Pluronic F108 (Sigma) for 2 hr, and rinsed three times with PBS before plating the cells. Cells on the micropatterned substrates were lysed and harvested using a cell scraper, and odontogenic induction was analyzed by qRT-PCR.
Statistical Analysis
Statistical significance was evaluated using an unpaired Student’s t test after ANOVA analysis (all data are presented as mean ± SEM).
Acknowledgments
This work was supported by grants from NIH (RL1DE019023 to D.E.I, 1R56DE022803-01A1 to D.E.I.) and DoD (W81XWH-10-1-0565 to D.E.I), Boston Children’s Hospital Faculty Career Development Fellowship (to T.M. and A.M.), AHA (to A.M.), American Brain Tumor Association (to A.M.) and the Wyss Institute for Biologically Inspired Engineering at Harvard University. We thank Heiko Peters for helpful discussion on expression pattern of Pax9 in the developing tooth.
References
- Baldock C, Sherratt MJ, Shuttleworth CA, Kielty CM. The supramolecular organization of collagen VI microfibrils. J Mol Biol. 2003;330:297–307. doi: 10.1016/s0022-2836(03)00585-0. [DOI] [PubMed] [Google Scholar]
- Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RR, Press W, Engvall E, Timpl R, Gross J. Type VI collagen in extracellular, 100-nm periodic filaments and fibrils: identification by immunoelectron microscopy. J Cell Biol. 1986;103:393–404. doi: 10.1083/jcb.103.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15:369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Dassah M, Almeida D, Hahn R, Bonaldo P, Worgall S, Hajjar KA. Annexin A2 mediates secretion of collagen VI, pulmonary elasticity and apoptosis of bronchial epithelial cells. J Cell Sci. 2014;127:828–844. doi: 10.1242/jcs.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvall E, Hessle H, Klier G. Molecular assembly, secretion, and matrix deposition of type VI collagen. J Cell Biol. 1986;102:703–710. doi: 10.1083/jcb.102.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro C, Braghetta P, Girotto D, Piccolo S, Volpin D, Bressan GM. Cell type-specific transcription of the alpha1(VI) collagen gene. Role of the AP1 binding site and of the core promoter. J Biol Chem. 1999;274:1759–1766. doi: 10.1074/jbc.274.3.1759. [DOI] [PubMed] [Google Scholar]
- Frenz DA, Akiyama SK, Paulsen DF, Newman SA. Latex beads as probes of cell surface-extracellular matrix interactions during chondrogenesis: evidence for a role for amino-terminal heparin-binding domain of fibronectin. Dev Biol. 1989a;136:87–96. doi: 10.1016/0012-1606(89)90132-2. [DOI] [PubMed] [Google Scholar]
- Frenz DA, Jaikaria NS, Newman SA. The mechanism of precartilage mesenchymal condensation: a major role for interaction of the cell surface with the amino-terminal heparin-binding domain of fibronectin. Dev Biol. 1989b;136:97–103. doi: 10.1016/0012-1606(89)90133-4. [DOI] [PubMed] [Google Scholar]
- Goto T, Matsui Y, Fernandes RJ, Hanson DA, Kubo T, Yukata K, Michigami T, Komori T, Fujita T, Yang L, Eyre DR, Yasui N. Sp1 family of transcription factors regulates the human alpha2 (XI) collagen gene (COL11A2) in Saos-2 osteoblastic cells. J Bone Miner Res. 2006;21:661–673. doi: 10.1359/jbmr.020605. [DOI] [PubMed] [Google Scholar]
- Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–147. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hamalainen ER, Kemppainen R, Pihlajaniemi T, Kivirikko KI. Structure of the human lysyl oxidase gene. Genomics. 1993;17:544–548. doi: 10.1006/geno.1993.1369. [DOI] [PubMed] [Google Scholar]
- Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci. 2013;126:403–413. doi: 10.1242/jcs.109447. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Integrins, tensegrity, and mechanotransduction. Gravit Space Biol Bull. 1997;10:49–55. [PubMed] [Google Scholar]
- Ingber DE, Jamieson JD. In: Cells as tensegrity structures: architectural regulation of histodifferentiation by physical forces tranduced over basement membrane. Andersson LC, Gahmberg CG, Ekblom P, editors. 1985. [Google Scholar]
- Izu Y, Ansorge HL, Zhang G, Soslowsky LJ, Bonaldo P, Chu ML, Birk DE. Dysfunctional tendon collagen fibrillogenesis in collagen VI null mice. Matrix Biol. 2011;30:53–61. doi: 10.1016/j.matbio.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamajski S, Oldberg A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010;29:248–253. doi: 10.1016/j.matbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Keene DR, Engvall E, Glanville RW. Ultrastructure of type VI collagen in human skin and cartilage suggests an anchoring function for this filamentous network. J Cell Biol. 1988;107:1995–2006. doi: 10.1083/jcb.107.5.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CB, Toole BP. Changes in the pericellular matrix during differentiation of limb bud mesoderm. Dev Biol. 1985;112:308–318. doi: 10.1016/0012-1606(85)90401-4. [DOI] [PubMed] [Google Scholar]
- Kulyk WM, Upholt WB, Kosher RA. Fibronectin gene expression during limb cartilage differentiation. Development. 1989;106:449–455. doi: 10.1242/dev.106.3.449. [DOI] [PubMed] [Google Scholar]
- Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14:2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee C, Nurminskaya M, Faverman L, Galera P, Linsenmayer TF. SP3/SP1 transcription activity regulates specific expression of collagen type X in hypertrophic chondrocytes. J Biol Chem. 2005;280:25331–25338. doi: 10.1074/jbc.M412549200. [DOI] [PubMed] [Google Scholar]
- Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279:26323–26330. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- Mammoto A, Ingber DE. Cytoskeletal control of growth and cell fate switching. Curr Opin Cell Biol. 2009;21:864–870. doi: 10.1016/j.ceb.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Mammoto A, Mammoto T, Ingber DE. Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci. 2012 doi: 10.1242/jcs.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development. 2010;137:1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto T, Mammoto A, Ingber DE. Mechanobiology and Developmental Control. Annu Rev Cell Dev Biol. 2013;29:27–61. doi: 10.1146/annurev-cellbio-101512-122340. [DOI] [PubMed] [Google Scholar]
- Mammoto T, Mammoto A, Torisawa YS, Tat T, Gibbs A, Derda R, Mannix R, de Bruijn M, Yung CW, Huh D, Ingber DE. Mechanochemical control of mesenchymal condensation and embryonic tooth organ formation. Dev Cell. 2011;21:758–769. doi: 10.1016/j.devcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraldi NM, Sabatelli P, Columbaro M, Zamparelli A, Manzoli FA, Bernardi P, Bonaldo P, Merlini L. Collagen VI myopathies: from the animal model to the clinical trial. Adv Enzyme Regul. 2009;49:197–211. doi: 10.1016/j.advenzreg.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Moore KA, Polte T, Huang S, Shi B, Alsberg E, Sunday ME, Ingber DE. Control of basement membrane remodeling and epithelial branching morphogenesis in embryonic lung by Rho and cytoskeletal tension. Dev Dyn. 2005;232:268–281. doi: 10.1002/dvdy.20237. [DOI] [PubMed] [Google Scholar]
- Oberlender SA, Tuan RS. Expression and functional involvement of N-cadherin in embryonic limb chondrogenesis. Development. 1994;120:177–187. doi: 10.1242/dev.120.1.177. [DOI] [PubMed] [Google Scholar]
- Risteli M, Ruotsalainen H, Salo AM, Sormunen R, Sipila L, Baker NL, Lamande SR, Vimpari-Kauppinen L, Myllyla R. Reduction of lysyl hydroxylase 3 causes deleterious changes in the deposition and organization of extracellular matrix. J Biol Chem. 2009;284:28204–28211. doi: 10.1074/jbc.M109.038190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Hoffman LM, Beckerle MC. LIM proteins in actin cytoskeleton mechanoresponse. Trends Cell Biol. 2014;24:575–583. doi: 10.1016/j.tcb.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Slot-Verhoeven AJ, van Dura EA, Attema J, Blauw B, Degroot J, Huizinga TW, Zuurmond AM, Bank RA. The type of collagen cross-link determines the reversibility of experimental skin fibrosis. Biochim Biophys Acta. 2005;1740:60–67. doi: 10.1016/j.bbadis.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Rossert J, Mauviel A. Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J Invest Dermatol. 2001;116:755–763. doi: 10.1046/j.1523-1747.2001.01326.x. [DOI] [PubMed] [Google Scholar]
- Vindevoghel L, Chung KY, Davis A, Kouba D, Kivirikko S, Alder H, Uitto J, Mauviel A. A GT-rich sequence binding the transcription factor Sp1 is crucial for high expression of the human type VII collagen gene (COL7A1) in fibroblasts and keratinocytes. J Biol Chem. 1997;272:10196–10204. doi: 10.1074/jbc.272.15.10196. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010;8:47. doi: 10.1186/1741-7007-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]