

Abstract
Myeloperoxidase (MPO), lactoperoxidase (LPO) and eosinophil peroxidase (EPO) play a central role in oxidative damage in inflammatory disorders by utilizing hydrogen peroxide and halides/pseudo halides to generate the corresponding hypohalous acid. The catalytic sites of these enzymes contain a covalently modified heme group, which is tethered to the polypeptide chain at two ester linkages via the methyl group (MPO, EPO and LPO) and one sulfonium bond via the vinyl group (MPO only). Covalent cross-linking of the catalytic site heme to the polypeptide chain in peroxidases is thought to play a protective role, since it renders the heme moiety less susceptible to the oxidants generated by these enzymes. Mass-spectrometric analysis revealed the following possible pathways by which hypochlorous acid (HOCl) disrupts the heme-protein cross-linking: (1) the methyl-ester bond is cleaved to form an alcohol; (2) the alcohol group undergoes an oxygen elimination reaction via the formation of an aldehyde intermediate or undergoes a demethylation reaction to lose the terminal CH2 group; and (3) the oxidative cleavage of the vinyl-sulfonium linkage. Once the heme moiety is released it undergoes cleavage at the carbon-methyne bridge either along the δ-β or a α-γ axis to form different pyrrole derivatives. These results indicate that covalent cross-linking is not enough to protect the enzymes from HOCl mediated heme destruction and free iron release. Thus, the interactions of mammalian peroxidases with HOCl modulates their activity and sets a stage for initiation of the Fenton reaction, further perpetuating oxidative damage at sites of inflammation.
1. Introduction
Myeloperoxidase (MPO), eosinophil peroxidase (EPO), and lactoperoxidase (LPO) are homologous enzymes, all belonging to the heme peroxidase superfamily [1-5]. Although they differ from each other with respect to their sites of expression, primary sequences, and substrate specificities, they share between 50 and 70% overall protein amino acid sequence homology [6-11]. Additionally, all three contain a covalently linked heme prosthetic group in their active site, with a central iron atom coordinated to the four nitrogen atoms of the porphyrin ring, and also to a nitrogen atom provided by the proximal histidine residue [12]. Substrate binding to the catalytic site, which in these cases is the sixth coordinate position on the distal side of the heme group, is limited by the close proximity of surrounding amino acids [12]. The heme moiety of mammalian peroxidases is covalently bound to the protein, and uses hydrogen peroxide (H2O2) as the electron acceptor in the catalysis of halides and pseudohalides to produce the corresponding hypohalous acid [1, 12-18]. As shown in Fig. 1, the heme prosthetic of MPO is covalently attached to the protein through glutamate (R1), methionine (R2), and aspartate (R3) residues, while LPO and EPO are covalently attached to the protein through glutamate (R1) and aspartate (R3) residues only [19-24]. MPO x-ray structure analysis, like EPO and LPO, showed a non-planar configuration of the heme moiety in which pyrrole rings B and D are nearly co-planar, while rings A and C are tilted toward the distal side (Fig. 1 B) [20]. MPO is found in the azurophilic granules of the cells of myeloid origin, such as neutrophils and monocytes [11, 25]. It is composed of two identical subunits each containing a light chain with a molecular mass of 15 kDa and heavy chain with molecular mass of 60 kDa [11, 26]. The two subunits are joined to each other via a single disulfide bridge manipulating the architecture of the heme pocket and permitting the active dimeric form to be generated [11]. The heavy-chain contains the active site modified iron protoporphyrin IX, which is covalently connected to the heavy chain polypeptide [24, 27, 28]. The secreted MPO typically uses H2O2 and chloride (Cl−) as substrates to generate the potent oxidant hypochlorous acid (HOCl). Hypochlorous acid plays an important role in the innate immune response and aids in killing invading pathogens in the phagolysosome [29]. However, sustained high levels of HOCl result in tissue damage [1, 30-34]. EPO is a monomeric molecule comprised of a light chain and a heavy chain with molecular masses of 15.5 and 50 kDa, respectively [35]. The enzyme is stored in eosinophil granules and catalyzes the formation of antimicrobial species from the oxidation of Br– and SCN– [35-37]. LPO is a monomeric single polypeptide chain with a molecular mass of 78.5 kDa [38-41] and is implicated in the pathogenesis of lung diseases such as asthma [42-44]. LPO has been identified as an antimicrobial agent within exocrine gland secretions such as milk, saliva, and tears through the oxidation of thiocyanate by H2O2 to yield the intermediary oxidation product hypothiocyanite (OSCN–) [45, 46]. This enzyme has been linked to host defense and is believed to mediate infection risk in lung diseases such as cystic fibrosis [5, 47].
Figure 1.

A) Structure of heme cofactor of the three proteins used in this study (modified from [19-24]). B) Non planar heme porphyrin ring of myeloperoxidase and its covalent attachments to Asp94, Glu242 and Met243. The figure was constructed using the coordinates deposited in the Protein Data Bank (accession code 3F9P), using PyMOL (www.pymol.org).
Despite the potential significance of mammalian peroxidases to both human health and disease, little is known about the factors that govern their heme destruction and subsequent free iron release. Previously, we elucidated the remarkable ability of HOCl to oxidatively destroy the heme moiety of mammalian peroxidases and other related hemoproteins through a mechanism that involves the formation of a transient Fe(III)-OCl complex which was subsequently oxidized to ferryl intermediates, , either as an Fe(IV)=O porphyrin π-cation radical containing species (Compound I), or as an Fe(IV)=O (Compound II), which ultimately leads to heme degradation [48-52]. These processes have been extensively characterized in an in vitro cell free system such as free heme [50], purified mammalian peroxidases[51, 52], and purified hemoglobin (Hb) [48] and also as a model for the physiological system in human red blood cells [48]. Recent studies by Maitra et. al. have demonstrated that MPO may be regulated by feedback inhibition via HOCl [49, 51, 52]. HOCl reacts with MPO heme iron at nearly diffusion-controlled rates suggesting that the enzyme is a potential target for HOCl feedback destruction during steady state catalysis [52] [Maitra et. al., submitted for publication, unpublished observations]. Heme degradation products are extremely toxic to different cells and organs leading to serious pathological consequences [53]. Their toxicity is mainly due to their capacity to participate in the generation of reactive oxygen species that mediate cellular mitochondria poisoning, lipid peroxidation, and oxidative phosphorylation uncoupling [54-57]. Free iron damages blood vessels and produces vasodilation with increased vascular permeability, leading to hypotension and metabolic acidosis [57, 58].
In the present study, we utilize a H2O2-selective electrode and absorbance spectroscopy to determine if HOCl can mediate MPO, EPO and LPO heme destruction and augment the relative rate of enzyme activity for those enzymes. Then, we utilized liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS) to characterize HOCl interactions with MPO, EPO and LPO. Our results indicate that HOCl can mediate free Fe release through a mechanism that involves heme destruction, ultimately resulting in oxidative cleavage of the heme moiety, generating fluorescent and nonfluorescent porphyrin derivatives. This mechanism may contribute to vascular endothelial dysfunction that is induced by oxidative stress in various inflammatory diseases. A comprehensive chemical model that describes the possible pathways in which HOCl-mediated MPO, EPO and LPO heme destruction and the disruption of the heme-protein covalent crosslinking of mammalian peroxidases is presented. On the basis of the present results and recent published findings [48, 50-52, 59], we hypothesize that a complex bidirectional relationship exists between peroxidase activity and HOCl levels at sites of inflammation. HOCl serves to modulate peroxidase catalytic activity, while peroxidases serve as a catalytic sink for HOCl, influencing its bioavailability and function.
2. Material and Methods
2.1 Materials
All the materials used were of highest grade purity and used without further purification. Sodium hypochlorite (NaOCl), L-methionine, ascorbic acid, bovine Hb, dimethylformamide, methanol, and trifluoroacetic acid (TFA)—HPLC grade were obtained from Sigma Aldrich (St. Louis, MO, USA). HPLC-grade acetonitrile (CH3CN) was obtained from EMD Chemicals (Gibbstown, NJ, USA).
2.2 Enzyme purification
MPO was purified from detergent extracts of human leukocytes as previously described [60]. Trace levels of contaminating eosinophil peroxidase were then removed by passage over a sulfopropyl-Sephadex column [61]. The sample was judged pure by SDS-PAGE analysis and had a corresponding Reinheitszahl (RZ) value of >0.85 (A430/A280). Additionally in-gel tetramethylbenzidine peroxidase staining was used to confirm no observable contaminating eosinophil peroxidase activity [62]. EPO was purified from porcine whole blood as previously described [63]. The sample was judged pure by SDS-PAGE analysis and had a corresponding RZ value of >0.9 (A415/A280) [64]. Enzyme concentrations were determined spectrophotometrically utilizing molar absorptivities of 89,000 and 112,000 M–1 cm–1 per heme of MPO (429 nm) [62] and EPO (413 nm) [65, 66], respectively. Bovine LPO was obtained from Worthington Bio-Chemistry Corp. (Lakewood, NJ, USA) and used with further purification by sepharose column as previously reported [67] . The sample was judged pure by SDS-PAGE analysis and had a corresponding RZ value of >0.78 (A415/A280), [68]. LPO concentration was determined spectrophotometrically by utilizing an molar absorptivity of 112,000 M–1 cm–1 at 412 nm [65].
2.3 H2O2-selective electrode measurements
Hypochlorous acid measurements were carried out using an H2O2-selective electrode (Apollo 4000 free radical analyzer; World Precision Instruments, Sarasota, FL, USA). Experiments were performed at 25 °C by immersing the electrode in 3 mL of 0.2 M sodium phosphate buffer at pH 7.0. Prior to H2O2 addition, the electrode was immersed in the buffer solution, and after equilibration for at least 15 min, the signal was adjusted to zero as a baseline. Addition of 100 mM Cl− (final) or 40 nM MPO (final) alone or in combined had no effect on the zero baseline.
Where indicated, 10 μL MPO (40 nM final) was added to a continuously stirred buffer solution supplemented with 100 mM Cl− followed by the addition of MPO (40 nM final) pre-incubated with increasing concentration of HOCl (20 - 60 μM). The rise and fall of the H2O2 concentration was continuously monitored (the default sampling rate is 10 samples per second).
2.4 Absorbance measurements
The absorbance spectra were recorded using a Cary 100 Bio UV–visible spectrophotometer, at 25 °C and pH 7.0. Experiments were performed in 1 mL phosphate buffer solution supplemented with a fixed amount of MPO (1.0 μM) and increasing concentrations of HOCl. After five minutes of incubation for reaction completion, methionine (five-fold the final HOCl concentration) was added to eliminate excess HOCl and absorbance changes were recorded from 300 to 700 nm.
2.5 Mass spectrometric analysis of heme degradation products
After treatment of the enzymes with HOCl the reaction mixture was filtered through an Amicon Ultra-15 centrifugal filter unit with Ultracel-10 membrane (from Millipore) with a 3-kDa cut-off by centrifuging at 14,000 relative centrifugal force (rcf) for 30 min at 4 °C. Mass spectrometry experiments were performed using an Agilent 6410 triple-quadrupole mass spectrometer coupled with an Agilent 1200 HPLC system (Agilent Technologies, New Castle, DE, USA), equipped with an electrospray source. A Waters symmetry C18 column (particle size 3.5 μm; 2.1 × 100 mm) (Milford, MA, USA) was used to separate reaction products. Solvent A was H2O with 0.1% formic acid and solvent B was acetonitrile with 0.1% formic acid. The column was equilibrated with 80% solvent A and 20% solvent B. The gradient was 20–95% solvent B over 10 min, 95% solvent B for 10 min, 95–20% solvent B for 1 min, and 80% solvent A for 14 min. Five microliters of the sample was injected at a flow rate of 0.25 ml/min. LC-ESI–MS in the positive mode was performed using the following parameters: spray voltage 4000 V, drying gas flow 10 L/min, drying gas temperature 325 °C, and nebulizer pressure 40 psi. Fragmentor voltage was optimized using flow-injection analysis of hematin (Sigma Aldrich). Optimal fragmentor voltage was 210 V in MS2 scan mode. Mass range between m/z 150 and m/z 1450 was scanned to obtain full-scan mass spectra.
2.6 Solution preparation
HOCl preparation
HOCl was prepared as previously described with some modification [69]. Briefly, a stock solution of HOCl was prepared by adding 1 mL of NaOCl solution to 40 mL of 154 mM NaCl and the pH was adjusted to ~3 by adding HCl. The concentration of active total chlorine species in solution, expressed as [HOCl]T (where [HOCl]T = [HOCl] + [Cl2] + [Cl3–] + [OCl–]) in 154 mM NaCl, was determined by converting all the active chlorine species to OCl– by adding a bolus of 40 μL of 5 M NaOH followed by the measurement of the concentration of OCl–. The concentration of OCl– was determined spectrophotometrically at 292 nm (ε = 362 M–1 cm–1) [70]. As HOCl is unstable, the stock solution was freshly prepared on a daily basis, stored on ice, and used within 1 h of preparation. For further experimentation, dilutions were made from the stock solution using 200 mM phosphate buffer, pH 7, to make working solutions of lower HOCl concentrations.
2.7 HOCl treatment
MPO, LPO, and EPO (1-1.5 μM) were incubated with increasing concentration of HOCl (0, 100, 200 and 400 μM) for 1 hour for reaction completion. Five-fold molar excess of methionine with respect to H2O2 was added to stop the reaction. The reaction mixture was then filtered through an Amicon Ultra-15 centrifugal filter unit with Ultracel-10 membrane (from Millipore) with a 3-kDa cutoff by centrifuging at 14,000 rcf for 30 min at 4 °C. The filtrate was used for LC-MS analyses to detect heme degradation products.
3. Results
3.1 HOCl inhibited MPO catalytic activity in a dose dependent manner
Our initial experiments utilized a H2O2-selective electrode to determine whether exogenous HOCl can inhibit MPO catalytic activity. Catalytic amounts of MPO-Fe(III) (40 nM) were first incubated with increasing concentrations of HOCl (10-60 μM) for reaction completion (five minutes), and then the effect of HOCl on the enzyme catalytic activity was determined utilizing an H2O2-selective electrode. Addition of H2O2 (10 μM final) to a continuously stirred buffer solution supplemented with 100 mM Cl− caused rapid rise in the H2O2 signal, achieved a maximum after ~30 s, and fell gradually as H2O2 was depleted by disproportionation (2H2O2 → 2H2O +O2) (Fig. 2, left trace). Subsequent addition of the untreated MPO sample to the reaction mixture caused a rapid decay in the level of free H2O2 (Fig. 2, left trace), indicating that the enzyme is active and H2O2 is consumed as a substrate by MPO during steady-state catalysis. As shown in Fig. 2, slower rates of H2O2 consumption (longer duration of H2O2 signals) were observed when the same experiment was repeated by adding MPO samples pre-incubated with increasing concentrations of HOCl to the H2O2/Cl– mixture. Collectively, the H2O2 – selective electrode measurements revealed that exogenously added HOCl displayed the capacity to partially or completely inhibit MPO catalytic activity in a HOCl concentration dependent manner.
Figure 2.

Exogenous HOCl inhibited MPO catalytic activity in a dose-dependent manner. Following addition of 10 μM H2O2 (as indicated by the arrows) to a stirred 0.2 M sodium phosphate buffer supplemented with 100 mM Cl− (pH 7.0) at 25 °C showed a rapid buildup in H2O2 signal which then saturates indicating the stability of H2O2 solution (dashed line). Subsequent addition of 40 nM MPO shows rapid consumption of H2O2. When the same reaction was repeated by the addition of fixed amount of MPO (40 nM) pre-incubated with increasing concentrations of HOCl (for 5 mins), it showed a significant decrease in MPO activity. Tracings shown are from a typical experiment performed in triplicate or greater.
3.2 HOCl-mediated MPO, EPO, and LPO heme destruction
We next utilized absorbance spectroscopy to determine the mechanism by which HOCl inhibited MPO activity. MPO-Fe(III) displays a Soret absorbance peak centered at 430 nm and additional visible peaks at 573, 630, and 694 nm (Fig. 3, left panel), while LPO–Fe(III) (Fig. 3, right panel) and EPO-Fe(III) display Soret absorbance peaks at 413 nm and additional visible peaks at 502, 544, and 635 nm (data not shown). Addition of saturated amounts of HOCl (140 μM, final) to the ferric forms of MPO caused immediate heme destruction, as judged by a complete loss of the Soret feature (Fig. 3, left panel). Similar results were observed when saturated amounts of HOCl were added to LPO (Fig. 3, right panel) and EPO (data not shown). Our results indicated that peroxidase inhibition, mediated by HOCl, is due to heme degradation/destruction.
Figure 3.

HOCl inhibits MPO/LPO catalytic activity through a mechanism that involves heme destruction. The panels show the absorbance spectrum of MPO (1.2 μM)/LPO (0.8 μM) before (blue line) and after (red line) the addition of 200 μM HOCl.
3.3 LC-ESI-MS of heme fragmentation products
LC-ESI-MS in the positive mode was then utilized to elucidate the HOCl-mediated reaction products of MPO, EPO, and LPO. Fixed amounts of each enzyme were incubated with increasing concentrations of HOCl for 5 min (reaction completion). For each sample, the heme degradation products were extracted and subjected to LC-ESI-MS analysis. The majority of the products were identified by detecting the molecular ions [M + H]+. The methods we used are widely acceptable in the field and typical used by others. For example He et. al and Schaefer et. al. previously used these methods for the characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme [71, 72]. Table 1 summarizes the structures of the different heme degradation products that were identified for all the enzymes samples treated with increasing HOCl concentrations. Table 2 shows the relative fragment abundance as a function of HOCl concentration for each of the enzymes studied. We observed low levels of fragments for MPO m/z 349, 327 and 305 which did not meet our criteria signal-noise threshold.
Table 1.
Proposed structure molecular formula and m/z values of the heme degradation products as identified from LC-MS studies.
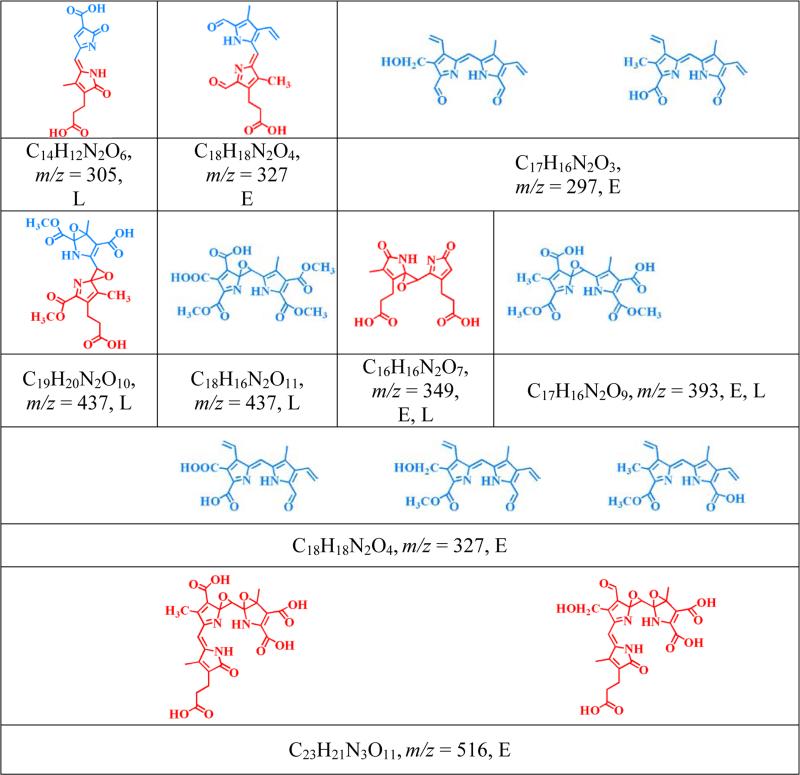
|
Single letter abbreviations, (M = MPO, E = EPO and L = LPO) denote the enzyme from which each product was detected following HOCl treatment.
Table 2.
Table of relative fragment abundance as a function of HOCl concentration for each of the enzymes studied.
| AUC under different HOCl concentrations (μM) | |||||||
|---|---|---|---|---|---|---|---|
| m/z | Retention time (min) | 25 | 50 | 100 | 200 | 400 | Enzyme |
| 305 | 3.10 | 410361 | 178206 | ND | ND | ND | LPO |
| 327 | 2.944 | 494201 | 75478 | 269720 | 392143 | EPO | |
| 437 | 10.74 | 40446 | 76157 | ND | ND | ND | LPO |
| 349 | 4.57 | 96507 | 61260 | ND | ND | ND | LPO |
| 2.95 | 477644 | 106787 | 234896 | 359623 | EPO | ||
| 297 | 2.692 | 98081 | 17189 | 56441 | 71033 | EPO | |
| 393 | 5.23 | 4224 | ND | ND | ND | ND | LPO |
| 3.262 | 268307 | 59775 | 140642 | 157746 | EPO | ||
| 516 | 3.597 | 1160964 | 177697 | 738459 | 1119200 | EPO | |
The area under the curve (AUC) was derived from the EIC of the LC-MS-ESI experiments. ‘ND’ = not detected.
The identified heme degradation products could be grouped into two different categories based on their pattern of cleavage. A proposed scheme that describes the possible pathways of generation of these fragments by the action of HOCl on the heme moiety is shown in Fig. 4. Group 1 consists of tripyrrole derivatives formed by the successive cleavage of the heme-protein covalent bonds and the cleavage of one pyrrole ring from the parent tetrapyrrole ring. Two structural/functional isomers were assigned to m/z 516. Fig. 5 depicts the EIC (extracted ion chromatogram) and MS spectrum for m/z 516 detected from the EPO-HOCl reaction mixture. The oxidative cleavage of the γ carbon-methyne bridge led to the formation of ketone (–C=O), while the β carbon-methyne bridge was sequentially oxidized, first to aldehyde (–CHO) and then to the –COOH group. The difference between the two isomers is at the modification at the R1 and R2 positions. In one isomer the R2 group was oxidized to –COOH and the R1 group was first hydrolyzed to –OH and then subsequently the oxygen atom was eliminated to form the –CH3 derivative. While in the second isomer, R2 was oxidized to –CHO and the R1 ester was hydrolyzed to –OH.
Figure 4.

Schematic representation for the route of the generation of different heme degradation products following the action of HOCl on the heme moiety. The structures are color coded with darker and lighter colors to represent which segments of the tetrapyrrole ring are cleaved to generate specific fragments. Structural/functional isomers corresponding to the same m/z values/molecular weights are represented by ‘or’.
Figure 5.

Formation of tripyrrole derivatives by the action of HOCl on the active site heme moiety. EPO-HOCl reaction mixture was separated by reverse-phase HPLC and subjected to ESI/MS in the positive ion mode as described under the Materials and Methods section. The major reaction product produced an intense peak at 3.8 min. Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 516. The extracted ion chromatogram (A) and the MS spectrum of this peak (B) are depicted. See Table 1 for the assigned molecular formula and structure corresponding to this product.
Group 2 consists of dipyrrole derivatives and could be divided into two sub-groups: 2A and 2B. Group 2A had dipyrrole compounds that were generated by the cleavage of the tetrapyrrole ring along the δ-β axis. Mass spectral analytes m/z 297, 327, 349, 393 and 437 are examples of this category. While m/z 327 and 349 were observed from all the three enzymes, m/z 297 was only formed from EPO and m/z 437 from LPO. The dipyrrole derivative m/z 393 was formed in EPO and LPO extracts. Three different functional isomers are proposed for m/z 327, which differ in the nature of the modification at the R1 position and the final oxidized form at the δ carbon-methyne bridge. While two of the isomers display a –OCH3 modification at the δ carbon-methyne bridge, the third one displays a –COOH modification. The modification at the R1 ester bond shows a wide range of variation, from –OH, -COOH to –CH3. The dipyrrole derivative, m/z 297 also displays two functional isomers, with respect to the modification at R1 and δ carbon-methyne bridge. The amino acid-heme ester bond at R1 was hydrolyzed to form an –OH and the δ carbon-methyne bridge was oxidized to –CHO in one of the isomers. While in the second isomer, the R1 was modified to form a –CH3 and the δ carbon-methyne bridge was oxidized to –COOH. The EIC and MS spectrum of two representative members of this group, m/z 349 and m/z 297, are shown in Fig. 6. Group 2B consisted of dipyrrole derivatives generated by cleavage along the α-γ axis. Mass spectral analytes m/z 305, 327, and 437 are examples of this class of compound. The mass spectral analyte m/z 327 was observed from MPO and EPO, m/z 305 was observed in MPO and LPO, while m/z 437 was observed in LPO only. The EIC and MS spectrum of two representative members of this group, namely m/z 327 and m/z 305 are shown in Fig. 7.
Figure 6.

HOCl-mediated cleavage of the heme moiety along the δ-β axis to generate dipyrrole derivatives. Mass spectral analyte m/z 349 was detected in MPO, EPO and LPO-HOCl reaction mixtures, while, m/z 297 was detected only in EPO-HOCl reaction mixture. The reaction mixtures were separated by reverse-phase HPLC and subjected to ESI/MS in the positive ion mode as described under Materials and Methods. The major reaction product produced an intense peak at 4.2 min (A) and 2.8 min (C). Examination of the mass spectrum revealed that the [M + H]+ ion had a m/z 349 (B) and 297 (D). The extracted ion chromatograms (A) and (C) and the MS spectra (B) and (D) are depicted. See Table 1 for the assigned molecular formula and structure corresponding to this product.
Figure 7.

HOCl-mediated cleavage of the heme moiety along the α-γ axis to generate dipyrrole derivatives. Mass spectral analytes m/z 327 was detected in MPO, EPO and LPO-HOCl reaction mixtures, while, m/z 305 was detected only in MPO and LPO-HOCl reaction mixtures. The reaction mixtures were separated by reverse-phase HPLC and subjected to ESI/MS in the positive ion mode as described under Materials and Methods. The major reaction product produced an intense peak at 3 min (A) and 2.5 min (C). Examination of the MS spectrum revealed that the [M + H]+ ion had a m/z 327 (B) and 305 (D). The extracted ion chromatograms (A) and (C) and the mass spectra (B) and (D) are depicted. See Table 1 for the assigned molecular formula and structure corresponding to this product.
4. Discussion
MPO, EPO, and LPO, are receiving increasing attention due to their roles in host defenses and their contribution to the pathogenesis of tissue injury in inflammatory disorders [1-5, 73]. These enzymes operate in their ferric forms to utilize H2O2 and halides (Cl−, Br−)/pseudo halides (SCN−) to produce the corresponding hypohalous acids (HOCl, HOBr, HOSCN) [74]. During inflammation the enzymes catalytic sites, which have the heme prosthetic group, are target of a multitude of reactive oxidants and diffusible radical species either as substrates or ligands. Given the extreme sensitivity of the heme moiety to these oxidants [48, 50, 51, 75], the enzymes might adopt a protective mechanism to prolong their lifetime despite their exposure to these oxidants [76]. This assumption is built on the previous mutational analysis by Huang et. al. on horseradish peroxidase which demonstrated that the heme-protein covalent cross-linking performs this protective role [76]. This covalent tethering of the heme to the polypeptide chain, renders the carbon-methyne bridges less susceptible to the electrophilic/nucleophilic attack of these reactive species. Our current and recently published results have shown that HOCl, the final product of MPO, not only mediates oxidative destruction of free heme, but also mediates the destruction of the heme moiety bound to the protein covalently (e.g. MPO and LPO) or noncovalently (e.g. hemoglobin) [48, 50]. The present study elucidates a global chemical mechanism by which HOCl-mediates disruption of the heme-protein covalent cross-linking in mammalian peroxidases (MPO, EPO and LPO) and releases redox active free iron.
Hydrogen peroxide-selective electrode and absorbance measurements indicated that HOCl down regulates MPO catalytic activity through a mechanism that involves MPO heme destruction. LC-MS analysis confirms this notion and indeed a total of twelve heme degradation products were identified from MPO, EPO, and LPO treated with increasing concentrations of HOCl. The heme modification and fragmentations were tentatively identified from their m/z values, the comparison with the previously identified products, and on the chemical reactivity of HOCl with carbon–carbon double bonds [48, 50, 59, 75, 77]. For simplicity, a schematic representation for the generation of different heme degradation products following the action of HOCl on the heme moiety that applies for all three enzymes is shown is Fig. 4. Heme release requires the liberation of the axial ligand and the oxidative cleavage of all the three heme-protein covalent bonds (R1-R3). However, the release of free iron does not necessarily require oxidative cleavage of all the three heme-protein covalent bonds, but does require the opening or degradation of heme moiety. HOCl can either attack the covalent tethering points leading to the release of the intact tetrapyrrole ring and/or randomly attack any of the four carbon–methyne bridges [19-24]. At the HOCl concentrations used in this study, the intact tetrapyrrole ring was not detected, but instead one tripyrrole, and eleven dipyrroles were identified. These resulted from further oxidative cleavage of the heme moiety following MPO, EPO and LPO treatment with HOCl. The tripyrrole (m/z 516) derivative with no iron was detected from the extracts of the EPO-HOCl mixture. The population of this fragment increased with increasing HOCl concentrations (Table 2). The separation of the pyrrole ring C from the heme moiety indicates the γ and β carbon-methyne bridges are more exposed to solution and more susceptible to HOCl attacks. The x-ray structure analysis revealed that the heme moiety in mammalian peroxidases adopts a nonplannar configuration in which pyrrole rings B and D are nearly co-planar, while ring A and, to a lesser degree, ring C are tilted toward the distal side (see Fig. 1, B) [78]. Because of these distortions to the porphyrin ring, the α, β, γ, and δ carbon-methyne bridges become more susceptible to HOCl attack, which in turn negatively affects the affinity of the heme iron toward the axial ligands [19-24]. The majority of the twelve identified compounds are dipyrrole derivatives formed by HOCl mediated cleavage of the tetrapyrrole ring either through the δ-β axis or through the α-γ axis. Only one tripyrrole (m/z 516) derivative were detected (see Fig. 4 and Table 1). A careful analysis of the heme degradation products revealed that cleavage of the heme-protein covalent bonding led to five different types of modification, including hydrolysis of the ester bond to form an alcohol (–OH), oxidation of –OH to carboxylic acid (–COOH) which was subsequently converted to methyl ester (–OCH3), demethylation of –OH bound methyl group (–CH3), elimination of oxygen from –OH to form a –CH3 group and cleavage of the sulfonium linkage. The oxidative cleavage of the C=C of the tetrapyrrole ring carbon-methyne bridges and terminal vinyl group followed the same pattern as previously reported [48, 50], namely epoxide, diols, aldehyde (–CHO), ketone (–C=O), –COOH, and –OCH3 formation.
Three dipyrrole derivatives of molecular weights m/z 371, 349 and 327 were detected when MPO, EPO and LPO were treated with HOCl. As mentioned earlier, MPO is covalently bound to three different points along the heme ring, while EPO and LPO are bound through two heme-amino acid ester bonds. Thus it could be deduced that MPO would be less susceptible to HOCl-mediated cleavage compared to EPO and LPO. Indeed, when the relative amount of m/z 349 and 327 (relative quantification by the area under the curve in the EIC) were compared to each other it was observed that at all HOCl concentrations tested, there was a higher amount of the heme degradation products in EPO and LPO when compared to MPO (see the Results section and Table 2).
In general, the mechanism of oxidative cleavage of the C=C bonds in the carbon-methyne bridge and the terminal vinyl group is similar to that previously reported [48, 50]. In addition to oxidation of C=C bonds, the present work extends prior observation and proposes novel structures generated by the modification of the heme-protein covalent cross-links.
As shown in our results, four major types of modifications are observed depending on the position involved. The first three types are observed at positions R1 and R3 which are amino acid-heme ester bonds. The first proposed mechanism for hydrolysis is depicted in Fig. 8 (I). We believe that protonation, generated by hydrolysis of HOCl, leads to a diol-like intermediate, which by the action of further protonation leads to intermolecular electron rearrangement formimg –OH and –COOH functional groups. In the proceeding step, as shown in Fig. 8 (II), the –OH most likely aids in a demethylation process as has been previously described by our group [77]. Alternatively the –OH group generated from ester hydrolysis might undergo a process of elimination with conversion to a –CH3 substituent (see Fig. 8 (III)). We believe that the –OH group is oxidized to form a –CHO, which by the action of HOCl, is converted to an –OH and –OCl tetrahedral intermediate. This intermediate, by the action of another molecule of HOCl (on the C–OH bond) and electron rearrangement of the C–OCl bond, is converted to a C=OCl species and then to two –OCl bound species. This might lead to the release of an –OCl molecule to form a radical, which abstracts a proton from another molecule of HOCl, (H atom shown in red) [48, 50, 59]. This intermediate forms a radical, which then abstracts a proton from HOCl (shown in blue) to form the –CH3 group. In the case of MPO, the R2 position is occupied by the sulfhydryl bond between the methionine-heme macrocycle. As shown in Fig. 8 (IV), the sulfonium bond is cleaved by HOCl. We believe that this process is initiated by the heterolytic cleavage of the HOCl molecule and the attack of the OH moiety on the sulfur bound –CH3 group. This most likely leads to abstraction of the –CH3 group, which combines with the OH to form a CH3-OCl (MeO-Cl) moiety. After the formation of a chlorinated sulfur intermediate, we believe that the bond is broken by the •OH radical generated from the action of HOCl and free iron. Two resonant stabilized intermediates are formed and react with a proton generated from HOCl to form the sulfonium product. In addition, we note that some of the structures show –OCH3 modifications. One likely explanation is that the amino acid ester bond is hydrolyzed to –OH (Fig. 8 (I)) which is then sequentially oxidized to form –COOH (as previously reported [48, 50, 59]. It is possible that oxidation of the terminal C=C bond leads to the formation of formaldehyde. This single-carbon aldehyde could then be oxidized to formic acid by addition of HOCl. Thus, the formaldehyde generated is used for the formation of –OCH3 as seen in some of the structures. Collectively, through these simultaneous mechanisms, HOCl mediates the cleavage of the heme tetrapyrrole macrocycle from the peptide chain to generate free heme.
Figure 8.

Proposed chemical mechanism for (I) hydrolysis of amino acid esters to alcohol, (II) demethylation of alcohol, (III) elimination of oxygen from alcohol and (IV) sulfonium bond cleavage.
Activated neutrophils generate around 150−425 μM HOCl/hr, whereas at sites of inflammation, the HOCl levels are estimated to reach as high as 5 mM [79, 80]. At these concentrations, HOCl is sufficient to destroy the heme moiety of mammalian peroxidases. Indeed, the current findings and recently published results showed that HOCl, exogenously added or self-generated during steady-state catalysis of MPO, is capable of destroying the heme moiety of MPO and other hemoproteins [48, 50-52]. Our finding is inconsistent with that previously reported by Montellano and co-workers who suggested that covalent linkages between the prosthetic heme and the protein in mammalian peroxidases serve to protect the heme moiety from modification by the reactive HOX (where X = Cl−, Br−, and SCN−) species generated during the catalytic process [76]. Their assumption was built on the concept that the horseradish peroxidase (HRP) F41E mutant, which forms one covalent bond between the heme and the protein, is resistant to modification by autocatalytically generated HOBr. Whereas HOBr modified the heme of native HRP in which no heme-protein covalent bond exists. Additionally, the LPO E375D mutant, which only retained one of the two normal heme-protein covalent bonds, remained completely resistant to heme modification by autocatalytically generated HOBr [76]. These rather contradictory results may be due to the fact that HOCl is a stronger acid than HOBr, and is not only capable of causing the destruction of the heme–protein bonds but also the heme moiety of mammalian peroxidases.
Free heme is extremely toxic by virtue of its ability to mediate protein oxidation, covalent cross-linking, and aggregate formation (see [56] for a detailed review). Free heme also acts as a pro-inflammatory molecule and heme-induced inflammation is involved in the pathology of diverse conditions such as renal failure, atherosclerosis, complications after artificial blood transfusion, peritoneal endometriosis, and heart transplant failure [56]. Additionally, given the hydrophobic nature of heme, it can easily permeate and intercalate the cell membrane lipid bilayer [81, 82]. Reactive oxygen species then act on the membrane incorporated heme to release the redox active free iron which can catalyze lipid peroxidation and DNA damage [56, 81-83]. Given the relatively stable nature of free heme it could be transported through the circulation and elicit a systemic response. We used pathophysiologically relevant concentrations of HOCl for these experiments. Thus even if the HOCl concentration is not high enough to mediate release of free iron, release of free heme might act through a ‘delayed release’ mechanism to amplify and prolong the toxicity of HOCl. On the other hand, at higher HOCl concentrations (which could occur at sites of inflammation), the heme moiety will be destroyed to release free iron. Free iron is toxic since it can act as the Fenton reagent and generate highly reactive free radicals such as hydroxyl and peroxyl, which mediate oxidative damage to a variety of biomolecules [54, 55, 57]. Free iron could also be released through the incomplete cleavage of the heme ring from the protein chain such as through the generation of dipyrrole derivatives. Thus, this might be a potential mechanism for perpetuating oxidative damage in inflammatory disorders.
5. Table of Abbreviations
- MPO
Myeloperoxidase
- EPO
Eosinophil Peroxidase
- LPO
Lactoperoxidase
- Hb
Hemoglobin
- LC-ESI-MS
Liquid Chromatography Electrospray Ionization Mass Spectrometry
- TFA
Trifluoroacetic Acid
- RZ
Reinheitszahl
- RCF
Relative centrifugal force
- EIC
Extracted ion chromatogram
- HRP
Horseradish Peroxidase
References
- 1.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Antioxid Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 2.Klebanoff SJ. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 3.Mitra SN, Slungaard A, Hazen SL. Redox Rep. 2000;5:215–224. doi: 10.1179/135100000101535771. [DOI] [PubMed] [Google Scholar]
- 4.Weiss SJ, Test ST, Eckmann CM, Roos D, Regiani S. Sci J. 1986;234:200–203. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 5.Gerson C, Sabater J, Scuri M, Torbati A, Coffey R, Abraham JW, Lauredo I, Forteza R, Wanner A, Salathe M, Abraham WM, Conner GE. Am J Respir Cell Mol Biol. 2000;22:665–671. doi: 10.1165/ajrcmb.22.6.3980. [DOI] [PubMed] [Google Scholar]
- 6.Belding ME, Klebanoff SJ, Ray CG. Sci J. 1970;167:195–196. doi: 10.1126/science.167.3915.195. [DOI] [PubMed] [Google Scholar]
- 7.Kimura S, Ikeda-Saito M. Protein J. 1988;3:113–120. doi: 10.1002/prot.340030206. [DOI] [PubMed] [Google Scholar]
- 8.Petrides PE. J Mol Med. 1998;76:688–698. doi: 10.1007/s001090050269. [DOI] [PubMed] [Google Scholar]
- 9.Ten RM, Pease LR, McKean DJ, Bell MP, Gleich GJ. J Exp Med. 1989;169:1757–1769. doi: 10.1084/jem.169.5.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda T, Sakamaki K, Kuroki T, Yano I, Nagata S. Eur J Biochem. 1997;243:32–41. doi: 10.1111/j.1432-1033.1997.0032a.x. [DOI] [PubMed] [Google Scholar]
- 11.Nauseef WM, Malech HL. Blood. 1986;67:1504–1507. [PubMed] [Google Scholar]
- 12.Zederbauer M, Furtmuller PG, Brogioni S, Jakopitsch C, Smulevich G, Obinger C. Nat Prod Rep. 2007;24:571–584. doi: 10.1039/b604178g. [DOI] [PubMed] [Google Scholar]
- 13.Abu-Soud HM, Hazen SL. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 14.Abu-Soud HM, Khassawneh MY, Sohn JT, Murray P, Haxhiu MA, Hazen SL. Biochemistry. 2001;40:11866–11875. doi: 10.1021/bi011206v. [DOI] [PubMed] [Google Scholar]
- 15.Galijasevic S, Saed GM, Hazen SL, Abu-Soud HM. Biochemistry. 2006;45:1255–1262. doi: 10.1021/bi051438k. [DOI] [PubMed] [Google Scholar]
- 16.Marquez LA, Dunford HB, Van Wart H. J Biol Chem. 1990;265:5666–5670. [PubMed] [Google Scholar]
- 17.Kettle AJ, Winterbourn CC. Redox Rep. 1997;3:3–15. doi: 10.1080/13510002.1997.11747120. [DOI] [PubMed] [Google Scholar]
- 18.Marquez LA, Huang JT, Dunford HB. Biochemistry. 1994;33:1447–1454. doi: 10.1021/bi00172a022. [DOI] [PubMed] [Google Scholar]
- 19.Battistuzzi G, Bellei M, Vlasits J, Banerjee S, Furtmuller PG, Sola M, Obinger C. Arch Biochem Biophys. 2010;494:72–77. doi: 10.1016/j.abb.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 20.Fiedler TJ, Davey CA, Fenna RE. J Biol Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 21.Oxvig C, Thomsen AR, Overgaard MT, Sorensen ES, Hojrup P, Bjerrum MJ, Gleich GJ, Sottrup-Jensen L. J Biol Chem. 1999;274:16953–16958. doi: 10.1074/jbc.274.24.16953. [DOI] [PubMed] [Google Scholar]
- 22.Rae TD, Goff HM. J Biol Chem. 1998;273:27968–27977. doi: 10.1074/jbc.273.43.27968. [DOI] [PubMed] [Google Scholar]
- 23.Sheikh IA, Singh AK, Singh N, Sinha M, Singh SB, Bhushan A, Kaur P, Srinivasan A, Sharma S, Singh TP. J Biol Chem. 2009;284:14849–14856. doi: 10.1074/jbc.M807644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng J, Fenna RE. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 25.Klebanoff SJ, Clark RA. The neutrophil: function and clinical disorders. 1978:1–810. [Google Scholar]
- 26.Nauseef WM, Cogley M, McCormick S. J Biol Chem. 1996;271:9546–9549. doi: 10.1074/jbc.271.16.9546. [DOI] [PubMed] [Google Scholar]
- 27.Dugad LB, La Mar GN, Lee HC, Ikeda-Saito M, Booth KS, Caughey WS. The J Biol Chem. 1990;265:7173–7179. [PubMed] [Google Scholar]
- 28.Taylor KL, Strobel F, Yue KT, Ram P, Pohl J, Woods AS, Kinkade JM., Jr. Arch Biochem Biophys. 1995;316:635–642. doi: 10.1006/abbi.1995.1083. [DOI] [PubMed] [Google Scholar]
- 29.Prokopowicz ZM, Arce F, Biedron R, Chiang CL, Ciszek M, Katz DR, Nowakowska M, Zapotoczny S, Marcinkiewicz J, Chain BM. Open J Immunol. 2010;184:824–835. doi: 10.4049/jimmunol.0902606. [DOI] [PubMed] [Google Scholar]
- 30.Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. J Biol Chem. 1999;274:34621–34628. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 31.Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, Jackson-Lewis V, Vila M, Vonsattel JP, Heinecke JW, Przedborski S. J Neurosci. 2005;25:6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss SJ. New Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 33.Pennathur S, Heinecke JW. Curr Diab Rep. 2007;7:257–264. doi: 10.1007/s11892-007-0041-3. [DOI] [PubMed] [Google Scholar]
- 34.Vivekanadan-Giri A, Wang JH, Byun J, Pennathur S. Rev Endocr Metab Dis. 2008;9:275–287. doi: 10.1007/s11154-008-9093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolscher BG, Zoutberg GR, Cuperus RA, Wever R. Biochim Biophys Acta. 1984;784:189–191. doi: 10.1016/0167-4838(84)90127-4. [DOI] [PubMed] [Google Scholar]
- 36.Arlandson M, Decker T, Roongta VA, Bonilla L, Mayo KH, MacPherson JC, Hazen SL, Slungaard A. J Biol Chem. 2001;276:215–224. doi: 10.1074/jbc.M004881200. [DOI] [PubMed] [Google Scholar]
- 37.Caulfield JP, Korman G, Butterworth AE, Hogan M, David JR. J Cell Biol. 1980;86:46–63. doi: 10.1083/jcb.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cals MM, Mailliart P, Brignon G, Anglade P, Dumas BR. Eur J Biochem. 1991;198:733–739. doi: 10.1111/j.1432-1033.1991.tb16073.x. [DOI] [PubMed] [Google Scholar]
- 39.Carlstrom A. Acta Chem Scand. 1969;23:185–202. doi: 10.3891/acta.chem.scand.23-0185. [DOI] [PubMed] [Google Scholar]
- 40.Dull TJ, Uyeda C, Strosberg AD, Nedwin G, Seilhamer JJ. DNA Cell Biol. 1990;9:499–509. doi: 10.1089/dna.1990.9.499. [DOI] [PubMed] [Google Scholar]
- 41.Ferrari RP, Laurenti E, Cecchini PI, Gambino O, Sondergaard I. J Inorg Biochem. 1995;58:109–127. doi: 10.1016/0162-0134(94)00041-8. [DOI] [PubMed] [Google Scholar]
- 42.Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Free Radic Biol Med. 2003;35:213–225. doi: 10.1016/s0891-5849(03)00278-8. [DOI] [PubMed] [Google Scholar]
- 43.Wedes SH, Wu W, Comhair SA, McDowell KM, DiDonato JA, Erzurum SC, Hazen SL. J Pediatr. 2011;159:248–255. e241. doi: 10.1016/j.jpeds.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu W, Samoszuk MK, Comhair SA, Thomassen MJ, Farver CF, Dweik RA, Kavuru MS, Erzurum SC, Hazen SL. TJ Clin Invest. 2000;105:1455–1463. doi: 10.1172/JCI9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reiter B, Perraudin JP. Everse J, Everse KE, Grisham MB, editors. Peroxidases in Chemistry and Biology. 1991. pp. 143–180.
- 46.Wolfson LM, Sumner SS. J. Food Prot. 1993;56:887–892. doi: 10.4315/0362-028X-56.10.887. [DOI] [PubMed] [Google Scholar]
- 47.Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. FEBS Lett. 2007;581:271–278. doi: 10.1016/j.febslet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maitra D, Byun J, Andreana PR, Abdulhamid I, Diamond MP, Saed GM, Pennathur S, Abu-Soud HM. Free Radic Biol Med. 2011;51:374–386. doi: 10.1016/j.freeradbiomed.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maitra D, Abdulhamid I, Diamond MP, Saed GM, Abu-Soud HM. J Pineal Res. 2012;53:198–205. doi: 10.1111/j.1600-079X.2012.00988.x. [DOI] [PubMed] [Google Scholar]
- 50.Maitra D, Byun J, Andreana PR, Abdulhamid I, Saed GM, Diamond MP, Pennathur S, Abu-Soud HM. Free Radic Biol Med. 2011;51:364–373. doi: 10.1016/j.freeradbiomed.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souza CE, Maitra D, Saed GM, Diamond MP, Moura AA, Pennathur S, Abu-Soud HM. PloS One. 2011;6:e27641. doi: 10.1371/journal.pone.0027641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maitra D, Shaeib F, Abdulhamid I, Abdulridha RM, Saed GM, Diamond MP, Pennathur S, Abu-Soud HM. Free Radic Biol Med. 2013;63C:90–98. doi: 10.1016/j.freeradbiomed.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Pharmacol Therapeuti. 2006;111:327–345. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 54.Clark RA. J Invest Dermatol. 2008;128:2361–2364. doi: 10.1038/jid.2008.257. [DOI] [PubMed] [Google Scholar]
- 55.Crichton RR, Wilmet S, Legssyer R, Ward RJ. J Inorg Biochem. 2002;91:9–18. doi: 10.1016/s0162-0134(02)00461-0. [DOI] [PubMed] [Google Scholar]
- 56.Kumar S, Bandyopadhyay U. Toxicol Lett. 2005;157:175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 57.Ong WY, Halliwell B. Ann N Y Acad Sci. 2004;1012:51–64. doi: 10.1196/annals.1306.005. [DOI] [PubMed] [Google Scholar]
- 58.Trinder D, Fox C, Vautier G, Olynyk JK. Gut. 2002;51:290–295. doi: 10.1136/gut.51.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pennathur S, Maitra D, Byun J, Sliskovic I, Abdulhamid I, Saed GM, Diamond MP, Abu-Soud HM. Free Radic Biol Med. 2010;49:205–213. doi: 10.1016/j.freeradbiomed.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rakita RM, Michel BR, Rosen H. Biochemistry. 1990;29:1075–1080. doi: 10.1021/bi00456a033. [DOI] [PubMed] [Google Scholar]
- 61.Wever R, Plat H, Hamers MN. FEBS Lett. 1981;123:327–331. doi: 10.1016/0014-5793(81)80320-1. [DOI] [PubMed] [Google Scholar]
- 62.Agner K. Acta Chem Scand. 1963;17 [Google Scholar]
- 63.Jorg A, Pasquier JM, Klebanoff SJ. Biochim Biophys Acta. 1982;701:185–191. doi: 10.1016/0167-4838(82)90112-1. [DOI] [PubMed] [Google Scholar]
- 64.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Biochem J. 1997;327(Pt 2):487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolscher BG, Plat H, Wever R. Biochim Biophys Acta. 1984;784:177–186. doi: 10.1016/0167-4838(84)90125-0. [DOI] [PubMed] [Google Scholar]
- 66.Carlson MG, Peterson CG, Venge P. Open J Immunol. 1985;134:1875–1879. [PubMed] [Google Scholar]
- 67.Atasever A, Ozdemir H, Gulcin I, Irfan Kufrevioglu O. Food Chem. 2013;136:864–870. doi: 10.1016/j.foodchem.2012.08.072. [DOI] [PubMed] [Google Scholar]
- 68.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 69.Wang L, Bassiri M, Najafi R, Najafi K, Yang J, Khosrovi B, Hwong W, Barati E, Belisle B, Celeri C, Robson MC. J Burns Wounds. 2007;6:e5. [PMC free article] [PubMed] [Google Scholar]
- 70.Furman CS, Margerum DW. Inorg Chem. 1998;37:4321–4327. doi: 10.1021/ic980262q. [DOI] [PubMed] [Google Scholar]
- 71.Schaefer WH, Harris TM, Guengerich FP. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 72.He K, Bornheim LM, Falick AM, Maltby D, Yin H, Correia MA. Biochemistry. 1998;37:17448–17457. doi: 10.1021/bi9808464. [DOI] [PubMed] [Google Scholar]
- 73.Nicholls SJ, Hazen SL. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 74.Davies MJ. J Clin Biochem Nutr. 2011;48:8–19. doi: 10.3164/jcbn.11-006FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagababu E, Rifkind JM. Antioxid Redox Signal. 2004;6:967–978. doi: 10.1089/ars.2004.6.967. [DOI] [PubMed] [Google Scholar]
- 76.Huang L, Wojciechowski G, Ortiz de Montellano PR. J Biol Chem. 2006;281:18983–18988. doi: 10.1074/jbc.M602307200. [DOI] [PubMed] [Google Scholar]
- 77.Abu-Soud HM, Maitra D, Byun J, Souza CE, Banerjee J, Saed GM, Diamond MP, Andreana PR, Pennathur S. Free Radic Bio Med. 2012;52:616–625. doi: 10.1016/j.freeradbiomed.2011.10.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furtmuller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Arch Biochem Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 79.Kettle AJ, Winterbourn CC. Methods Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- 80.Weiss SJ. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 81.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Proc Natl Acad Sci U S A. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Belcher JD, Beckman JD, Balla G, Balla J, Vercellotti G. Antioxid Redox Signal. 2010;12:233–248. doi: 10.1089/ars.2009.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Halliwell B, Gutteridge JM. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]