

Abstract
Lung cancer is the most common cause of cancer mortality worldwide. Non-small-cell lung carcinomas (NSCLCs), which represent around 80% of lung tumors, exhibit poor prognosis and are usually refractory to conventional chemotherapy. Elucidating the molecular and cellular mechanisms that are dysregulated in NSCLCs may lead to new possibilities for targeted therapy or enhanced efficacy of current therapies. Here we demonstrate Wnt pathway activation in around 50% of human NSCLC cell lines and primary tumors, through different mechanisms, including autocrine Wnt pathway activation involving upregulation of specific Wnt ligands. Downregulation of activated Wnt signaling inhibited NSCLC proliferation and induced a more differentiated phenotype. Together, our findings establish importance of activated Wnt signaling in human NSCLCs and offer the possibility of targeting upregulated Wnt signaling as a new therapeutic modality for this disease.
Keywords: autocrine, lung cancer, wnt
Introduction
The canonical Wnt/β catenin pathway has an important function in the proliferation and differentiation of stem/progenitor cells in a variety of adult epithelial tissues (van de Wetering et al., 2002; Reya and Clevers, 2005; Clevers, 2006). This ability is exploited by cancer cells to promote distinct aspects of self-renewal such as survival, proliferation and inhibition of differentiation (Reya and Clevers, 2005). In the same tissues where Wnt signaling normally maintains stem/progenitor cells, constitutive activation of this pathway due to dysregulation or genetic aberrations of key components underlies tumorigenesis. This has been best demonstrated in the intestinal crypt, where Wnt signaling normally regulates the stem cells at the bottom of the crypt (Reya and Clevers, 2005; Clevers, 2006). Aberrant Wnt signaling activation caused by mutations in adenomatous polyposis coli (APC) or β-catenin results in uncontrolled expansion of cells that are unable to appropriately differentiate and can ultimately lead to colorectal cancer (CRC) (Reya and Clevers, 2005; Clevers, 2006; Polakis, 2007; Klaus and Birchmeier, 2008). In fact, APC or β-catenin mutations are observed in greater than 90% of CRCs (Morin et al., 1997; Polakis, 2007).
Lung cancer is the most common cause of cancer mortality worldwide for both men and women (Minna et al., 2002). Despite some improvements in therapy over the past 30 years, the prognosis is generally poor with 85–90% patients dying from the disease (Minna et al., 2002). Lung cancers are divided into two histopathological types, non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC), which represent approximately 80 and 20% of tumors, respectively (Minna et al., 2002). SCLC have neuroendocrine features and arise mainly from the central airways, whereas lung adenocarcinomas, the most frequent form of NSCLC, usually originate in the peripheral lung and arise from progenitor cells located in the bronchioles (Clara cells) or alveoli (AT2 cells).
Recent studies have demonstrated the importance of Wnt/β-catenin signaling in regulating the balance between normal lung bronchioalveolar stem cells (BASCs) growth and differentiation during early lung development (Reynolds et al., 2008; Zhang et al., 2008). Hyperactivation of β-catenin in lung epithelium of genetically engineered mice leads to defective epithelial differentiation, increased proliferation, expansion of BASCs and can result in lung tumor formation (Okubo and Hogan, 2004; Mucenski et al., 2005; Reynolds et al., 2008; Zhang et al., 2008). In fact, NSCLCs have been reported to exhibit increased levels of cytosolic or nuclear β-catenin as visualized by increased immunostaining (Shigemitsu et al., 2001; Ohgaki et al., 2004). However, mutations of β-catenin or APC, the most common mechanism of aberrant Wnt pathway activation, are relatively rare (Shigemitsu et al., 2001; Sunaga et al., 2001; Ohgaki et al., 2004; Ding et al., 2008). As yet, there has been no systematic investigation of the frequency of functional Wnt pathway activation or the biological effects of its disruption on NSCLC phenotype. Our present studies identify Wnt pathway aberrations including autocrine Wnt activation at high frequency in such tumors. Moreover, we show that inhibition of upregulated Wnt signaling in NSCLC cells inhibited their growth and induced a more differentiated phenotype through a mechanism involving c-Myc.
Results
Constitutive Wnt pathway activation in human NSCLC cell lines
To investigate Wnt pathway activation in human lung carcinoma, we analysed uncomplexed and total β-catenin levels in lysates of a large panel of NSCLC and SCLC cell lines as well as two immortalized, non-tumorigenic human lung epithelial lines, normal Human Bronchial Epithelial (NHBE) and NL20, as controls (Supplementary Table S1). Figure 1a shows that the majority of NSCLC lines exhibited high levels of uncomplexed β-catenin, reflecting its transcriptionally active form, as detected by a glutathione S-transferase (GST) pull-down assay using recombinant E-cadherin (Bafico et al., 1998). Most of these lines represented the adenocarcinoma type of NSCLC. In contrast, NHBE and NL20 cells, which showed comparable levels of total β-catenin to these NSCLC tumor lines, demonstrated only very low amounts of uncomplexed β-catenin. Of note, we also observed undetectable or very low levels of uncomplexed and total β-catenin in A549 and H460, two NSCLC cell lines that were previously reported to exhibit Wnt pathway activation (He et al., 2004; You et al., 2004). Of the positives, A427 and HCC15 were previously reported to harbor activating mutations in β-catenin (Shigemitsu et al., 2001; Sunaga et al., 2001). Sequencing of β-catenin exon 3 revealed no additional activating β-catenin mutations in any of the other positive tumor lines (data not shown). In a series of seven SCLC lines analysed, we observed no detectable elevation of uncomplexed β-catenin (Table S1), suggesting that Wnt pathway activation is infrequent in SCLC.
Figure 1.

Wnt signaling activation in human non-small-cell lung carcinoma (NSCLC) cell lines. (a) Total cell lysates (1 mg) were subjected to precipitation with a glutathione S-transferase (GST)-E cadherin fusion protein (Bafico et al., 1998). Total cell lysates (25 µg) and the GST-E-cadherin precipitates were subjected to immunoblot analysis with an mAb directed against β-catenin. (b) Fluorescence-activated cell sorting (FACS) analysis, phase contrast and fluorescence images of H460 (upper panel) and H23 (lower panel) NSCLC cells infected with TOP or FOP TCF-GFP reporter lentiviruses or with enhanced green fluorescent protein (EGFP) expressing lentivirus (LV-GFP). BF, bright field; FL, fluorescence. (c) Lentivirus-mediated TCF-GFP reporter activity in human NSCLC cells. Results are depicted as the ratio TOP/FOP GFP mean fluorescence intensity (MFI). Results from two independent experiments are shown.
To confirm that the elevated levels of uncomplexed β-catenin observed in NSCLC cells resulted in Wnt pathway activation, we developed a lentivirus-based reporter system for T-cell factor (TCF)-dependent transcription in which seven wild-type (TOP) or mutant (FOP) TCF-binding sites (Veeman et al., 2003) were used to drive expression of either enhanced green fluorescent protein (EGFP) or luciferase. Whereas no activation was seen in H460 cells, H23 cells showed a strong increase in TOP-GFP mean fluorescence intensity (MFI) in comparison to FOP-GFP (Figure 1b). As a control, cell lines were infected with similar efficiency using a lentivirus expressing GFP driven by a constitutive phosphoglycerate kinase (PGK) promoter (LV-GFP). Figure 1c shows the results of two independent TCF-GFP reporter screens in a series of NSCLC cell lines. Of note, all four lines that showed low or undetectable levels of uncomplexed β-catenin (NHBE, NL20, H460 and A549) also showed a low TOP/FOP ratio (less than twofold), and were, thus, considered negative for Wnt pathway activation. Using this criterion, we observed elevated levels of TCF-GFP reporter activity in 9 of 16NSCLC lines (Figure 1c; Supplementary Table S1), which generally correlated well with expression levels of uncomplexed β-catenin (Figure 1a).
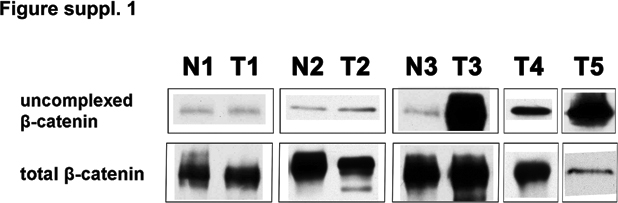
In an effort to extend these findings to primary tumors, we screened five human NSCLCs of the adenocarcinoma subtype for expression of uncomplexed β-catenin. As shown in Supplementary Figure S1, out of three matched cases of normal and tumor samples from the same patients, two showed similar low levels of uncomplexed β-catenin in both the normal and tumor tissues, whereas the third tumor showed a striking increase in uncomplexed β-catenin. Increased levels of uncomplexed β-catenin were also observed in two other tumor samples (T4 and T5) as compared to normal lung tissue samples analysed under identical conditions (N1–N3). Sequencing of the tumor DNAs revealed no detectable β-catenin activating mutations (data not shown). Collectively, these findings demonstrate upregulation of uncomplexed β-catenin, without genetic alteration of β-catenin, in a high fraction of human NSCLC primary tumors and tumor cell lines.
Wnt cell-surface antagonists reveal autocrine Wnt signaling in human NSCLC lines
We have previously demonstrated an autocrine mechanism for Wnt pathway activation in human ovarian and breast cancer cell lines associated with high levels of uncomplexed β-catenin in the absence of detectable mutations afflicting either β-catenin or APC (Bafico et al., 2004). To test the possibility of an autocrine Wnt signaling mechanism in NSCLCs, we took advantage of FRP1 and DKK1 antagonists, which inhibit Wnt ligand–receptor interactions (Kawano and Kypta, 2003). Because these inhibitors specifically inhibit Wnt signaling at the cell surface, they can distinguish between extracellular and intracellular pathway activating mechanisms (Bafico et al., 2004). We generated lentiviral vectors expressing hemagglutinin (HA)-tagged FRP1 or Flag-tagged DKK1 under the control of either a constitutive or tetracycline (Tet-off) regulatable promoter and tested their ability to decrease the levels of uncomplexed β-catenin and TCF reporter activity. As shown in Figure 2a, stable expression of FRP1 or DKK1 in H1819 NSCLC cells resulted in a marked decrease in uncomplexed β-catenin level. To further explore the effects of FRP1 or DKK1 on uncomplexed β-catenin levels, we used inducible FRP1 or DKK1 expression in several other NSCLC lines. As shown in Figure 2b, FRP1 caused a significant reduction in uncomplexed β-catenin levels in H23 and A1146tumor lines, even under conditions of low FRP1 expression levels in the presence of doxycycline (dox) due to leakiness of the inducible system. Similarly, low DKK1 expression levels in the presence of dox were also associated with a decrease in uncomplexed β-catenin levels in H23 and A1146cell s, which were further reduced on full induction (Figure 2a). In contrast, FRP1 or DKK1 showed no effect on uncomplexed β-catenin levels in H2347, H358 or A427 cells, whereas only A427 cells harbored a β-catenin mutation.
Figure 2.

Effects of FRP1 and DKK1 inhibition on Wnt/β-catenin signaling and growth of human non-small-cell lung carcinoma (NSCLC) cells. (a) Effects of constitutive expression of FRP1 and DKK1 on uncomplexed β-catenin in H1819 NSCLC cell line. FRP1 and DKK1 expression was determined by immunoblot analysis as described in Materials and methods. (b) Analysis of NSCLC lines for uncomplexed β-catenin using regulatable expression of hemagglutinin (HA)-tagged FRP1 (upper panel) and Flag-tagged DKK1 (lower panel). NSCLC cells expressing Tet regulatable FRP1-HA or DKK1-Flag were generated as described in Supplementary materials and methods. Expression of FRP1-HA or DKK1-Flag was induced by removal of dox from the culture medium. Cells expressing tetracycline Trans-activator (tTa) were used as control. Analysis of uncomplexed β-catenin was performed as described in Materials and methods using 1mg total cell lysates, except for A427 cells, where 0.1 mg cell lysate was used. FRP1 and DKK1 expression was determined by immunoblot analysis as described in Materials and methods. (c) FRP1- and DKK1-mediated inhibition of TCF luciferase reporter activity in NSCLC cell lines. Luciferase reporter activity was calculated by dividing the TOP/RL ratio by the FOP/RL ratio. Results were normalized to the results with vector-transduced cultures. The values represent the mean ± s.d. from two independent experiments. (d) Real-time PCR quantification of FRP1 and DKK1 effects on axin2 mRNA expression. H23, A1146 and H1819 cells were infected with vector (VEC), FRP1-HA or DKK1-Flag lentiviruses. qRT–PCR was performed as described in Supplementary materials and methods. Relative mRNA expression levels were quantified using the ΔΔC(t) method (Pfaffl, 2001). (e) Effects of DKK1 on cell growth. A549, H23, H1819 and A427 cells were infected with lentiviruses expressing vector (VEC) or DKK1-Flag and 2 × 104 cells were plated into 60mm tissue culture dishes. Cultures were visualized using crystal violet staining 2–3 weeks after plating. Expression of Flag-tagged DKK1 was assessed by immunoblot analysis as described in Materials and methods.
Both antagonists significantly inhibited TCF reporter activity in H1819, H23 and A1146tumor lines, corroborating the observed decrease in uncomplexed β-catenin levels (Figure 2c). In contrast, TCF reporter activity in A427 cells was unaffected by these antagonists consistent with their lack of effects on β-catenin levels in these cells (Figure 2a). As shown in Figure 2d, each antagonist also significantly downregulated expression of axin2, a prototypic Wnt target gene (Jho et al., 2002), in H1819, H23 and A1146tumor lines. Taken together, these results demonstrate constitutive autocrine Wnt activation in these NSCLC lines. Conversely, the lack of effects of these Wnt antagonists on H2347 and H358 tumor lines, which showed increased uncomplexed β-catenin levels and increased TCF reporter activity in the absence of β-catenin mutations, implies that other mechanisms were responsible for Wnt signaling activation in these NSCLC lines.
Wnt signaling promotes proliferation and altered cell growth properties (Bafico et al., 1998). To study the effects of Wnt pathway inhibition on tumor cell proliferation, we stably expressed DKK1 in several NSCLC cell lines. As shown in Figure 2e, DKK1 exerted antiproliferative effects on H23 and H1819 tumor cells in comparison to vector control cells. To confirm that these effects were due to Wnt activity inhibition, we compared the effects of DKK1 on A549 cells, which showed no evidence of Wnt pathway activation, and on β-catenin mutant A427 cells. As expected, expression of DKK1 in these NSCLC lines was not associated with any detectable growth inhibition. We confirmed similar expression levels of Flagtagged DKK1 in each of these cell lines by immunoblot analysis (Figure 2e). Taken together, these results strongly support involvement of constitutive Wnt pathway activation in promoting the proliferation of Wnt autocrine NSCLC cells.
Identification of canonical Wnts involved in autocrine activation of human NSCLC lines
We next attempted to identify Wnt ligands, which might be overexpressed in these tumor cells. Semiquantitative RT–PCR for expression of 19 Wnts revealed that some were ubiquitously expressed in both β-catenin positive and negative NSCLC lines (Wnt2b, Wnt7a and Wnt9a), whereas Wnt2 and Wnt3a mRNAs were overexpressed primarily in the tumor lines exhibiting autocrine signaling (Supplementary Figure S2). No other canonical or non-canonical Wnts were detected using this method. We used real-time PCR to more accurately quantitate Wnt2 and Wnt3a expression levels in a panel of nine NSCLC and the immortalized NHBE and NL20 lines. Figures 3a and b show that Wnt2 mRNA expression levels in H23 and A1146cells were more than 300-fold and 30-fold, respectively, above that of NHBE or NL20 cells, and Wnt3a expression in H1819 tumor cells was almost 40-fold higher than in NHBE cells.
To establish whether these Wnts are involved in the constitutive autocrine signaling in H23 and H1819 cells, we used an shRNA knockdown approach. As shown in Figure 3c, Wnt 2 and Wnt3a shRNA constructs efficiently knocked down the expression of their corresponding mRNA targets (80% for Wnt2 and 70% for Wnt3a). Knockdown of Wnt2 in H23 cells and Wnt3a in H1819 cells resulted in each case in a significant decrease in both TCF-mediated luciferase reporter activity and axin2 expression (Figures 3d and e). These results provide strong evidence that constitutive autocrine Wnt signaling in H23 and H1819 NSCLC cells involves Wnt2 and Wnt3a, respectively.
Figure 3.

Overexpression of Wnt2 and Wnt3a contributes to constitutive Wnt activation in autocrine non-small-cell lung carcinoma (NSCLC) cells. (a, b) Real-time PCR quantification of Wnt2 (a) and Wnt3a (b) expression in H23 and H1819 cells, respectively. To visualize relative expression levels of Wnt2 and Wnt3a, qPCR reactions were removed before saturation and PCR products were separated on 1.5% agarose gel and stained with ethidium bromide. (c) shRNA knockdown quantification of Wnt2 and Wnt3a. H23 and H1819 cells were infected with lentiviruses expressing shRNA targeting green fluorescent protein (GFP), Wnt2 or Wnt3a. (d) Effect of shRNA knockdown of Wnt2 and Wnt3a on TCF reporter activity. Luciferase reporter activity was calculated by dividing the TOP/RL ratio by FOP/RL ratio. Each column represents the mean ± s.d. of two independent experiments. (e) Effect of shRNA knockdown of Wnt2 and Wnt3a on axin2 mRNA expression. H23 and H1819 cells were infected with lentiviruses expressing shRNA targeting GFP, Wnt2 or Wnt3a.
Dominant-negative TCF-4 induces p21-associated cell-cycle arrest of NSCLC cells with constitutive Wnt pathway activation
To compare the biological effects of Wnt pathway downregulation in NSCLC lines with Wnt autocrine and β-catenin activating mutations as well as mechanisms involved, we used constitutive or Tet regulatable expression of a dominant-negative TCF-4 (DNTCF-4), which lacks the first 32 amino acids and is unable to bind β-catenin but retains its DNA-binding ability (van de Wetering et al., 2002). This approach was used previously to investigate the effects of Wnt pathway inhibition in colon carcinoma cells with APC or β-catenin oncogenic mutations (van de Wetering et al., 2002). We generated lentiviral constructs expressing two versions of either constitutive or inducible DNTCF-4 (designated DN) or DNTCF-4 fused to mOrange (designated DN-mO). To assess the ability of DN and DN-mO to inhibit Wnt activation, we tested their effects on TCF luciferase reporter activity. As shown in Supplementary Figure 3SA, both DNTCFs strongly inhibited the constitutively high levels of TCF reporter activity in HCC15 tumor cells harboring a β-catenin mutation, as well as in Wnt autocrine H23 and H1819 NSCLC lines. Supplementary Figure 3SB shows that DNTCFs expression exerted no effect on the growth of NL20 or A549 cells without any detectable Wnt pathway activation (Figure 1). In contrast, constitutive expression of both DNTCFs resulted in obvious growth inhibition of Wnt autocrine H23 and H1819 tumor cells (Supplementary Figure S3C). DN-mO was more potent, presumably because the DN-mO fusion product exhibited an extended half-life (Supplementary Figure 3SC). DN-mO also inhibited to a lesser extent the growth of HCC15 cells, which contained a β-catenin mutation (Supplementary Figure 3SC).
To further investigate the effects of DNTCF expression, we established mass populations of H23 and H1819 cells expressing Tet-inducible versions of DNTCF-4 and DN-mO as well as control lentivector (designated VEC). Low expression levels of the two DN forms were observed even in the presence of dox due to leakiness of the system (Figure 4a), and strong induction was observed on dox removal. To determine the extent to which the low and high DNTCF expression levels inhibited Wnt pathway activation in H23 and H1819 cells, we analysed expression of axin2 (Jho et al., 2002) by real-time PCR. As shown in Figure 4b, even low expression levels of the DN forms in the presence of dox were sufficient to cause some reduction in axin2 mRNA expression levels. Full induction of the two DN forms resulted in further reduction in levels of this prototypic Wnt target gene. Although leaky expression of the two DN forms in the presence of dox retarded cell growth, full induction of DN or DN-mO resulted in strong G1 arrest as measured by fluorescence-activated cell sorting (FACS) at 72 h. Of note, there was no detectable increase in apoptosis under the same conditions (Figure 4c). An example of the expression of DN-mO in the presence or absence of dox, and its effects on proliferation of H23 cells, is shown in Figure 4d. As shown in Figure 4e, leaky expression of the DNTCFs in the presence of dox was associated with decreased colony-forming ability as compared to VEC cells. However, full induction of DN and DN-mO exerted more profound growth inhibition.
Figure 4.

Effects of inducible dominant-negative TCF-4 (DNTCF-4) on growth of non-small-cell lung carcinoma (NSCLC) autocrine cells. (a) Immunoblot analysis of DNTCFs expression. H23 and H1819 cells were infected with lentiviruses expressing DNTCF-4 (DN), DN-mOrange (DN-mO) and vector (VEC) under the control of a tetracycline-inducible promoter and selected with puromycin in the presence of dox. After washing, cells were divided into separate cell culture dishes in the presence or absence of dox and analysed by immunoblot 3 days after induction. Expression of DNTCF-4 proteins was detected using an antibody to TCF-4. Lower molecular weight immunoreactive DNTCF species were also observed. Molecular weights in kilodaltons are indicated (kDa). (b) Effect of DNTCFs on axin2 mRNA expression. RNA was extracted from H23 and H1819 cells infected as in (a) and maintained in the presence or absence of dox for 3 days. (c) Effect of DNTCFs on cell-cycle profile. Propidium iodide (PI) analysis of H23 and H1819 cells infected as in (a) and maintained in the presence or absence of dox at 3 days after induction. Numbers indicate the percentage of cells in G1 or S phase for each cell line analysed. Results are representative of at least two independent experiments. (d) Effects on proliferation of H23 cells expressing inducible DN-mO in the presence or absence of dox, observed at 3 days after induction. BF, bright field; FL, fluorescence. (e) Effects on growth of H23 and H1819 at 2–3 weeks following expression of VEC, DN and DN-mO. A total of 2 × 104 cells were plated into 60mm plates in the presence or absence of dox. Cultures were visualized by crystal violet staining. (f) Effects of DNTCFs on expression of c-Myc, cyclin D1 and p21. Protein lysates from H23 and H1819 cells, infected with VEC, DN and DN-mO, and grown in the presence or absence of dox for 3 days, were analysed by immunoblot. Dox, doxycycline.
Clevers and colleagues have shown that inhibition of TCF signaling in clonally selected human CRC lines with APC or β-catenin mutations by regulatable expression of DNTCF-4 induced cell-cycle arrest associated with decreased expression of c-Myc and increased expression of the cell-cycle inhibitor, p21 (van de Wetering et al., 2002). These authors showed further that c-Myc normally inhibits p21 transcription, so that reduced c-Myc expression resulting from TCF signaling inhibition releases p21 to mediate cell-cycle arrest and differentiation effects in these cells (van de Wetering et al., 2002). Thus, we analysed expression levels of these cell-cycle regulators by immunoblot analysis in NSCLC cells expressing DNTCFs or VEC in the presence or absence of dox. As shown in Figure 4f, leaky expression of the two DNTCFs in mass cell populations decreased c-Myc expression, especially with the more potent DN-mO, and full induction led to a dramatic inhibition of c-Myc expression. On full DNTCF induction, we observed strong upregulation of p21 levels as well. Cyclin D1 protein levels were not significantly affected by the expression of either form of DNTCF-4. The reduction in c-Myc expression levels with concomitant increase in p21 correlated well with cell-cycle profile analysis and the effects observed on cell growth (Figure 4c–f). Taken together, these findings in NSCLC cells strongly support previous findings that c-Myc is a key mediator of cell proliferation induced by Wnt signaling through a mechanism involving p21 repression (van de Wetering et al., 2002).
Canonical Wnt signaling has been shown to maintain lung epithelial cells in an undifferentiated stem-/progenitor-like state (Reynolds et al., 2008; Zhang et al., 2008). Thus, we also examined whether expression of DNTCF in H23 and H1819 cells altered their differentiation state. Real-time PCR analysis showed that induction of DN and DN-mO led to an upregulation of several differentiation markers known to be expressed in differentiated bronchiolar (Clara-cell-specific protein, CCSP) or alveolar type 2 (AT2) cells (A1AT, ICAM-1 and MUC-1) (Supplementary Figure S4; Braga et al., 1992; Guzman et al., 1994; Venembre et al., 1994; Nakamura et al., 2006). These results demonstrate that inhibition of autocrine Wnt signaling by DNTCF-4 leads to increased expression of differentiation markers associated with both alveolar (AT2) and bronchial (Clara) lineages.
Discussion
Our present findings establish that constitutive Wnt signaling activation, as demonstrated by increased levels of uncomplexed β-catenin, occurs at high frequency in NSCLC cell lines and in primary NSCLCs. Upregulated TCF reporter activity was found to correlate generally well with increased levels of uncomplexed β-catenin and provided confirmation of Wnt pathway activation in the tumor lines. Of nine positive NSCLC lines, two contained mutations in β-catenin, the most frequently reported genetic aberration in tumors other than CRC, where APC loss-of-function mutations are generally more prevalent (Clevers, 2006; Polakis, 2007). The high incidence of Wnt signaling upregulation in NSCLCs by other mechanisms argues that this pathway is a much more frequent target than has been previously recognized in this common epithelial tumor. Our findings were also specific to NSCLC as a survey of human SCLC lines revealed no evidence of Wnt pathway activation in this type of lung cancer.
Wnt antagonists, FRP1 and DKK1, which inhibit Wnt signaling at the cell surface (Kawano and Kypta, 2003), caused dramatic downregulation of uncomplexed β-catenin levels, TCF reporter activity and expression of the prototypic Wnt target gene axin2 in around 30% of Wnt-activated NSCLC lines, strongly implicating a Wnt autocrine mechanism. We observed further that either Wnt2 or Wnt3a was specifically overexpressed and that its specific shRNA knockdown decreased TCF reporter activity and axin2 expression in these tumor lines. The Wnt2 gene resides on the long arm of chromosome 7 in proximity to a number of proto-oncogenes including c-MET, which can be amplified in lung tumors. However, real-time PCR analysis of H23 and H1819 cells showed no evidence of either Wnt2 or Wnt3a gene amplification in these Wnt autocrine tumor lines (data not shown). Hence, the underlying mechanism responsible for the specific overexpression of either Wnt2 or Wnt3a in enforcing a Wnt autocrine loop in NSCLCs remains to be elucidated.
Previous reports have suggested autocrine Wnt signaling activation in certain NSCLC lines in which either Wnt1 or Wnt2 expression was detected by antibodies, which could also induce the same tumor cells to undergo apoptosis (He et al., 2004; You et al., 2004). These studies chiefly focused on A549 and H460 tumor lines, in which we observed no detectable expression of Wnt 1, and Wnt2 was also undetectable in A549 cells by sensitive reverse transcription (RT)–PCR techniques. Of note, we found that these tumor lines exhibited only very low or undetectable levels of uncomplexed as well as total β-catenin and also lacked evidence of upregulated TCF reporter activity. Moreover, we observed no detectable biological effects of known Wnt antagonists or DNTCF on A549 cells. In contrast, the Wnt autocrine NSCLC lines identified by us exhibited growth inhibition in the absence of detectable apoptosis in response to these same inhibitors under conditions in which they also caused downregulation of β-catenin and TCF reporter activity. Our findings are analogous to results in CRC (van de Wetering et al., 2002) and breast/ovarian tumor cells (Bafico et al., 2004), respectively, where downregulation of Wnt signaling resulted in cell growth inhibition rather than apoptosis. Thus, the results of He et al. (2004) and You et al. (2004) with A549, a tumor line established by us (Giard et al., 1973) as well as with H460, unlikely reflect a mechanism involving upregulated Wnt signaling.
We also identified several Wnt pathway-activated NSCLC lines, which exhibited no detectable evidence of Wnt autocrine signaling or β-catenin mutations. Further studies are needed to identify the mechanisms involved in Wnt signaling upregulation in these tumor lines. Nonetheless, our findings imply the existence of at least three distinct mechanisms that together account for the high frequency of constitutive Wnt activation in human NSCLCs.
Recent studies suggest that BASCs may be the cells of origin of murine lung adenocarcinoma (Kim et al., 2005). Notably, BASCs show Wnt signaling activation (Zhang et al., 2008) and can give rise to progeny with either Clara cell or AT2 cell phenotype (Kim et al., 2005). The cell-cycle arrest induced by DNTCF-4 initiated a differentiation program toward both Clara (CCSP) and AT2 (AIAT, ICAM-1 and MUC-1) cell lineages. This suggests that a high proportion of human adenocarcinomas may originate from Wnt-positive BASCs or, alternatively, that aberrant activation of Wnt signaling in more differentiated progenitors may endow them with stem/progenitor properties including enhanced proliferative capacity and cell-survival properties.
We showed that downregulation of TCF signaling in autocrine NSCLC cells resulted in decreased c-Myc levels concomitant with increased p21 expression. These findings are consistent with the observed effects of TCF downregulation on c-Myc and p21 levels in CRC lines mutant for APC or β-catenin (van de Wetering et al., 2002). In fact, the oncogenic effects conferred by loss of APC on the mouse small intestine were shown to be almost entirely dependent on functional c-Myc as simultaneous deletion of APC and c-Myc rescued the APC knockout tumor phenotype (Sansom et al., 2007). Of note, c-Myc is frequently overexpressed in lung cancer (Richardson and Johnson, 1993). Although gene amplification can explain its deregulation in a subset of tumors and cell lines, c-Myc overexpression is seen in a much higher percentage of cases in the absence of gene amplification (Richardson and Johnson, 1993; Bernasconi et al., 2000). Thus, the high prevalence of Wnt pathway activation observed by us in NSCLC cell lines and primary tumors may help to account for the high frequency of c-Myc overexpression in NSCLC.
Materials and methods
Cell culture
Human NSCLC cell lines A1146, A549 and A427 were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Grand Island, NY, USA). NSCLC lines H23, H1819, H1355, H2347, HCC193, HCC515, H358, H1171, HCC461, HCC827, H1299, HCC15, H460 and SCLC cell lines H128, H82, H209, H2081, H1184, H889 and H249 were grown in RPMI medium (Invitrogen) supplemented with 10% FBS (Invitrogen). Immortalized human bronchial epithelial cell line, NL20 (Schiller et al., 1992) was purchased from ATCC (American Type Culture Collection) and grown in a specific growth medium as recommended. The NHBE cells were purchased from Lonza (Allendale, NJ, USA) and cultured in the recommended medium (Lonza). All cells were cultured at 37 °C in 5% CO2.
Analysis of uncomplexed β-catenin and immunoblot analysis
GST-E-cadherin binding and immunoblot analysis was performed as previously described (Bafico et al., 1998). Concentration of serum-free conditioned medium obtained from vector or DKK1-expressing cells was performed using Amicon Ultra-15 centrifugal filters (Millipore, Ireland). Expression of FRP1-HA was detected in cell lysates using anti-HA antibody. For immunoblot analysis the following primary antibodies were used: HA, Flag and c-Myc (9E10; Hybridoma Center, Mount Sinai School of Medicine, New York, NY, USA), β-catenin and p21 (BD Pharmingen, San Jose, CA, USA), cyclin D1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Antimouse immunoglobulin G (IgG) or anti-rabbit IgG secondary antibodies conjugated to horseradish peroxidase or Alexa Fluor 680 were purchased from Amersham Bioscience (GE Healthcare, UK) or from Molecular Probes (Eugene, OR, USA), respectively. Quantification of signal immunoreactivity was obtained using enhanced chemiluminescence detection system (Amersham, Piscataway, NJ, USA) or the Licor Odyssey Imaging System (LI-COR, Lincoln, Nebraska, USA).
FACS analysis of TCF-mediated GFP reporter activity
Cells infected with TOP or FOP-EGFP reporter lentiviruses were transferred to polystyrene tubes (Falcon, NJ, USA) and subjected to FACS analysis (FACScan; Becton Dickinson, NJ, USA) using CellQuest 3.2 software (Becton Dickinson, San Jose, CA, USA).
Quantification of TCF-mediated luciferase reporter activity
At 24 h before infection, stable reporter cell lines expressing TOP or FOP TCF luciferase and renilla luciferase were plated in six-well plates at 1 × 105 cells per well. The following day cells were infected with lentiviruses expressing different Wnt antagonists, selected with 2 µg/ml puromycin for 3 days, lysed and processed for luciferase reporter assay using the dual luciferase reporter kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Luciferase reporter activity was calculated by dividing the ratio TOP/RL by the FOP/RL ratio. Results were normalized to the results with vectortransduced cultures.
FACS analysis of cell cycle and Annexin-PI
For DNA content analysis, cells were trypsinized, combined with floating cells, washed with phosphate-buffered saline (PBS), stained with propidium iodide (PI), using the CycleTEST Plus DNA reagent kit (Becton Dickinson) following the manufacturer’s instructions, and subjected to FACS analysis. For Annexin V-PI cells were treated as for cell-cycle analysis, stained with Annexin-PI using the Annexin V-FITC apoptosis detection kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions and subjected to FACS analysis. Results were analysed using CellQuest 3.2 software (Becton Dickinson).
Cell growth analysis assay
Transduced and marker selected cells were trypsinized, counted and 1 to 2 × 104 cells were plated into 60mm tissue culture dishes. At 2–3 weeks, cells were washed with PBS, fixed in 10% methanol/acetic acid solution and stained with 1% crystal violet.
Statistical analysis
Statistical analysis was performed using two-way analysis of variance with Bonferroni multiple testing corrections using the Prism 5 software (GraphPad Software, San Diego, CA, USA). A P-value o0.05 was considered statistically significant. Values are represented as arithmetical mean ± s.d.
Supplementary data
Detailed description of lentiviral constructs, production of lentiviruses, RT–PCR, shRNA sequences, tissue specimens and primer lists can be found in supplementary data.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grant number 5R01CA071672 from the National Cancer Institute. SV is supported by consecutive post-doctoral fellowships from New York State Department of Health and American Urological Association. We are grateful to Dr J Minna and Dr A Gazdar (University of Texas Southwestern Medical Center, Dallas, TX, USA) for providing us with some NSCLC cell lines. We thank Dr Stefano Rivella (Weill Medical College of Cornell University, New York, NY, USA) for the generous gift of the lentiviral vector, pRRL-SIN-cPPT-PGK-GFP. We also thank Dr Robert Hannigan (Mount Sinai School of Medicine, New York, NY, USA) and Professor Roger Tsien (Howard Hughes Medical Institute, University of California, San Diego, CA, USA) for kindly providing us with the cDNA encoding mOrange. We especially thank Randy Arroyave for excellent technical assistance and Martina Kracikova and Ioana Rus for critical reading of this paper.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Bafico A, Gazit A, Wu-Morgan SS, Yaniv A, Aaronson SA. Characterization of Wnt-1 and Wnt-2 induced growth alterations and signaling pathways in NIH3T3 fibroblasts. Oncogene. 1998;16:2819–2825. doi: 10.1038/sj.onc.1201797. [DOI] [PubMed] [Google Scholar]
- Bafico A, Liu G, Goldin L, Harris V, Aaronson SA. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004;6:497–506. doi: 10.1016/j.ccr.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Bernasconi NL, Wormhoudt TAM, Laird-Offringa IA. Post-transcriptional deregulation of myc genes in lung cancer cell lines. Am J Respir Cell Mol Biol. 2000;23:560–565. doi: 10.1165/ajrcmb.23.4.4233. [DOI] [PubMed] [Google Scholar]
- Braga VM, Pemberton LF, Duhig T, Gendler SJ. Spatial and temporal expression of an epithelial mucin, Muc-1, during mouse development. Development. 1992;115:427–437. doi: 10.1242/dev.115.2.427. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. in vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- Guzman J, Izumi T, Nagai S, Costabel U. ICAM-1 and integrin expression on isolated human alveolar type II pneumocytes. Eur Respir J. 1994;7:736–739. doi: 10.1183/09031936.94.07040736. [DOI] [PubMed] [Google Scholar]
- He B, You L, Uematsu K, Xu Z, Lee AY, Matsangou M, et al. A monoclonal antibody against Wnt-1 induces apoptosis in human cancer cells. Neoplasia. 2004;6:7–14. doi: 10.1016/s1476-5586(04)80048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Mucenski ML, Nation JM, Thitoff AR, Besnard V, Xu Y, Wert SE, et al. beta-Catenin regulates differentiation of respiratory epithelial cells in vivo. Am J Physiol Lung Cell Mol Physiol. 2005;289:L971–L979. doi: 10.1152/ajplung.00172.2005. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Kobayashi K, Nakamoto M, Kohno T, Sasaki H, Matsuno Y, et al. Identification of tumor markers and differentiation markers for molecular diagnosis of lung adenocarcinoma. Oncogene. 2006;25:4245–4255. doi: 10.1038/sj.onc.1209442. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kros JM, Okamoto Y, Gaspert A, Huang H, Kurrer MO. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 2004;207:197–203. doi: 10.1016/j.canlet.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Okubo T, Hogan BL. Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. J Biol. 2004;3:11. doi: 10.1186/jbiol3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Reynolds SD, Zemke AC, Giangreco A, Brockway BL, Teisanu RM, Drake JA, et al. Conditional stabilization of beta-catenin expands the pool of lung stem cells. Stem Cells. 2008;26:1337–1346. doi: 10.1634/stemcells.2008-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson GE, Johnson BE. The biology of lung cancer. Semin Oncol. 1993;20:105–127. [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Schiller JH, Bittner G, Oberley TD, Kao C, Harris C, Meisner LF. Establishment and characterization of a SV40 T-antigen immortalized human bronchial epithelial cell line. in vitro Cell Dev Biol. 1992;28A:461–464. doi: 10.1007/BF02634125. [DOI] [PubMed] [Google Scholar]
- Shigemitsu K, Sekido Y, Usami N, Mori S, Sato M, Horio Y, et al. Genetic alteration of the beta-catenin gene (CTNNB1) in human lung cancer and malignant mesothelioma and identification of a new 3p21.3 homozygous deletion. Oncogene. 2001;20:4249–4257. doi: 10.1038/sj.onc.1204557. [DOI] [PubMed] [Google Scholar]
- Sunaga N, Kohno T, Kolligs FT, Fearon ER, Saito R, Yokota J. Constitutive activation of the Wnt signaling pathway by CTNNB1 (beta-catenin) mutations in a subset of human lung adenocarcinoma. Genes Chromosomes Cancer. 2001;30:316–321. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1097>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The [beta]-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Venembre P, Boutten A, Seta N, Dehoux MS, Crestani B, Aubier M, et al. Secretion of alpha 1-antitrypsin by alveolar epithelial cells. FEBS Lett. 1994;346:171–174. doi: 10.1016/0014-5793(94)80695-0. [DOI] [PubMed] [Google Scholar]
- You L, He B, Xu Z, Uematsu K, Mazieres J, Mikami I, et al. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene. 2004;23:6170–6174. doi: 10.1038/sj.onc.1207844. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K, et al. A Gata6–Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet. 2008;40:862–870. doi: 10.1038/ng.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.