

SUMMARY
Differentiating pluripotent epiblast cells in eutherians undergo random X-inactivation, which equalizes X-linked gene expression between the sexes by silencing one of the two X-chromosomes in females. Tsix RNA is believed to orchestrate the initiation of X-inactivation, influencing the choice of which X remains active by preventing expression of the antisense Xist RNA, which is required to silence the inactive-X. Here we profile X-chromosome activity in Tsix-mutant (XΔTsix) mouse embryonic epiblasts, epiblast stem cells, and embryonic stem cells. Unexpectedly, we find that Xist is stably repressed on the XΔTsix in both sexes in undifferentiated epiblast cells in vivo and in vitro, resulting in stochastic X-inactivation in females despite Tsix-heterozygosity. Tsix is instead required to silence Xist on the active-X as epiblast cells differentiate in both males and females. Thus, Tsix is not required at the onset of random X-inactivation; instead, it protects the active-X from ectopic silencing once X-inactivation has commenced.
Keywords: EpiSCs, ESCs, embryonic epiblasts, Tsix, Xist, X-inactivation
Graphical Abstract
INTRODUCTION
X-chromosome inactivation results in the mitotically-stable transcriptional silencing of genes along one of the two X-chromosomes in female mammals (Lyon, 1961). In the pluripotential mouse epiblast cells, which will form the embryo proper, the selection of which X to inactivate is random. Molecularly, random X-inactivation is posited to be controlled in cis by a pair of oppositely-transcribed X-linked long non-coding (lnc) RNAs, Xist and Tsix (Barakat and Gribnau, 2012). Xist RNA is believed to initiate epigenetic silencing of genes in cis by physically coating the X-chromosome from which it is transcribed and recruiting proteins that catalyze heterochromatin formation (Payer and Lee, 2008). Tsix transcription across the Xist promoter, conversely, is proposed to inhibit Xist expression (Lee, 2000; Lee and Lu, 1999; Luikenhuis et al., 2001; Navarro et al., 2005; Sado et al., 2005; Sado et al., 2001). Because of its ability to repress Xist, the Tsix locus is postulated to be the site where molecular signals converge to help ensure that one X-chromosome remains active in both males and females (Clerc and Avner, 1998; Cohen et al., 2007; Debrand et al., 1999; Gontan et al., 2012; Lee, 2005; Luikenhuis et al., 2001; Morey et al., 2004; Navarro et al., 2010; Stavropoulos et al., 2005; Vigneau et al., 2006).
Investigations of mutations that reduce or abrogate Tsix RNA expression, however, have resulted in disparate outcomes. In differentiating male embryonic stem cells (ESCs), a cell culture model of X-inactivation, some Tsix mutations display ectopic Xist induction, consistent with Tsix serving to inhibit Xist and thereby X-inactivation (Clerc and Avner, 1998; Debrand et al., 1999; Luikenhuis et al., 2001; Morey et al., 2004; Sado et al., 2002; Vigneau et al., 2006). Other Tsix-mutant male ESCs, though, do not exhibit Xist expression upon differentiation (Cohen et al., 2007; Lee, 2000; Lee and Lu, 1999; Minkovsky et al., 2013). The differences observed between the mutant ESC lines may reflect residual Tsix expression due to the incomplete ablation of Tsix or differences in the protocols employed to differentiate ESCs.
Whereas ectopic X-inactivation may or may not occur in Tsix-mutant males, the choice of which X to inactivate appears absolutely biased in Tsix-heterozygous females (Cohen et al., 2007; Kalantry and Magnuson, 2006; Lee, 2000; Sado et al., 2001). In these animals, the Tsix-mutant X-chromosome is inactive in all cells of the differentiating epiblast lineage, which would otherwise undergo random X-inactivation. This bias in choice has been explained by the preferential induction of Xist from the Tsix-mutant X-chromosome prior to or at the onset of X-inactivation in the epiblast lineage.
Despite the proposed models of Tsix function, the significance of Tsix RNA remains unclear in both males and females. In the course of a previous study, we noticed that the epiblast in XΔTsixY post-implantation embryos appeared to ectopically express Xist in the absence of Tsix (Maclary et al., 2014). We therefore hypothesized that Tsix-heterozygous females might also aberrantly express Xist during development. Thus, an alternative explanation for the apparent lack of ectopic Xist expression and skewed X-inactivation in Tsix-heterozygotes is that a secondary cell-selection effect rapidly removes cells with two inactive-Xs from the population. Due to the tight coupling of X-inactivation with epiblast differentiation (Monk and Harper, 1979), ectopic silencing of the previously active XΔTsix may occur concurrently with or shortly after the initiation of random X-inactivation. Inactivation of both Xs in females would render the cells effectively nullizygous for many X-linked genes, thus compromising proliferation and viability. Later-stage epiblast and ESC derivatives would therefore consist only of cells with an active WT X-chromosome. Here, we investigate Tsix function by profiling embryos harboring a Tsix-null allele at the onset of random X-inactivation; and, by deriving Tsix hemizygous male and heterozygous female EpiSC and ESC lines.
RESULTS
Tsix Absence Results in Ectopic Xist RNA Expression and Coating in Male Embryonic Epiblasts
Random X-inactivation initiates in epiblast cells between embryonic day (E) 4.5–6.5 in mice, just as the pluripotential epiblast cells begin to differentiate (Gardner and Lyon, 1971; Kalantry and Magnuson, 2006; McMahon et al., 1983; Rastan, 1982). To examine the role of Tsix RNA at the onset of X-inactivation, we generated embryonic day (E) 5.25 post-implantation stage embryos that inherit either a WT or a Tsix-null maternal X-chromosome from Tsix-heterozygous females. The previously described Tsix mutation, TsixAA2Δ1.7 (herein referred to as XΔTsix) (Sado et al., 2001), terminates the Tsix transcript in exon 2 and also deletes the critical DXPas34 repeat thought to serve as a platform to drive Tsix expression (Fig. 1A) (Cohen et al., 2007; Maclary et al., 2014; Navarro et al., 2010; Stavropoulos et al., 2005; Vigneau et al., 2006). Since transcription across the Xist promoter region is required for the Tsix RNA to inhibit Xist expression (Navarro et al., 2005; Sado et al., 2005), XΔTsix is a bona fide null Tsix mutation (Fig. 1B) (Maclary et al., 2014; Sado et al., 2001). We first tested if the absence of Tsix RNA led to Xist induction in male epiblasts by RT-PCR. Whereas WT E5.25 XY epiblasts exhibited Tsix but not Xist expression, XΔTsixY epiblasts displayed the opposite pattern (Fig. 1B). We next independently assessed Xist induction and X-inactivation in E5.25 XY and XΔTsixY epiblast cells by immunofluorescence (IF) coupled with RNA fluorescence in situ hybridization (RNA FISH). We first marked epiblast cells via IF detection of NANOG, which distinguishes the epiblast from the extra-embryonic cells (Fig. 1C). In the same samples, using strand-specific RNA FISH probes we also assayed expression of Tsix and Xist RNAs. In WT XY epiblasts, Tsix RNA signal but not Xist RNA coating was detectable from the sole X-chromosome (Fig. 1C). In contrast, in XΔTsixY mutant embryos ~34% of the nuclei displayed Xist RNA coating (Fig. 1C). Moreover, Xist coating resulted in the accumulation of histone H3 lysine 27 trimethylation (H3-K27me3), a chromatin mark catalyzed by the Polycomb repressive complex 2 that is associated with the inactive-X heterochromatin (Fig. 1D) (Plath et al., 2003; Silva et al., 2003), and accompanied silencing of the X-linked Pgk1 gene (Fig. 1E). Thus, Tsix absence leads to Xist RNA induction, coating, and gene silencing on the single X-chromosome in male epiblast cells.
Figure 1. Xist is induced from the XΔTsix in E5.25 male epiblast cells.

(A) Diagram illustrating WT Xist and Tsix loci and the ΔTsix mutation. Dotted lines indicate the locations of strand-specific (ss) RNA FISH probes. Filled arrowheads mark the locations of RT-PCR primer pairs. 1 (orange arrowheads), Xist RT-PCR amplicon; 2 (blue arrowheads), Tsix exon 4 RT-PCR amplicon; 3 (purple arrowheads), Tsix RT-PCR amplicon spanning exons 2–4. (B) RT-PCR amplification of Tsix (exon 4) and Xist RNAs in E5.25 epiblasts. M, marker; NTC, no template control; +, reaction with reverse transcriptase (RT); −, no RT control lane. (C) Strand-specific RNA FISH detection of Xist RNA (green) and Tsix RNA (red) coupled with IF staining for NANOG (purple) in isolated epiblasts and extra-embryonic ectoderm, which serves as a negative control for NANOG expression. Nuclei are stained blue with DAPI. Scale bar, 10 μm. Right, quantification of Xist and Tsix expression in NANOG-positive epiblast nuclei. The X-axis of each graph represents the average percentage of nuclei per embryo in each class (n = 3 embryos/genotype; 49–68 nuclei/embryo). Diagrams along the Y-axis depict all observed expression patterns. Error bars represent the standard deviation of data from 3 different embryos. *, p ≤ 0.001 (Chi-square test). (D) Xist RNA coating (green) and H3-K27me3 enrichment (purple) in E5.25 XΔTsixY epiblast nuclei. (E) Silencing of the X-linked gene Pgk1 (red) in E5.25 XΔTsixY epiblast nuclei upon ectopic Xist RNA coating (green).
Ectopic Xist Induction in Differentiating But Not Undifferentiated XΔTsixY Epiblast Stem Cells
To further explore the requirement of Tsix in the epiblast, we derived WT XY and mutant XΔTsixY epiblast stem cells (EpiSCs; Fig. S1A–C; Table S1). EpiSCs are thought to represent an early phase of X-inactivation (Bernemann et al., 2011; Brons et al., 2007; Han et al., 2011; Pasque et al., 2011a; Pasque et al., 2011b; Tesar et al., 2007). If Tsix negatively regulates Xist in undifferentiated epiblast cells, EpiSCs lacking Tsix are expected to display aberrant Xist activation. In assaying Xist expression by RT-PCR, we found that Xist RNA was undetectable in the WT XY EpiSC lines (Fig. 2A). In XΔTsixY EpiSC lines, however, Xist RNA was expressed at minimally detectable levels (Fig. 2A). This low level of Xist expression may reflect the induction of Xist in the small fraction of differentiated cells that are often found in stem cell cultures. This notion prompted us to test if Xist would be induced to high levels if we actively differentiated XΔTsixY EpiSCs (Fig. S1D). Indeed, Xist expression in XΔTsixY but not XY cells increased markedly upon differentiation (Fig. 2A).
Figure 2. Ectopic Xist RNA induction in differentiated but not undifferentiated XΔTsixY EpiSCs and ESCs.

(A) RT-PCR amplification of Xist and Tsix RNAs in undifferentiated and differentiated XY and XΔTsixY EpiSC lines (2 and 4 cell lines, respectively). β-actin amplification serves as control. M, marker; +, reaction with reverse transcriptase (RT); −, no RT control lane. (B) RNA FISH detection of Xist RNA (green) and Tsix RNA (red) in representative undifferentiated and day (d) 10 differentiated EpiSC lines (XY line number [no.] 1; XΔTsixY line no. 2). Nuclei are stained blue with DAPI. Scale bar, 10 μm. (C) Quantification of Xist RNA coated nuclei in undifferentiated (d0) and d5 and d10 differentiated EpiSC lines. Scale bar, 10 μm. Only cells with a single Xist locus detected by DNA FISH (left) following RNA FISH were counted; n=100 nuclei/cell line. (D) RNA FISH detection of Xist (green) combined with IF detection of H3-K27me3 (red) in d10 differentiated XΔTsixY EpiSCs. Data from two different lines are shown. (E) Silencing of Pgk1 (red) upon Xist RNA (green) coating in representative d10 differentiated XΔTsixY EpiSCs. (F–G) RT-PCR (F) and RNA FISH (G) detection of Xist and Tsix RNAs in undifferentiated or embryoid body-differentiated XY and XΔTsixY ESC lines (2 and 3 lines, respectively). Scale bar, 10 μm. (H) Quantification of Xist RNA coated nuclei in the differentiated ESC lines. Only cells with one Xist locus detected by DNA FISH (left) following RNA FISH were counted; n=100 nuclei/cell line. Scale bar, 10 μm. See also Figs. S1–S2 and Table S1.
To examine if the ectopic Xist expression coincided with coating of the X-chromosome, we performed Xist RNA FISH on undifferentiated and differentiated EpiSCs. As expected, neither undifferentiated nor differentiated XY EpiSC lines exhibited any Xist RNA coated X-chromosomes (Fig. 2B). In all four of the XΔTsixY EpiSC lines, we observed a similar lack of Xist RNA coating in the undifferentiated cells (Fig. 2B). However, upon differentiation a significant percentage of the mutant cells displayed Xist RNA coating (29–35%; Fig. 2B–C). As in E5.25 mutant epiblast cells, many Xist RNA-coated XΔTsixY cells still expressed NANOG (38–42%) (Fig. S1E). Xist RNA coating also resulted in the accumulation of histone H3-K27me3 and silencing of Pgk1 on the XΔTsix in a vast majority of the mutant cells (84–94%) (Fig. 2D–E). Together, the RT-PCR and RNA FISH data from XΔTsixY EpiSCs prompt the conclusion that Tsix RNA does not participate in repressing Xist in undifferentiated male EpiSCs. Instead, Tsix is required to prevent ectopic Xist induction and X-linked gene silencing during the differentiation of male epiblast progenitor cells.
Ectopic Xist Induction in Differentiating XΔTsixY ESCs
That XΔTsixY EpiSCs displayed robust Xist induction only upon differentiation is incongruous with some previous studies with Tsix-mutant male ESCs. Tsix deficiency in male ESCs is suggested to either be innocuous in both undifferentiated and differentiated cells (Cohen et al., 2007; Lee, 2000; Lee and Lu, 1999; Minkovsky et al., 2013; Ohhata et al., 2006; Sado et al., 2002; Sado et al., 2001); or, conversely, result in ectopic Xist RNA coating of the Tsix-mutant X during differentiation (Debrand et al., 1999; Luikenhuis et al., 2001; Morey et al., 2004; Navarro and Avner, 2010; Vigneau et al., 2006). We therefore derived XY and XΔTsixY ESC lines (see Fig. S1) and tested Xist induction in both undifferentiated and differentiated cells by RT-PCR and RNA FISH. As with EpiSCs, we found that Xist remained silenced in undifferentiated XY as well as in XΔTsixY ESCs (Fig. 2F–G); however, upon differentiation Xist RNA was induced in XΔTsixY but not XY ESCs (Fig. 2F–H).
To distinguish if Xist induction in XΔTsixY ESCs occurred at the onset of differentiation or later, we transiently differentiated the ESCs into epiblast-like cells (EpiLCs) (Hayashi et al., 2011). EpiLCs arise early during ESC differentiation and share key features with EpiSCs (Fig. S2A–C) (Buecker et al., 2014). We found that the mutant EpiLCs displayed low-level Xist expression by RT-PCR, with only a few cells displaying Xist RNA coating (10%) (Fig. S2C–E). The Xist RNA-coated cells appeared to have differentiated beyond the EpiLC state, as suggested by reduced NANOG expression (Fig. S2E). When the EpiLCs were differentiated further, significantly more cells displayed Xist RNA coating (27–36%) (Fig. S2F), consistent with the EpiSC data.
Absence of Biased X-chromosome Choice in Tsix-heterozygous Female Epiblasts
We next examined the impact of the XΔTsix mutation in females. The two X-chromosomes in inbred XX epiblast cells are normally equally likely to undergo inactivation; in heterozygous Tsix-mutant epiblasts, however, previous work has concluded that only the XΔTsix X-chromosome is chosen for inactivation (Lee, 2000; Sado et al., 2001). This model of biased inactivation in favor of the XΔTsix is borne out by allele-specific Xist RT-PCR analyses of F1 hybrid WT and Tsix-heterozygous E6.5 epiblasts (Fig. 3A–B). The X-chromosomes in these embryos are derived from two divergent mouse strains and are polymorphic, thereby allowing allele-specific expression analysis. Both Sanger sequencing (Fig. 3A) and Pyrosequencing (Fig. 3B), which quantifies allele-specific expression, of the cDNAs revealed that Xist is transcribed from either X in WT XLabXJF1 and XJF1XLab embryos (by convention, the maternal allele precedes the paternal allele), whereas in XΔTsixXJF1 and XJF1XΔTsix epiblasts Xist is expressed almost exclusively from the XΔTsix.
Figure 3. Xist expression in E6.5 and E5.25 WT and Tsix-heterozygous female epiblast cells.

(A) Allele-specific RT-PCR detection of Tsix (exon 4) and Xist RNAs in epiblasts of three individual WT (XLabXJF1) and Tsix-heterozygous (XΔTsixXJF1) E6.5 embryos. M, marker; NTC, no template control; +, reaction with reverse transcriptase (RT); −, no RT control lane. Bottom, Sanger sequencing of the amplified cDNAs. Blue highlights mark a SNP that differs between the XLab / XΔTsix and XJF1 mouse strains. (B) RT-PCR followed by Pyrosequencing-based quantification of allelic Xist expression in epiblasts of individual E6.5 embryos. Error bars represent the standard deviation of data from 3 different embryos. (C–D) RNA FISH detection of Xist and Tsix RNAs coupled with IF detection of NANOG in isolated E6.5 (C) and E5.25 (D) epiblasts. Nuclei are stained blue with DAPI. Scale bars, 10 μm. Bottom, quantification of Xist and Tsix expression. The X-axis of each graph represents the average percentage of nuclei in each class (n = 3 embryos/genotype; 100 nuclei/E6.5 embryo and 45–71 nuclei/E5.25 embryo). Diagrams along the Y-axis depict all observed expression patterns. Error bars represent the standard deviation of data from 3 different embryos. *, p ≤0.01 (Chi-square test). (E) RT-PCR amplification of Tsix (exon 4) and Xist RNAs in WT and Tsix-heterozygous epiblasts. Bottom, Sanger sequencing of the Tsix and Xist cDNAs. (F) RT-PCR followed by Pyrosequencing-based quantification of allelic Xist expression in epiblasts of individual E5.25 embryos. Error bars represent the standard deviation of data from 3 different embryos. No significant differences in allelic Xist expression were observed between WT and Tsix-mutant embryos (p = 0.44, E5.25 XLabXJF1 vs. XΔTsixXJF1; p = 0.46, E5.25 XJF1XLab vs. XJF1XΔTsix; Welch’s two-sample T-test.). See also Figure S3.
To evaluate the expression of Xist and Tsix in XX, XΔTsixX, and XXΔTsix E6.5 epiblasts at the single cell resolution, we performed strand-specific RNA FISH. As with male embryos, we again confirmed the identity of epiblast cells by first assaying expression of NANOG by IF. We observed Xist RNA coating of both Xs by RNA FISH in a small fraction of XΔTsixX and XXΔTsix mutant (~2%), but not WT XX, E6.5 epiblast cells (Fig. 3C). Based on this observation and the hypothesis that cells with ectopic inactivation of the XΔTsix are eliminated, we reasoned that a higher percentage of cells in Tsix-heterozygotes may display Xist RNA coating of both X-chromosomes at an earlier stage of embryogenesis. We therefore assayed epiblast cells in E5.25 embryos by RNA FISH (Fig. 3D). Although most nuclei displayed Xist RNA accumulation, a proportion lacked Xist RNA coating but displayed nascent Xist and Tsix RNAs, suggesting that X-inactivation was just beginning in the epiblast. Of the Xist RNA coated nuclei, a small but significant percentage clearly displayed Xist RNA coating of both X-chromosomes in XΔTsixX epiblasts (12%) compared to XX epiblasts (0%), although one of the two Xist coats in the mutants was often comparatively weaker (Fig. 3D). To rule out a parent-of-origin effect, we also investigated E5.25 epiblasts with paternally-transmitted XΔTsix mutation. A similar percentage of XXΔTsix epiblast cells (11%) exhibited Xist RNA coating of both X-chromosomes (Fig. 3D). Xist RNA coating of both Xs coincided with H3-K27me3 enrichment and silencing of Pgk1 on both Xs in 80–90% of the nuclei, suggesting that both Xs were inactivated (Fig. S3A–B).
In addition to nuclei with two Xist RNA coats, E5.25 Tsix-heterozygotes lacked Tsix RNA expression from the active-X in a significant percentage of nuclei (24%). We suspected that in these cells the XΔTsix was chosen as the active-X, and, hence, the WT X as the inactive-X. Upon differentiation, this population of cells would ectopically induce Xist from and undergo inactivation of the XΔTsix. We therefore set out to test directly if the XΔTsix can be chosen as the active-X in E5.25 epiblasts, by exploiting the expression of a β-galactosidase cassette integrated into the mutant Tsix locus (see Fig. 1A); LacZ nascent transcripts uniquely mark the XΔTsix (Sado et al., 2001). Both unmodified Tsix and the mutated Tsix locus expressing LacZ are subject to X-inactivation and therefore are only transcribed when they reside on the active-X (Fig. S3C) (Maclary et al., 2014; Sado et al., 2001). Simultaneous probing of Xist, Tsix, and LacZ RNAs by FISH in WT XX E5.25 epiblasts, which do not carry the transgene, showed Tsix but not LacZ expression as expected (Fig. S3C). In XΔTsixX and XXΔTsix epiblasts, by contrast, a significant percentage of nuclei (30–39%) expressed LacZ but not Tsix (Fig. S3C). Thus, the XΔTsix can indeed be chosen as the active-X at the onset of random X-inactivation.
To interrogate X-chromosomal choice further, we assayed Xist expression via allele-specific RT-PCR followed by Sanger sequencing and Pyrosequencing in individual F1 hybrid E5.25 WT and Tsix-heterozygous epiblasts. Both sets of epiblasts displayed biallelic Xist expression by Sanger sequencing and negligible differences in allelic Xist expression by Pyrosequencing (Fig. 3E–F). The similarly unequal expression of the two Xist alleles in WT and Tsix-mutant epiblasts is consistent with differences in the X-controlling element (Xce) on the polymorphic X-chromosomes in F1 hybrid embryos (Chadwick et al., 2006; Johnston and Cattanach, 1981; Ohhata et al., 2008).
We next tested the inference that the paucity of ectopic Xist RNA coated cells in Tsix-heterozygous E6.5 epiblasts compared to E5.25 epiblasts is due to a failure of mitotic division of E5.25 epiblast cells with two inactive X-chromosomes. We found that the mitotic index, as measured by the presence of phosphorylated-histone H3, is significantly reduced in E5.25 XΔTsixX epiblast cells exhibiting Xist RNA coating and silencing of Pgk1 on both Xs, compared to cells with Xist RNA coating and silencing of Pgk1 on only one X (p = 0.003; Fig. S3D). Thus, the proliferative potential of cells with two inactive-Xs is compromised.
Tsix-heterozygous EpiSCs Undergo Ectopic X-inactivation Only Upon Differentiation
We next wished to investigate if ectopic Xist RNA induction from the XΔTsix in heterozygous females occurs at the onset of X-inactivation or is linked to epiblast differentiation as in XΔTsixY males. We therefore derived multiple WT XLabXJF1 and XJF1XLab and mutant XΔTsixXJF1 and XJF1XΔTsix EpiSC lines from F1 hybrid embryos (see Fig. S1 and Table S1). Although Tsix was expressed from both X-chromosomes in WT EpiSCs, in Tsix-heterozygotes only the WT XJF1 expressed Tsix (Fig. 4A–B). To quantify how often each of the two parental X-chromosomes were chosen for inactivation, we Pyrosequenced Xist cDNA and cDNAs from the X-inactivated genes Rnf12 and Atrx. The WT EpiSC lines displayed nearly equal levels of Xist expression from the two X-chromosomes, consistent with random choice (Fig. 4C). The Tsix-heterozygous EpiSC lines did not uniformly show Xist expression exclusively from the XΔTsix, nor were Rnf12 and Atrx expressed only from the WT XJF1 in all the mutant cell lines (Fig. 4C). The cell lines instead displayed a normal distribution of which of the two Xs was inactivated, demonstrating that X-inactivation is not skewed in favor of the XΔTsix. Although the variability was higher in the mutants, the mean as well as the median expression levels of Xist, Rnf12, and Atrx in parent-of-origin-matched WT and Tsix-mutant EpiSC lines were not significantly different (p>0.1 in all cases).
Figure 4. Lack of biased X-inactivation in undifferentiated Tsix-heterozygous EpiSC lines.

(A) RT-PCR amplification of Tsix RNA from WT and Tsix-heterozygous EpiSC lines. M, marker; NTC, no template control; +, reaction with reverse transcriptase (RT); −, no RT control lane. (B) Representative Sanger sequencing chromatograms of Tsix cDNAs. (C) RT-PCR followed by Pyrosequencing-based quantification of allelic expression of Xist and the X-linked genes Rnf12 and Atrx. Each bar represents an individual EpiSC line. Xm, maternal X-chromosome; Xp, paternal X-chromosome. Error bars represent the standard deviation of ≥3 independent results. The mean and median of allelic expression of Xist, Rnf12, and Atrx lack significant difference (p > 0.1, Welch’s two-sample T-test and Mood’s Median test) between parent-of-origin matched WT and mutant EpiSCs. See also Figure S1 and Table S1.
We next tested if the XΔTsix induced Xist in female EpiSCs as a function of differentiation, as it does in XΔTsixY EpiSCs. We therefore differentiated a subset of the WT and Tsix-mutant female cell lines (Fig. S4). The selected XΔTsixXJF1 and XJF1XΔTsix EpiSC lines encompassed different degrees of Xist mosaicism; the fraction of total Xist RNA transcribed from each of the two Xs varied between the cell lines, ranging from 0–100% of the total Xist expression in the undifferentiated EpiSCs (Fig. 5). Upon differentiation, the allelic ratio of Xist, Rnf12, and Atrx RNAs did not change in WT XLabXJF1 and XJF1XLab EpiSC lines (Figs. 5 and S5A). In differentiating XΔTsixXJF1 and XJF1XΔTsix EpiSC lines, however, we found distinct alterations in allelic Xist expression (Fig. 5). In the eight mutant EpiSC lines most highly mosaic for Xist, where the WT XJF1 accounted for ~25–75% of total Xist RNA output (XJF1XΔTsix line 4; and, XΔTsixXJF1 lines 4, 5, 6, 8, 10, 12, and 14), Xist expression became restricted to the XΔTsix mutant X-chromosome by the end of 20 days (d20) of differentiation (Fig. 5). Conversely, we found that Rnf12 and Atrx in these cell lines were increasingly expressed from the WT XJF1 over the course of differentiation (Fig. S5B–C). In two mutant EpiSC lines in which >90% of Xist RNA was expressed from the WT XJF1 at d0 (XJF1XΔTsix lines 5 and 6), Xist expression from the mutant XΔTsix increased only slightly by d20 of differentiation (Fig. 5). Rnf12 and Atrx displayed a correspondingly minimal decrease in expression from the XΔTsix in these cell lines (Fig. S5B). In the mutant cell lines in which Xist was expressed almost exclusively from the WT XJF1 or from the XΔTsix at d0 (XΔTsixXJF1 lines 15 and 2, respectively), the allelic expression profile of Xist, Rnf12, and Atrx did not change upon differentiation (Figs. 5 and S5C).
Figure 5. Change in allelic Xist expression in differentiating Tsix heterozygous EpiSC lines.

(A) RT-PCR followed by Pyrosequencing-based quantification of Xist expression in EpiSC lines (cell line numbers in parentheses) differentiated for 0, 5, 10, 15, and 20 days (d). Xm, maternal X-chromosome; Xp, paternal X-chromosome. Each bar represents an individual EpiSC line. Error bars represent the standard deviation of ≥3 independent results. See also Figures S4 and S5.
We observed a similar change in X-inactivation patterns by RNA FISH in the differentiating EpiSCs. As in E5.25 epiblasts, we exploited the mutually exclusive expression of Tsix and LacZ RNAs from the WT XJF1 and mutant XΔTsix, respectively, to determine which of the two X-chromosomes is chosen as the active-X. We profiled three EpiSC lines that displayed distinct and varied patterns of inactivation suggested by allele-specific Xist expression (XΔTsixXJF1 lines 2, 6, and 15; see Fig. 4C). Consistent with the inactivation pattern inferred by allele-specific Xist RT-PCR, XΔTsixXJF1 EpiSC line 2 lacked LacZ RNA FISH signal in all cells examined throughout differentiation (Fig. S6A). XΔTsixXJF1 EpiSC line 6 displayed nearly equal numbers of cells expressing LacZ and Tsix at d0, but during the course of differentiation this pattern gradually shifted to yield only cells with a Tsix RNA FISH signal by d20 (Fig. S6A). By contrast, although EpiSC XΔTsixXJF1 line 15 only exhibited cells with a LacZ signal at d0, consistent with the entire population being eligible to undergo ectopic inactivation, this pattern did not change appreciably even by d20 of differentiation (Fig. S6A).
Reduced Proliferation and Induced Cell Death Upon Ectopic X-inactivation in Tsix-heterozygous EpiSCs
The changes in X-inactivation patterns in differentiating XΔTsix mutant EpiSCs could arise from one of two possibilities. In the first, Xist expression switches from the WT XJF1 in favor of the mutant XΔTsix in individual cells. Alternatively, the ectopic induction of Xist from the XΔTsix results in two inactive-Xs in differentiating EpiSCs that had originally activated Xist from the WT XJF1. In this latter scenario, the deficiency in X-linked gene expression due to both Xs being inactivated would drive selection against these cells. The remaining population of cells would then be descendants of cells that had initially chosen to inactivate the XΔTsix, which do not undergo ectopic Xist induction from the WT XJF1.
To further distinguish amongst the two possibilities, we performed single-cell analysis of differentiating EpiSCs. We found that whereas XX EpiSCs displayed Xist RNA coating of only a single X-chromosome in undifferentiated and in d5 and d10 differentiated cells, XΔTsixX and XXΔTsix EpiSC lines exhibited Xist RNA coating of a single X in undifferentiated cells but of both Xs upon differentiation (Fig. 6A–C). A substantial percentage of the double Xist RNA-coated cells early in differentiation (d5) were also NANOG+ (32–35%; Fig. S6B), consistent with the data from embryos. Nearly all the nuclei with double Xist RNA coats also displayed enrichment of H3-K27me3 and silencing of Pgk1 on both Xs (both >90%; Fig. 6D–E). To examine if the cells with two inactive Xs are selected against, we compared the mitotic indices of cells with one inactive- vs. two inactive-Xs by staining for phosphorylated-histone H3. Differentiating EpiSCs with two Xist RNA coats appeared to divide significantly less often than with one Xist coat (p<0.001; Fig. 6F). We also evaluated cell death in differentiating EpiSCs, and found that cells with two Xist RNA coats were significantly more likely to be dead or dying compared to cells with one Xist coat (p<0.001; Fig. 6G).
Figure 6. Ectopic Xist RNA coating in differentiated Tsix-heterozygous EpiSC lines.

(A) RNA FISH detection of Xist RNA (green) and Tsix RNA (red) in representative undifferentiated and d10 differentiated WT and Tsix-heterozygous EpiSC lines (XJF1XLab cell line no. 1; XΔTsixXJF1 cell line no. 14). Nuclei are stained blue with DAPI. Scale bars, 10 μm. (B) RNA FISH detection of Xist RNA coat using an exonic probe (white) and nascent Xist RNA with an intronic probe (red), demonstrating that in cells with two Xist RNA coats both Xist alleles are transcribed. (C) Quantification of EpiSC nuclei displaying single vs. double Xist RNA coats during differentiation. Scale bar, 10 μm. Only cells with two Xist loci detected by DNA FISH (left) following RNA FISH were counted; n=100 nuclei/cell line. (D) Enrichment of H3-K27me3 on Xist RNA coated X-chromosomes in d10 differentiated XΔTsixXJF1 EpiSCs. Data from two different lines (nos. 5 and 14) are shown. (E) Silencing of Pgk1 (red) upon ectopic Xist RNA coating (green) in d10 differentiated XΔTsixXJF1 EpiSCs. (F) Reduced phospho-H3 staining, a marker of cell proliferation, in d10 differentiated XΔTsixXJF1 EpiSCs (cell line 14) with two Xist RNA coats compared to nuclei with a single Xist coat (p<0.001, Fisher’s exact test). (G) Increased death of cells with two inactive-Xs compared to cells with one inactive-X in d10 differentiated XΔTsixXJF1 EpiSCs (p<0.001, Welch’s two-sample T-test). The inactive-X is marked by H3-K27me3 accumulation (purple). Ethd-1 (red) marks dead cells and Calcein AM (green) marks live cells. (H) Reduced cell counts during differentiation of Tsix-heterozygous compared to WT EpiSCs. (I–J) Reduced viability of adherent (I) and non-adherent cells in suspension (J) during differentiation of Tsix-heterozygous compared to WT EpiSCs. See Table S2 for statistical comparisons; see also Figs. S6–S7.
The reduced proliferation of cells with two inactive-Xs would predict decreased cell numbers during differentiation of some but not other Tsix-heterozygous EpiSCs. Tsix-mutant EpiSC lines with few cells eligible to undergo ectopic inactivation are expected to display comparable cell counts to WT EpiSCs. Consistent with this scenario, XJF1XΔTsix EpiSC lines 1–3 and XΔTsixXJF1 EpiSC lines 1–2, which exhibit exclusive or almost exclusive inactivation of the XΔTsix and therefore lack cells that can undergo ectopic inactivation (Fig. 4C), have the highest cell counts throughout differentiation and are indistinguishable from WT EpiSCs (Fig. 6H and Table S2). Conversely, EpiSC lines that have completely or almost completely inactivated the WT XJF1 X-chromosome, XJF1XΔTsix lines 5 and 6 and XΔTsixXJF1 line 15 (see Fig. 4C), and thus harbor the highest percentage of cells that are able to undergo ectopic inactivation, have the lowest cell counts by d20 of differentiation (Fig. 6H and Table S2). Cell counts in XJF1XΔTsix line 4 and XΔTsixXJF1 lines 6 and 10 with intermediate percentages of cells subject to ectopic inactivation (~20–75%) again correlate with the available pool of cells eligible to ectopically inactivate the XΔTsix (Figs. 4C and 6H; Table S2). Thus, EpiSC lines with a higher percentage of cells that can ectopically induce Xist from the XΔTsix and thereby inactivate the second X display lower cell counts during differentiation (r = −0.94)
We also quantified cell viability in populations of differentiating EpiSCs. Consistent with the higher rate of death of cells with two inactive-Xs (see Fig. 6G), cell viability measurements showed that the higher the percentage of EpiSCs subject to ectopic X-inactivation the lower their viability during differentiation (Fig. 6I–J and Table S2; r = −0.95 for adherent viable cells and −0.99 for viable cells in suspension). The phospho-histone H3 staining and cell death results together with the cell count and viability data lead to the conclusion that ectopic Xist induction from the XΔTsix and the resultant inactivation of both Xs potently selects against cells via both reduced cell proliferation and induced cell death. Ultimately, the outcome is skewed X-inactivation in favor of cells that had chosen to initially inactivate the XΔTsix.
We also tested if ectopic Xist induction selects against XΔTsixY cells. Phospho-histone H3 staining suggested slightly if not significantly reduced proliferation of cells with an Xist RNA coated X-chromosome compared to those without (p=0.02; Fig. S6C). The ratio of live:dead cells, however was indistinguishable between cells with Xist RNA coating and those without (Fig. S6D). The cell numbers and viability through 30d of differentiation were reduced in the mutants, but mostly at d25 and d30 time points (Fig. S6E–G), which contrasts with the striking reduction in both measurements by d20 of differentiation in Tsix-heterozygous female EpiSCs. This difference potentially reflects a reduced level of ectopic Xist induction and X-linked gene silencing in mutant males compared to females (see Discussion).
Ectopic Xist Induction in Differentiating Tsix-heterozygous Female ESCs
As with XΔTsixY EpiSCs, we sought to test if our observations of Tsix-heterozygous EpiSCs also apply to mutant ESCs. Although both WT XLabXJF1 / XJF1XLab and mutant XΔTsixX JF1 / XJF1XΔTsix undifferentiated ESCs displayed a low level of Xist RNA expression by RT-PCR, all four genotypes induced Xist from either allele upon differentiation (Fig. S6H–I). In agreement with the RT-PCR results, both WT and mutant ESCs displayed Xist RNA coating only upon differentiation (Fig. S6J). A subset of the differentiating mutant (but not WT) cells, though, exhibited Xist RNA coating of both Xs (Fig. S6J–K).
We next differentiated the ESCs into EpiLCs to determine when during differentiation Tsix-heterozygous ESCs ectopically induced Xist (Fig. S7A–C). The two Xs in WT EpiLCs were nearly equally likely to be chosen as the inactive-X, as evidenced by the allelic expression profiles of Xist (Fig. S7D). Although the mutant EpiLC samples displayed a wide distribution of allelic Xist expression, the average expression ratios of the two Xist alleles matched closely that of the WT EpiLCs (Fig. S7D), recapitulating the pattern observed in EpiSCs (Fig. 4C). RNA FISH demonstrated that a vast majority of the Tsix-heterozygous EpiLCs harbored only one Xist RNA coated X-chromosome (96%; Fig. S7E–F). Upon further differentiation, the mutant cells displayed increasingly biased inactivation of the XΔTsix, consistent with selection favoring cells that had originally inactivated the XΔTsix (Fig. S7G).
DISCUSSION
Tsix repression of Xist at the onset of X-inactivation has been invoked previously to support a role for the Tsix locus in X-chromosome counting and/or choice (Clerc and Avner, 1998; Cohen et al., 2007; Debrand et al., 1999; Lee, 2000, 2005; Lee and Lu, 1999; Morey et al., 2004; Navarro et al., 2010; Sado et al., 2001; Vigneau et al., 2006). In the counting step, the cell senses the number of X-chromosomes; only if there are two or more Xs do the cells proceed to the choice and inactivation steps (Grumbach et al., 1963; Lyon, 1962). In the choice step, one of the two X-chromosomes is selected for silencing; only then does X-inactivation ensue (Rastan, 1983; Takagi, 1980). In this model of random X-inactivation, counting must precede choice, with the last step being inactivation itself. Thus, XY male epiblast cells do not undergo X-inactivation because the cells ‘count’ only one X-chromosome, which would preclude both the choice and inactivation steps.
Our data, however, rule out a function for Tsix in X-chromosome counting, in agreement with Monkhorst et al., (Monkhorst et al., 2008). In a diploid male or female cell, the counting process protects one X-chromosome from inactivation; a defect in counting is therefore expected to result in inactivation of the single X-chromosome in males at some frequency (Avner and Heard, 2001). The absolute absence of Xist RNA coating and X-inactivation in undifferentiated XΔTsixY EpiSCs is evidence that the Tsix RNA is not part of the counting mechanism. Xist is only induced when XΔTsixY EpiSCs differentiate. That not all differentiating XΔTsixY cells express Xist may reflect intercellular variability in the levels of an Xist activating factor (see below).
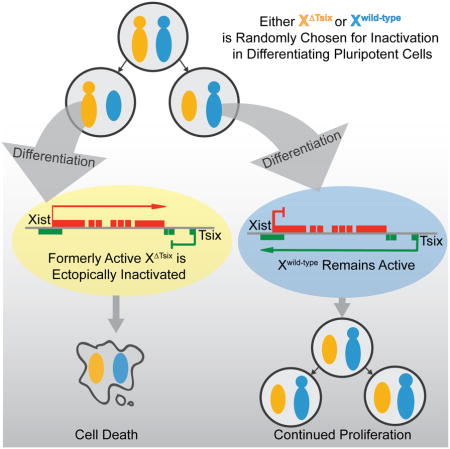
Our findings also exclude a primary role for Tsix in the choice of which X undergoes inactivation. Biased X-inactivation in Tsix-heterozygous cells occurs through a secondary cell selection effect, rather than through primary inactivation of the XΔTsix at the onset of X-inactivation (Fig. 7). Tsix therefore constitutes a failsafe mechanism that prevents ectopic Xist induction and inactivation of the active X-chromosome, but only after X-inactivation has initiated (Fig. 7). Thus, Tsix is required not to establish but to maintain the randomized pattern of X-inactivation. This protective function of Tsix in the epiblast lineage appears to be conserved in extra-embryonic cell types. Stem cells of the trophectoderm lineage, which undergoes imprinted X-inactivation of the paternal X-chromosome, similarly ectopically silence the XΔTsix only upon differentiation both in vivo and in vitro (Maclary et al., 2014).
Figure 7. A model of Tsix function in X-inactivation.

At the onset of X-inactivation, Tsix heterozygous epiblast cells undergo stochastic X-inactivation indistinguishable from WT epiblasts. Upon continued differentiation of the epiblast cells, the XΔTsix ectopically induces Xist RNA. In female cells that had originally inactivated the WT X-chromosome, ectopic Xist induction accompanies the initiation of X-inactivation a second time (of the XΔTsix), resulting in two inactive-Xs. As a result of a paucity of X-linked gene expression, these cells are selected away due both to reduced proliferation and induced cell death. Thus, the developing embryo is ultimately populated only with cells that had originally inactivated the XΔTsix.
Tsix is expressed in pluripotent cells, but it is only required to silence Xist as these cells differentiate. Tsix expression in epiblast precursor cells in E4.5 embryos as well as in EpiSCs and EpiLCs may prime the epiblast cells to forestall inactivation of the active X-chromosome upon impending differentiation. In support of this idea, Tsix is robustly expressed in ESCs yet its loss does not lead to ectopic Xist induction in pluripotent cells of either sex, as shown here and in earlier studies (Cohen et al., 2007; Debrand et al., 1999; Lee and Lu, 1999; Luikenhuis et al., 2001; Minkovsky et al., 2013; Morey et al., 2004; Ohhata et al., 2006).
If Tsix does not regulate X-chromosome counting or choice, then alternate mechanisms must explain why X-inactivation does not occur in males and does so randomly in females. We favor a parsimonious model of random inactivation whereby a dose-dependent X-linked activity triggers inactivation only in females. For example, the increased dosage of an X-linked factor in XX compared to XY cells at the onset of inactivation when both Xs are active may facilitate X-inactivation by stochastically and directly activating Xist on one of the two X-chromosomes in females, as has been proposed (but debated) for RNF12 (Barakat et al., 2011; Gontan et al., 2012; Jonkers et al., 2009; Shin et al., 2014). The lower level of such a factor may explain why Xist is ectopically induced from the mutant X in only some XΔTsixY embryonic cells, but in all XΔTsixX embryonic cells. Xist may also be expressed to a lesser extent in individual XΔTsixY cells compared to female cells, resulting in a comparatively reduced degree of X-linked gene silencing in males and potentially explaining why differentiating Tsix-mutant female but not male EpiSCs are subject to cell selection. Future work will clarify the underlying reasons for this difference.
Our work lends caution to the modeling of X-inactivation kinetics in differentiating ESCs. Depending on the ESC differentiation regimen, aberrantly inactivated cells may be rapidly outcompeted by appropriately inactivated ones, thus masking a defect in the initiation phase of X-inactivation. Conversely, errors in X-inactivation that manifest only during the maintenance phase are difficult to distinguish from those that occur at the onset due to the asynchronous differentiation of ESCs. Such a scenario may resolve the seemingly discordant observations of the Tsix-mutant X-chromosome appearing to be both susceptible and resistant to Xist induction in differentiating ESCs. Directed differentiation of ESCs into EpiLCs may be one route to capturing cells just after X-inactivation has initiated. Conversion of ESCs into EpiLCs, however, is also subject to key shortcomings. Not all ESCs differentiate into EpiLCs, thus resulting in a heterogeneous population of cells; and, when they do, the EpiLCs are only transiently present (Buecker et al., 2014; Hayashi et al., 2011).
Our data instead highlight the utility of EpiSCs as a model system to uncouple the onset of random X-inactivation from differentiation of pluripotent cells. A comparison of X-inactivation defects in Tsix-mutant EpiSCs with embryonic epiblasts suggests that EpiSCs can capture a window in differentiation of naïve pluripotent epiblast cells immediately after X-inactivation has initiated. Whereas Tsix-heterozygous embryonic epiblasts display ectopic Xist induction beginning at ~E5.25 stage of embryogenesis, sex and genotype-matched EpiSCs do not. Upon differentiation, however, these EpiSCs exhibit Xist RNA coating of both Xs, mimicking the pattern of ectopic Xist induction in the mutant epiblasts as the embryos develop from E5.25, just after random X-inactivation has commenced, to E6.5, a stage by which ectopic Xist induction is almost undetectable. By E6.5, Tsix heterozygote epiblasts are comprised almost exclusively of cells in which the XΔTsix is the inactive-X, due to rapid selection against cells that had originally chosen the WT X for silencing but subsequently ectopically induced Xist and underwent inactivation of the XΔTsix. Thus, the pattern of inactivation changes rapidly within ~1 day of development, at a stage of embryogenesis that is not easily accessible. By mirroring early epiblast cells just after they have undergone X-inactivation, EpiSCs are a valuable resource to tease apart defects in the initiation of X-inactivation from differentiation of the pluripotential epiblast cells.
EXPERIMENTAL PROCEDURES
Ethics Statement
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were handled according to protocols approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan (protocol #PRO00004007).
Derivation, Culture, Differentiation, and Characterization of Epiblast Stem Cell (EpiSC) Lines
EpiSCs were derived from pre-, peri-, and post-implantation stages essentially as described (Brons et al., 2007; Najm et al., 2011; Tesar et al., 2007). EpiSCs derived from different stages of embryogenesis (Table S1) did not display any noticeable differences in Xist induction and X-inactivation patterns. For derivation of EpiSCs from pre- and peri-implantation mouse embryos, individual embryos were plated on quiescent mouse embryonic fibroblast (MEF) feeder cells in K15F5 medium containing Knockout DMEM (GIBCO, #10829–018) supplemented with 15% Knockout Serum Replacement (KSR; GIBCO, #A1099201), 5% ESC-qualified fetal bovine serum (FBS; GIBCO, #104390924), 2 mM L-glutamine (GIBCO, #25030), 1X nonessential amino acids (GIBCO, #11140–050), and 0.1 mM 2-mercaptoethanol (Sigma, #M7522). After 5–6 days, blastocyst outgrowths were dissociated partially with 0.05% trypsin (Invitrogen, #25300–054). The partial dissociates were plated individually into a 1.9 cm2 well containing MEF feeder layer and cultured for an additional 4–6 days in K15F5 medium. The culture was then passaged by a brief exposure (2–3 min) to 0.05% trypsin/EDTA with gentle pipetting to prevent complete single-cell dissociation of pluripotent clusters, and plated into a 9.6 cm2 well containing MEF feeders in K15F5 medium. Morphologically distinct mouse EpiSC colonies became evident over the next 4–8 days and were subcloned from a mixed population of cells, including ESCs. EpiSC colonies were manually dissociated into small clusters using a glass needle and plated into 1.9 cm2 wells containing MEF feeders in EpiSC cell medium consisting of Knockout DMEM supplemented with 20% KSR, 2 mM Glutamax (GIBCO, #35050061), 1X nonessential amino acids, 0.1 mM 2-mercaptoethanol, and 10 ng/ml FGF2 (R&D Systems, #233-FB).
For derivation of EpiSCs from postimplantation mouse embryos, the epiblast layer was microdissected from E5.5 embryos and plated on MEF cells in EpiSC medium and cultured for 3–4 days to form a large EpiSC colony. EpiSC colonies were then manually dissociated into small clusters using a glass needle and plated into 1.9 cm2 wells containing MEF feeders in EpiSC cell medium. EpiSCs were passaged every third day using 1.5 mg/ml collagenase type IV (GIBCO, #17104–019) with pipetting into small clumps.
Differentiation of EpiSCs was achieved by growing the EpiSCs on gelatin-coated tissue culture dishes in EpiSC medium lacking FGF2. Expression of pluripotency markers Oct4, Nanog; mesodermal marker Brachyury; neuroectodermal marker β-III tubulin; and hepatocyte marker FoxA2 was assessed by RT-PCR using Invitrogen SuperScript III One-Step RT-PCR System (Invitrogen, #12574–026). Primer sequences were designed using the primer bank web software (http://pga.mgh.harvard.edu/primerbank/; PrimerBank ID: Oct4:356995852c2; Nanog: 153791181c2; Brachyury:118130357c1; β-III tubulin:12963615a1; FoxA2:153945803c1). Fgf5 forward primer: CTGTACTGCAGAGTGGGCATCGG; Fgf5 reverse primer: GACTTCTGCGAGGCTGCGACAGG. Cer1 forward primer: CTCTGGGGAAGGCAGACCTAT; Cer1 reverse primer: CCACAAACAGATCCGGCTT. Rex1 forward primer: TGGAAGCGAGTTCCCTTCTC; Rex1 reverse primer: GCCGCCTGCAAGTAATGAG. All primer pairs except Tsix (exon 4) spanned an intron, thereby distinguishing cDNA from genomic DNA amplification. Nevertheless, control reactions lacking reverse transcriptase for each sample were performed to rule out genomic DNA contamination.
For IF and/or RNA FISH, EpiSCs were cultured on gelatin-coated glass coverslips. The cells were then permeabilized through sequential treatment with ice-cold cytoskeletal extraction buffer (CSK:100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 10 mM PIPES buffer, pH 6. 8) for 30 sec, ice-cold CSK buffer containing 0.4% Triton X-100 (Fisher Scientific, #EP151) for 30 sec, followed twice with ice-cold CSK for 30 sec each. After permeabilization, cells were fixed by incubation in 4% paraformaldehyde for 10 min. Cells were then rinsed 3X in 70% ethanol and stored in 70% ethanol at −20°C prior to IF and/or RNA FISH.
Quantification of Allele-specific Expression
Allele-specific expression was quantified using Qiagen PyroMark sequencing platform. Amplicons containing SNPs were designed using the PyroMark Assay Design software. cDNAs were synthesized using Invitrogen SuperScript III One-Step RT-PCR System (Invitrogen,#12574-026). Following the PCR reaction, 5 ul of a total of 25 uL of reaction was run on a 3% agarose gel to assess the efficacy of amplification. The samples were then prepared for pyrosequencing according to the standard recommendations for use with the PyroMark Q96 ID sequencer. For Xist, the following primers were used: forward, CAAGAAGAAGGATTGCCTGGATTT; reverse, 5′-biotin-GCGAGGACTTGAAGAGAAGTTCTG; sequencing, CAAACAATCCCTATGTGA. For Atrx, the following primers were used: forward: ATAGCTTCAGATTCTGATGAAACC; reverse: 5′-biotin-ACATCGTTGTCACTGCCACTT; sequencing: taagctcagatgaaaaga. For Rnf12, the following primers were used: forward: 5′-Biotin-TGCAGCCAACAAGTGAAATTCC; reverse: TATCTGCTGTCTCAGGGTCACATG; sequencing: tagaacttccttcaggc. All three amplicons span intron(s), thus permitting discrimination of RNA vs. any contaminating genomic DNA amplification due to size differences. Control reactions lacking reverse transcriptase for each sample were also performed to rule out genomic DNA contamination.
Microscopy
Samples were imaged using a Nikon Eclipse TiE inverted microscope with a Photometrics CCD camera. The images were deconvolved and uniformly processed using NIS-Elements software.
Statistics
p=0.01 was used as the cutoff for statistical significance. Tests used to calculate statistical significance are indicated in the corresponding figure legends.
Supplementary Material
Acknowledgments
We thank members of the Kalantry lab for discussions and critical review of the manuscript. We thank Mrinal K. Sarkar for contributing to the initial genotyping and RT-PCR of some EpiSC lines. We also thank Angela Andersen of Pickersgill and Andersen, Life Science Editors, for editing services. We acknowledge the services of the University of Michigan Sequencing Core Facility, supported in part by the University of Michigan Comprehensive Cancer Center. This work was funded by an NIH National Research Service Award 5-T32-GM07544 from the National Institute of General Medicine Sciences (E.M.); a University of Michigan Reproductive Sciences Program training grant; an NIH National Research Service Award 1F31HD080280-01 from the National Institute of Child Health and Human Development (E.M.); a Rackham Predoctoral Fellowship from the University of Michigan (E.M.); an NIH Director’s New Innovator Award (DP2-OD-008646-01) (S.K.); a March of Dimes Basil O’Connor Starter Scholar Research Award (5-FY12-119) (S.K.); and the University of Michigan Endowment for Basic Sciences.
Footnotes
AUTHOR CONTRIBUTIONS
S.G., E.M., and S.K. conceived the study and designed the experiments. S.G. derived, characterized, and analyzed EpiSC, ESC, and EpiLC lines. E.M. performed dissections and analysis of post-implantation embryos. E.B. developed strand-specific RNA FISH probes. M.H. developed and optimized Pyrosequencing assays. S.G., E.M., and S.K. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2:59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- Barakat TS, Gribnau J. X chromosome inactivation in the cycle of life. Development. 2012;139:2085–2089. doi: 10.1242/dev.069328. [DOI] [PubMed] [Google Scholar]
- Barakat TS, Gunhanlar N, Pardo CG, Achame EM, Ghazvini M, Boers R, Kenter A, Rentmeester E, Grootegoed JA, Gribnau J. RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet. 2011;7:e1002001. doi: 10.1371/journal.pgen.1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernemann C, Greber B, Ko K, Sterneckert J, Han DW, Arauzo-Bravo MJ, Scholer HR. Distinct developmental ground states of epiblast stem cell lines determine different pluripotency features. Stem Cells. 2011;29:1496–1503. doi: 10.1002/stem.709. [DOI] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Bryja V, Bonilla S, Arenas E. Derivation of mouse embryonic stem cells. Nat Protoc. 2006;1:2082–2087. doi: 10.1038/nprot.2006.355. [DOI] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell. 2014;14:838–853. doi: 10.1016/j.stem.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick LH, Pertz LM, Broman KW, Bartolomei MS, Willard HF. Genetic control of X chromosome inactivation in mice: definition of the Xce candidate interval. Genetics. 2006;173:2103–2110. doi: 10.1534/genetics.105.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Avner P. Role of the region 3′ to Xist exon 6 in the counting process of X- chromosome inactivation [see comments] Nat Genet. 1998;19:249–253. doi: 10.1038/924. [DOI] [PubMed] [Google Scholar]
- Cohen DE, Davidow LS, Erwin JA, Xu N, Warshawsky D, Lee JT. The DXPas34 repeat regulates random and imprinted X inactivation. Dev Cell. 2007;12:57–71. doi: 10.1016/j.devcel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Debrand E, Chureau C, Arnaud D, Avner P, Heard E. Functional analysis of the DXPas34 locus, a 3′ regulator of Xist expression. Mol Cell Biol. 1999;19:8513–8525. doi: 10.1128/mcb.19.12.8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RL, Lyon MF. X chromosome inactivation studied by injection of a single cell into the mouse blastocyst. Nature. 1971;231:385–386. doi: 10.1038/231385a0. [DOI] [PubMed] [Google Scholar]
- Gontan C, Achame EM, Demmers J, Barakat TS, Rentmeester E, van IW, Grootegoed JA, Gribnau J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012;485:386–390. doi: 10.1038/nature11070. [DOI] [PubMed] [Google Scholar]
- Grumbach MM, Morishima A, Taylor JH. Human Sex Chromosome Abnormalities in Relation to DNA Replication and Heterochromatinization. Proc Natl Acad Sci U S A. 1963;49:581–589. doi: 10.1073/pnas.49.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DW, Greber B, Wu G, Tapia N, Arauzo-Bravo MJ, Ko K, Bernemann C, Stehling M, Scholer HR. Direct reprogramming of fibroblasts into epiblast stem cells. Nat Cell Biol. 2011;13:66–71. doi: 10.1038/ncb2136. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011;146:519–532. doi: 10.1016/j.cell.2011.06.052. [DOI] [PubMed] [Google Scholar]
- Hogan B. Manipulating the mouse embryo : a laboratory manual. 2. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Hopfl G, Gassmann M, Desbaillets I. Differentiating embryonic stem cells into embryoid bodies. Methods Mol Biol. 2004;254:79–98. doi: 10.1385/1-59259-741-6:079. [DOI] [PubMed] [Google Scholar]
- Johnston PG, Cattanach BM. Controlling elements in the mouse. IV. Evidence of non-random X-inactivation. Genet Res. 1981;37:151–160. doi: 10.1017/s0016672300020127. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Barakat TS, Achame EM, Monkhorst K, Kenter A, Rentmeester E, Grosveld F, Grootegoed JA, Gribnau J. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell. 2009;139:999–1011. doi: 10.1016/j.cell.2009.10.034. [DOI] [PubMed] [Google Scholar]
- Kalantry S, Magnuson T. The Polycomb group protein EED is dispensable for the initiation of random X-chromosome inactivation. PLoS Genet. 2006;2:e66. doi: 10.1371/journal.pgen.0020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry S, Mills KC, Yee D, Otte AP, Panning B, Magnuson T. The Polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat Cell Biol. 2006;8:195–202. doi: 10.1038/ncb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry S, Purushothaman S, Bowen RB, Starmer J, Magnuson T. Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature. 2009;460:647–651. doi: 10.1038/nature08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell. 2000;103:17–27. doi: 10.1016/s0092-8674(00)00101-x. [DOI] [PubMed] [Google Scholar]
- Lee JT. Regulation of X-chromosome counting by Tsix and Xite sequences. Science. 2005;309:768–771. doi: 10.1126/science.1113673. [DOI] [PubMed] [Google Scholar]
- Lee JT, Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99:47–57. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- Luikenhuis S, Wutz A, Jaenisch R. Antisense transcription through the Xist locus mediates Tsix function in embryonic stem cells. Mol Cell Biol. 2001;21:8512–8520. doi: 10.1128/MCB.21.24.8512-8520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Lyon MF. Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet. 1962;14:135–148. [PMC free article] [PubMed] [Google Scholar]
- Maclary E, Buttigieg E, Hinten M, Gayen S, Harris C, Sarkar MK, Purushothaman S, Kalantry S. Differentiation-dependent requirement of Tsix long non-coding RNA in imprinted X-chromosome inactivation. Nat Commun. 2014;5:4209. doi: 10.1038/ncomms5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A, Fosten M, Monk M. X-chromosome inactivation mosaicism in the three germ layers and the germ line of the mouse embryo. J Embryol Exp Morphol. 1983;74:207–220. [PubMed] [Google Scholar]
- Minkovsky A, Barakat TS, Sellami N, Chin MH, Gunhanlar N, Gribnau J, Plath K. The pluripotency factor-bound intron 1 of xist is dispensable for x chromosome inactivation and reactivation in vitro and in vivo. Cell reports. 2013;3:905–918. doi: 10.1016/j.celrep.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, Harper MI. Sequential X chromosome inactivation coupled with cellular differentiation in early mouse embryos. Nature. 1979;281:311–313. doi: 10.1038/281311a0. [DOI] [PubMed] [Google Scholar]
- Monkhorst K, Jonkers I, Rentmeester E, Grosveld F, Gribnau J. X inactivation counting and choice is a stochastic process: evidence for involvement of an X-linked activator. Cell. 2008;132:410–421. doi: 10.1016/j.cell.2007.12.036. [DOI] [PubMed] [Google Scholar]
- Morey C, Navarro P, Debrand E, Avner P, Rougeulle C, Clerc P. The region 3′ to Xist mediates X chromosome counting and H3 Lys-4 dimethylation within the Xist gene. EMBO J. 2004;23:594–604. doi: 10.1038/sj.emboj.7600071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm FJ, Chenoweth JG, Anderson PD, Nadeau JH, Redline RW, McKay RD, Tesar PJ. Isolation of epiblast stem cells from preimplantation mouse embryos. Cell Stem Cell. 2011;8:318–325. doi: 10.1016/j.stem.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Avner P. An embryonic story: analysis of the gene regulative network controlling Xist expression in mouse embryonic stem cells. Bioessays. 2010;32:581–588. doi: 10.1002/bies.201000019. [DOI] [PubMed] [Google Scholar]
- Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, Schoorlemmer J, Rougeulle C, Chambers I, Avner P. Molecular coupling of Tsix regulation and pluripotency. Nature. 2010;468:457–460. doi: 10.1038/nature09496. [DOI] [PubMed] [Google Scholar]
- Navarro P, Pichard S, Ciaudo C, Avner P, Rougeulle C. Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: implications for X-chromosome inactivation. Genes Dev. 2005;19:1474–1484. doi: 10.1101/gad.341105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohhata T, Hoki Y, Sasaki H, Sado T. Tsix-deficient X chromosome does not undergo inactivation in the embryonic lineage in males: implications for Tsix-independent silencing of Xist. Cytogenet Genome Res. 2006;113:345–349. doi: 10.1159/000090851. [DOI] [PubMed] [Google Scholar]
- Ohhata T, Hoki Y, Sasaki H, Sado T. Crucial role of antisense transcription across the Xist promoter in Tsix-mediated Xist chromatin modification. Development. 2008;135:227–235. doi: 10.1242/dev.008490. [DOI] [PubMed] [Google Scholar]
- Pasque V, Gillich A, Garrett N, Gurdon JB. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 2011a;30:2373–2387. doi: 10.1038/emboj.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V, Halley-Stott RP, Gillich A, Garrett N, Gurdon JB. Epigenetic stability of repressed states involving the histone variant macroH2A revealed by nuclear transfer to Xenopus oocytes. Nucleus. 2011b;2:533–539. doi: 10.4161/nucl.2.6.17799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- Rastan S. Timing of X-chromosome inactivation in postimplantation mouse embryos. J Embryol Exp Morphol. 1982;71:11–24. [PubMed] [Google Scholar]
- Rastan S. Non-random X-chromosome inactivation in mouse X-autosome translocation embryos--location of the inactivation centre. J Embryol Exp Morphol. 1983;78:1–22. [PubMed] [Google Scholar]
- Sado T, Hoki Y, Sasaki H. Tsix silences Xist through modification of chromatin structure. Dev Cell. 2005;9:159–165. doi: 10.1016/j.devcel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Sado T, Li E, Sasaki H. Effect of TSIX disruption on XIST expression in male ES cells. Cytogenet Genome Res. 2002;99:115–118. doi: 10.1159/000071582. [DOI] [PubMed] [Google Scholar]
- Sado T, Wang Z, Sasaki H, Li E. Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development. 2001;128:1275–1286. doi: 10.1242/dev.128.8.1275. [DOI] [PubMed] [Google Scholar]
- Shin J, Wallingford MC, Gallant J, Marcho C, Jiao B, Byron M, Bossenz M, Lawrence JB, Jones SN, Mager J, et al. RLIM is dispensable for X-chromosome inactivation in the mouse embryonic epiblast. Nature. 2014 doi: 10.1038/nature13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell. 2003;4:481–495. doi: 10.1016/s1534-5807(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Stavropoulos N, Rowntree RK, Lee JT. Identification of developmentally specific enhancers for Tsix in the regulation of X chromosome inactivation. Mol Cell Biol. 2005;25:2757–2769. doi: 10.1128/MCB.25.7.2757-2769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi N. Primary and secondary nonrandom X chromosome inactivation in early female mouse embryos carrying Searle’s translocation T(X; 16)16H. Chromosoma. 1980;81:439–459. doi: 10.1007/BF00368155. [DOI] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RD. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Vigneau S, Augui S, Navarro P, Avner P, Clerc P. An essential role for the DXPas34 tandem repeat and Tsix transcription in the counting process of X chromosome inactivation. Proc Natl Acad Sci U S A. 2006;103:7390–7395. doi: 10.1073/pnas.0602381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.