

Summary
The tethering factor p115 (known as Uso1p in yeast) has been shown to facilitate Golgi biogenesis and membrane traffic in cells in culture. However, the role of p115 within an intact animal is largely unknown. Here, we document that depletion of p115 by using RNA interference (RNAi) in C. elegans causes accumulation of the 170 kD soluble yolk protein (YP170) in the body cavity and retention of the yolk receptor RME-2 in the ER and the Golgi within oocytes. Structure–function analyses of p115 have identified two homology regions (H1 and H2) within the N-terminal globular head and the coiled-coil 1 (CC1) domain as essential for p115 function. We identify a new C-terminal domain of p115 as necessary for Golgi ribbon formation and cargo trafficking. We show that p115 mutants that lack the fourth CC domain (CC4) act in a dominant-negative manner to disrupt Golgi and prevent cargo trafficking in cells containing endogenous p115. Furthermore, using RNAi of p115 and the subsequent transfection with p115 deletion mutants, we show that CC4 is necessary for Golgi ribbon formation and membrane trafficking in cells depleted of endogenous p115. p115 has been shown to bind a subset of ER-Golgi SNAREs through CC1 and CC4 domains (Shorter et al., 2002). Our findings show that CC4 is required for p115 function, and suggest that both the CC1 and the CC4 SNARE-binding motifs participate in p115-mediated membrane tethering.
KEY WORDS: Golgi complex, p115, Trafficking, Uso-1
Introduction
Secretion in eukaryotic cells involves the passage of cargo through a linear assembly of membrane-bound compartments and is mediated by vesicular carriers. Transport requires pairing of transport vesicles with the appropriate acceptor membrane. Recognition between a vesicle and a target membrane is mediated by tethering factors and soluble N-ethylmaleimide (NEM)-sensitive factor (NSF) attachment protein (SNAP) receptors (SNAREs). Tethering factors appear to mediate initial, loose association of vesicles and target membranes, which is followed by a tighter pairing facilitated by the SNAREs. Despite extensive inquiry, the exact mechanisms of tethering remain poorly characterized.
One of the best-studied tethers is the general vesicular transport factor p115 and its yeast homologue Uso1p. Initial studies identified Uso1p as an ER-Golgi transport factor because the temperature sensitive mutant uso1-1 blocks traffic of yeast invertase to the Golgi (Nakajima et al., 1991). Subsequently, Uso1p has been shown to regulate sorting of select proteins into COPII vesicles in vivo (Morsomme et al., 2003; Morsomme and Riezman, 2002) and to mediate COPII vesicle tethering to Golgi membranes in vitro (Barlowe, 1997).
Mammalian p115 has been implicated in COPII and COPI vesicle tethering. p115 is detected on COPII vesicles and COPII vesicles do not tether to Golgi membranes in the presence of anti-p115 antibodies (Allan et al., 2000; Alvarez et al., 2001). In mammalian cells, COPII vesicles may fuse with each other to form larger structures – perhaps vesicular tubular clusters (VTCs) – and p115 appears to be required for this step because removal of p115 when carrying out an in vitro assay prevents fusion of COPII vesicles to generate larger intermediates (Bentley et al., 2006).
p115 was initially identified as a cytosolic factor that is required for COPI-vesicle-mediated intra-Golgi transport (Clary and Rothman, 1990; Sapperstein et al., 1995; Waters et al., 1992; Wilson et al., 1992). In agreement, p115 has been detected on isolated COPI vesicles (Malsam et al., 2005); it also promotes tethering of COPI vesicles to Golgi membranes in vitro (Sonnichsen et al., 1998).
Findings from in vitro assays were analyzed together with results from in vivo analyses in insect and mammalian cells. Depletion of p115 in insect cells causes fragmentation of Golgi cisternae (Kondylis and Rabouille, 2003), whereas inactivation of p115 with antibodies or siRNA-mediated depletion of p115 from mammalian cells causes fragmentation of Golgi ribbon and the formation of Golgi mini-stacks adjacent to ER exit sites (Alvarez et al., 1999; Guo et al., 2008; Holloway et al., 2007; Nelson et al., 1998; Puthenveedu and Linstedt, 2001; Puthenveedu and Linstedt, 2004; Smith et al., 2009; Sohda et al., 2007; Sohda et al., 2005).
The requirement for p115 in protein trafficking is varied. Mammalian cells depleted of p115 show inhibition of vesicular stomatitis virus glycoprotein (VSV-G) traffic during exit from the ER (Puthenveedu and Linstedt, 2004), but trafficking of the transmembrane ligand Delta (ligand of Notch) to the surface of S2 insect cells (Kondylis and Rabouille, 2003) appears unaffected. Similarly, secretion of soluble proteins is delayed but not inhibited in p115-depleted mammalian cells (Sohda et al., 2007; Sohda et al., 2005). Thus, it appears that p115 exerts a modest effect on trafficking of some proteins and has a more pronounced effect on trafficking of other cargoes, such as VSV-G. Here, we assess p115 function in intact C. elegans, and show that depletion of p115 by using RNA interference (RNAi) does not inhibit secretion of the 170 kD soluble yolk protein (YP170) from the intestine, but affects the trafficking of the transmembrane yolk receptor RME-2 in oocytes.
p115 has an N-terminal globular domain that contains two homology regions (H1 and H2), which show a high degree of amino acid conservation in all p115 orthologs. The globular domain is followed by a region that is predicted to form four coiled-coil domains (CC1, CC2, CC3, CC4) and one C-terminal acidic domain (AD) (see Fig. 4A, B). p115 exists in vivo as a parallel homodimer (Sapperstein et al., 1995). p115 binds a number of cellular proteins, such as Rab1 GTPase through a region that requires residue R39 in H1 (An et al., 2009; Beard et al., 2005; Shorter et al., 2002), β-COP through a region that requires E19 in H1 (Guo et al., 2008), COG2 through a region that requires amino acids 200-247 in H2 (Sohda et al., 2007), SNAREs through CC1 and CC4 (Shorter et al., 2002), and GM130 and giantin through the AD (Alvarez et al., 2001; Linstedt et al., 2000; Nakamura et al., 1997; Nelson et al., 1998; Sonnichsen et al., 1998). Structure-function studies, in which endogenous p115 had been replaced with p115 that were mutated in specific domains, show that mutations in H1 or H2, and deletions of H2 or CC1 inhibit p115 function in Golgi biogenesis and/or cargo traffic (Guo et al., 2008; Puthenveedu and Linstedt, 2004; Sohda et al., 2007). Thus, H1, H2 and CC1 represent functional domains.
Fig. 4.
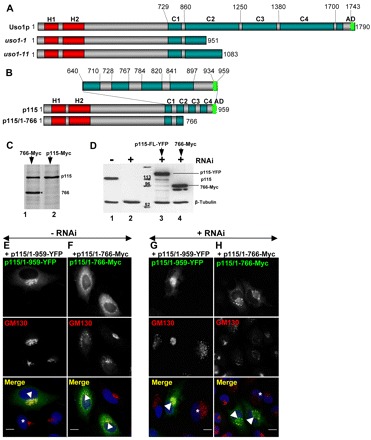
p115/1-766 mutant disrupts Golgi ribbon. (A,B) Diagram of full-length Uso1p and Uso1p encoded by uso1-1 and uso1-11, full-length p115 and p115/1-766. H, homology regions between the yeast and mammalian proteins. C, coiled-coil regions. AD, acidic domain. (C) Control HeLa cells (lane 2) or HeLa cells expressing p115/1-766 (lane 1) were labeled with 35S-Met/Cys for 18 hours, lysed and lysates immunoprecipitated with anti-p115 antibodies. Precipitates were processed by SDS-PAGE and fluorography. Similar levels of endogenous p115 and of exogenous p115/1-766 are detected. (D) Control HeLa cells (lane 1) or HeLa cells silenced with anti-p115 siRNA for 3 days (lanes 2–4) were either mock transfected (lane 2), transfected with YFP-p115/1-959 (lane 3) or Myc-p115/1-766 (lane 4) and cultured for additional 18 hours. Cells were lysed and lysates were processed by SDS-PAGE and immunoblotted with indicated antibodies. Cells transfected with anti-p115 RNA oligonucleotides show substantial depletion of endogenous p115 (lane 2). p115-depleted cells transfected with constructs show robust expression of YFP-p115/1-959 (lane 3) or Myc-p115/1-766 (lane 4). (E,F) HeLa cells transfected with GFP-tagged p115/1-959 or p115/1-766 were analyzed by immunofluorescence with indicated antibodies. Expression of p115/1-959 does not alter Golgi morphology (E). Expression of p115/1-766 disrupts Golgi ribbons (F). Scale bars: 10 µm. (G,H) HeLa cells silenced with anti-p115 siRNA for 3 days were transfected with YFP-p115/1-959 or Myc-p115/1-766, cultured for 18 hours and analyzed by immunofluorescence with anti-GM130 and either YFP fluorescence (G) or anti-Myc antibodies (H). Depletion of p115 fragments Golgi ribbon (cells marked with *). Expression of full-length p115 reverses Golgi disruption (G, cell marked with arrowhead). Expression of p115/1-766 does not reverse Golgi disruption (H, cells marked with arrowheads). Scale bars: 10 µm.
However, the temperature sensitive yeast mutants uso1-1 and uso1-11, which contain the entire globular head and the CC1 domain (Fig. 4) are functionally compromised (Seog et al., 1994; Yamakawa et al., 1996). This suggests that domains CC2, CC3 and CC4, as well as the AD – all of which are missing in uso1-1 and uso1-11 – are important for the function of Uso1p and, perhaps, p115. In support, in vivo deletion of the C-terminal region from bovine p115 has been reported to inhibit exocytic traffic (Satoh and Warren, 2008).
Here, we assessed the ability of mutant p115 that lack various C-terminal domains to sustain Golgi ribbon formation and cargo traffic, using overexpression and ‘replacement’ strategy. We show that deletion of the AD does not influence p115 function, in support of a previous report (Puthenveedu and Linstedt, 2004). We document that p115 constructs that miss the C-terminal CC3–CC4–AD or only the CC4–AD are compromised in function. Furthermore, we show that p115 that lacks only CC4 is unable to support Golgi ribbon formation and cargo trafficking. Our findings suggest that CC4 represents a so-far-unknown functional domain in p115. Because CC4 has been shown to bind a subset of SNAREs that are involved in traffic between the ER and the Golgi, our findings suggest a new model for p115-mediated membrane tethering.
Results
Effect of p115 depletion on the Golgi ribbon and cargo traffic
Two p115-targeting siRNA sequences (siRNA #9 and siRNA#12) effectively (>75%) depleted p115 from HeLa cells (Fig. 1A). siRNA #9 was used in subsequent experiments. RNAi experiments appear to have been specifically targeting p115 because scrambled RNA of the same nucleotide composition does not cause p115 depletion (Fig. 1A), and normal levels of non-target GM130, Sly1 and calreticulin are present in p115-depleted cells (Fig. 1B).
Fig. 1.
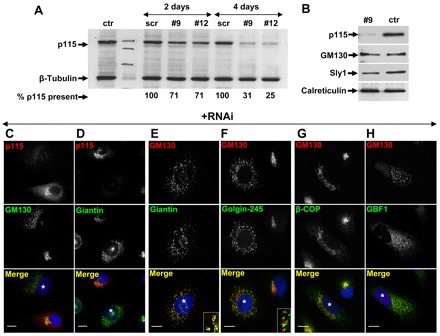
Effects of p115 depletion on Golgi ribbon. (A) HeLa cells mock transfected (ctr), transfected with scrambled (scr) RNA or with siRNA targeting p115 (#9 and #12) were cultured for 2 or 4 days, lysed, and the lysates immunoblotted with the indicated antibodies. Blots were quantified by densitometry to assess p115 levels (numbers below panel). (B) HeLa cell lysates from mock-transfected cells (ctr) or cells silenced with anti-p115 siRNA for 4 days were immunoblotted using the antibodies indicated. (C–H) HeLa cells silenced with anti-p115 siRNA for 4 days were analyzed by immunofluorescence using the antibodies indicated. p115 depletion causes disruption of the Golgi complex (C–D). Golgi fragments show polarized localization of GM130, giantin and golgin-245 (E–F). Insets show higher magnification views. GBF1 and COPI are recruited to Golgi fragments (G–H). p115 depleted cells are marked with *. Scale bars: 10 µm.
p115 depletion causes fragmentation of the Golgi ribbon, as evidenced by the relocation of GM130 and giantin from a perinuclear structure to punctate elements dispersed throughout the cell (Fig. 1C,D, cells marked with *). In most images we selected fields that contain at least one non-depleted cell among the vast majority of p115-depleted cells. The Golgi fragments appear polarized as shown by the localization of the cis-Golgi marker GM130 relative to giantin (Fig. 1E) or the TGN marker golgin-245 (Fig. 1F). This is in agreement with EM images showing stacks of individual Golgi cisternae in p115-depleted cells (Sohda et al., 2005).
Golgi polarity is believed to arise and be maintained through continuous COPI-mediated recycling of Golgi proteins from distal to proximal cisterna (Losev et al., 2006). In p115-depleted cells, Golgi fragments contain the GBF1 guanine nucleotide exchange factor, an ARF activator shown to regulate COPI function at the ER–Golgi interface (Fig. 1H) (Garcia-Mata et al., 2003) and the β-COP component of COPI (Fig. 1G). This suggests that COPI-mediated recycling occurs in p115-depleted cells.
p115 has been shown to be required for trafficking of VSV-G (Alvarez et al., 1999; Puthenveedu and Linstedt, 2004). We confirmed this finding by expressing ts045VSV-G in control cells and in p115-depleted cells at the non-permissive temperature, and by assessing VSV-G traffic following the shift to the permissive temperature. VSV-G is misfolded and retained within the ER at non-permissive temperature in control cells and p115-depleted cells (Fig. 2A,B). After shifting control cells to the permissive temperature for 2 hours, VSV-G is detected within the Golgi and on the plasma membrane (Fig. 2C). By contrast, in p115-depleted cells, VSV-G is still predominantly detected within the ER (Fig. 2D). Only after 12 hours at the permissive temperature, VSV-G exits the ER and is delivered to Golgi fragments and the plasma membrane (Fig. 2E).
Fig. 2.
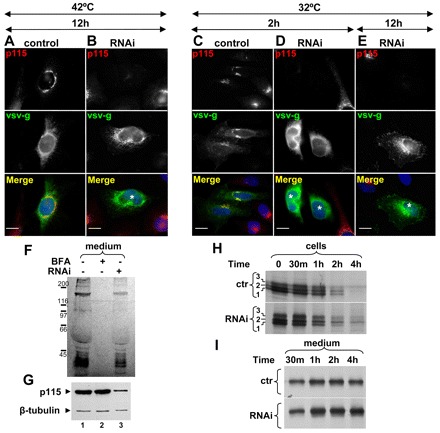
Effect of p115 depletion on cargo traffic. (A–E) Mock-transfected HeLa cells (control) or cells transfected with anti-p115 siRNA for 3 days (RNAi) were transfected with ts045-VSV-G and cultured for additional 12 hours at 42°C (A–B). Cells were shifted to 36°C for indicated times and analyzed by immunofluorescence with the antibodies indicated. In control cells, VSV-G translocates to the Golgi and the plasma membrane (C). In p115-depleted cells, VSV-G is largely retained within the ER (D). In p115-depletd cells after 12 hours at 36°C, VSV-G is detected in internal punctate fragments and the plasma membrane (E). p115-depleted cells are marked with *. Scale bars: 10 µm. (F,G) Untreated cells, cells treated with BFA for 30 minutes and cells silenced with anti-p115 siRNA for 4 days were pulsed with 35S-Met/Cys for 30 minutes and chased with non-radioactive medium (with or without BFA) for 1 hour. Equivalent amounts of cell lysates and media were processed by SDS-PAGE and fluorography. Secretion occurs from control and p115-depleted cells, but not from BFA-treated cells (F). p115 depletion was confirmed by immunoblot of cell lysates with indicated antibodies (G). (H,I) Control cells and cells silenced with anti-p115 siRNA for 3 days were transfected with Myc-tagged cochlin for 24 hours, pulsed with 35S-Met/Cys for 30 minutes and chased with non-radioactive medium for indicated times. At each time point, media were collected and cells were lysed and subjected to immunoprecipitation with anti-Myc. Precipitates were analyzed by SDS-PAGE and fluorography. Cochlin is processed and secreted from control and p115-depleted cells.
p115 depletion has minimal effect on the trafficking of a soluble form of the transmembrane protein dipeptidyl peptidase IV (named sDPPIV) (Sohda et al., 2005). However, sDPPIV </emph>might not represent an optimal soluble secretory cargo. Thus, we assessed trafficking of a bona-fide secretory protein. We selected cochlin, an extracellular matrix glycoprotein that is efficiently synthesized and secreted from HeLa cells (Grabski et al., 2003). We first show that control cells and p115-depleted cells secrete analogous subsets of radiolabeled proteins in a pulse-chase experiment (Fig. 2F, lanes 1 and 3). As a negative control, BFA-treated cells that synthesize but not secrete proteins were used (Fig. 2F, lane 2). An immunoblot confirms efficient p115 depletion within this experiment (Fig. 2G).
Cochlin trafficking was explored in control cells and p115-depleted cells by pulse-chase experiments. In control cells, cochlin is synthesized as a 60 kD peptide (Fig. 2H, control panel, band 1) that is rapidly modified into two additional cochlin forms (bands 2 and 3). The protein visible in band 3 is resistant to Endo-H digestion (Grabski et al., 2003; data not shown), indicating Golgi-mediated carbohydrate processing. Fully glycosylated cochlin is released from control cells within 30 minutes of pulse, and release continues during the chase (Fig. 2I). The majority (>75%) of cochlin present at the start of the chase is released from cells within 2 hours of chase.
The same three cochlin protein bands are detected in p115-depleted cells at the beginning of the chase (Fig. 2H, RNAi panel). Processing of cochlin in p115-depleted cells might be slower, and the partially glycosylated protein from band 2 is still detectable after 4 hours of chase, at a time when it is not present in control cells. Thus, ER to Golgi trafficking of cochlin might be delayed in p115-depleted cells. Nevertheless, cochlin is detected in the culture medium of p115-depleted cells within 30 minutes of chase (Fig. 2I, RNAi panel), and the release continues during the chase. As in control cells, the majority (>78%) of cochlin present at the start of the chase is secreted within 2 hours of chase. Thus, it appears that trafficking of soluble secretory proteins, such as cochlin, is minimally influenced by the depletion of p115. Together, the Golgi biogenesis and the cargo traffic studies define the baseline for examining p115 function in intact animals and in replacement approaches to identify new functional domains within p115.
Effect of p115 depletion in the intact animal
Previous studies in Arabidopsis thaliana p115 null-mutants have shown viable but dwarf plants (Takahashi et al. 2010). By contrast, genetic ablation of p115 in the fly Drosophila melanogaster causes embryonic lethality (FlyBase). This suggests that the complex morphogenic events that are necessary for the development of the animal require p115, and that p115 function in animals can only be examined by silencing the gene encoding p115 in select tissues or at select times. Thus, we used RNAi to deplete the p115 homologue in C. elegans, uso-1 (Kamath and Ahringer, 2003). RNAi in worms most effectively silences gene expression in the intestine and oocytes (Kamath et al., 2003). We first tested whether depletion of C. elegans p115 affects secretion of YP170 from the intestine by using a GFP-tagged version of YP170 (YP170–GFP). In wild-type or RNAi control worms, YP170–GFP can be detected in the intestine – where it is synthesized, in oocytes – which endocytose it, and in the developing embryos – where it is used as a nutrient (Fig. 3A, left image). The majority of YP170-GFP in control worms is detected within oocytes. In uso-1-depleted worms we observed increased accumulation of YP170-GFP in the intestine (Fig. 3A, middle image, arrows). When worms were left to feed on uso-1 double-stranded RNA for longer >4 days, YP170-GFP progressively accumulated in the body cavity (Fig. 3A, right image). Abnormal accumulation of YP170-GFP in the body cavity suggests that YP170-GFP is secreted from intestinal cells, but is not efficiently endocytosed into oocytes.
Fig. 3.
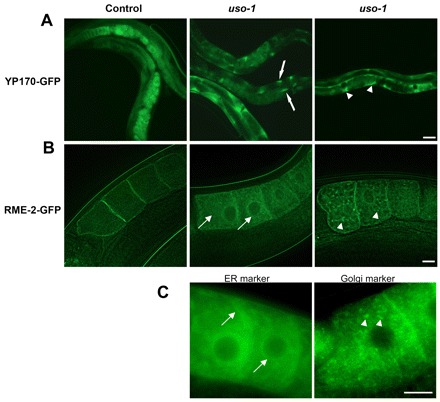
Effect of p115 depletion in C. elegans. (A) Localization of YP170–GFP in control and uso-1 siRNA-treated animals. YP170–GFP in RNAi control worms is observed in the intestine, the oocytes and embryos. uso-1-depleted animals show abnormal accumulation of YP170–GFP in the intestine (middle image, arrows) and body cavity (right image, arrowheads). Scale bar: 50 µm. (B) Localization of RME-2–GFP in RNAi control worms and uso-1 siRNA-treated animals. In control worms RME-2–GFP shows predominantly cell surface localization. uso-1-depleted animals show abnormal accumulation of RME-2–GFP in the ER (middle image, arrows) and the Golgi (right image, arrowheads). Scale bar: 10 µm. (C) Localization of the GFP-labeled ER marker protein SP12 and Golgi marker protein UGTP-1 in oocytes. Scale bar: 10 µm.
YP170 is endocytosed into oocytes by the RME-2 yolk receptor. Thus, we tested trafficking of GFP-tagged RME-2 (RME-2-GFP) in control worms and those that were uso-1-depleted. In control oocytes, RME-2–GFP is predominantly localized at the plasma membrane and in cortical endosomes (Fig. 3B, left image). Depletion of uso-1 in RNAi experiments resulted in two types of characteristic trafficking phenotype (Balklava et al., 2007). In the majority of uso-1-depleted worms, RME-2–GFP increasingly accumulated in the endoplasmic reticulum, which is dispersed throughout the cytoplasm of the oocyte (Fig. 3B, middle image). The RME-2-GFP pattern resembled that of the GFP-labeled ER marker protein SP12 (Fig. 3C, left image). A substantial number of worms also showed possible Golgi accumulation in punctate structures that were dispersed in the cytoplasm of oocytes (Fig. 3B, right image). This pattern is analogous to the pattern of the GFP-labeled Golgi marker protein UGTP-1 (Fig. 3C, right image). The intracellular accumulation of RME-2-GFP correlated with reduced levels of RME-2–GFP at the cortex and the cell surface. Thus, it appears that – like in mammalian cells – p115 regulates protein transport in worms. Furthermore, in both mammals and worms, p115 depletion appears to inhibit trafficking of transmembrane proteins more than that of soluble proteins.
Effect of p115/1-766 on the Golgi ribbon
H1, H2 and CC1 are essential for p115 function (An et al., 2009; Guo et al., 2008; Puthenveedu and Linstedt, 2004; Sohda et al., 2007; Takahashi et al., 2010). A possible role for CC2–CC4–AD was suggested by the uso1-1 and uso1-11 yeast mutants that contain intact H1, H2 and CC1 (Fig. 4A) but are functionally compromised (Seog et al., 1994), and the finding that deletion of the C-terminal region in bovine p115 inhibits exocytic traffic (Satoh and Warren, 2008). To identify functional domains within the C-terminal region of p115, we generated a deletion mutant p115/1-766 that is similar to the yeast uso1-11 (Fig. 4B) in that it lacks the CC3, CC4 and AD regions. Expression of p115/1-766 in HeLa cells containing endogenous p115 caused disruption of the Golgi ribbon into punctate fragments (Fig. 4F). Only 32% of HeLa cells that express p115/1-766 contained morphologically normal Golgi ribbons. This compares with >90% of HeLa cells that express full-length p115 and contain Golgi ribbons (Fig. 4E).
The approximate ratio of p115/1-766 to endogenous p115 in transfected HeLa cells was assessed and, as shown in Fig. 4C, approximately equivalent levels were detected. Within this experiment, immunofluorescence analysis indicates an ∼65% transfection rate (not shown). This suggests that, within transfected cells, the amount of p115/1-766 exceeds that of endogenous p115 ∼1.5-fold. This relatively low level of overexpression is sufficient for the dominant-negative effect of p115/1-766. Thus, eliminating CC3-CC4-AD appears to inhibit p115 function in Golgi ribbon formation.
The function of p115/1-766 was further explored in replacement assays. The baseline for replacement experiments was established by assessing the phenotype of HeLa cells in which endogenous p115 was replaced with full-length rat p115 (our siRNA inhibits synthesis of human but not rat p115). As shown in Fig. 4G, the Golgi is fragmented in p115-depleted cells (cell marked with *), which can be rescued when endogenous p115 in such cells is replaced with full-length p115 (cell marked with arrowhead). Replacement of p115-depleted cells with p115/1-959 rescued Golgi ribbon in 83% of cells. By contrast, replacing p115-depleted cells with p115/1-766 does not rescue Golgi architecture (Fig. 4H) and only 38% of cells that express p115/1-766 contain Golgi ribbons.
The ability of the full-length p115 to support Golgi biogenesis and the failure of the p115/1-766 mutant to do so is not owing to differences in expression levels. As shown in Fig. 4D, full-length p115 and p115/1-766 are expressed in similar amounts. These results suggest that the CC3-CC4-AD region that is absent in p115/1-766 is important in p115 function.
Effect of p115/1-766 on VSV-G trafficking
The effect of p115/1-766 on VSV-G traffic was first assessed in HeLa cells containing endogenous p115. In control cells, VSV-G accumulates within the ER at 42°C (Fig. 2A) and, after shifting the cells to 32°C for 2 hours, is detected at the Golgi and the plasma membrane (Fig. 2C). In cells expressing p115/1-766, VSV-G is also detected in the ER after 12 hours at 42°C (Fig. 5A). However, shifting such cells to 32°C for 1 hour or 2 hours results in VSV-G trafficking to punctate structures that are clustered in the perinuclear region (Fig. 5B,C). VSV-G is not detected on the plasma membrane in cells expressing p115/1-766 even after 2 hours, suggesting that p115/1-766 is functionally compromised.
Fig. 5.
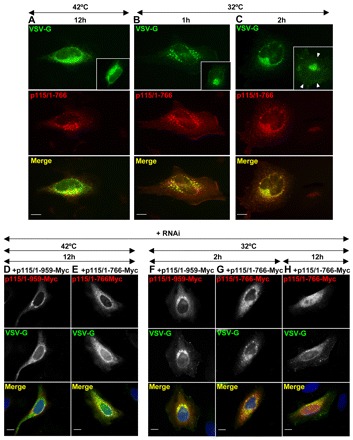
p115/1-766 inhibits VSV-G traffic. (A–C) HeLa cells transfected with GFP-p115/1-766 for 24 hours were transfected with ts045-VSV-G at 42°C, cultured for additional 12 hours and either analyzed directly (A) or shifted to 36°C for 1 hour (B) or 2 hours (C) and processed for immunofluorescence to detect VSV-G and p115/1-766. VSV-G is largely retained within punctate fragments after 1 hour (B) and 2 hours at 36°C (C). Scale bars: 10 µm. (D–H) HeLa cells silenced with anti-p115 siRNA for 3 days were co-transfected at 42°C with ts045VSV-G and either Myc-p115/1-959 (D,F) or Myc-p115/1-766 (E,G–H), cultured for additional 12 hours, shifted to 36°C for indicated times and analyzed by immunofluorescence. In p115-depleted cells replaced with full-length p115, VSV-G traffics to the Golgi and the plasma membrane (F). In p115-depleted cells replaced with p115/1-766, VSV-G is retained within punctate fragments after 2 hours (G) and after 12 hours (H). Scale bars: 10 µm.
The effect of p115/1-766 on VSV-G traffic without interference from endogenous p115 was assessed in p115-depleted HeLa cells, and VSV-G traffic in HeLa cells in which endogenous p115 had been replaced with rat p115 provided the experimental baseline for this replacement. VSV-G is present within the ER of HeLa cells incubated at 42°C (Fig. 5D). After incubating the cells at 32°C for 2 hours, VSV-G is detected within the Golgi and at the plasma membrane (Fig. 5F). In p115-depleted cells replaced with p115/1-766 and incubated at 42°C, VSV-G is present within the ER (Fig. 5E). After shifting the cells to 32°C for 2 hours, VSV-G is detected within punctate Golgi fragments, but not on the plasma membrane (Fig. 5G). VSV-G can be detected on the plasma membrane after 12 hours at 32°C (Fig. 5H). Thus, p115/1-766 is compromised in VSV-G traffic, which suggests that CC3-CC4-AD is important for p115 function.
Effect of p115 constructs with C-terminal deletions on Golgi architecture
p115-1/766 lacks the CC3, CC4 and AD regions. To dissect the importance of these domains we generated p115/1-820 and p115/1-934 that lack CC4 and AD, and the AD regions, respectively (Fig. 6A). The functionality of p115/1-820 was tested first. As shown in Fig. 6D, this construct acts in a dominant-negative manner to disrupt the Golgi when expressed in HeLa cells. This phenotype (of a disrupted Golgi complex) is not caused by the GFP tag added to the N-terminus of p115/1-820 because expression of full-length p115 tagged with yellow fluorescent protein (YFP) has no effect on Golgi architecture (Fig. 4E).
Fig. 6.
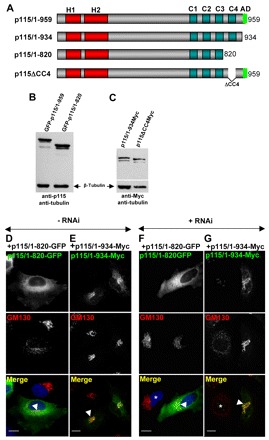
C-terminal region is required for p115 function. (A) Diagram of full-length and C-terminal p115 mutants. (B,C) HeLa cells transfected with GFP-tagged p115/1-959 or p115/1-820 (B) or Myc-tagged p115/1-934 or p115ΔCC4 (C) for 18 hours were lysed and the lysates were immunoblotted with anti-p115 and anti-β-tubulin antibodies (B) or with anti-Myc and anti-β-tubulin antibodies (C). All constructs express the appropriate proteins. (D,E) HeLa cells transfected with GFP-p115/1-820 or Myc-p115/1-934 for 18 hours were analyzed by immunofluorescence with indicated antibodies. Expression of p115/1-820 disrupts Golgi ribbon (D, cell marked with arrowhead). Expression of p115/1-934 has no visible effect on Golgi architecture (E, cell marked with arrowhead). Scale bars: 10 µm. (F,G) HeLa cells silenced with anti-p115 siRNA for 3 days were transfected with GFP-p115/1-820 or Myc-p115/1-934 for 18 hours, and analyzed by immunofluorescence with indicated antibodies. p115 depletion fragments the Golgi (cell marked with *). Expression of p115/1-820 does not reverse Golgi disruption (F, cell marked with arrowhead). Expression of p115/1-934 reverses Golgi disruption (G, cell marked with arrowhead). Scale bars: 10 µm.
p115/1-820 function was further explored in HeLa cells that has been depleted of endogenous p115. When full-length p115 is expressed in HeLa cells depleted of p115, the Golgi ribbon structure reformed (Fig. 4G). By contrast, when p115/1-820 is expressed in cells depleted of p115 the Golgi ribbon structure remains fragmented (Fig. 6F). The difference in (re)formation of Golgi ribbon structure is not due to expression levels of full-length p115 or mutant p115/1-820, because immunoblotting detected both constructs in similar amounts (Fig. 6B). These results imply that p115/1-820 compromises the formation of Golgi ribbons.
One study described that removal of the AD of p115 inhibits cargo traffic (Satoh and Warren, 2008), but another found that function of p115 was not affected upon deletion of the AD (Puthenveedu and Linstedt, 2004). To test the role of the AD in our system, we used the AD deletion construct p115/1-934 (Fig. 6A) that had been shown not to bind GM130 and giantin (Linstedt et al., 2000). Expression of p115/1-934 in cells containing endogenous p115 does not affect Golgi architecture (Fig. 6E). Introducing p115/1-934 in p115-depleted cells rescued Golgi ribbon formation (Fig. 6G). Expression of p115/1-934 was confirmed by immunoblotting (Fig. 6C). Thus, it appears appears that eliminating the AD is not detrimental to p115 function. The finding that p115/1-934, but not p115/1-820 supports Golgi ribbon formation suggests that the CC4 region, which is present in p115//1-934 but absent in p115/1-820, is functionally important.
Effect of p115ΔCC4 on Golgi architecture and cargo traffic
To assess the role of CC4 in p115 function, we generated the deletion mutant p115ΔCC4 that lacks CC4 (Fig. 6A). Expression of p115ΔCC4 was confirmed by immunoblotting (Fig. 6C). This construct acts in a dominant-negative manner and disrupts the Golgi complex when expressed in cells that contain endogenous p115 (Fig. 7B). Similarly, when p115ΔCC4 is introduced in p115-depleted cells, the Golgi complex remains largely fragmented, with many elements remaining scattered throughout the cell (Fig. 7D). Disruption of the Golgi complex was quantified in HeLa cells transfected with scrambled RNA (scr), in p115-depleted cells (RNAi), and in p115-depleted cells replaced with full-length p115 (+1-959), p115ΔCC4 (+ΔCC4), p115/1-820 (+1-820) or p115/1-766 (+1-766). As shown in Fig. 7E, only 10% of cells transfected with scrambled RNA have a fragmented Golgi complex. By contrast, 69% of cells transfected with anti-p115 siRNA have a fragmented Golgi complex. The ∼30% of cells that show normal Golgi complexes probably contain residual endogenous p115 because our depletion methods are only ∼70% effective (Fig. 1A). Introduction of full-length p115 in p115-depleted HeLa cells almost completely rescues the fragmentation of the Golgi complex, with only 18% of cells showing a disruption of the Golgi. By contrast, transfection of p115/1-766 does not rescue Golgi fragmentation, and the Golgi remains fragmented in 62% of cells. Transfection of p115/1-820 or p115ΔCC4 shows an intermediate phenotype, with partial but incomplete rescue. The phenotype of a disrupted Golgi complex is reduced from 69% to 43% by p115/1-820 and from 69% to 31% by p115ΔCC4 but, in both cases, does not approach the almost complete rescue mediated by transfection of full-length p115 (reduction from 69% to 18%). Thus, p115/1-820 and p115ΔCC4 appear compromised in their ability to sustain Golgi ribbon formation.
Fig. 7.
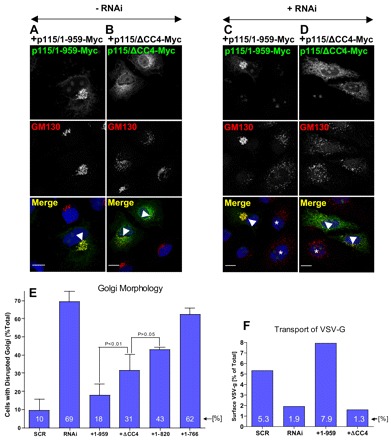
CC4 is important for p115 function. (A,B) HeLa cells transfected with Myc-tagged p115 or p115ΔCC4 for 18 hours were analyzed by immunofluorescence with indicated antibodies. Expression of p115-Myc has no visible effect on Golgi architecture (A, cell marked with arrowhead). Expression of p115ΔCC4-Myc disrupts Golgi ribbon (B, cell marked with arrowhead). Scale bars: 10 µm. (C,D) HeLa cells silenced with anti-p115 siRNA for 3 days were transfected with Myc-tagged p115 or p115ΔCC4 for 18 hours, and analyzed by immunofluorescence with indicated antibodies. p115 depletion fragments the Golgi (cell marked with *). Expression of p115-Myc reverses Golgi disruption (C, cell marked with arrowhead). Expression of p115ΔCC4-Myc does not reverse Golgi disruption (D, cell marked with arrowhead). Scale bars: 10 µm. (E) Golgi disruption was quantified in control cells treated with scrambled RNA (scr), in cells depleted of endogenous p115 (RNAi) and in cells that were depleted of endogenous p115 and expressed either Myc-tagged full-length p115 (+1-959), p115ΔCC4 (+ΔCC4), p115/1-820 (+1-820) or p115/1-766 (+1-766). The values represent the averages of three independent experiments, with more than 50 cells counted each time. (F) VSV-G traffic was quantified in HeLa cells treated with scrambled RNA (scr), depleted of endogenous p115 (RNAi) and in cells that were depleted of endogenous p115 and expressed either Myc-tagged p115/1-959 (+1-959) or p115ΔCC4 (+ΔCC4). The levels of VSV-G present at the plasma membrane after a 2-hour shift to permissive temperature is represented as the percentage of total cellular VSV-G. The values represent the averages of two independent experiments.
We tested the ability of p115ΔCC4 to support VSV-G traffic in p115-depleted cells. Surface biotinylation was used to quantify the amount of VSV-G transported to the plasma membrane within 2 hours during the shift from 42°C to 32°C in cells transfected with scrambled RNA or siRNA targeting p115, and in p115-depleted cells transfected with p115/1-959 or p115ΔCC4. Cells transfected with scrambled RNA transported 5.3% of cellular VSV-G to cell surface, whereas cells transfected with siRNA targeting p115 transported only 1.9% (Fig. 7F). Transfection of p115-depleted cells with p115/1-959 rescues VSV-G trafficking and 7.9% of VSV-G reaches the plasma membrane. By contrast, transfection with p115ΔCC4 does not rescue VSV-G trafficking and only 1.3% of VSV-G is detected on the plasma membrane. Together, our findings show that p115ΔCC4 is compromised in Golgi ribbon formation and VSV-G trafficking, and suggest that CC4 is important for p115 function.
Interaction of p115 mutants with cellular proteins
The dominant-negative effects of p115/1-766, p115/1-820 and p115ΔCC4 could be due to heterodimerization of mutant p115 with endogenous p115 to form inactive complexes. To test heterodimerization, we expressed GFP-p115/1-959 in HeLa cells, immunoprecipitated the lysates with anti-GFP antibodies and analyzed the precipitates for GFP-p115/1-959 and endogenous p115. GFP-p115/1-959 and endogenous p115 are detected in cell lysates (Fig. 8A, lane 1) and GFP-p115/1-959 is efficiently immunoprecipitated (lane 2). Importantly, only GFP-p115/1-959 is recovered during immunoprecipitation (Fig. 8A, lane 3). Immunoprecipitation with non-immune IgG (Fig. 8A, lanes 4 and 5) confirms specificity of this assay. Thus, it appears that GFP-p115/1-959 and endogenous p115 do not heterodimerize. A possible explanation is that dimerization occurs co-translational. An mRNA encoding p115 or GFP-p115/1-959 will be simultaneously translated by numerous ribosomes attached approximately every 100 base pairs, with proteins on adjacent ribosomes ∼30 amino acids apart. Thus, while the C-terminus is still being translated, nascent p115 on adjacent ribosomes has CC1–CC2 regions available for dimerization and are likely to dimerize before being released from the ribosome. The co-translational model of dimerization implies that monomeric p115 is unlikely to be an important cellular component. This, indeed, appears to be the case because p115 monomers have not been detected in cells (Sapperstein et al., 1995). This also suggests that, once formed, homodimers remain stable and do not undergo monomer exchange.
Fig. 8.
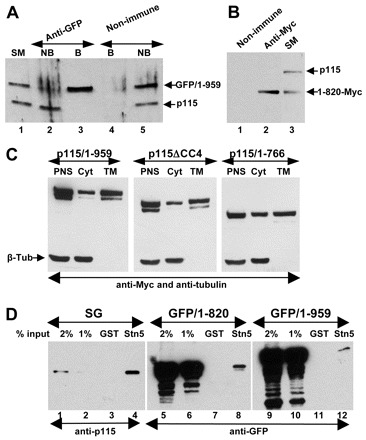
Interactions of mutant p115 with cellular proteins. (A) HeLa cells transfected with GFP-p115/1-959 for 18 hours were lysed and lysates immunoprecipitated with non-immune or anti-GFP antibodies. The starting material (SM), non-bound fractions (NB) and bound precipitates (B) were analyzed by SDS-PAGE and immunoblotted with anti-p115 antibodies. Similar levels of endogenous p115 and GFP-p115/1-959 are present in the SM (lane 1). GFP-p115/1-959 is depleted from the NB fraction (lane 2). Only GFP-p115/1-959 is recovered in the precipitate (lane 3). (B) HeLa cells transfected with Myc-p115/1-820 for 18 hours were lysed and the lysates immunoprecipitated with unspecific (lane 1) or anti-Myc (lane 2) antibodies. The starting material (lane 3) and precipitates were analyzed by SDS-PAGE and immunoblotted with anti-p115 antibodies. Similar levels of endogenous p115 and Myc-p115/1-820 are present in SM (lane 3). Only Myc-p115/1-820 is recovered in the precipitate (lane 2). (C) HeLa cells transfected with Myc-tagged p115/1-959, p115ΔCC4 or p115/1-766 for 18 hours were fractionated to generate a post-nuclear supernatant (PNS) which was subsequently fractionated into cytosol (Cyt) and total membranes (TM). Similar levels of p115/1-959, p115ΔCC4 and p115/1-766 associate with membranes. (D) GST or GST with the cytoplasmic domain of syntaxin-5 were incubated with solubilized stacked Golgi (SG) fraction (lanes 1–4), lysate from HeLa cells transfected with GFP-p115/1-820 (lanes 5–8) or with GFP-p115/1-959 (lanes 9–12). Bound proteins were eluted and analyzed by SDS-PAGE and immunoblotting with anti-p115 antibodies. The starting material for each binding is shown (lanes 1–2, 5–6, and 9–10). Full-length p115/1-959 and p115/1-820 bind to beads containing syntaxin-5 (lanes 4, 8 and 12).
The lack of heterodimerization between GFP-p115/1-959 and endogenous p115 also suggested that exogenously expressed mutants do not dimerize with endogenous p115. We assessed this hypothesis experimentally, by testing heterodimer formation between p115/1-820-Myc and endogenous p115. As shown in Fig. 8B, lane 3, p115/1-820-Myc and endogenous p115 are detected in cell lysates. However, only p115/1-820-Myc is recovered after immunoprecipitation with anti-Myc antibodies (Fig. 8B, lane 2). Immunoprecipitation with non-immune IgG does not recover p115 or p115/1-820 (Fig. 8B, lane 1). Thus, exogenously expressed p115 mutants do not form heterodimers with endogenous p115, suggesting that the dominant-negative effects of p115 mutants occur without heterodimerization. Instead, it is possible that p115 mutants compete with endogenous p115 for binding to cellular proteins that are required for trafficking. p115 is a cytoplasmic protein that rapidly cycles on and off membranes in a process regulated by the binding to SNAREs (Brandon et al., 2006). Thus, it is possible that mutant p115 competes with endogenous p115 for binding to SNAREs. This model predicts that p115 mutants associate with membranes and interact with SNAREs.
Membrane association of p115/1-959, p115/1-766 and p115ΔCC4 was tested using cell fractionation. As shown in Fig. 8C, all these mutant constructs are detected in a post-nuclear supernatant of transfected HeLa cells and, after fractionation, are recovered in fractions of the cytosol and the total membrane. Importantly, all p115 constructs appear to associate with membranes to a similar extent, suggesting that – like p115/1-959 – p115/1-766 and p115ΔCC4 bind efficiently to membrane ‘receptors’, perhaps SNAREs.
p115 interacts with the SNAREs GOS-28, membrin, Ykt6p and syntaxin-5 through CC1 (Shorter et al., 2002). p115/1-766, p115/1-820 and p115ΔCC4 each contain CC1. To assess whether these proteins exert their dominant-negative effects by binding SNAREs, we tested whether p115/1-820 binds to syntaxin-5. We first show that endogenous p115 within an isolated stacked Golgi fraction can be recovered by using GST–syntaxin beads but not by using GST beads (Fig. 8D, lanes 1–4). Similarly, GFP-p115/1-959 binds to beads that contain GST–syntaxin-5 (Fig. 8D, lanes 9–12). Importantly, GFP-p115/1-820 also binds GST–syntaxin-5 beads (Fig. 8D, lanes 5–8). Thus, p115/1-820 might exert its dominant-negative effect by competing with endogenous p115 for binding to p115 partners, such as syntaxin-5.
Discussion
We have characterized Golgi architecture and cargo traffic in p115-depleted cells in order to set a baseline to assess p115 function in intact C. elegans, and utilized RNAi and the subsequent transfection of HeLa cells with p115 deletion mutants to identify new functional domains within p115.
The role of p115 in Golgi ribbon formation
We and others have shown that the Golgi complex fragments into polarized dispersed structures in p115-depleted cells (Holloway et al., 2007; Puthenveedu and Linstedt, 2001; Puthenveedu and Linstedt, 2004; Smith et al., 2009; Sohda et al., 2007; Sohda et al., 2005). A plausible model for this phenotype is that COPII vesicles continue to bud and fuse to generate larger structures (perhaps VTCs) adjacent to ER exit sites, but that those structures do not mature into transport-competent VTCs. Instead, each VTC remains close to specialized ER exit sites (ERES) and differentiates into a Golgi mini-stack in a manner similar to that observed in nocodazole-treated cells (Miles et al., 2001; Rhee et al., 2005; Storrie, 2005). This model has several implications regarding the tethering function of p115. Transmembrane Golgi proteins continuously cycle between the Golgi and the ER (Miles et al., 2001; Rhee et al., 2005) in a process mediated by COPII vesicles (Peng et al., 1999; Aridor et al., 2001; Powers and Barlowe, 2002; Zeuschner et al., 2006). The formation of Golgi mini-stacks in p115-depleted cells suggests that COPII vesicles can fuse to generate VTCs in the absence of p115. The subcompartment organization of the Golgi depends on proteins being recycled from distal to proximal cisternae by COPI vesicles (Glick and Nakano, 2009; Storrie, 2005). COPI vesicle traffic between medial- and cis-Golgi appears to involve p115 interactions with the GM130 and giantin tethers (Sonnichsen et al., 1998). The polarized distribution of proteins within Golgi fragments in p115-depleted cells suggests COPI-mediated recycling. This is supported by the presence of the ARF activator GBF1 and β-COPI on Golgi fragments. Thus, p115 might not be absolutely required for COPII and COPI vesicle tethering. We cannot exclude that incomplete p115 depletion provides sufficient p115 to facilitate tethering, but studies in Arabidopsis also suggest that p115 is dispensable for tethering (which is believed to be an essential process). In Arabidopsis, genetic ablation of p115 results in dwarf, but viable plants (Takahashi et al. 2010). Thus, p115-mediated tethering might improve the efficiency of trafficking by facilitating more rapid SNARE encounters and more efficient membrane docking and fusion.
Interestingly, findings from in vivo studies differ from those using in vitro assays, in which p115 is absolutely required for fusion of COPII vesicles with Golgi membranes or with each other, and for COPI-mediated intra-Golgi traffic. A possible explanation for this discrepancy may be that the described in vivo phenotypes occur following long-term treatments with siRNA or the expression of dominant-negative p115 mutants that allow cells to develop compensatory mechanisms. It is also possible that other tethers, such as TRAPP and COG, substitute for p115 function in vivo. Alternatively, in vivo studies monitor localization of endogenous Golgi proteins, such as giantin and GalNacT, whereas in all in vitro studies VSV-G was used to measure p115 function. p115 depletion strongly inhibits VSV-G trafficking (see below) and this may have influenced the interpretation of in vitro results.
The role of p115 in cargo traffic
Trafficking of soluble proteins including sDPPIV (Sohda et al., 2007; Sohda et al., 2005) and cochlin (this study) seems minimally affected by p115 depletion. Both sDPPIV and cochlin show slightly delayed but otherwise normal glycosylation, consistent with the correct differentiation of Golgi subdomains. Disruption in glycosyltransferase localization (for example by inhibiting COG function) leads to altered glycosylation of cargo proteins (Zolov and Lupashin, 2005). That p115 depletion has a limited effect on the secretion of soluble cargoes is also supported by findings that the soluble YP170 appears efficiently secreted in p115-depleted C. elegans.
A number of transmembrane proteins including giantin, GS27, mannosidase 2, GalNacT, ERGIC53 (Sohda et al., 2007; Sohda et al., 2005) and ATP7A (Holloway et al., 2007) localize to Golgi mini-stacks in p115-depleted cells. However, transport kinetics for those proteins have not been defined yet, and might be delayed. Here, we show that p115 depletion affects trafficking of the transmembrane RME-2 yolk receptor in intact C. elegans.
p115 depletion has a limited effect on the secretion of soluble proteins and a moderate effect on some transmembrane proteins, which contrasts with the strong inhibition in VSV-G traffic. Most sensitive to p115 depletion is exit of proteins from the ER. This is consistent with the requirement for Uso1p in sorting selected cargo proteins for ER exit in yeast (Belden and Barlowe, 1996; Morsomme et al., 2003; Morsomme and Riezman, 2002). The extent to which p115 regulates trafficking of other proteins requires to define the p115-dependent traffic proteome. Overall, it appears that p115 has a differential effect on protein trafficking, and that ER–Golgi transit of select cargoes is more sensitive to p115 depletion. Thus, p115 might have a function in the sorting of some cargo proteins, in addition to its general role in membrane tethering.
Mapping a new functional domain within p115
Until now, only the H1, H2 and CC1 have been implicated in p115 function (An et al., 2009; Guo et al., 2008; Puthenveedu and Linstedt, 2004; Sohda et al., 2007). The role of the AD is controversial, with one study suggesting that it is required for cargo trafficking (Satoh and Warren, 2008), whereas another study documents that the AD is dispensable (Puthenveedu and Linstedt, 2004). Our findings that p115/1-934 supports Golgi ribbon formation in p115-depleted cells supports a limited role for AD. The discrepancy might be owing to the size of the AD truncation. Our p115-1-934 construct removes only the AD, whereas the construct in the Satoh and Warren study generates p115/1-886 and removes not only the AD but also 11 amino acids from CC4 (Fig. 4B, see diagram). Considering the essential role of CC4 (see below), it is possible that the observed traffic defect is due to disruption of CC4, rather than elimination of AD.
We identified CC4 as a so-far-unknown functional p115 domain. Our studies were prompted by the functionally compromised uso1-1 and uso1-11 mutants (Seog et al., 1994; Yamakawa et al., 1996), and the finding that CC4 interacts with a subset of ER-Golgi SNAREs (Shorter et al., 2002). We generated three C-terminally truncated p115 mutants (p115/1-766, p115/1-820 and p115/1-934) and examined their effect on Golgi ribbon architecture. We show that p115/1-766 and p115/1-820, but not p115/1-934, act as dominant-negatives and disrupt Golgi ribbons in cells that contain endogenous p115. Furthermore, p115/1-766 and p115/1-820 do not support Golgi ribbon formation when expressed in p115-depleted cells, whereas p115/1-934 sustains Golgi ribbon formation. These results suggested that CC4 is important for p115 function. In support, we showed that p115ΔCC4 acts as a dominant-negative regulator, causing Golgi fragmentation in cells that contain endogenous p115 and does not support Golgi ribbon formation in p115-depleted cells. We also explored the role of p115 C-terminus in cargo traffic. We document that p115/1-766 and p115ΔCC4 are unable to support VSV-G trafficking in cells depleted of endogenous p115. Together, our data suggest that CC4 is essential for p115 function in Golgi ribbon formation and cargo trafficking.
The Golgi disruption and traffic arrest caused by p115ΔCC4 appear similar to those caused by p115ΔCC1 (Puthenveedu and Linstedt, 2004). This similarity suggests that CC1 and CC4 function at the same stage of trafficking, perhaps to engage partner proteins in a molecular event that results in membrane tethering and fusion. For CC1, these partners might be Rab1 (Beard et al., 2005), Sly1 and the SNARE proteins syntaxin-5, GOS28, membrin, Ykt6, Sec22, Bet1 and GS15 (Shorter et al., 2002). By contrast, CC4 does not bind Rab1 or Sly1, and interacts with only a subset of SNAREs, namely GOS28, membrin, Ykt6, Bet1 and GS15. A speculative but plausible model for CC1 and CC4 function is to tether membranes by binding SNARE proteins on opposite membranes. In this model, p115 bridges membranes by committing one CC domain to bind the donor membrane while the other CC domain binds the target membrane. The initial linking function might be followed by p115 promoting SNARE assembly by releasing inhibition or catalyzing the formation of a SNARE pin. p115 appears to preferentially bind free SNARE proteins in vitro (Bentley et al., 2006) and in vivo (Brandon et al., 2006), which is consistent with a function in the assembly of the SNARE complex. The four CC regions are separated by proline-rich ‘hinge’ regions, which might facilitate rotation relative to the polypeptide backbone. The presence of the hinges has been proposed to facilitate an ‘accordion-like’ collapse of the tether in order to bring the donor and acceptor membranes into proximity (Yamakawa et al., 1996). Thus, a bridge between two membranes through binding of CC1 to a SNARE on the donor membrane and CC4 to a SNARE on the target membrane might collapse to bring the two membranes into close proximity. Our discovery that the SNARE-binding CC4 has a key role in p115 function will enable future studies aimed at understanding p115-mediated trafficking.
Materials and Methods
Worm strains
ojIs37 (pie-1p::GFP::ugtp-1) was obtained from the Caenorhabditis Genetics Centre, pwIs23(vit-2::GFP) was a gift from Barth D. Grant (Molecular Biology and Biochemistry, Rutgers University, NJ), pie-1p::GFP::SP12 (Poteryaev et al., 2005) and pwIs116(rme-2::GFP) (Balklava et al., 2007) have been described previously.
RNA interference in worms
An Uso-1 RNAi clone from the Ahringer library (Kamath and Ahringer, 2003) was fed to worms as described previously (Kamath et al., 2003), with few modifications. Overnight cultures of control (empty L4440 vector) and L4440-uso-1-containing bacteria were seeded into dishes that contained NGM-lite agar medium supplemented with 2 mM IPTG and 25 µg ml–1 carbenicillin, and were induced overnight at room temperature. 30–50 eggs were transferred to the induced plates after 4 days at 20°C animals were scored as young adults in the P0 generation. Live worms were mounted on 2% agarose pads with 10 mM tetramisole and imaged using a fully motorized Zeiss Axiovert 200M fluorescence microscope (Carl Zeiss Ltd., Welwyn Garden City, UK) and a Hamamatsu Orca camera driven by Volocity software (Improvision, Coventry, UK).
Reagents and antibodies
Restriction enzymes and molecular reagents were from Promega (Madison, WI) or New England BioLabs (Beverly, MA). SuperSignal West Pico Chemiluminescence Substrate, EZ-Link Sulfo-NHS-Biotin reagent and NeutrAvidin Agarose were from Thermo Fisher Scientific (Rockford, IL).
Rabbit polyclonal antibodies against p115, GM130 and GBF1 have been described previously (Barroso et al., 1995; Garcia-Mata and Sztul, 2003; Nelson et al., 1998). Monoclonal anti-giantin G1/133 antibody (Linstedt and Hauri, 1993) was from Hans-Peter Hauri (University of Basel, Basel, Switzerland). Sly1 polyclonal antibody was from Jesse Hay (The University of Montana). The following commercially available antibodies were used: rabbit polyclonal anti-Myc (Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal anti-Myc and anti-GFP (Invitrogen, Carlsbad, CA), goat anti-rabbit and goat anti-mouse conjugated to Alexa-Fluor-488 and Alexa-Fluor-594 (Molecular Probes, Eugene, OR), HRP labeled goat anti-rabbit, goat anti-mouse and monoclonal anti-transferrin receptor (Zymed, San Francisco, CA), rabbit polyclonal anti-β-COP and rabbit polyclonal anti-calreticulin (Affinity BioReagents, Golden, CO), monoclonal anti-GM130 (BD Transduction Laboratories, Lexington, KY), monoclonal anti-golgin-245 (BD Pharmingen, San Diego, CA), monoclonal anti-β-tubulin (Upstate, Lake Placid, NY) and monoclonal anti-VSV-G (Abcam, Cambridge, MA). 35S-Met/Cys was from MP Biomedicals (Irvine, CA).
Transfection and immunofluorescence microscopy
DNA transfection was carried out using TransIT-LT1 Polyamine transfection reagents (Mirus Corporation, Madison, WI) according to the manufacturer's protocol. RNA oligonucleotides were transfected with SiLentFect reagent (BioRad, Hercules, CA) according to the manufacturer's protocol. HeLa cells were cultured and processed for immunofluorescence as described before (Grabski et al., 2003). Fluorescence patterns were visualized using a Leitz Orthoplan epifluorescence microscope (Wetzlar, Germany). Optical sections were captured with a CCD high-resolution camera equipped with a camera and/or computer interface. Images were analyzed with a power Mac using IPLab Spectrum software (Scanalytics Inc., Fairfax, VA).
DNA and RNA constructs
The siRNA sequences that were used for RNA interference of human p115 were designed by comparing human and rat p115 cDNA. The sequence of siRNA 9 (hereafter referred to as siRNA#9) is 23 nucleotides long and targets nucleotides 509-531 of human p115 cDNA (gATTgATGGACTTaCTaGCgGAT; lowercase letters indicate differences in human and rat cDNA); this sequence was used to synthesize the sense and anti-sense RNA oligonucleotides (IDT, Coralville, IA).
YFP-tagged p115 was generated by PCR and cloned into the KpnI–BamHI restriction sites of pEYFP-N1 (Clontech Laboratories, Mountain View, CA). GFP-tagged p115/1-820 (lacking the CC4 and AD regions) and p115-959 (which is similar to Uso1p wild type) were cloned into the XhoI-BamHI restriction sites of pEGFP-C2 (Clontech). GFP-tagged p115/1-766 lacks the CC3, CC4 and AD regions and was cloned into the KpnI–BamHI restriction sites of pEGFP-N2. The Myc-tagged p115/1-766 was cloned into pcDNA4/TO/Myc-His-A (Invitrogen, Carlsbad, CA) using BamHI and XhoI restriction enzymes. The Myc-tagged cochlin was described previously (Grabski et al., 2003). The p115/ΔCC4 (p115 lacking CC4) was generated using the p115/1-820 construct as a backbone. The 75-nucleotide long p115 acidic-tail fragment was added using overlapping PCR, and cloned into the pcDNA4/TO/Myc-His-A vector using BamHI and XhoI restriction enzymes. p115/1-934 lacks the AD region, was generated by PCR and cloned into the pcDNA4/TO/Myc-His-A vector using BamHI and XhoI restriction enzymes.
Pulse-chase metabolic labeling
HeLa cells silenced for 3 days with anti-p115 RNA oligonucleotides were co-transfected with Myc-tagged cochlin and YFP-p115/1-959 or GFP-p115/1-766. After 24 hours, cells were subjected to 1 hour starvation with medium lacking methionine (Met) and cysteine (Cys) (Mediatech Inc, Birmingham, AL), followed by a 30-minute pulse with S35-Met/Cys (MP Biomedicals, Irvine, CA), and chasing for indicated times. Cells were lysed and medium was collected at indicated time points. In some experiments, HeLa cells silenced for 4 days with anti-p115 RNA oligonucleotides were treated with BFA (5 µg/ml, Sigma Chemical Co., St Louis, MO) for 30 minutes, followed by a pulse-chase as above in the presence of BFA.
Immunoprecipitation, SDS-PAGE and immunoblotting
HeLa cells were solubilized and immmunoprecipitated with anti-Myc or anti-GFP antibodies as in (Grabski et al., 2003). Precipitates were analyzed by SDS-PAGE followed by fluorography or transferred to NitroPure nitrocellulose membrane (Micron Separations Inc., Westborough, MA), and subjected to immunoblotting as described (Gao et al., 1998).
GST-binding assay
Bacterially expressed GST or a chimera of GST and the cytoplasmic domain of syntaxin-5 were immobilized on SH beads (Amersham Bioscience AB, Uppsala, Sweden). Stacked Golgi fraction prepared as described previously (Alvarez et al., 1999) or HeLa cells transfected with GFP-tagged p115/1-959 or p115/1-820 for 24 hours were lysed in HKM buffer (25 mM HEPES pH 7.4, 125 mM potassium acetate, 5 mM magnesium acetate, 5% glycerol, 0.5% Triton X-100, 0.1 mM DTT, protease inhibitor cocktail). Beads and lysates were incubated 1 hour at 4°C, centrifuged at 750 g, 4°C for 5 minutes and beads washed ten times with HKM buffer. Starting material and bound proteins were separated by SDS-PAGE and immunoblotted using antibodies against p115.
Traffic of VSV-G protein
HeLa cells were transfected with anti-p115 RNA oligonucleotides for 3 days and then transfected with ts045VSV-G-GFP and Myc-tagged p115/1-959 or p115/1-766 at non-permissive temperature of 42°C and incubated for ∼18 hours. Cells were shifted to the permissive temperature of 32°C and incubated for 2 hours or 12 hours, fixed and processed for immunofluorescence.
Evaluation of Golgi morphology
HeLa cells transfected with anti-p115 or scrambled RNA oligonucleotides for 3 days were transfected with Myc-tagged p115/1-959, p115/ΔCC4, p115/1-820 or p115/1-766. After 24 hours, cells were processed for immunofluorescence with anti-GM130 and anti-Myc antibodies. Triplicates of two microscope coverslips for each experimental setup were used to count 600 cells per expressed p115 construct. Golgi morphology was evaluated and analyzed using GraphPad Prism Software (GraphPad Software, Inc., La Jolla, CA). The obtained results were processed using one-way ANOVA statistical analysis.
Surface biotinylation of VSV-G protein
HeLa cells transfected with anti-p115 or scrambled RNA oligonucleotides for 3 days were co-transfected with ts045VSV-G-GFP and Myc-tagged p115/1-959 or p115/ΔCC4, and incubated at non-permissive temperature for ∼18 hours. For each condition cells were processed at t = 0 hours and t = 2 hours after incubation at permissive temperature. Surface biotinylation was at 4°C (according to the manufacturer's protocol) to prevent protein trafficking. After biotinylation, cells were lysed in Buffer A (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 1 mM EGTA, protease inhibitor cocktail), and 50 µg of each lysate was processed for precipitation of biotinylated proteins with 50 µl of NeutrAvidin agarose (Thermo Fisher Scientific) equilibrated in Buffer A. After washing, the fraction bound to NeutrAvidin agarose was released in sample buffer, and processed for SDS-PAGE and immunoblotting with antibodies against VSV-G and transferrin receptor. The intensity of the VSV-G band was measured using LabWorks software (UVP, Inc., Upland, CA), normalized to the intensity of the signal for transferrin receptor, and presented as the percentage of surface VSV-G relative to total cellular VSV-G.
Cell fractionation
HeLa cells transfected with Myc-tagged p115/1-959, p115/ΔCC4 or p115/1-766 constructs for 2 days, were fractionated as in described previously (Szul et al., 2005). The post-nuclear supernatant, cytosol and total membrane fractions were analysed by SDS-PAGE followed by immunoblotting using anti-Myc and anti β-tubulin antibodies.
Acknowledgments
We thank Hans-Peter-Hauri, Jesse Hay, Jennifer Lippincott-Schwartz and Sharon Tooze for gifts of antibodies and plasmids and Anne Theibert for constructive comments.
Footnotes
Funding
This work was supported by American Heart Association Grant in Aid (to E.S.). Z.B. was supported by a Royal Society Dorothy Hodgkin Fellowship.
References
- Allan B. B., Moyer B. D., Balch W. E. (2000). Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science 289, 444–448. 10.1126/science.289.5478.444 [DOI] [PubMed] [Google Scholar]
- Alvarez C., Fujita H., Hubbard A., Sztul E. (1999). ER to Golgi transport: Requirement for p115 at a pre-Golgi VTC stage. J. Cell Biol. 147, 1205–1222. 10.1083/2Fjcb.147.6.1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C., Garcia-Mata R., Hauri H. P., Sztul E. (2001). The p115-interactive proteins GM130 and giantin participate in endoplasmic reticulum-Golgi traffic. J. Biol. Chem. 276, 2693–2700. 10.1074/2Fjbc.M007957200 [DOI] [PubMed] [Google Scholar]
- An Y., Chen C. Y., Moyer B., Rotkiewicz P., Elsliger M. A., Godzik A., Wilson I. A., Balch W. E. (2009). Structural and functional analysis of the globular head domain of p115 provides insight into membrane tethering. J. Mol. Biol. 391, 26–41. 10.1016/2Fj.jmb.2009.04.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M., Fish K. N., Bannykh S., Weissman J., Roberts T. H., Lippincott-Schwartz J., Balch W. E. (2001). The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 152, 213–229. 10.1083/2Fjcb.152.1.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balklava Z., Pant S., Fares H., Grant B. D. (2007). Genome-wide analysis identifies a general requirement for polarity proteins in endocytic traffic. Nat. Cell Biol. 9, 1066–1073. 10.1038/2Fncb1627 [DOI] [PubMed] [Google Scholar]
- Barlowe C. (1997). Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J. Cell Biol. 139, 1097–1108. 10.1083/2Fjcb.139.5.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso M., Nelson D. S., Sztul E. (1995). Transcytosis-associated protein (TAP)/p115 is a general fusion factor required for binding of vesicles to acceptor membranes. Proc. Natl. Acad. Sci. USA 92, 527–531. 10.1073/2Fpnas.92.2.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard M., Satoh A., Shorter J., Warren G. (2005). A cryptic rab1-binding site in the p115 tethering protein. J. Biol. Chem. 280, 25840–25848. 10.1074/2Fjbc.M503925200 [DOI] [PubMed] [Google Scholar]
- Belden W. J., Barlowe C. (1996). Erv25p, a component of COPII-coated vesicles, forms a complex with Emp24p that is required for efficient endoplasmic reticulum to Golgi transport. J. Biol. Chem. 271, 26939–26946. 10.1074/2Fjbc.271.43.26939 [DOI] [PubMed] [Google Scholar]
- Bentley M., Liang Y., Mullen K., Xu D., Sztul E., Hay J. C. (2006). SNARE status regulates tether recruitment and function in homotypic COPII vesicle fusion. J. Biol. Chem. 281, 38825–38833. 10.1074/2Fjbc.M606044200 [DOI] [PubMed] [Google Scholar]
- Brandon E., Szul T., Alvarez C., Grabski R., Benjamin R., Kawai R., Sztul E. (2006). On and off membrane dynamics of the endoplasmic reticulum-golgi tethering factor p115 in vivo. Mol. Biol. Cell 17, 2996–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary D. O., Rothman J. E. (1990). Purification of three related peripheral membrane proteins needed for vesicular transport. J. Biol. Chem. 265, 10109–10117. [PubMed] [Google Scholar]
- Gao Y. S., Alvarez C., Nelson D. S., Sztul E. (1998). Molecular cloning, characterization, and dynamics of rat formiminotransferase cyclodeaminase, a Golgi-associated 58-kDa protein. J. Biol. Chem. 273, 33825–33834. 10.1074/2Fjbc.273.50.33825 [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R., Szul T., Alvarez C., Sztul E. (2003). ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol. Biol. Cell 14, 2250–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick B. S., Nakano A. (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25, 113–132. 10.1146/2Fannurev.cellbio.24.110707.175421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabski R., Szul T., Sasaki T., Timpl R., Mayne R., Hicks B., Sztul E. (2003). Mutations in COCH that result in non-syndromic autosomal dominant deafness (DFNA9) affect matrix deposition of cochlin. Hum. Genet. 113, 406–416. [DOI] [PubMed] [Google Scholar]
- Guo Y., Punj V., Sengupta D., Linstedt A. D. (2008). Coat-tether interaction in Golgi organization. Mol. Biol. Cell 19, 2830–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway Z. G., Grabski R., Szul T., Styers M. L., Coventry J. A., Monaco A. P., Sztul E. (2007). Activation of ADP-ribosylation factor regulates biogenesis of the ATP7A-containing trans-Golgi network compartment and its Cu-induced trafficking. Am. J. Physiol. Cell Physiol. 293, C1753–C1767. 10.1152/2Fajpcell.00253.2007 [DOI] [PubMed] [Google Scholar]
- Kamath R. S., Ahringer J. (2003). Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321. [DOI] [PubMed] [Google Scholar]
- Kamath R. S., Fraser A. G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M. et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. 10.1038/2Fnature01278 [DOI] [PubMed] [Google Scholar]
- Kondylis V., Rabouille C. (2003). A novel role for dp115 in the organization of tER sites in Drosophila. J. Cell Biol. 162, 185–198. 10.1083/2Fjcb.200301136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt A. D., Hauri H. P. (1993). Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 4, 679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt A. D., Jesch S. A., Mehta A., Lee T. H., Garcia-Mata R., Nelson D. S., Sztul E. (2000). Binding relationships of membrane tethering components. The giantin N terminus and the GM130 N terminus compete for binding to the p115 C terminus. J. Biol. Chem. 275, 10196–10201. 10.1074/2Fjbc.275.14.10196 [DOI] [PubMed] [Google Scholar]
- Losev E., Reinke C. A., Jellen J., Strongin D. E., Bevis B. J., Glick B. S. (2006). Golgi maturation visualized in living yeast. Nature 441, 1002–1006. 10.1038/2Fnature04717 [DOI] [PubMed] [Google Scholar]
- Malsam J., Satoh A., Pelletier L., Warren G. (2005). Golgin tethers define subpopulations of COPI vesicles. Science 307, 1095–1108. 10.1126/2Fscience.1108061 [DOI] [PubMed] [Google Scholar]
- Miles S., McManus H., Forsten K. E., Storrie B. (2001). Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155, 543–555. 10.1083/2Fjcb.200103104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsomme P., Riezman H. (2002). The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev. Cell 2, 307–317. [DOI] [PubMed] [Google Scholar]
- Morsomme P., Prescianotto-Baschong C., Riezman H. (2003). The ER v-SNAREs are required for GPI-anchored protein sorting from other secretory proteins upon exit from the ER. J. Cell Biol. 162, 403–412. 10.1083/2Fjcb.200212101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H., Hirata A., Ogawa Y., Yonehara T., Yoda K., Yamasaki M. (1991). A cytoskeleton-related gene, uso1, is required for intracellular protein transport in Saccharomyces cerevisiae. J. Cell Biol. 113, 245–260. 10.1083/2Fjcb.113.2.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N., Lowe M., Levine T. P., Rabouille C., Warren G. (1997). The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell 89, 445–455. [DOI] [PubMed] [Google Scholar]
- Nelson D. S., Alvarez C., Gao Y. S., Garcia-Mata R., Fialkowski E., Sztul E. (1998). The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 143, 319–331. 10.1083/2Fjcb.143.2.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R., Grabowski R., De Antoni A., Gallwitz D. (1999). Specific interaction of the yeast cis-Golgi syntaxin Sed5p and the coat protein complex II component Sec24p of endoplasmic reticulum-derived transport vesicles. Proc. Natl. Acad. Sci. USA 96, 3751–3756. 10.1073/2Fpnas.96.7.3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D., Spang A. (2005). A role of SAND-family proteins in endocytosis. Biochem. Soc. Trans. 33, 606–608. 10.1042/2FBST0330606 [DOI] [PubMed] [Google Scholar]
- Powers J., Barlowe C. (2002). Erv14p directs a transmembrane secretory protein into COPII-coated transport vesicles. Mol. Biol. Cell 13, 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu M. A., Linstedt A. D. (2001). Evidence that Golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and GM130. J. Cell Biol. 155, 227–238. 10.1083/2Fjcb.200105005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu M. A., Linstedt A. D. (2004). Gene replacement reveals that p115/SNARE interactions are essential for Golgi biogenesis. Proc. Natl. Acad. Sci. USA 101, 1253–1256. 10.1073/2Fpnas.0306373101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S. W., Starr T., Forsten-Williams K., Storrie B. (2005). The steady-state distribution of glycosyltransferases between the Golgi apparatus and the endoplasmic reticulum is approximately 90:10. Traffic 6, 978–990. [DOI] [PubMed] [Google Scholar]
- Sapperstein S. K., Walter D. M., Grosvenor A. R., Heuser J. E., Waters M. G. (1995). p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc. Natl. Acad. Sci. USA 92, 522–526. 10.1073/2Fpnas.92.2.522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A., Warren G. (2008). In situ cleavage of the acidic domain from the p115 tether inhibits exocytic transport. Traffic 9, 1522–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seog D. H., Kito M., Yoda K., Yamasaki M. (1994). Uso1 protein contains a coiled-coil rod region essential for protein transport from the ER to the Golgi apparatus in Saccharomyces cerevisiae. J. Biochem. 116, 1341–1345. [DOI] [PubMed] [Google Scholar]
- Shorter J., Beard M. B., Seemann J., Dirac-Svejstrup A. B., Warren G. (2002). Sequential tethering of Golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J. Cell Biol. 157, 45–62. 10.1083/2Fjcb.200112127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. D., Willett R., Kudlyk T., Pokrovskaya I., Paton A. W., Paton J. C., Lupashin V. V. (2009). The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic 10, 1502–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohda M., Misumi Y., Yoshimura S., Nakamura N., Fusano T., Sakisaka S., Ogata S., Fujimoto J., Kiyokawa N., Ikehara Y. (2005). Depletion of vesicle-tethering factor p115 causes mini-stacked Golgi fragments with delayed protein transport. Biochem. Biophys. Res. Commun. 338, 1268–1274. 10.1016/2Fj.bbrc.2005.10.084 [DOI] [PubMed] [Google Scholar]
- Sohda M., Misumi Y., Yoshimura S., Nakamura N., Fusano T., Ogata S., Sakisaka S., Ikehara Y. (2007). The interaction of two tethering factors, p115 and COG complex, is required for Golgi integrity. Traffic 8, 270–284. [DOI] [PubMed] [Google Scholar]
- Sonnichsen B., Lowe M., Levine T., Jamsa E., Dirac-Svejstrup B., Warren G. (1998). A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol. 140, 1013–1021. 10.1083/2Fjcb.140.5.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie B. (2005). Maintenance of Golgi apparatus structure in the face of continuous protein recycling to the endoplasmic reticulum: making ends meet. Int. Rev. Cytol. 244, 69–94. [DOI] [PubMed] [Google Scholar]
- Szul T., Garcia-Mata R., Brandon E., Shestopal S., Alvarez C., Sztul E. (2005). Dissection of Membrane Dynamics of the ARF-Guanine Nucleotide Exchange Factor GBF1. Traffic. 6, 374–385. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Tamura K., Takagi J., Koumoto Y., Hara-Nishimura I., Shimada T. (2010). MAG4/Atp115 is a golgi-localized tethering factor that mediates efficient anterograde transport in Arabidopsis. Plant Cell Physiol. 51, 1777–1787. 10.1093/2Fpcp%2Fpcq137 [DOI] [PubMed] [Google Scholar]
- Waters M. G., Clary D. O., Rothman J. E. (1992). A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 118, 1015–1026. 10.1083/2Fjcb.118.5.1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. W., Whiteheart S. W., Wiedmann M., Brunner M., Rothman J. E. (1992). A multisubunit particle implicated in membrane fusion. J. Cell Biol. 117, 531–538. 10.1083/2Fjcb.117.3.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa H., Seog D. H., Yoda K., Yamasaki M., Wakabayashi T. (1996). Uso1 protein is a dimer with two globular heads and a long coiled-coil tail. J. Struct. Biol. 116, 356–365. 10.1006/2Fjsbi.1996.0053 [DOI] [PubMed] [Google Scholar]
- Zeuschner D., Geerts W. J., van Donselaar E., Humbel B. M., Slot J. W., Koster A. J., Klumperman J. (2006). Immuno-electron tomography of ER exit sites reveals the existence of free COPII-coated transport carriers. Nat. Cell Biol. 8, 377–383. 10.1038/2Fncb1371 [DOI] [PubMed] [Google Scholar]
- Zolov S. N., Lupashin V. V. (2005). Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 168, 747–759. 10.1083/2Fjcb.200412003 [DOI] [PMC free article] [PubMed] [Google Scholar]