

Abstract
Primary myelofibrosis is characterized by clonal myeloproliferation, dysmegakaryopoiesis, extramedullary hematopoiesis associated with myelofibrosis and altered stroma in the bone marrow and spleen. The expression of CD9, a tetraspanin known to participate in megakaryopoiesis, platelet formation, cell migration and interaction with stroma, is deregulated in patients with primary myelofibrosis and is correlated with stage of myelofibrosis. We investigated whether CD9 participates in the dysmegakaryopoiesis observed in patients and whether it is involved in the altered interplay between megakaryocytes and stromal cells. We found that CD9 expression was modulated during megakaryocyte differentiation in primary myelofibrosis and that cell surface CD9 engagement by antibody ligation improved the dysmegakaryopoiesis by restoring the balance of MAPK and PI3K signaling. When co-cultured on bone marrow mesenchymal stromal cells from patients, megakaryocytes from patients with primary myelofibrosis displayed modified behaviors in terms of adhesion, cell survival and proliferation as compared to megakaryocytes from healthy donors. These modifications were reversed after antibody ligation of cell surface CD9, suggesting the participation of CD9 in the abnormal interplay between primary myelofibrosis megakaryocytes and stroma. Furthermore, silencing of CD9 reduced CXCL12 and CXCR4 expression in primary myelofibrosis megakaryocytes as well as their CXCL12-dependent migration. Collectively, our results indicate that CD9 plays a role in the dysmegakaryopoiesis that occurs in primary myelofibrosis and affects interactions between megakaryocytes and bone marrow stromal cells. These results strengthen the “bad seed in bad soil” hypothesis that we have previously proposed, in which alterations of reciprocal interactions between hematopoietic and stromal cells participate in the pathogenesis of primary myelofibrosis.
Introduction
Primary myelofibrosis (PMF) is a Philadelphia chromosome-negative myeloproliferative neoplasm characterized by clonal myeloproliferation, dysmegakaryopoiesis and extramedullary hematopoiesis associated with myelofibrosis and altered bone marrow (BM)/splenic stroma.1 The myeloproliferative process features an increased number of CD34+ hematopoietic stem/progenitor cells with hypersensitivity to cytokines, which have been attributed to the presence of mutations including Jak2V617F and MPL515L/K.2,3 More recently, various other mutations affecting epigenetics,4,5 the spliceo-some6 and metabolism7 have been discovered and have been correlated with a worse prognosis8 and with leukemic transformation.4 The myeloproliferation is associated with massive mobilization of CD34+ hematopoietic stem/progenitor cells, including megakaryocyte progenitors, from the BM to the spleen, which was suggested to be partly due to down-regulation of the expression of CXCR4, one of the two CXCL12 receptors.9 PMF megakaryocytes are characterized by prominent proliferation, a dysplastic appearance with a plump nucleus and altered nuclear/cytoplasmic maturation. They also have changes in the process of apoptosis depending on the stromal context. Indeed, a para-apoptotic process was observed in BM biopsies,10 contrasting with data from molecular studies11 and CD34+ hematopoietic stem/progenitor cell cultures,12 which showed a reduction of the apoptotic process. Furthermore, evidence is accumulating that altered stromal cells in the BM and spleen of PMF patients may contribute to the hematopoietic clone emergence/development through mutually dependent interactions with clonal hematopoietic cells.1
CD9, a four transmembrane glycoprotein that belongs to the tetraspanin family,13 has recently been reported to be deregulated in PMF. It is strongly expressed on platelets14 and was first cloned from megakaryocyte libraries.15 Treatment of K562 cells with tetradecanoylphorbol-13-acetate induces megakaryocytic differentiation associated with up-regulation of CD9 expression which precedes the appearance of GPIIb/IIIa.16 We have previously demonstrated that CD9 participates in normal megakaryopoiesis and platelet formation through its action on megakaryocyte demarcation membrane separation.17 In PMF patients, CD9 molecular expression is increased in CD34+ cells,18 as well as in megakaryocytes microdissected from BM biopsies, and is reported to be correlated with the stage of BM fibrosis.19 Beside its role in megakaryopoiesis, CD9 is suggested to regulate interactions with the microenvironment by promoting the recruitment of numerous molecular partners grouped in lipid-rich microdomains, including integrins that are receptors for extracellular matrix components such as collagen, laminin and fibronectin.13 CD9 also participates in cell adhesion/motility20 and, in CD34+ cells, the CD9-mediated mobilization involves the CXCL12/CXCR4 axis.21
Taking into account the role of CD9 in megakaryopoiesis and in BM stromal interactions, we analyzed the role of CD9 in the pathogenesis of PMF. Herein, we show that CD9 takes part in the alterations affecting the survival, differentiation and CXCL12/CXCR4-mediated migration of PMF megakaryocytes. We also demonstrate that CD9 is involved in the altered interactions between megakaryocytes and BM mesenchymal stromal cells (MSC) isolated from patients, participating in the dysmegakaryopoiesis of PMF.
Methods
Peripheral blood and bone marrow samples
Peripheral blood and BM samples from PMF patients (n=174) and non-mobilized healthy donors (HD; n=76) were obtained from the French/European networks, A.M. Vannucchi (Florence, Italy), and the Blood Transfusion Center of Armies (Clamart, France) according to the Helsinki declaration and with approval from Institutional Ethics Committees.
CD34+ cell selection and bone marrow mesenchymal stromal cells
CD34+ cells were selected from mononuclear cells from peripheral blood or BM samples from PMF patients and HD using the Miltenyi miniMACS system.22
BM MSC were obtained from BM biopsies (PMF patients) or hip surgery (HD) by incubating BM mononuclear cells (5×106/mL) for 48–72 h in plastic flasks. Adherent MSC were cultured in DMEM + 10% SVF in 5% CO2 for three passages.
Phenotypic analysis
Cells (5×104) were labeled with 2 μg/mL monoclonal antibodies: CD105-PE/clone-166707, CD9-FITC/clone-209306, HLA-DR-FITC/clone-L203 (R&D systems); CD73-PE/clone-AD2, CD62P-PE/clone-AC1.2, CD90-PE/clone-5E10, CXCR4-PE/clone-12G5 (BD Biosciences); CD41-FITC/CD41-PE (clone-P2, IOTest Beckman-Coulter); and CD9-biotinyled/clone-SYB (a kind gift from E. Rubinstein and C. Boucheix). Membrane antigen fluorescence was quantified using Cellquest software on a FACScalibur (Becton-Dickinson); 5×103 live cells were analyzed.
Megakaryocytes derived from CD34+ cultures
CD34+ cells (5×104/500 μL/well) were cultured in SYN.H medium containing recombinant human (rh) interleukin (IL)-3 (2 ng/mL), rhIL-6 (1 ng/mL), rhIL-11 (40 ng/mL), and rh thrombopoietin (Tpo) (50 ng/mL) (Preprotec, France) giving rise to more than 85% of megakaryocytes after 10 to 14 days of culture. In some experiments, cultures were treated with monoclonal antibodies (anti-CD9/clone-SYB: 10 μg/mL). For RNA silencing, cells were cultured for 6 days and distributed in 250 μL 24-well plates with or without scrambled control or specific short interfering (si) RNA (1 μg/106 cells) and MISSION II vector (1/50; Sigma).
Colony-forming units-megakaryocyte
Circulating CD34+ cells (5×103) were seeded in Megacult C containing type-I bovine collagen (Stem Cell), rhIL-3 (1 μg/mL), rhIL-6 (10 μg/mL), and rhTpo (10 μg/mL). Colony-forming units-megakaryocyte (CFU-Mk) were quantified, on the basis of CD41 immunostaining, after 10 days of culture at 37°C in 5% CO2.
Analysis of intracellular phosphorylation
Cells were fixed for 1 h in formol-2% and overnight in ethanol-70% (4°C). After washes in PBS-0.5%+BSA-Triton-0.25X, cells were incubated with phosphorylated protein monoclonal antibodies (Cell Signaling, USA), washed and incubated with a secondary anti-rabbit Alexa-fluor-488nm coupled antibody (Invitrogen, France). Live cells (104) were analyzed on FACScalibur with Cellquest software.
Western blotting
Cell lysates (105 cells), obtained as described previously,22 were subjected to sodium dodecylsulfate polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes and blotted using primary monoclonal antibodies (Cell Signaling, USA). Membranes were revealed with fluorescent anti-mouse or anti-rabbit IgG. Fluorescence signals were detected on an Odyssey System (Li-cor, USA) and quantified by using ImageJ version 1.43u.
In vitro migration assay
Transwell migration assays were performed as described elsewhere.22 Cells (105) were loaded on the top chamber in RPMI/0.5% BSA and CXCL12 (100 ng/mL, R&D System) was added or not to the bottom chamber. Cells were incubated for 24 h or 48 h (siRNA treatment); the migration percentage was calculated after quantification of live cells in both chambers.
Quantitative real-time polymerase chain reaction analysis
Samples of total RNA were treated with RNase-free-DNase and converted into cDNA using the Reverse-Transcription kit (Applied-Biosystem). cDNA (2 μL) was added to a Quantitech SybrGreen amplification reaction (Qiagen, Courtaboeuf, France) in a final volume of 20 μL containing 10 pmol of each primer (Online Supplementary Table S1). Relative quantification was normalized to RPL38 using the 2ΔΔCT method.23
Statistical analysis
Results are expressed as mean ± standard deviation. Statistical differences between patients and controls or between conditions were validated by unpaired or paired t-tests, respectively, with P<0.05 being considered statistically significant. Paired data are represented in dot plots with lines between significant conditions.
Results
CD9 expression is deregulated during megakaryopoiesis in primary myelofibrosis
CD9 is reported to participate in megakaryopoiesis and in platelet formation. We therefore evaluated whether its expression was modulated during PMF megakaryopoiesis.
As previously described,18,19 Figure 1A illustrates that CD9 mRNA was increased 2.54-fold in PMF circulating CD34+ cells compared to CD34+ cells from unmobilized HD [(1.531±0.7413 arbitrary units (AU), n=12 versus 0.6019±0.0928 AU, n=7, respectively; P=0.0023]. Surprisingly, when quantified by flow-cytometry, the level of expression of CD9 on the membrane of circulating CD34+ cells from patients was heterogeneous compared to that on circulating CD34+ or CD42b−CD34+ cells from non-mobilized HD controls (259.5±58.54 AU, n=14 and 218.5±47.02 AU, n=10, respectively; Figure 1B). Heterogeneity in the CD9 expression on CD34+ cells between patients was ranked, discriminating three groups according to the level of CD9 expression (544.8±195.5 AU, n=9; 234.9±44.38 AU, n=14 and 97.09±33.18 AU, n=23) (Figure 1B). Figure 1C shows that most of the patients with a low number of platelets had low expression of CD9 on CD34+ cells. When quantified on platelets, the global CD9 membrane expression level was significantly lower in patients than in HD (116.1±150.4 AU, n=3 versus 406.4±83.73 AU, n=4, respectively; P=0.01; Figure 1D).
Figure 1.
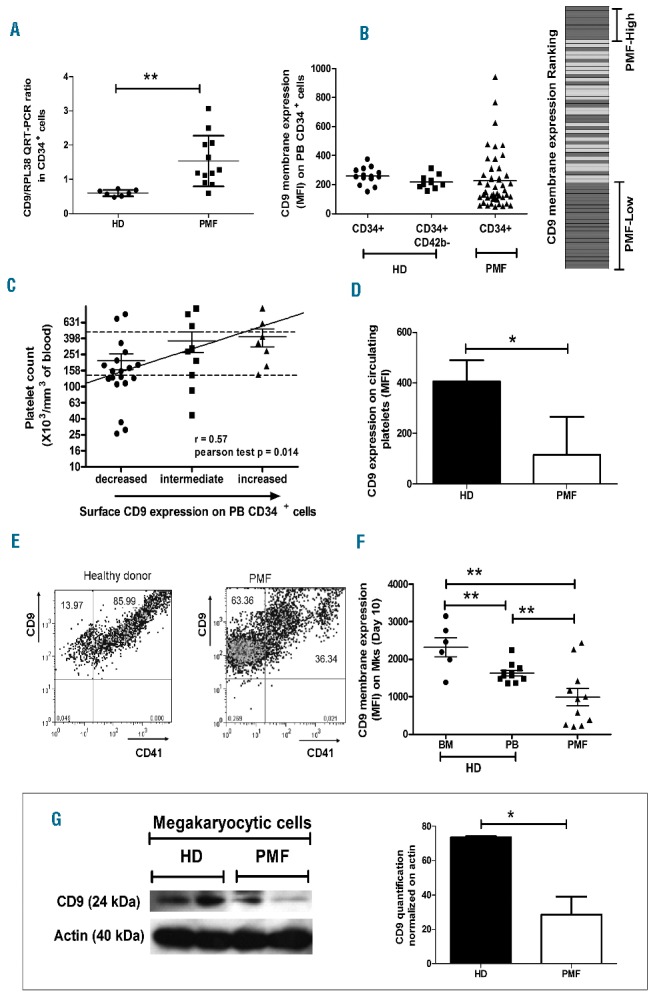
CD9 expression is deregulated during PMF dysmegakaryopoiesis. (A) Expression of CD9 transcripts normalized to RPL38 by QRT-PCR in CD34+ cells from healthy donors (HD) and primary myelofibrosis (PMF) patients. (B) CD9 membrane expression (MFI) by flow cytometry on CD34+ cells purified from the peripheral blood (PB) of HD and PMF patients and CD9 membrane expression ranking (gray bars: PMF patients; white bars: HD). (C) Correlation between the CD9 membrane expression level on PMF CD34+ cells and the patient’s platelet count. (D) CD9 expression level on HD and PMF circulating platelets determined by flow cytometry (MFI). (E) Co-expression of CD9 and CD41 on PMF megakaryocytes derived from HD and PMF circulating CD34+ cells (D10 of culture). (F) Membrane expression analysis of CD9 on megakaryocytes (Mks) derived from CD34+ cells purified from the bone marrow (BM) or PB of HD and PMF patients (D10 of culture) by flow cytometry. (G) Western blot analysis and quantification of CD9 expression in Mks derived from circulating CD34+ cells from HD and PMF patients (D10 of culture).
We further analyzed CD9 expression during in vitro megakaryocyte differentiation. At the end of in vitro megakaryocyte differentiation on day (D) 10, a lower proportion of megakaryocytes derived from PMF circulating CD34+ cells co-expressed CD9 and CD41 compared to the proportion from HD with a different expression profile (Figure 1E). The higher proportion of CD9+ cells that did not co-express CD41 in PMF patients is most likely mainly due to megakaryocyte progenitors (Online Supplementary Figure S1).
Actually, CD9 expression on megakaryocytes derived from PMF CD34+ cells was significantly reduced (988.2±788.2 AU, n=11) as compared to the expression on those derived from HD cells, independently of whether these cells were isolated from BM (2322±617 AU, n=6) or peripheral blood (1628±252.68.3 AU, n=11) (versus HD BM: P=0.0014 and versus HD peripheral blood: P=0.0092) (Figure 1F). The reduction of CD9 protein expression on PMF megakaryocytes was further confirmed by western blot analysis (HD: 73.73±0.51 versus PMF: 28.54±10.50; P=0.01) (Figure 1G). Taken together, our results show deregulation of CD9 expression during PMF megakaryopoiesis.
CD9 participates in dysmegakaryopoiesis in primary myelofibrosis
The differential CD9 expression during megakaryopoiesis incited us to investigate the potential role of CD9 in the dysmegakaryopoiesis that characterizes PMF.
Figure 2A shows that cell surface CD9 engagement by antibody ligation (Syb clone, 10 μg/mL) during in vitro megakaryocyte differentiation from PMF CD34+ cells stimulated megakaryocyte differentiation, as evidenced by an increased number of CFU-Mk (189.3±108.7, n=8 versus 99.63±63.91 for anti-CD9-treated cells and IgG-treated control cells, respectively, for 5,000 CD34+ seeded cells; P=0.002) at D10 of culture. This was associated with an increase in CFU-Mk size and with the presence of large megakaryocytes in colonies. A similar effect of Syb monoclonal antibody was observed in liquid culture, with the presence of mature megakaryocytes with multi-lobulated nuclei and large granular cytoplasm in Syb-treated cultures, contrasting with dysplastic immature micromegakaryocytes with hypolobulated nuclei observed in untreated PMF cells. This differentiating effect was also associated with increased expression of megakaryocyte differentiation markers such as CD41 (1030±308.8 AU for anti-CD9-treated cells versus 769.5±228.3 AU for IgG-treated control cells, n=4, P=0.01) and CD62p (91.75±24.02 AU for anti-CD9-treated cells versus 60.0±11.34 AU for IgG-treated control cells, n=4, P=0.04) (Figure 2B).
Figure 2.
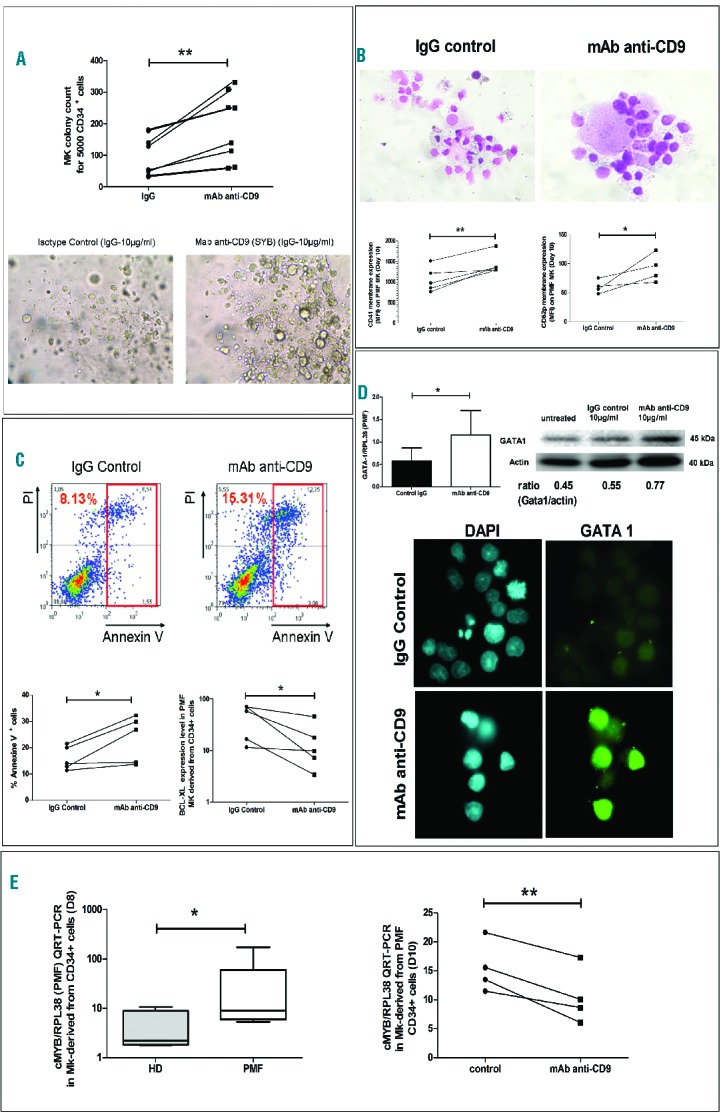
CD9 is involved in PMF dysmegakaryopoiesis. Effect of monoclonal antibody (mAb) ligation of surface CD9 (clone-SYB; 10 μg/mL) on (A) PMF megakaryocyte (Mk) colonies obtained from PMF circulating CD34+ cells in semi-solid collagen culture and (B) in liquid cultures (D8–10 of culture) on cell maturation (Giemsa) as well as CD41 and CD62p membrane expression (MFI) determined by flow cytometry, (C) on percentage of apoptotic annexin V+ cells and Bcl-XL expression level (MFI) determined by flow cytometry, (cells were gated on megakaryocytic cells derived from PMF CD34+ cell culture in the presence of a cocktail of cytokines including IL3, IL6, IL11 and TPO; a red quadrant corresponding to the addition of annexin V+ and pre-aptototic propidiumiodide− (IP) cells and of annexin V+ and post-apoptotic IP+ cells was drawn to define the total proportion of apoptotic cells), (D) on GATA-1 expression as shown by QRT-PCR and western blot analyses and by immunofluorescence microscopy and (E) on modulation of c-myb expression by QRT-PCR analysis.
Antibody ligation of CD9 on megakaryocytes derived from PMF CD34+ cells also increased the percentage of PMF annexin-V+ apoptotic cells associated with megakaryocyte maturation and reduced the level of anti-apoptotic Bcl-xL expression compared to those of IgG-treated control cells (15.92±11.51%. versus 23.45±8.81%, n=5, P=0.02 and 45.48±29.10 AU versus 16.8±16.95 AU, n=5, P=0.03, respectively) (Figure 2C).
We further studied the effect of cell surface CD9 engagement by antibody ligation on GATA-1 and c-Myb (two transcriptional regulators whose expression has opposing effects on megakaryocyte differentiation) during in vitro megakaryocyte differentiation of PMF CD34+ cells (D8–D10). Figure 2D shows that the molecular and protein expression of GATA-1, reported to be down-regulated in PMF megakaryocytes,24 was increased after anti-CD9 monoclonal antibody treatment compared to that of IgG-treated control cells (1.153±0.5487 versus 0.5744±0.2921, P=0.04, n=5, respectively in quantitative real-time polymerase chain reaction (QRT-PCR) experiments and 0.77 versus 0.55 for the Gata-1/Actin ratio in western blot analysis). The increase in GATA-1 protein expression after CD9 ligation was also confirmed by immunocytochemistry (Figure 2D). In contrast, the expression of c-Myb was 8.32-fold higher in PMF megakaryocytes than in HD megakaryocytes (37.26±66.44 versus 4.47±3.89, n=6; P=0.03) and its over-expression was reduced after CD9 antibody ligation (15.53±4.40 versus 10.5±4.82, n=4, P=0.007) in all tested patients (Figure 2E).
Altogether our results show that targeting CD9 through cell surface ligation with Syb monoclonal antibody restores the in vitro maturation of PMF megakaryocytes and suggest that CD9 participates in the dysmegakaryopoieis observed in patients with PMF.
CD9 controls Akt/MAPK balance during dysmegakaryopoiesis in primary myelofibrosis
A balance between Akt (Akt, GSK3β) and MAPK (JNK, p38, p42/p44) signaling pathways is reported to be critical for megakaryopoiesis. We, therefore, studied whether cell surface CD9 engagement by antibody ligation (Syb clone, 10 μg/mL) modified the Akt/MAPK balance in megakaryocytes derived from PMF CD34+ cultures (D6–8) in the presence of Tpo.
Figure 3A shows that cell surface CD9 engagement by antibody ligation reduced the phosphorylation level of Akt in PMF megakaryocytes, as evaluated by flow cytometry [mean fluorescence intensity (MFI) normalized to IgG-treated control cells over time: −11.93±7.215 AU, P=0.02; −7.865±3.524 AU, P=0.007; −3.495±1.724 AU, P=0.007, n=4] and western blot analysis (phospho-Akt Thr308 and Ser473 normalized to total Akt: 0.28 versus 0.80 for anti-CD9 monoclonal antibody-treated cells versus IgG-treated control cells). It also slightly decreased PTEN expression normalized to actin expression (0.51 versus 0.64 for anti-CD9 monoclonal antibody-treated cells versus IgG-treated control cells; data not shown) and decreased the phosphorylation level of downstream GSK3β on serine 9 in PMF megakaryocytes (phospho-GSK3β Ser9 normalized to total GSK3β: 0.31 versus 0.56 for anti-CD9 monoclonal antibody-treated cells versus IgG-treated control cells). In contrast, as shown by flow cytometry, a similar concentration of anti-CD9 monoclonal antibody induced an increase phosphorylation of: (i) JNK (MFI normalized to IgG-treated control cells over time: 12.01±8.48 AU, P=0.04; 15.21±9.82 AU, P=0.03; 25.59±18.68 AU, P=0.04; n=4), and (ii) p38 MAPK (MFI normalized to IgG-treated control cells over time: 3.563±1.449 AU, P=0.02; 11.12±7.55 AU, P=0.04; 14.39±10.78 AU, P=0.04; n=4) (Figure 3B). This increased phosphorylation was confirmed by immunocytology and by western blot analysis of PMF megakaryocytes (phospho-p38α Thr180 and Tyr182 normalized on total p38α: 0.64 versus 0.41 anti-CD9 monoclonal antibody-treated cells versus IgG-treated control cells). We also observed a 2-fold increase in the expression of Jun/AP1, a transcription factor downstream of p38 and JNK, as shown by QRT-PCR (0.064 ± 0.057 versus 0.031±0.339; P=0.02, n=7, fold change = +2.03) (Figure 3B); phosphorylation of ser73 Jun normalized to total Jun was also found to be increased (0.81 versus 0.52, Figure 3B). In contrast, a similar anti-CD9 treatment did not modulate the level of phosphorylation of MAPK p42/p44 (data not shown). Collectively, our results show that CD9 controls the balance of the Akt/MAPK signaling pathways during PMF dysmegakaryopoiesis.
Figure 3.
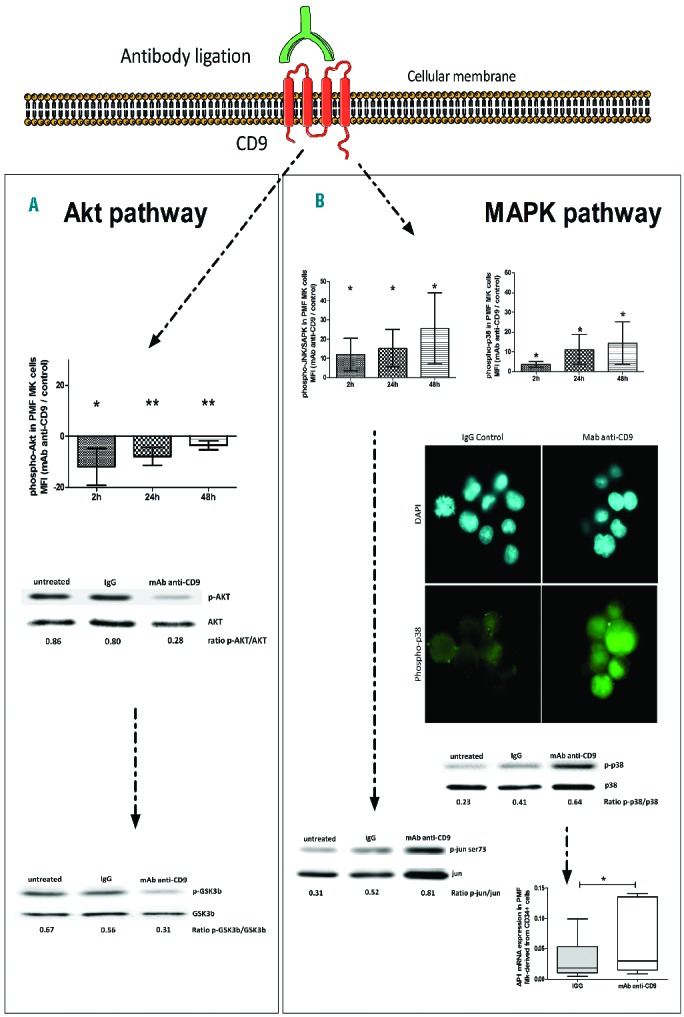
CD9 controls Akt/MAPK balance during PMF dysmegakaryopoiesis. Analysis by cytometry, microscopy and western blot of (A) phospho-Akt (Thr308 and Ser473), - GSK3β (Ser9) levels (MFI), or (B) phospho-p38 (Thr180 and Tyr182) and phospho-JNK levels (MFI) in megakaryocytes (Mk) derived from PMF circulating CD34+ cells (D6 of culture in the presence of Tpo) at 2, 24, and 48 h after anti-CD9 monoclonal antibody (mAb) (clone-SYB; 10 μg/mL) treatment. (A and B) Data were normalized either to IgG control values or to total protein level. (B) QRT-PCR analysis of AP1 transcript expression level in Mk derived from PMF circulating CD34+ cells, treated or not with anti-CD9 mAb (clone-SYB; 10 μg/mL) and normalized to actin level.
CD9 mediates megakaryocyte-mesenchymal stromal cell interactions in primary myelofibrosis
Taking into account the role of MSC in megakaryocyte proliferation and migration,25 we further studied whether CD9 influenced interactions between megakaryocytes and BM MSC and consequently played a role in megakaryocyte deregulation.
Tetraspanin CD9 has been shown to interact with stromal cells through cell surface molecules including CD44, immunoglobulin-like domain containing receptors and integrins within the teraspanin web, named the interactome (“Human Protein Reference Database” and “BioGRID 3.1” interactions on the website of the National Institute of Health).13,26,27 We, therefore, first studied whether the CD9 interactome genes involved in hematopoietic cell and MSC interactions were deregulated in patients. To that purpose, we isolated MSC from the BM of PMF patients (PMF-MSC) and HD (HD-MSC) and verified that cells from both origins expressed the canonical CD73+CD105+CD90+ phenotypic profile of MSC and that they did not express CD45, CD34 hematopoietic markers or HLA-DR (Online Supplementary Figure S2). Then, based on a gene set established on the NCBI database CD9 interactome (Online Supplementary Table S2), we applied gene set enrichment analysis to compare transcriptomes between BM PMF-MSC and HD-MSC (GEO submission number GSE44426), and evidenced a deregulation of CD9 interactome genes in PMF-MSC compared to HD-MSC (Online Supplementary Table S2 and Online Supplementary Figure S3). This deregulation was associated with a down-regulation of CD9 expression on PMF-MSC as compared to HD-MSC, both at transcriptional level (QRT-PCR normalized to the RPL38 housekeeping gene: 14.36±6.028, n=11 versus 21.87±12.56, n=9 for PMF patients and HD, respectively; P=0.05) and protein level (MFI by flow cytometry: 7.38±0.76 AU, n=3 versus 13.07±2.63 AU, n=4 for PMF patients and HD, respectively; P=0.008) (Figure 4A).
Figure 4.
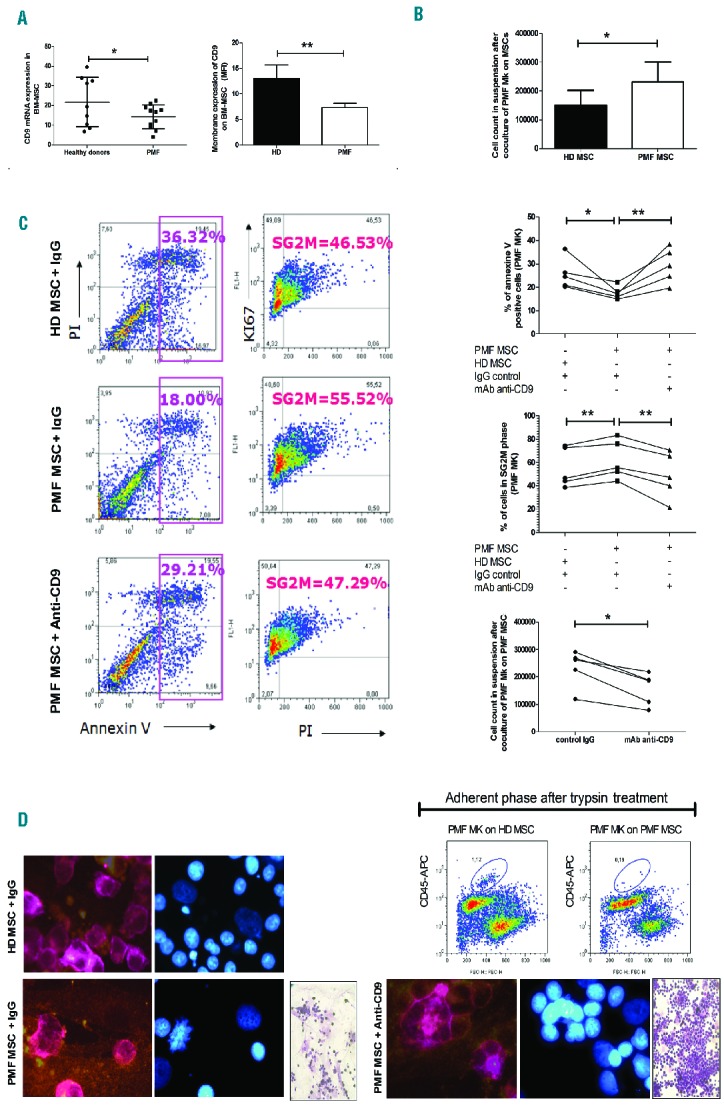
CD9 mediates PMF megakaryocyte-mesenchymal stromal cell interactions. (A) Analysis of CD9 transcript (QRT-PCR) and membrane expression (flow cytometry) levels in MSC isolated from the bone marrow of PMF patients (BM-MSC). (B) Effect of 2 days of co-culture between PMF megakaryocytes (Mk) derived from CD34+ culture (at D6 of Mk culture) and BM-MSC on the number of PMF Mk in suspension. (C) Effect of surface CD9 ligation by anti-CD9 monoclonal antibody (mAb) (clone-SYB; 10 μg/mL) on the proportion of PMF Mk derived from CD34+ culture in apoptosis (annexin V+ cells) and in the cell cycle phases (S+G2M cells) as well as on the cell count of non-adherent PMF Mk in the co-culture with BM MSC. (D) Effect of surface CD9 ligation by anti-CD9 mAb on the proportion of adherent PMF CD45+ hematopoietic cells obtained after co-culture on BM MSC isolated from HD or PMF patients and on adherent PMF Mk in co-culture with MSC isolated from the BM of either HD or PMF patients as shown by Giemsa staining and immunostaining (CD41+ Mk, in pink and CD105+ MSC, in orange).
Subsequently, we compared the effects of PMF-MSC and HD-MSC on the survival and proliferation of megakaryocyte progenitors derived from PMF CD34+ cells in megakaryocyte culture medium (at D6) and analyzed the effect of CD9 membrane ligation by Syb on these processes (from D6 to D8–D10). Figure 4B shows that the number of non-adherent PMF megakaryocytes was higher when PMF hematopoietic cells were co-cultured on PMF-MSC rather than on HD-MSC (from 100,000 implanted hematopoietic cells/mL to 232,600±68,197/mL and 149,800±52,060/mL at D6 of co-culture, respectively; n=5, P=0.03) (Figure 4B).
The higher number of non-adherent PMF megakaryocytes recovered after co-culture on PMF-MSC resulted from: (i) a lower percentage of annexin V+ apoptotic cells (17.68±2.79%, n=5 versus 25.65±6.47%; n=5; P=0.01) (Figure 4C); and (ii) a higher percentage of Ki67+IP+ cycling (S+G2M) cells (63.27±16.62%, n=5 versus 55.23±16.97% n=5, P=0.002) (Figure 4C) which was associated with the presence of PMF CD41+ megakaryocytes in mitosis (Figure 4D). CD9 ligation using Syb monoclonal antibody reduced the number of PMF megakaryocytes in suspension in all tested patients (232,600±68,197 versus 155,600±26,604, n=5; P=0.04; Figure 4C), demonstrating the participation of CD9 in this process. As expected, this was associated with an increased percentage of apoptotic cells (29.38±7.55% versus 17.68±2.79%, n=5; P=0.006) and a reduced proportion of Ki67+IP+ cycling (S+G2M) cells (49.01±19.01% versus 62.27±16.62%, n=5; P=0.003) (Figure 4C).
The higher number of PMF megakaryocytes in suspension is also compatible with their differential adhesion to PMF-MSC or HD-MSC. Cytometric analysis showed that, after trypsin treatment, a lower fraction of CD45+ hematopoietic cells was recovered in the adherent phase of co-cultures with MSC from patients than MSC from HD, suggesting that PMF megakaryocytes were less adhesive to PMF-MSC than to HD-MSC (Figure 4D). Here again, the altered adhesion of megakaryocytes to PMF-MSC was inhibited after CD9 ligation with Syb monoclonal antibody (Figure 4D), confirming the involvement of CD9 in the deregulated in vitro megakaryocyte-MSC interactions in PMF.
CD9 mediates megakaryocyte migration in response to CXCL12 in primary myelofibrosis
The CD9-mediated altered PMF megakaryocyte adhesion to stroma incited us to study the involvement of CD9 in the increased megakaryocyte mobilization/migration observed in the peripheral blood of patients with PMF. Phenotypic analysis performed on megakaryocyte cultures at D10 showed that all CD41 megakaryocytes from PMF patients co-expressed CXCR4 (Figure 5A). Actually, like megakaryocyte CD41+ cells from HD BM, megakaryocytes from PMF patients conserved a high level expression of CXCR4 receptor responsible for CXCL12-induced migration. Figure 5B shows that the percentage of cells co-expressing CD9 and CXCR4 was higher in the CD41high population than in the CD41low one, suggesting an increased co-expression of CD9 and CXCR4 during PMF megakaryocyte differentiation. Interestingly, cell surface CD9 engagement by Syb antibody ligation reduced the mRNA expression of both CXCR4 (0.028±0.015, n=4 versus 0.048±0.16, n=4; P=0.04) and CXCL12 (0.0044±0.0026, n=4 versus 0.0096±0.0013, n=4; P=0.03) in megakaryocytes derived from PMF CD34+ cultures as compared to control IgG-treated cells (Figure 5C), suggesting the possible role of CD9 in the CXCL12/CXCR4-mediated process of megakaryocyte migration. As ligation of CD9 by Syb monoclonal antibody induced cell aggregates,26 we further investigated the functional participation of CD9 on PMF megakaryocyte migration in response to CXCL12 by treating megakaryocytes with siRNA CD9. We first verified that addition of siRNA CD9 down-regulated the CD9 expression in megakaryocytes derived from PMF CD34+ cells (D9 of differentiation) at both transcript level (2.32±2.78 AU, n=6, versus 5.78±6.66 AU, n=6; P=0.04; fold change = −2.49, in siRNA CD9 versus siRNA control treated cells, respectively) and protein level (50% reduction 72 h after lipofection) (Figure 5D). We further demonstrated that a 2-fold reduction of CD9 expression inhibited the CXCL12-dependent migration of PMF megakaryocytes derived from CD34+ cells, as shown by a strong reduction of the percentage of migrating cells after siCD9 treatment as compared to siRNA control treated cells (115.5±20.9%, n=3 and 474.7±169.7%, n=3; respectively; P=0.03) (Figure 5D). Silencing CD9 also significantly reduced the Tyr576/577 phosphorylation level of FAK, the focal adhesion kinase participating in megakaryocyte migration, as compared to siRNA control treated cells (15.42±10.54 MFI, n=5 and 24.12±15.22 MFI, n=5; respectively; P=0.04).
Figure 5.
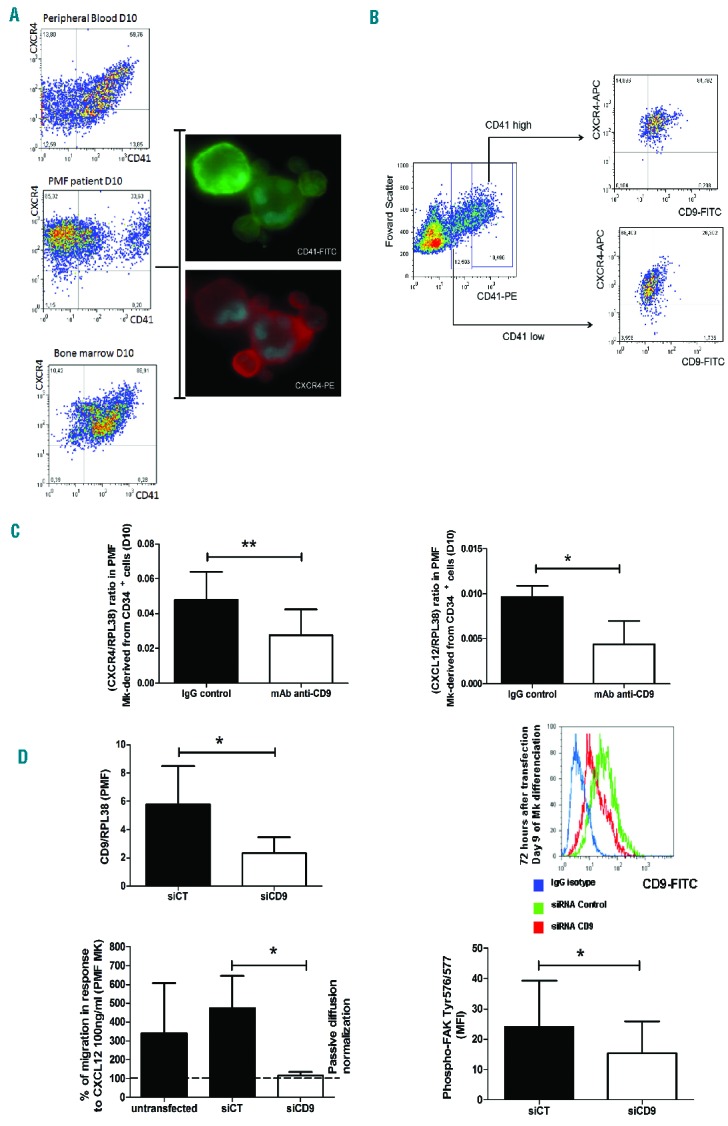
CD9 mediates PMF megakaryocyte migration in response to CXCL12. (A) Expression of CXCR4 on CD41+ PMF megakaryocytes (Mk) derived from PMF circulating CD34+ cells (D8–10 of culture) by flow cytometry and microscopy as compared to Mk derived from healthy donors (unmobilized peripheral blood or bone marrow). (B) CD9 and CXCR4 coexpression on CD41low and CD41high PMF cells. (C) Effect of surface CD9 ligation by anti-CD9 monoclonal antiboby (mAb) (clone-SYB; 10 μg/mL) on CXCR4 and CXCL12 transcript expression normalized to RPL38 by QRT-PCR in Mk derived from PMF circulating CD34+ cells (D8–10 of culture). (D) Effect of silencing CD9 using siRNA CD9 (1 μg/106 cells) in Mk derived from PMF circulating CD34+ cells (D8–10 of culture) on the CD9 transcript and protein expression levels analyzed by QRT-PCR (at 48 h) and by flow cytometry (at 72 h), respectively, and on the percentage of migrating PMF Mk in a Boyden chamber in response to CXCL12 (100 ng/mL) as compared to passive diffusion and on the phospho-FAK (Tyr 576/577) expression level (MFI).
Discussion
PMF is a chronic myeloproliferative neoplasm with numerous alterations of megakaryocytes,27 which are reported to play key roles in the pathophysiological process. Tetraspanin CD9, known to participate in normal megakaryocytic differentiation,17 has been described to be deregulated in myeloproliferative neoplasms and especially in PMF.18 In the present study, we analyzed the potential contribution of CD9 to PMF dysmegakaryopoiesis. Our results show that CD9 (i) is deregulated during PMF megakaryopoiesis; (ii) participates in PMF dysmegakaryopoiesis through modulation of megakaryocyte maturation, apoptosis and Akt/MAPK signaling pathway balance; and (iii) mediates altered megakaryocyte interactions with MSC from the BM of patients as well as altered megakaryocyte migration in response to CXCL12 chemokine.
We found that the CD9 gene was highly expressed in CD34+ cells from PMF patients. As described by Guglielmelli et al., such a high CD9 gene expression appears to be related to PMF patients since it allows their discrimination from healthy subjects and from patients with polycythemia vera and essential thrombocythemia.18 Interestingly, however, we showed that the mean level of CD9 membrane expression is heterogeneous on PMF CD34+ cells and is reduced during in vitro megakaryocyte differentiation to be lower on platelets from patients than on those from HD, suggesting a deregulation of CD9 during PMF megakaryopoieisis. PMF dysmegakaryopoieis is characterized by a defect of megakaryocyte maturation and apoptosis; cell surface CD9 engagement by antibody ligation restored the in vitro PMF megakaryocyte maturation and the apoptotic process known be involved in megakaryocyte differentiation.28 As reported for cancer cells,29 targeting CD9 restores apoptosis in PMF megakaryocytes and is accompanied by a normalization of the anti-apoptotic Bcl-xL protein expression abnormally over-expressed in PMF megakaryocytes.12 Ligation of CD9 on PMF megakaryocytes also induces increased phosphorylation of JNK and p38 MAPK associated with up-regulation of downstream AP1 as well as with a reduction of Akt/GSK3β phosphorylation, two signaling pathways the balance of which is reported to drive megakaryopoiesis.30 The expression of c-myb, which also participates in megakaryopoiesis,31,32 is decreased during megakaryocyte differentiation33 and is regulated by GSK3β34 and p38/JNK.35 In PMF, the increased c-myb expression observed in megakaryocyte derived from CD34+ cells is therefore consistent with reduced megakaryocyte maturation. Consequently, the reduction of c-myb expression after CD9 ligation is compatible with its participation in PMF dysmegakaryopoiesis through CD9 deregulation.
PMF is characterized by the egress of CD34+ progenitors and of micro-megakaryocytes from BM into peripheral blood.1,36,37 By participating in the tetraspanin network, CD9 can interact with molecules within rich lipid microdomains where the G protein-coupled receptors are regulated.13 As an example, CD9 is reported to participate in CXCL12-dependent CD34+ cell migration through modulation of CXCR4 receptor expression.21 As evidenced by bioinformatics prediction (Online Supplementary Figure S4), myb regulatory sites are present in the promoter regions of CXCL12 and CXCR4 genes, confirming that they are both target genes of c-myb.38,39 In PMF, our results showing that CD9 regulates CXCR4 and CXCL12 transcriptional expression (Figure 5) and that this process is reversed by silencing c-myb (data not shown), suggest a link between CD9, the CXCL12/CXCR4 axis and c-myb in the deregulated process of megakaryocyte migration. Thus, the reduced CD9 expression during PMF megakaryopoieisis may render megakaryocytes less sensitive to the CXCL12 chemotactic gradient present in the BM, contributing to their mobilization. This hypothesis is in agreement with the presence of micro-megakaryocytes in the peripheral blood of PMF patients36 and with the role of reduced CXCR4 expression in the increased PMF CD34+ cell migration.9
In patients with myeloproliferative neoplasms, while hematopoietic cells may have acquired similar mutations such as JAK2V617F, MPL or CALR,7,40,41 alterations in megakaryopoiesis differ according to the disease phenotype,42 suggesting that other factors, such as environmental ones, likely participate in the dysmegakaryopoiesis. In PMF, the BM microenvironment is impaired and splenic stromal cells are reported to exhibit altered differentiation and functional characteristics.43 Our present data, showing altered expression of CD9 on hematopoietic and stromal cells from PMF patients as well as modulated expression on osteoblasts in advanced stages of the disease,19 suggest that CD9 and the tetraspanin web participate in the altered interplay between hematopoietic and stromal cells in patients. The reduced adhesion of megakaryocyte to PMF-MSC associated with increased survival and proliferation in co-cultures with stroma, which was corrected after cell surface CD9 ligation, strengthens our hypothesis.
In conclusion, while the underlying mechanism for alterations in CD9 expression in PMF is still unknown, this study indicates that CD9 plays a role in PMF dysmegakaryopoiesis and affects interactions between megakaryocytes and BM stromal cells. Our results strengthen the “bad seed in bad soil” hypothesis that we have proposed in which alterations of the interplays between hematopoietic and stromal cells participate in the pathogenesis of the disease.1 Taking into account the role of megakaryocytes and stromal cells in the pathogenesis of PMF, they are of importance with regards to the recent development of new therapeutic strategies. Indeed, by regulating the apoptotic process, targeting CD9 may improve the dysmegakaryopoiesis that characterizes PMF, as demonstrated in human cancer cells.44 In the same way, the role of the Akt pathway driven by CD9 in PMF megakaryocytes is consistent with the findings of a recent study that established Akt as a rational therapeutic target for the treatment of patients with myeloproliferative neoplasms.45 In brief, our results indicate original therapeutic avenues targeting hematopoietic cell-stroma interactions in PMF.
Acknowledgments
The authors are indebted to Prof Y Masse, Dr. C Blondeau and Dr. A Touma (Hôpital Intercommunal, Aulnay-Sous-Bois, France), to Prof. JV Malfuson (HIA Percy, Clamart, France), to Ms Chantal Tisseuil, IDE Recherche Clinique (CHU Dupytren, Limoges, France) and to Ms Corinne Grafl from the Department of Internal Medicine I, Division of Hematology and Hemostaseology (Medical University of Vienna, Austria) for supplying samples from heathy donors and patients.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by grants from: the Association "Vaincre le Cancer-Nouvelles Recherches Biomédicales", Convention de Recherche INCa n. PL054 and n. R06031LP; the Association pour la Recherche contre le Cancer (ARC, 9806), “Laurette Fugain” Association (ALF/n. 06-06, project # R06067LL), the “Contrat d’Interface” with Paul Brousse Hospital, the "Groupement d’Intérêt Scientifique (GIS)-Institut des Maladies Rares 03/GIS/PB/SJ/n. 35, the European Union-EUMNET Project (QLRT-2001-01123), INCa (PL054 and 2007-1-PL5-Inserm 11-1), and the "Ligue Contre le Cancer" (Equipe labellisée “LA LIGUE 2010”). The work in Florence was supported by a special grant from Associazione Italiana per la Ricerca sul Cancro-“AIRC 5 per Mille”- to AGIMM, “AIRC-Gruppo Italiano Malattie Mieloproliferative” (#1005).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Lataillade JJ, Pierre-Louis O, Hasselbalch HC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112(8): 3026–3035. [DOI] [PubMed] [Google Scholar]
- 2.James C, Ugo V, Le Couedic JP, Staerk J, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. [DOI] [PubMed] [Google Scholar]
- 3.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476. [DOI] [PubMed] [Google Scholar]
- 4.Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217–1219. [DOI] [PubMed] [Google Scholar]
- 5.Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasho TL, Finke CM, Hanson CA, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia. 2012;26(5):1135–1137. [DOI] [PubMed] [Google Scholar]
- 7.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Eng J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. [DOI] [PubMed] [Google Scholar]
- 9.Bogani C, Ponziani V, Guglielmelli P, et al. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells. 2008;26(8):1920–1930. [DOI] [PubMed] [Google Scholar]
- 10.Florena AM, Tripodo C, Di Bernardo A, et al. Different immunophenotypical apoptotic profiles characterise megakaryocytes of essential thrombocythaemia and primary myelofibrosis. J Clin Pathol. 2009;62(4):331–338. [DOI] [PubMed] [Google Scholar]
- 11.Theophile K, Hussein K, Kreipe H, Bock O. Expression profiling of apoptosis-related genes in megakaryocytes: BNIP3 is down-regulated in primary myelofibrosis. Exp Hematol. 2008;36(12):1728–1738. [DOI] [PubMed] [Google Scholar]
- 12.Ciurea SO, Merchant D, Mahmud N, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110(3):986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J. 2009;420(2):133–154. [DOI] [PubMed] [Google Scholar]
- 14.Boucheix C, Perrot JY, Mirshahi M, et al. A new set of monoclonal antibodies against acute lymphoblastic leukemia. Leuk Res. 1985;9(5):597–604. [DOI] [PubMed] [Google Scholar]
- 15.Boucheix C, Benoit P, Frachet P, et al. Molecular cloning of the CD9 antigen. A new family of cell surface proteins. J Biol Chem. 1991;266(1):117–122. [PubMed] [Google Scholar]
- 16.Le Naour F, Francastel C, Prenant M, Lantz O, Boucheix C, Rubinstein E. Upregulation of CD9 expression during TPA treatment of K562 cells. Leukemia. 1997;11(8):1290–1297. [DOI] [PubMed] [Google Scholar]
- 17.Clay D, Rubinstein E, Mishal Z, et al. CD9 and megakaryocyte differentiation. Blood. 2001;97(7):1982–1989. [DOI] [PubMed] [Google Scholar]
- 18.Guglielmelli P, Zini R, Bogani C, et al. Molecular profiling of CD34+ cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem Cells. 2007;25(1):165–173. [DOI] [PubMed] [Google Scholar]
- 19.Bock O, Muth M, Theophile K, et al. Identification of new target molecules PTK2, TGFBR2 and CD9 overexpressed during advanced bone marrow remodelling in primary myelofibrosis. Br J Haematol. 2009;146(5):510–520. [DOI] [PubMed] [Google Scholar]
- 20.Rubinstein E, Le Naour F, Lagaudriere-Gesbert C, Billard M, Conjeaud H, Boucheix C. CD9, CD63, CD81, and CD82 are components of a surface tetraspan network connected to HLA-DR and VLA integrins. Eur J Immunol. 1996;26(11):2657–2665. [DOI] [PubMed] [Google Scholar]
- 21.Leung KT, Chan KY, Ng PC, Lau TK, Chiu WM, Tsang KS, et al. The tetraspanin CD9 regulates migration, adhesion, and homing of human cord blood CD34+ hematopoietic stem and progenitor cells. Blood. 2011;117(6):1840–1850. [DOI] [PubMed] [Google Scholar]
- 22.Desterke C, Bilhou-Nabera C, Guerton B, et al. FLT3-mediated p38-MAPK activation participates in the control of megakaryopoiesis in primary myelofibrosis. Cancer Res. 2011;71(8):2901–2915. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannucchi AM, Pancrazzi A, Guglielmelli P, et al. Abnormalities of GATA-1 in megakaryocytes from patients with idiopathic myelofibrosis. Am J Pathol. 2005;167(3):849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng L, Qasba P, Vanguri P, Thiede MA. Human mesenchymal stem cells support megakaryocyte and pro-platelet formation from CD34(+) hematopoietic progenitor cells. J Cell Physiol. 2000;184(1):58–69. [DOI] [PubMed] [Google Scholar]
- 26.Lagaudriere-Gesbert C, Le Naour F, Lebel-Binay S, et al. Functional analysis of four tetraspans, CD9, CD53, CD81, and CD82, suggests a common role in costimulation, cell adhesion, and migration: only CD9 upregulates HB-EGF activity. Cell Immunol. 1997;182(2):105–112. [DOI] [PubMed] [Google Scholar]
- 27.Thiele J, Kvasnicka HM, Zankovich R, Diehl V. Clinical and morphological criteria for the diagnosis of prefibrotic idiopathic (primary) myelofibrosis. Ann Hematol. 2001;80(3): 160–165. [DOI] [PubMed] [Google Scholar]
- 28.De Botton S, Sabri S, Daugas E, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100(4):1310–1317. [DOI] [PubMed] [Google Scholar]
- 29.Nakamoto T, Murayama Y, Oritani K, et al. A novel therapeutic strategy with anti-CD9 antibody in gastric cancers. J Gastroenterol. 2009;44(9):889–896. [DOI] [PubMed] [Google Scholar]
- 30.Guerriero R, Parolini I, Testa U, et al. Inhibition of TPO-induced MEK or mTOR activity induces opposite effects on the ploidy of human differentiating megakaryocytes. J Cell Sci. 2006;119(Pt 4):744–752. [DOI] [PubMed] [Google Scholar]
- 31.Ono M, Matsubara Y, Shibano T, Ikeda Y, Murata M. GSK-3beta negatively regulates megakaryocyte differentiation and platelet production from primary human bone marrow cells in vitro. Platelets. 2011;22(3):196–203. [DOI] [PubMed] [Google Scholar]
- 32.Hilton DJ, Kile BT, Alexander WS. Mutational inhibition of c-Myb or p300 ameliorates treatment-induced thrombocytopenia. Blood. 2009;113(22):5599–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barroga CF, Pham H, Kaushansky K. Thrombopoietin regulates c-Myb expression by modulating micro RNA 150 expression. Exp Hematol. 2008;36(12):1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitagawa K, Kotake Y, Hiramatsu Y, et al. GSK3 regulates the expressions of human and mouse c-Myb via different mechanisms. Cell Div. 2010;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolaides NC, Correa I, Casadevall C, Travali S, Soprano KJ, Calabretta B. The Jun family members, c-Jun and JunD, transactivate the human c-myb promoter via an Ap1-like element. J Biol Chem. 1992;267(27):19665–19672. [PubMed] [Google Scholar]
- 36.Castro-Malaspina H. Pathogenesis of myelofibrosis: role of ineffective megakaryopoiesis and megakaryocyte components. Prog Clin Biol Res. 1984;154:427–454. [PubMed] [Google Scholar]
- 37.Barosi G, Grossi A, Comotti B, Musto P, Gamba G, Marchetti M. Safety and efficacy of thalidomide in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;114(1):78–83. [DOI] [PubMed] [Google Scholar]
- 38.Quintana AM, Liu F, O’Rourke JP, Ness SA. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer. 2011; 11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Xu S, Zeng X, et al. c-myb activates CXCL12 transcription in T47D and MCF7 breast cancer cells. Acta Biochim Biophys Sin. 2010;42(1):1–7. [DOI] [PubMed] [Google Scholar]
- 40.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia. 2014;28(7):1407–1413. [DOI] [PubMed] [Google Scholar]
- 42.Balduini A, Badalucco S, Pugliano MT, et al. In vitro megakaryocyte differentiation and proplatelet formation in ph-negative classical myeloproliferative neoplasms: distinct patterns in the different clinical phenotypes. PloS one. 2011;6(6):e21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brouty-Boye D, Briard D, Azzarone B, et al. Effects of human fibroblasts from myelometaplasic and non-myelometaplasic hematopoietic tissues on CD34+ stem cells. Int J Cancer. 2001;92(4):484–488. [DOI] [PubMed] [Google Scholar]
- 44.Murayama Y, Miyagawa J, Oritani K, et al. CD9-mediated activation of the p46 Shc iso-form leads to apoptosis in cancer cells. J Cell Sci. 2004;117(Pt 15):3379–3388. [DOI] [PubMed] [Google Scholar]
- 45.Khan I, Huang Z, Wen Q, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia. 2013;27(9):1882–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]