

Significance
Immune escape by continuous genetic diversification is commonly described using a metaphor of “arms race” between virus and host. Here we show, however, that hepatitis C virus (HCV) genetic heterogeneity facilitates a stable state of immune adaptation rather than perpetuates an arms race. We developed a mathematical model that considers variation in breadth of immunoreactivity among antibodies and antigens and the contribution of immune memory to humoral response against HCV. The model applied to a complex network of cross-immunoreactivity suggests antigenic cooperation among variants as a mechanism of mitigating the immune pressure on certain HCV variants. Cooperative action of antigens results from their differential capability to bind or elicit highly polyspecific antibodies. Antigenic cooperation is a new target for therapeutic- and vaccine-development strategies.
Keywords: hepatitis C, viral quasispecies, cross-immunoreactivity, complex network, intrahost adaptation
Abstract
Hepatitis C virus (HCV) has the propensity to cause chronic infection. Continuous immune escape has been proposed as a mechanism of intrahost viral evolution contributing to HCV persistence. Although the pronounced genetic diversity of intrahost HCV populations supports this hypothesis, recent observations of long-term persistence of individual HCV variants, negative selection increase, and complex dynamics of viral subpopulations during infection as well as broad cross-immunoreactivity (CR) among variants are inconsistent with the immune-escape hypothesis. Here, we present a mathematical model of intrahost viral population dynamics under the condition of a complex CR network (CRN) of viral variants and examine the contribution of CR to establishing persistent HCV infection. The model suggests a mechanism of viral adaptation by antigenic cooperation (AC), with immune responses against one variant protecting other variants. AC reduces the capacity of the host’s immune system to neutralize certain viral variants. CRN structure determines specific roles for each viral variant in host adaptation, with variants eliciting broad-CR antibodies facilitating persistence of other variants immunoreacting with these antibodies. The proposed mechanism is supported by empirical observations of intrahost HCV evolution. Interference with AC is a potential strategy for interruption and prevention of chronic HCV infection.
Hepatitis C virus (HCV) causes chronic infection in of infected people, who become at risk for developing severe liver diseases (1). The virus establishes chronic infection by using several molecular mechanisms for averting innate immunity and attenuating effectiveness of adaptive immune responses (2). HCV is one of the most heterogeneous viruses infecting humans and exists in each infected host as a population of genetically related variants (3, 4). Substantial heterogeneity and drastic changes in genetic composition of the intrahost HCV population observed during chronic infections have been interpreted as evidence of a continuous immune escape via random mutations, thereby generating increasing genetic diversity of viral populations in infected individuals (5–7).
The observed cross-immunoreactivity (CR) of HCV variants from earlier stages of infection with antibodies (Abs) from later stages and ineffectiveness of Abs to immunoreact with variants from the same stage of infection (7) seemingly support the hypothesis of immune escape as a mechanism of intrahost evolution that contributes to establishment of persistent infections. However, several recent observations are incompatible with this hypothesis. First, the intrahost HIV population diversifies and diverts continuously from acute state to chronic infection until, at the onset of immunodeficiency, it starts losing heterogeneity and eventually stops diverting (8). Surprisingly, a similar temporal pattern of diversity and diversion was observed for intrahost HCV populations (9, 10). Furthermore, for HCV, the consistent increase in negative selection during chronic infection was observed (9–13). The late-stage HCV populations were shown to remain constant and homogeneous under the strong negative selection for years, indicating a high level of intrahost adaptation (9). Certain intrahost HCV variants were observed to persist in infected hosts for up to 16 y (9, 14, 15). These observations suggest that intrahost HCV subpopulations can remain unaffected by the immune system for years over the course of infection.
Second, complex dynamics of HCV populations were observed in infected hosts. The density of intrahost subpopulations was found to fluctuate significantly in the course of chronic HCV infection, with some subpopulations persisting at low frequency for years until becoming dominant or reemerging at later stages of infection after being undetectable for a long time (9, 10, 15, 16).
Third, the HCV hypervariable region 1 (HVR1) contains neutralizing antigenic epitopes (17, 18). Significant genetic variation of HVR1 during chronic infection was hypothesized to facilitate escape from neutralizing antibodies (17, 18). However, recent genetic and immunological analyses showed that HVR1 antigenic diversity is extensively convergent and effectively limited, with HVR1 variants from different genotypes and subtypes being broadly cross-immunoreactive (19–21).
Interactions of intrahost viral variants with the host immune system are highly complex and nonlinear (22) and were subjects of mathematical modeling with the goal to understanding the mechanisms that lead to chronic infection. Previously developed mathematical models of interaction between HIV (23–25) or HCV (22) and the immune system showed that immune escape is associated with increase in diversity of the viral population. However, the continuous immune escape predicted by these models is inconsistent with the aforementioned observations, particularly for HCV. Unlike HIV, HCV lacks the ability to induce systemic immune suppression, suggesting a different mechanism of immune adaptation. Here, we develop a model that takes into consideration broad CR among viral variants (18, 19, 26–28) and disparity between CR and neutralization (19, 29). The model predicts antigenic cooperation (AC) among HCV variants that results in protection rather than continuous escape of the HCV population from the neutralizing Ab, thus suggesting a mechanism of intrahost evolution that leads to chronic infection.
Model
The model considers a population of n viral antigenic variants inducing n immune responses in the form of Ab and memory B cells. Viral variants exhibit CR as defined by a CR network (CRN), which is a directed weighted graph , with vertices corresponding to viral variants and arcs connecting CR variants. Considering that not all interactions with Ab lead to neutralization (30–32), CRN has two weight functions, which are defined by immune neutralization and immune stimulation matrices and , where ; is a coefficient representing the binding affinity of Ab to j () with the ith variant; and is a coefficient reflecting strength of stimulation of Ab to j () by the ith variant. Immune response against variant is neutralizing; i.e., . The population dynamics are described by the following system of ordinary differential equations (ODEs):
| [1] |
| [2] |
Here, variants replicate at rates and are eliminated by immune responses at rates . Immune responses are stimulated by jth variants at rates , where represents the probability of stimulation of immune response by variant . It is assumed that preferentially stimulates preexisting immune responses capable of binding to with a relatively high affinity (33), and thus is calculated as
| [3] |
Immune response decays at a rate b in the absence of stimulation. In the absence of CR among variants, i.e., , the system [1] and [2] reduces to the model previously developed for a heterogeneous viral population (34).
We mainly focus on the study of effects caused by the topological structure of the CRN. We consider a special case of the model [1] and [2], where all viral variants have the same replication rate f, and, for every pair of CR-variants i and j, the corresponding immune stimulation and neutralization coefficients are equal to constants α and β, respectively. In this case, immune neutralization and stimulation matrices are completely defined by the structure of the CRN; i.e.,
| [4] |
where A is the adjacency matrix of . Considering that neutralization of viruses from the family requires binding K molecules of the Ab to a single virion (35–37), it is reasonable to assume that .
The relationship between the total viral population and immune response is described by the equation . The equilibrium solutions and are associated as follows:
| [5] |
Results
Antigenic Cooperation.
The model described by Eqs. 1 and 2 with CR-matrices [4]; parameters , , , , , , and (same values as for analogous parameters in ref. 34); and initial conditions , was simulated on 360 random CRNs with vertices, using Matlab (MathWorks). The CRN was modeled as a scale-free network with a power-law degree exponent (according to ref. 19), using the Complex Networks Package (38). Viral variants and immune responses were assumed abolished once their values fell below their initial conditions.
Without CR (), all viral variants are eliminated by the immune system, indicating that the only way for a virus to persist is via a constant generation of new variants escaping immune responses. However, broad CR () fundamentally changes the simulated outcome of infection. Although the majority of viral variants are eradicated by the immune system, on average of variants persist at the system equilibrium (Table 1, row a).
Table 1.
| Rows | K = 2 | K = 5 | K = 10 |
| a) | 9.224 (1.705) | 11.321 (2.089) | 11.124 (2.3) |
| b) | 1.132 (0.187) | 1.221 (0.146) | 1.223 (0.142) |
| c) | 0.918 (2.018) | 0.084 (0.186) | 0.004 (0.008) |
| d) | 99.082 (2.018) | 99.916 (0.186) | 99.996 (0.008) |
| e) | 18.256 (2.836) | 21.53 (3.058) | 21.132 (3.436) |
| f) | 0.082 (0.187) | 0.063 (0.146) | 0.095 (0.194) |
| g) | 98.236 (3.29) | 98.475 (2.21) | 98.349 (2.329) |
| h) | 99.487 (1.125) | 99.686 (0.715) | 99.564 (0.917) |
| i) | 50.028 (2.268) | 52.130 (2.721) | 52.072 (2.720) |
| j) | 99.083 (2.017) | 99.917 (0.186) | 99.996 (0.008) |
| k) | 12.241 (1.184) | 12.398 (1.224) | 12.386 (1.219) |
Rows a and b, percentages of persistent and altruistic variants; rows c and d, total frequencies of immune responses associated with persistent and altruistic variants; rows e and f, probabilities (in percent) of persistence for variants adjacent and nonadjacent to altruistic variants; row g, percentage of persistent variants without specific immune responses; rows h and i, percentages of persistent variants and all variants adjacent to altruistic variants; row j, total frequency of variants adjacent to altruistic variants; and row k, ratio of average in-degrees of altruistic and nonaltruistic variants. Values in parentheses are SDs.
The state of the immune response to persistent variants can be described as “local” immunodeficiency because specific immune responses against these variants constitute only a small fraction of the overall equilibrium immune response ( on average; Table 1, row c). Most of these variants (; Table 1, row g) do not produce any specific immune responses. Persistent variants formed an independent set in all CRNs; i.e., no CR exists among them.
The model suggests differential roles for intrahost viral variants. In addition to the persistent variants, the model identifies a group of variants that cannot be found at equilibrium but whose specific immune responses persist through the infection ( and ). Although these variants constitute a small fraction of the viral population (), immune responses against them are dominant at equilibrium and constitute (Table 1, rows b and d). Existence of these variants stems from Eq. 5. Abs against these variants cannot efficiently neutralize persistent variants. Dominance of immune responses coupled with lack of these variants at equilibrium indicates their altruistic role in supporting the continuous existence of persistent variants (Fig. 1).
Fig. 1.

CRN of 100 viral variants. Altruistic variants are shown in green, persistent variants in red, and others in yellow. Network was visualized using Pajek (56).
The relationship between altruistic and persistent variants becomes more clear after consideration of properties of these variants in the CRN. Altruistic variants are in-hubs, i.e., vertices with high in-degree, with the average in-degree of an altruistic variant being times greater than the average in-degree of all other variants (Table 1, row k).
Variants adjacent to altruistic variants constitute, on average, of the total viral load at equilibrium (Table 1, row j). Almost all persistent variants ( on average) are adjacent to altruistic variants (Table 1, row h), whereas the probability of being adjacent to an altruistic variant for an arbitrary node is (Table 1, row i). The average survival probability for in-neighbors of altruistic variants is times greater than for all other variants (Table 1, rows e and f).
These observations suggest that persistent variants achieve immune adaptation via specific interactions with altruistic variants as described by their properties in the CRN. If a persistent variant u is adjacent to an altruistic variant v, then an immune response competes for a stimulation signal with a response . Considering that is stimulated by many variants, it should be of high value, readily outcompeting , which results in a total lack or an extremely low level of . This interaction can be considered as a form of cooperation between variants, with altruistic variants losing their fitness by significantly contributing to the fitness of persistent variants. However, the majority of variants () do not persist and do not generate persistent immune responses (Fig. 1).
The above predictions are robust to small variations of parameters (SI Text, section S2).
Indirect Interactions in the CRN.
Interactions among viral variants in the CRN are complex, with variants significantly influencing fitness of not only adjacent variants but also variants that have no direct immunological links in the CRN, irrespective of their genetic relatedness, resulting in elimination of distant variants or boosting of their frequency. These effects can be studied by analyzing relations between equilibrium solutions and . Let , , , . For every we have . Solutions and satisfy the systems of equations
| [6] |
| [7] |
where . System [7] shows that the weighted sum of equilibrium populations of a closed neighborhood for each variant (variant plus its CRN neighbors) is fixed. This means that decline or increase of a population of a certain variant (or variants) results in a corresponding increase or decline of the population of other variants from the same closed neighborhood. Consequently, these changes propagate through the CRN to other closed neighborhoods. Thus, system [7] describes complex relationships among viral variants that belong to a single connected component of the CRN, reflecting chains of interactions spreading through the network, and reveals dependence of variant persistence on a global structure of the CRN.
This mechanism can be demonstrated using two examples of CRNs (Fig. 2 A and B, ). In both cases, there are equilibrium solutions with , and system [7] transforms into
| [8] |
| [9] |
for each CRN, respectively. For the first CRN, the solution is , , indicating elimination of variant u; whereas for the second CRN, we have , , , indicating persistence of u due to the effect of variant w. Thus, enhancement of u by w, which can be considered as a form of cooperation, is mediated by variant v, emphasizing the role of indirect interactions among variants in the CRN.
Fig. 2.
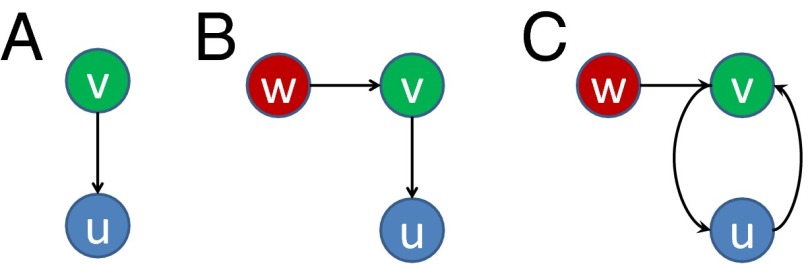
Examples of CRNs (A–C) with two and three variants.
The temporal appearance of viral variants involved in such interactions may lead to significant changes in population structure as illustrated in Fig. 3.
Fig. 3.

Changes in population structure induced by emergence of a new variant. (A and C) Variant dynamics; (B and D) dynamics of immune responses. Parameters: , , , .
Fig. 3 A and B shows dynamics of a system of two viral variants u and v with CR matrices and , with v (green) emerging at time . Immune response is stimulated by u even after v is eliminated. Because is nonneutralizing against variant u, the neutralization efficiency of the overall immune response against u decreases, contributing to an increase in population size of this variant, which eventually converges to a higher level at equilibrium. Thus, after declining to a low, potentially undetectable, level, u may reemerge, owing to a short-lived v.
Fig. 3 C and D shows system dynamics for a population of three viral variants with the CRN shown in Fig. 2C, with and . The initial population consists of variant u (blue). After emergence of variant v (green) at time , declines sharply. Emergence of variant w (red) at time leads to decline of v and consequent increase of u, which becomes dominant after . This example illustrates interactions among variants belonging to overlapping closed neighborhoods and resembles the observed frequency fluctuations and reemergence of HCV subpopulations at late stages of infection (9, 15, 16).
Model of Two Variants.
Reduction of the model [1] and [2] to two viral variants u and v with rates and and the CRN shown in Fig. 2A allows for analytic examination of dynamic relationships. Without CR (), the system converges to equilibrium
| [10] |
(Fig. S1). When , the system has several equilibrium solutions (SI Text, section S1.1). The family
| [11] |
where and , describes AC between u and v. The expression for indicates that v converges to a greater population size than in [10] by using response associated with u.
When , the system reaches equilibrium [11] with (Fig. S2). In this case, , which indicates that v achieves a greater population size by using the replicative ability of u (in contrast, depends only on ). Solution stability, if , is proved in SI Text, section S1.1.
If , then the system is degenerate and a stronger form of AC is observed (Fig. S3). The value of h here depends on the initial conditions and the parameters of [1] and [2]. Variant u is completely eliminated, but, under the same initial conditions, the population of v achieves a higher equilibrium level than in the case of (Fig. S4). These solutions are unstable if h is low ().
A solution describes a situation without AC (Fig. S5). It is stable, if (i.e., the replication rate of u is sufficient for outcompeting v and overcoming the effect of CR), and unstable, if . Several other equilibria of the system [1] and [2] are studied in SI Text, section S1.1.
Analysis of effects of replication rates and initial conditions on solutions [11] suggests that small replication rate and initial population size may be beneficial for a variant with an advantageous position in the CRN (SI Text, section S1.2).
Discussion
Intrahost viral evolution is a nonlinear dynamical process involving complex interactions between viral variants and the host’s immune system. Accurate modeling of viral evolution requires comprehensive accounting for complexity of these interactions. We developed and studied a model based on four key assumptions: (i) broad CR among viral variants, which form a complex scale-free CRN; (ii) disparity between CR activation and neutralization; (iii) variation in profiles of immune dominance; and (iv) threshold neutralization. Although models with pairwise CR were proposed earlier (39), to the best of our knowledge, there are no models that accommodate all these assumptions. However, all of them contribute significantly to intrahost evolution of viruses, particularly of HCV. Thus, in addition to CR, the model takes into consideration that neutralization requires simultaneous binding of a certain number of Ab molecules to each virion (threshold neutralization), which can be achieved only with high-affinity Abs. Additionally, antigenic variants differ in affinity binding to broad-CR Abs, with affinity being sufficient to recruit specific memory B cells by antigen binding to B-cell receptors but largely insufficient for neutralization, which results in disparity between stimulation and neutralization. The model also assumes that a temporal presentation of CR variants primarily results in stimulation of preexisting immune responses. It makes efficacy of specific immune responses against these variants dependent on previous exposures to other variants, which changes profiles of immune dominance for emerging epitopic variants.
The validity of these assumptions for HCV is supported with data. First, CR is known to be widespread among HCV HVR1 variants, with many of them showing a strict immune specificity and a fraction of the variants showing very broad CR (18, 19, 26–28). It results in organization of CR among HVR1 variants in a complex, scale-free network (19). HCV HVR1 variants can bind Abs obtained against genetically distant variants sharing only 20 of amino acid sites with the immunogen. Findings of the HVR1 antigenic diversity being extensively convergent and effectively limited (19) indicate frequent CR even among genetically distant HCV variants.
Second, although CR is essential for neutralization, Ab binding does not always lead to protection against infection, resulting in low correlation between neutralization and CR among HCV variants (30). Abs from many chronically HCV-infected patients or persons vaccinated with experimental vaccines were shown to bind to HCV envelope glycoproteins without neutralizing infection (31, 32). Some human serum and monoclonal Abs were even found to enhance HCV/vesicular stomatitis virus-pseudotype infections (40, 41). Accordingly, although other models (48) consider immune stimulation and neutralization matrices U and V as being identical, the model developed here assumes that V and U differ, with ; i.e., the stimulation of an Ab response to j by i is more probable than neutralization of i by an Ab to j.
Third, there is a fundamental difference between antigenicity (capacity to bind an Ab) and immunogenicity (capacity to elicit an Ab) (19, 29), with the first being a simple biochemical reaction and the second being a complex biological process involving several cell types and competition among cells capable of binding or responding to a specific immunogen (42–47). Such competition results in the immune system preferentially using memory cells in responding to the presentation of an immunogen immunoreactive with the memory B-cell receptors (33). Besides providing a rapid secondary immune response to reinfections with same pathogen, this mechanism is the basis for the phenomenon of original antigenic sin (OAS) (48), also known as heterologous immunity (49) or repertoire freeze (50). OAS results from stimulation of B-cell clones activated during the prior infection after exposure to an antigenically similar but not identical variant of the same pathogen. This effect is well characterized for influenza and dengue viruses (51, 52). Recruitment of preexisting immune responses against new intrahost viral variants may result in producing Abs with a lower affinity of binding to these variants and a poorer neutralization capability (50). These properties are accommodated in the model by coefficients which describe the rate of stimulation of immune responses by the jth variants. Similar to repertoire freeze (50), this mechanism acts among intrahost viral variants. However, in contrast to repertoire freeze, the specific contribution of viral variants to intrahost evolution depends on their position in the CRN and temporal emergence during infection. Ab-dependent enhancement of viral infectivity (40) has a potential of strengthening this mechanism, although it is not a part of our model.
Fourth, Ab-mediated neutralization of viruses from the Flaviviridae family requires a stoichiometric Ab binding that exceeds a certain threshold (35–37). This threshold was estimated to be up to 30 Ab molecules per virion for West Nile virus (53, 54) and is represented in our model with the relation between the probabilities and of neutralization and immune stimulation. The neutralization effectiveness of the Ab may be further hampered by dilution with low-affinity Abs resulting from boosting of different memory B cells by a viral variant or from polyclonal antigen-independent activation of naive B cells by HCV (55).
Immune escape by random mutations is considered to drive intrahost HCV evolution to establish chronic infection (5–7). However, the model proposed here suggests that the extensive genetic heterogeneity, usually observed among intrahost HCV variants, promotes establishment of a complex CRN resulting in AC among viral variants, which facilitates survival of some variants in the presence of neutralizing antibody. Although AC entails generation of intrahost variants of neutralizing antigenic epitopes, it differs fundamentally from the immune-escape mechanism in that that it engenders conditions of immune tolerance toward viral variants with advantageous positions in the CRN, e.g., variants shown in red in Fig. 1, rather than conditions favoring reduced antigenic CR to preceding variants. Differences in replicative fitness or frequency among variants at the initiation of infection and in specificity and breadth of CR influence succession of viral variants during infection. Variants having advantageous positions in the CRN, underrepresented at early stages of infection, thereby become dominant at later stages.
Whereas other models of host–viral interaction predict a constant increase in genetic heterogeneity of the intrahost viral population as a result of continuous immune escape (22–25), the AC model predicts a different outcome of the intrahost HCV evolution: reduction in heterogeneity of the persistent viral population. This prediction is strongly supported by experimental observations of increasing negative selection during intrahost HCV evolution, long-term persistence of viral variants, and substantial heterogeneity loss associated with a strong negative selection after years of infection (9–14).
The two most important conceptual outcomes from the proposed model are AC and differential roles of individual viral variants in establishing persistent infection. Cooperative action of antigens results from their differential capability to bind to or elicit a polyspecific antibody. Both properties have been shown to be differently distributed among HCV HVR1 variants (19). AC is a mechanism of reduction in the neutralizing capacity of the host immune system toward certain viral variants or local immunodeficiency. A major condition for AC is the existence of a complex CRN, which determines roles for each viral variant in adaptation to the host. Persistence and clearance of each variant are not solely defined by innate properties of a variant but by its position in the CRN. Some variants develop a polyspecific (broad-CR) Ab response that interferes with development of specific immune responses against other variants immunoreactive with these Abs. In the CRN, the former are hubs with a high in-degree (in-hubs), whereas the latter are vertices adjacent to in-hubs. Adjacent variants boost the in-hub-specific memory B cells, which results in producing neutralizing Abs against in-hubs rather than against adjacent variants themselves. Thus, in-hub variants can be viewed as playing an altruistic role in AC because they improve fitness of adjacent variants at their own fitness cost, whereas adjacent variants are selfish because they gain fitness at the expense of in-hub variants. These selfish variants remain in existence without eliciting strong specific immune responses and are predicted to persist under negative selection as was recently observed (9, 11–15). In contrast, altruistic variants are efficiently eliminated from the viral population while their immune responses are being continuously sustained. However, the majority of variants are eliminated and their specific immune responses become abrogated over time.
Immunological interactions essential for persistence of viral variants are determined not only by immediate neighbors but also by the entire structure of the CRN as described in Eqs. 6 and 7. Complex interactions among viral variants through the network may lead to an effect when an emergence of CR variants attenuates the stimulation of the neutralizing Ab against distant variants, thus improving their chances of persistence (Fig. 3), potentially resulting in modifying the entire structure of the viral population. This phenomenon may explain complex fluctuations in frequencies of viral subpopulations and reemergence of subpopulations after years of infection (9, 10, 15, 16).
In conclusion, the mathematical model developed here posits that intrahost evolution increases HCV immune adaptation during infection rather than perpetuates an “arms race” with the host’s immune system. This mechanism of immune adaptation is fundamentally different from immune escape, during which surviving variants continuously evolve away from the “cloud” of Abs elicited against the preceding variants. In immune adaptation, the intrahost evolution directs viral variants into the cloud of Abs, with variants, which do not have any additional antigenic specificity compared with the preceding variants, being the ones establishing persistent infection. The state of immune adaptation is attained by AC, which is based on the differential capacity of variants to bind to and elicit broadly specific Abs. The model explains recent observations not compatible with the immune-escape model, although it does not consider contribution of other branches of innate and adaptive immunity to the establishment of persistent HCV infections. It can be applied to any heterogeneous virus with broad CR. Organization of intrahost viral variants into a complex CRN is important for establishing AC. It is conceivable that the CRN structure is affected by attributes of epitopes, restrictions of Ab responses within the 1F7 idiotype as observed for HIV and HCV (20, 21, 48), or any other factors. Understanding all of these factors warrants further research. Although the arms race model is not instructive for the development of therapeutics and vaccines, the model presented here identifies AC as a target for therapeutic and vaccine approaches, suggesting the use of a specific Ab to transform the CRN from supporting to discouraging AC. Understanding the structure of CRNs should inform development of novel strategies for preventing or interrupting viral infections in naive or infected hosts, correspondingly, by undermining immune adaptation via immunization with specially selected variants of neutralizing epitopes that are capable of disrupting AC.
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422942112/-/DCSupplemental.
References
- 1.Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10(9):553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 2.Sklan EH, Charuworn P, Pang PS, Glenn JS. Mechanisms of HCV survival in the host. Nat Rev Gastroenterol Hepatol. 2009;6(4):217–227. doi: 10.1038/nrgastro.2009.32. [DOI] [PubMed] [Google Scholar]
- 3.Domingo E, et al. Viruses as quasispecies: Biological implications. Curr Top Microbiol Immunol. 2006;299:51–82. doi: 10.1007/3-540-26397-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: Patterns and determinants. Nat Rev Genet. 2008;9(4):267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 5.Farci P, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288(5464):339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 6.Farci P. Hepatitis C virus. The importance of viral heterogeneity. Clin Liver Dis. 2001;5(4):895–916. doi: 10.1016/s1089-3261(05)70200-2. [DOI] [PubMed] [Google Scholar]
- 7.von Hahn T, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132(2):667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 8.Shankarappa R, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73(12):10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran S, et al. Temporal variations in the hepatitis C virus intrahost population during chronic infection. J Virol. 2011;85(13):6369–6380. doi: 10.1128/JVI.02204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gismondi MI, et al. Dynamic changes in viral population structure and compartmentalization during chronic hepatitis C virus infection in children. Virology. 2013;447(1-2):187–196. doi: 10.1016/j.virol.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Li H, et al. Genetic diversity of near genome-wide hepatitis C virus sequences during chronic infection: Evidence for protein structural conservation over time. PLoS ONE. 2011;6(5):e19562. doi: 10.1371/journal.pone.0019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campo DS, et al. Next-generation sequencing reveals large connected networks of intra-host HCV variants. BMC Genomics. 2014;15(Suppl 5):S4. doi: 10.1186/1471-2164-15-S5-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu L, et al. HCV selection and HVR1 evolution in a chimpanzee chronically infected with HCV-1 over 12 years. Hepatol Res. 2008;38(7):704–716. doi: 10.1111/j.1872-034X.2008.00320.x. [DOI] [PubMed] [Google Scholar]
- 14.Palmer BA, et al. Insertion and recombination events at hypervariable region 1 over 9.6 years of hepatitis C virus chronic infection. J Gen Virol. 2012;93(Pt 12):2614–2624. doi: 10.1099/vir.0.045344-0. [DOI] [PubMed] [Google Scholar]
- 15.Palmer BA, et al. Analysis of the evolution and structure of a complex intrahost viral population in chronic hepatitis C virus mapped by ultradeep pyrosequencing. J Virol. 2014;88(23):13709–13721. doi: 10.1128/JVI.01732-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray RR, Salemi M, Klenerman P, Pybus OG. A new evolutionary model for hepatitis C virus chronic infection. PLoS Pathog. 2012;8(5):e1002656. doi: 10.1371/journal.ppat.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eckels DD, Zhou H, Bian TH, Wang H. Identification of antigenic escape variants in an immunodominant epitope of hepatitis C virus. Int Immunol. 1999;11(4):577–583. doi: 10.1093/intimm/11.4.577. [DOI] [PubMed] [Google Scholar]
- 18.Puntoriero G, et al. Towards a solution for hepatitis C virus hypervariability: Mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO J. 1998;17(13):3521–3533. doi: 10.1093/emboj/17.13.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campo DS, et al. Hepatitis C virus antigenic convergence. Sci Rep. 2012;2:267. doi: 10.1038/srep00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hattori M, et al. Broadly reactive antibodies to hypervariable region 1 in hepatitis C virus-infected patient sera: Relation to viral loads and response to interferon. Hepatology. 1998;27(6):1703–1710. doi: 10.1002/hep.510270633. [DOI] [PubMed] [Google Scholar]
- 21.Yoshioka K, et al. Humoral immune response to the hypervariable region of hepatitis C virus differs between genotypes 1b and 2a. J Infect Dis. 1997;175(3):505–510. doi: 10.1093/infdis/175.3.505. [DOI] [PubMed] [Google Scholar]
- 22.Wodarz D. Hepatitis C virus dynamics and pathology: The role of CTL and antibody responses. J Gen Virol. 2003;84(Pt 7):1743–1750. doi: 10.1099/vir.0.19118-0. [DOI] [PubMed] [Google Scholar]
- 23.Nowak MA, May RM, Anderson RM. The evolutionary dynamics of HIV-1 quasispecies and the development of immunodeficiency disease. AIDS. 1990;4(11):1095–1103. doi: 10.1097/00002030-199011000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Nowak MA, May RM. Mathematical biology of HIV infections: Antigenic variation and diversity threshold. Math Biosci. 1991;106(1):1–21. doi: 10.1016/0025-5564(91)90037-j. [DOI] [PubMed] [Google Scholar]
- 25.Nowak MA, et al. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254(5034):963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 26.Scarselli E, et al. Occurrence of antibodies reactive with more than one variant of the putative envelope glycoprotein (gp70) hypervariable region 1 in viremic hepatitis C virus-infected patients. J Virol. 1995;69(7):4407–4412. doi: 10.1128/jvi.69.7.4407-4412.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lesniewski RR, et al. Hypervariable 5′-terminus of hepatitis C virus E2/NS1 encodes antigenically distinct variants. J Med Virol. 1993;40(2):150–156. doi: 10.1002/jmv.1890400213. [DOI] [PubMed] [Google Scholar]
- 28.da Silva LC, et al. Long term follow-up and patterns of response of ALT in patients with chronic hepatitis NANB/C treated with recombinant interferon-alpha. Rev Inst Med Trop Sao Paulo. 1995;37(3):239–243. doi: 10.1590/s0036-46651995000300010. [DOI] [PubMed] [Google Scholar]
- 29.Van Regenmortel MH. Basic research in HIV vaccinology is hampered by reductionist thinking. Front Immunol. 2012;3:194. doi: 10.3389/fimmu.2012.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarr AW, et al. Hepatitis C patient-derived glycoproteins exhibit marked differences in susceptibility to serum neutralizing antibodies: Genetic subtype defines antigenic but not neutralization serotype. J Virol. 2011;85(9):4246–4257. doi: 10.1128/JVI.01332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagging LM, et al. Neutralization of pseudotyped vesicular stomatitis virus expressing hepatitis C virus envelope glycoprotein 1 or 2 by serum from patients. J Infect Dis. 2002;185(8):1165–1169. doi: 10.1086/339679. [DOI] [PubMed] [Google Scholar]
- 32.Ray R, et al. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J Infect Dis. 2010;202(6):862–866. doi: 10.1086/655902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nara PL, et al. How can vaccines against influenza and other viral diseases be made more effective? PLoS Biol. 2010;8(12):e1000571. doi: 10.1371/journal.pbio.1000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nowak MA, May RM. Virus Dynamics: Mathematical Principles of Immunology and Virology. Oxford Univ Press; New York: 2001. [Google Scholar]
- 35.Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog. 2011;7(6):e1002111. doi: 10.1371/journal.ppat.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dowd KA, Pierson TC. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology. 2011;411(2):306–315. doi: 10.1016/j.virol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabo MC, et al. Hepatitis C virus epitope exposure and neutralization by antibodies is affected by time and temperature. Virology. 2012;422(2):174–184. doi: 10.1016/j.virol.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muchnik L. 2013. Complex Networks Package for MatLab (Version 1.6). Available at www.levmuchnik.net/Content/Networks/ComplexNetworksPackage.html.
- 39.Iwasa Y, Michor F, Nowak M. Some basic properties of immune selection. J Theor Biol. 2004;229(2):179–188. doi: 10.1016/j.jtbi.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Meyer K, Ait-Goughoulte M, Keck ZY, Foung S, Ray R. Antibody-dependent enhancement of hepatitis C virus infection. J Virol. 2008;82(5):2140–2149. doi: 10.1128/JVI.01867-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer K, Banerjee A, Frey SE, Belshe RB, Ray R. A weak neutralizing antibody response to hepatitis C virus envelope glycoprotein enhances virus infection. PLoS ONE. 2011;6(8):e23699. doi: 10.1371/journal.pone.0023699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freitas AA, Rosado MM, Viale A-C, Grandien A. The role of cellular competition in B cell survival and selection of B cell repertoires. Eur J Immunol. 1995;25(6):1729–1738. doi: 10.1002/eji.1830250636. [DOI] [PubMed] [Google Scholar]
- 43.McLean AR, Rosado MM, Agenes F, Vasconcellos R, Freitas AA. Resource competition as a mechanism for B cell homeostasis. Proc Natl Acad Sci USA. 1997;94(11):5792–5797. doi: 10.1073/pnas.94.11.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacLennan ICM. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 45.Tarlinton D. B-cell memory: Are subsets necessary? Nat Rev Immunol. 2006;6(10):785–790. doi: 10.1038/nri1938. [DOI] [PubMed] [Google Scholar]
- 46.Schwickert TA, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446(7131):83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 47.Pan K. Understanding original antigenic sin in influenza with a dynamical system. PLoS ONE. 2011;6(8):e23910. doi: 10.1371/journal.pone.0023910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Francis T., Jr On the doctrine of original antigenic sin. Proc Am Philos Soc. 1960;104(6):572–578. [Google Scholar]
- 49.Rehermann B, Shin EC. Private aspects of heterologous immunity. J Exp Med. 2005;201(5):667–670. doi: 10.1084/jem.20050220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsons MS, Muller S, Kohler H, Grant MD, Bernard NF. On the benefits of sin: Can greater understanding of the 1F7-idiotypic repertoire freeze enhance HIV vaccine development? Hum Vaccin Immunother. 2013;9(7):1532–1538. doi: 10.4161/hv.24460. [DOI] [PubMed] [Google Scholar]
- 51.Kim JH, Skountzou I, Compans R, Jacob J. Original antigenic sin responses to influenza viruses. J Immunol. 2009;183(5):3294–3301. doi: 10.4049/jimmunol.0900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Midgley CM, et al. An in-depth analysis of original antigenic sin in dengue virus infection. J Virol. 2011;85(1):410–421. doi: 10.1128/JVI.01826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierson TC, et al. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe. 2007;1(2):135–145. doi: 10.1016/j.chom.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mehlhop E, et al. Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe. 2009;6(4):381–391. doi: 10.1016/j.chom.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosa D, et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci USA. 2005;102(51):18544–18549. doi: 10.1073/pnas.0509402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Batagelj V, Mrvar A. In: Pajek - Analysis and Visualization of Large Networks, Graph Drawing Software. Juenger M, Mutzel P, editors. Springer; Berlin: 2003. pp. 77–103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.