

Abstract
Background
Overexpression or mutation of the epidermal growth factor receptor (EGFR) potently enhances the growth of many solid tumors. Tumor cells frequently display resistance to mechanistically-distinct EGFR-directed therapeutic agents, making it valuable to develop therapeutics that work by additional mechanisms. Current EGFR-targeting therapeutics include antibodies targeting the extracellular domains, and small molecules inhibiting the intracellular kinase domain. Recent studies have identified a novel prone extracellular tetrameric EGFR configuration, which we identify as a potential target for drug discovery.
Methods
Our focus is on the prone EGFR tetramer, which contains a novel protein-protein interface involving extracellular domain III. This EGFR tetramer is computationally targeted for stabilization by small molecule ligand binding. This study performed virtual screening of a Life Chemicals, Inc. small molecule library of 345,232 drug-like compounds against a molecular dynamics simulation of protein-protein interfaces distinct to the novel tetramer. One hundred nine chemically diverse candidate molecules were selected and evaluated using a cell-based high-content imaging screen that directly assessed induced internalization of the EGFR effector protein Grb2. Positive hits were further evaluated for influence on phosphorylation of EGFR and its effector ERK1/2.
Results
Fourteen hit compounds affected internalization of Grb2, an adaptor responsive to EGFR activation. Most hits had limited effect on cell viability, and minimally influenced EGFR and ERK1/2 phosphorylation. Docked hit compound poses generally include Arg270 or neighboring residues, which are also involved in binding the effective therapeutic cetuximab, guiding further chemical optimization.
Conclusions
These data suggest that the EGFR tetrameric configuration offers a novel cancer drug target.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-015-1415-6) contains supplementary material, which is available to authorized users.
Keywords: Epidermal growth factor receptor, Grb2, Protein multimerization, Extracellular domain
Background
For many solid tumors, overexpression or activating mutation of the epidermal growth factor receptor (EGFR) serves as an oncogenic driver. Even in some tumors in which EGFR is not overexpressed, it can serve as an essential intermediate in signaling required for cell growth and survival [1, 2], reflecting its role as a potent regulator of multiple downstream effector cascades, including PI3K/AKT/mTOR, JAK/STAT, and RAS/RAF/MEK. Because of this prominent role, multiple therapeutic agents targeting EGFR have been developed. These include monoclonal antibodies such as cetuximab and panitumumab, which target the extracellular domain, and small molecule inhibitors including erlotinib, gefitinib, lapatinib, and afatinib, which target the cytosolic kinase domain. These agents often provide significant clinical benefit, but are sometimes ineffective because of intrinsic or acquired tumor resistance factors. Mechanisms of resistance can include expression of an EGFRvIII variant [3], which eliminates the epitope for some monoclonal antibodies, or missense mutations involving the EGFR kinase domain (e.g., T790M), among others [4]. The frequent occurrence of these forms of EGFR-intrinsic resistance mutations in some cancers has motivated ongoing efforts to identify additional agents that use independent mechanisms to inhibit EGFR [5]. Further, the combination of multiple classes of inhibitory agents has been recently recognized to provide thorough blockade of EGFR signaling and can yield significant clinical benefit [6, 7]; this provides a strong motivation to enlarge the suite of mechanistic options for EGFR inhibition.
EGFR protein structure and protein interactions have been the subject of intensive study [8, 9]. As a transmembrane, ligand-dependent receptor tyrosine kinase, EGFR contains an extracellular N-terminal moiety comprised of four discrete domains (Domains I-IV), a transmembrane domain, an intracellular juxtamembrane domain, and a C-terminal intracellular kinase domain. Unliganded EGFR is predominantly monomeric, with very low propensity to dimerize. EGFR monomers exist in an equilibrium of conformations, at least one of which is competent to dimerize with a second EGFR monomer (estimated at ~5 % of the monomer population). Binding of EGF or other ligands to domains I and III stabilizes the protein conformer that is competent for dimerization, thus increasing its mole fraction within the monomer population and promoting dimerization [10, 11]. The dimerization-competent conformer contains domain II in an exposed, rather than buried environment. Dimeric EGFR binding to two molecules of EGF is characterized by high-affinity and low-affinity events, which are proposed to represent an initial high-affinity binding event that favors an asymmetric dimeric structure in which the second subunit is sterically prevented from binding ligand [12]. In the dimeric conformation, each EGF binding site is comprised of only one EGFR subunit. Binding of dimeric EGFR to two molecules of EGF favors a conformation of the kinase domain that activates intracellular auto-phosphorylation and initiates downstream signaling events. Subsequently, ligand-bound EGFR is internalized from the cell surface, limiting the duration of signaling [13].
In the physiologic context of the cell surface, this basic and elegant paradigm acquires additional nuance. A number of studies have demonstrated that patches or “rafts” on the cell surface, enriched in specific lipids such as cholesterol, allow concentration of EGFR in clusters that support accumulation of unliganded dimeric and higher-order oligomeric species, including tetramers [14–17]. These pre-formed dimers and multimers have been associated with specific activity states of EGFR (discussed in [18]). The oligomeric state of EGFR may also be relevant to oncogenic transformation, given that the overexpression of EGFR that is common in and driver for many solid tumors (reviewed in [19]) would be expected to promote oligomerization. While most studies have addressed the protein packing events associated with tetrameric conformations defined as “back-to-back” or “side-to-side”, in which EGFR protrudes at a 90° angle to the cell membrane, an intriguing group of studies have supported the existence of a physiological “head-to-head” tetramer, in which the extracellular domains of EGFR lie prone along the plasma membrane (Fig. 1a) [17, 20, 21]. The biological significance of an EGFR tetramer as a key signaling unit is well documented [22, 23]. However, it is possible that EGFR can form each of the proposed architecturally distinct tetrameric assemblies and that these multimers have distinct functions. This is consistent with the myriad functions that have been attributed to assemblies larger than a dimer. The four EGF binding sites in the “head-to-head” prone tetramer have an intrinsic asymmetry. Two of these sites are at the dimer-dimer interface; EGF binding affinity is strengthened by interactions from a second subunit, which has been proposed as an alternative explanation for the high affinity mode of EGF binding to EGFR. If so, targeting the unique structural features of the prone tetramer would be predicted to provide an entirely new approach to disrupting early stages of the EGFR activation process. The ability to form higher order oligomeric complexes is not restricted to EGFR. Landgraf and colleagues have elegantly demonstrated that heteromeric complexes of the EGFR family members Her2 and Her3 can form functional oligomers of heterodimers and that this complex formation can be modulated by a specific aptamer [24–27]. In the current study, we have combined molecular docking targeted at the EGFR tetramer interface with a high throughput microscopy based screen to identify compounds that influence EGFR internalization, either independently or contingent upon the presence of EGF.
Fig. 1.
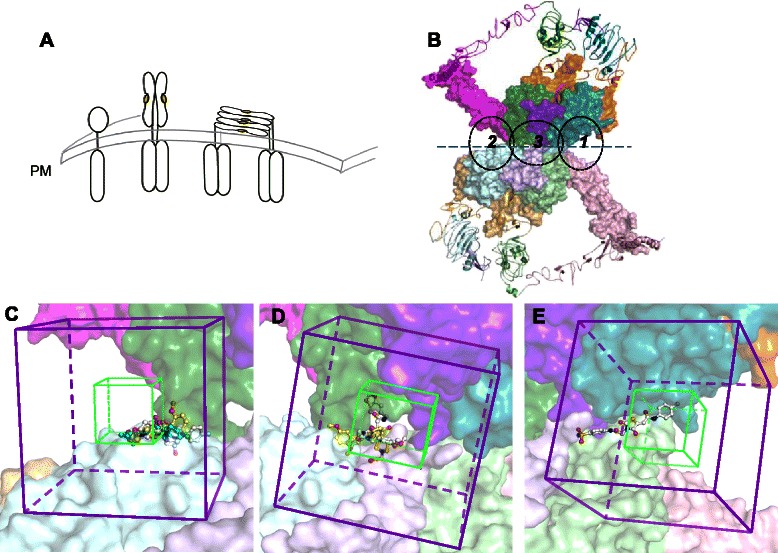
Virtual screening methods identify candidate EGFR multimer stabilizers. Panel a depicts a simple cartoon showing unliganded EGFR monomer in the context of the plasma membrane (PM), the EGF-bound EGFR upright dimer, and the EGF-bound prone tetramer. Panel b depicts a structural model of two canonical back-to-back EGF-bound EGFR dimers (extracellular portion, each with one monomer in ribbons and the other in spacefill) that further multimerize to form a tetramer along a novel head-to-head interface (dashed line). Each monomer is colored by domain: I–cyan, II–orange, III–green, IV–pink, bound EGF–purple, with the chains of one canonical dimer in dark shades and the other in light shades. Dashed ovals indicated the approximate positions of the three docking boxes (2, 3 and 1) that were explored to identify small molecule ligands proposed to bind to and stabilize the novel interface. c A detailed view of Box 2 illustrates the boundaries in which docked molecules must be contained (purple) and in which the center of the docked molecules must be located (green). Identified hits that docked in Box 2 are shown in ball-and-stick representation with the carbons colored in yellow (F5230-0424) or cyan (F2738-2186) or white (F2573-0380) and other atoms in CPK. Boxes 3 and 1 are similarly detailed in panels d and e; hits are shown with the carbons in white or yellow (Box 3: F0922-0900 and F3385-3143 respectively, Box 1: F3109-0096). The views in panels c-e have slightly different rotations to optimally show the orientation of the boxes and ligands. Molecular details and interactions for each of the ligands are shown in Fig. 5
Methods
Virtual docking and in silico compound selection
Coordinates for a theoretical prone head-to-head tetramer were obtained from the laboratory of Dr. Martyn Winn and are included as Additional file 1: Data file S1 [20]. The coordinates indicating the corners of the three docking boxes 1, 2, and 3 are included as Additional file 2: Data file S2; Additional file 3: Data file S3 and Additional file 4: Data file S4, respectively. The tetramer coordinates include EGF bound to EGFR, and EGF was retained in the coordinates for docking preparation and docking. The protein preparation wizard from Maestro (Schrödinger Suite 2010 Protein Preparation Wizard; Epik version 2.1, Schrödinger, LLC, New York, NY, 2010; Impact version 5.6, Schrödinger, LLC, New York, NY, 2010; Prime version 2.2, Schrödinger, LLC, New York, NY, 2010) was used to prepare the receptor model for docking and to define docking boxes.
A flow chart depicting the docking process is included as Additional file 5: Figure S1. Compounds from the in-stock library from Life Chemicals, Inc., comprising ~350,000 drug-like small molecules, were prepared for in silico docking using LigPrep version 2.5 and QikProp, version 3.3 (Schrödinger, LLC, New York, NY, 2010). An estimate of solubility (LogS) was generated for each compound using QikProp and compounds were filtered using a cutoff of −6.0 for LogS. All tautomers and stereoisomers as well as possible ionization states at pH 7.0 were generated for each compound in the filtered set. These compounds were first docked using the Glide version 5.6 software package (Schrödinger, LLC, New York, NY, 2010 [28–30]) in Standard Precision mode, which emphasizes goodness-of-fit of the drug-like small molecules to the defined protein surface cavities. The best scoring 10 % of compounds were then re-prepared using LigPrep, and again docked using Extra Precision mode, which emphasizes the energetics of the interaction between the small molecule and the protein (e.g., charge, hydrophobicity). The best scoring 10 % of compounds from the Extra Precision docking were combined for the three docking boxes (~5000 compounds per box) and divided into two sets based on whether or not the compound as docked was within 4Å of either EGF molecule in the dimer interface of the prone tetramer receptor model. Each set was filtered to remove duplicates, as well as different tautomers or stereoisomers of the same compound. The remaining compounds in each set were then filtered to improve chemical diversity. In the Life Chemicals libraries, the first four digits of the manufacturer ID reflects chemical similarity. Families of compounds with the same first four digit ID were further reduced to include only the highest scoring member. This brought both the “contacts EGF” and “does not contact EGF” sets to below 1000 molecules each. All compounds were then viewed as docked on the receptor and manual selection was based on docking position to maximize diversity of docking locations on the EGFR dimer. One hundred nine compounds were selected for functional evaluation, with the selection biased to approximately 75 % of the chosen compounds as not predicted to interact with EGF.
Grb2 internalization assay and compound selection metrics
Time points for assessment of Grb2 were selected based on studies of the recycling of the EGFR signaling complex [31–33], as well as empirical optimization of signaling in initial time course experiments. Drug concentrations for test compounds and cetuximab and erlotinib reference compounds were selected based on values in the literature used for compound screening and/or analysis of EGFR-related signaling [34, 35]. 6 × 103 SCC61 cells were plated in each well of a poly-lysine (cat # P482, Sigma, St. Louis, MO) coated 96-well plate 24 h prior to screening. Four hours prior to compound addition, cells were starved in DMEM/F12 and 0.2 % FBS. After starvation, compounds at 5 mM dissolved in DMSO or DMSO vehicle were added to cells by pin tool transfer on a CyBi-Well Vario equipped with a 100 nl pin tool (V and P Scientific, San Diego, CA) for a final concentration of 10 μM compound. After 5 min, EGF (10 ng/ml in serum free media) or vehicle was added. After 50 min, cells were washed once with PBS, and fixed for ten minutes at room temperature in 4 % PFA. Cells were washed 3 times with PBS, and permeabilized in 0.2 % Triton. Cells were incubated with primary Grb2 antibody (#3972, Cell Signaling, Danvers, MA used at 1:500 in 0.2 % BSA) for 2 h at room temperature. Cells were washed 3 times and subsequently incubated with secondary antibody (#A11008 anti-rabbit Alexa 488, Life Technologies, Carlsbad, CA) diluted in 0.2 % BSA for 2 h. Cells were washed as before and incubated with DAPI prior to imaging. Controls used to evaluate screen performance included wells treated with or without EGF, with added vehicle control, cetuximab (2 μM) or erlotinib (20 μM).
An average of twelve × 20 images per well were acquired utilizing the Image Xpress micro high content imaging system (Molecular Devices, Downingtown, PA) driven by Meta Xpress software. Images were analyzed with the Transfluor Meta Xpress image analysis module and results were displayed and exported utilizing the Acuity Xpress software package (Molecular Devices). Plates were ranked for inclusion based on the ratio of raw pit count per well for EGF versus untreated controls. Plates with ratios below 1.5 were excluded from analysis. Two normalization methods were used; the first method normalized each parameter to the mean-of-the plate same parameter; the second method normalized to mean of vehicle (per run) same parameter. The normalized data was used to calculate the ratio of +EGF to -EGF conditions. In the primary screen, hit selection was based on the parameter sum of pit count per well. Hits were ranked by FDR, and by the EGF-to-vehicle ratio for the parameter total pit count.
Immunoblot assays and analysis
SCC61 cells were plated at the density of 6000 cells/0.32 cm2 in 6-well plates. The next day cells were pre-treated with Vehicle (DMSO), controls (erlotinib at 20 μM, cetuximab at 4 μM, or drugs at 10 μM at 37 °C for 5 min, and then 10 ng/ml EGF (final concentration) was added to each well. After 15 min or 40 min at 37 °C as indicated, cell culture media was removed and cells were immediately washed once with cold PBS. These time points were taken to parallel the process of EGFR/Grb2 recycling, and correspond to time periods known to reflect maximal changes in activity following EGF stimulation [31–33]. Cells were then lysed in Mammalian Protein Extraction Reagent (MPER™, Thermo Scientific, Waltham, MA) for about 20 min on ice. Lysis buffer was supplemented with Halt™ Phosphatase and Protease Inhibitor Cocktail (Thermo Scientific). After collection, lysates were centrifuged at 17,000 × g at 4 °C for 15 min, and protein concentrations were determined using the BCA assay (Thermo Scientific, Waltham, MA). Samples were boiled in Lane Marker Reducing Sample Buffer (Thermo Scientific, Waltham, MA) for 5 min. Proteins were resolved on 8 % SDS-PAGE gels and transferred to PVDF membrane (EMD Millipore, Billerica, MA). Membranes were blocked in Odyssey Blocking Buffer, incubated overnight at 4 °C in primary antibody, followed by IRDye®-conjugated secondary antibody and signal detected with Odyssey Infrared Imaging System (all from LI-COR Biosciences, Lincoln, NE). Vinculin was used as loading control. Antibodies are used as follows: pMAPKT202/Y204 (#9101), MAPK (#4696), and EGFR (#4267) from Cell Signaling, Danvers, MA); pEGFRY1173 (#44794G, from Invitrogen, Carlsbad, CA); and vinculin (#A1978, from Sigma-Aldrich, St. Louis, MO). Immunoblots were quantified using Image Studio Software (http://www.licor.com/bio/products/software/). The ratio of phosphorylated to total protein was calculated and normalized to the value of the positive control for each experiment (vehicle only, in the presence of EGF).
Viability determination and drug synergy
Viability in response to compound alone or compounds in combination with erlotinib or cetuximab was determined with the CellTiterBlue assay (Promega, Madison, WI). Cells were plated (SCC61, 3000 cells/well; A431, 4000 cells/well) 24 h prior to addition of compounds, transferred into 96 well plates as described above. For these experiments, lower doses of cetuximab and erlotinib were used than for screening, to maximize the chance of identifying synergy with newly identified compounds. Cetuximab was added to 4 μM and erlotinib to 2 μM. Cells were incubated for 72 h after which 5 μl of CTB reagent was added. Fluorescence intensity was measured on an Envision Multilabel plate reader (Perkin Elmer, Waltham, MA) at 2 h after CTB addition.
Results
In silico selection of compounds targeting a prone EGFR tetramer
The structure of a membrane-aligned EGFR tetramer with bound EGF ligand (Fig. 1b) was generated using molecular dynamics simulation to yield the head-to-head prone tetramer (coordinates provided by Martyn Winn; see Additional file 1: Data S1) [20]. In contrast to the back-to-back, membrane-protruding EGFR dimer, the head-to-head configuration is uniquely predicted to contain a tetramer-specific interface between the 2 dimers that could potentially bind small molecules. Ligand binding along this interface is predicted to stabilize the tetramer, increase its mole fraction, and promote its function. To support virtual screening for small molecule agents that would specifically bind the tetramer, the interface was divided into three overlapping docking boxes (coordinates provided in Additional files 2, 3 and 4: Data S2–S4, illustrated in Fig. 1c, d and e, and quantification of the docking process is provided as a flow chart Additional file 5: Figure S1; computational limitations prevented our simultaneous docking along the entire interface. One box included the EGF ligands and two boxes flanked the EGF binding site, but did not include EGF.
The Life Chemicals, Inc. library of 345,232 small molecules was prescreened for drug-like properties; the resultant set of compounds was then re-expanded to consider all potential tautomeric isoforms, giving 496,398 candidate ligands. Using Glide software (see Methods), the candidate ligands were docked to each of the three boxes (~1.5 million docking processes). The top scoring 15,000 compounds were manually filtered to select compounds with chemically diverse scaffolds interacting with the greatest variety of discrete elements within the docking boxes. Based on this analysis, 109 candidate ligands were selected for purchase and in vitro analysis. All purchased compounds docked within 4Å of both EGFR molecules; 27 also docked within 4Å of at least one EGF molecule (Additional file 6: Table S1). None of the compounds are chemically similar to known small molecule inhibitors of the EGFR kinase domain, such as erlotinib.
Candidate compounds alter internalization of the EGFR effector Grb2
Following activation by EGF-induced dimerization and autophosphorylation, EGFR recruits a group of binding partners including the scaffolding protein Grb2 to transmit proliferative and other signals. EGFR activity is subsequently limited by the internalization of EGFR, Grb2, and associated signaling proteins through coated pits to early endosomes, followed by recycling to the membrane or trafficking to lysosomes for destruction (Fig. 2a). This process of internalization followed by recycling or destruction takes between 30 and 90 min, and is influenced by a number of feedback loops [31–33]. Clinically relevant inhibitors of EGFR influence its internalization, inhibit its autophosphorylation, or both [36–38]. For antibodies targeting the extracellular domain of EGFR and altering its oligomerization state, changes in internalization properties are readily apparent in cell culture. We posited that targeted small-molecule stabilization of the EGFR tetramer would also affect internalization and recycling.
Fig. 2.
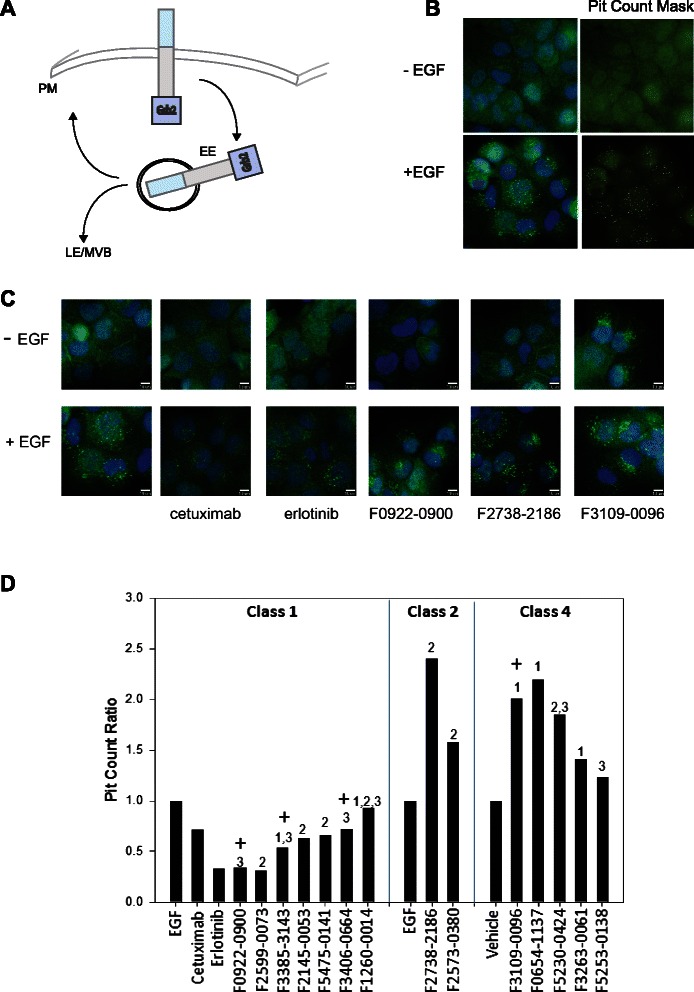
Evaluation of compounds for their effect on internalization of Grb2. a Schematic of EGFR internalization cycle through Grb2-positive early endosomes (EE) and late endosomes (LE) [23]. b SCC61 cells treated with vehicle or EGF, visualized with antibody to Grb2. Right panels represent image overlayed with object mask generated by the Transfluor module. c Representative fields for untreated versus EGF-treated cells pre-incubated with controls (vehicle, erlotinib, cetuximab) or indicated compounds. d Quantitation of changes in pit count ratio; numbers over the bars indicate the docking boxes for each compound; + indicates that docking pose predicts EGF interaction. Class 1 and 2 compounds are reported comparing EGF-to-vehicle ratio in the absence of compounds versus EGF-to-vehicle ratio in the presence of compound, representative screening data. Compound classes are indicated. Representative screening data for Class 4 compounds are reported comparing pit count ratio for compound alone normalized to vehicle indicating activity in the absence of EGF
We developed a high-content fluorescence microscopy-based screen to assess candidate compound activity in influencing internalization of the EGFR effector Grb2 in SCC61 cells, a head and neck squamous cell carcinoma cell line that expresses abundant EGFR [39]. We chose to use Grb2 as a surrogate marker because it was unclear how EGFR activity state, reflected by auto-phosphorylation, would be affected by addition of compounds, while EGFR dimerization was typically associated with internalization of its signaling partners. Grb2 localization was determined utilizing an imaging approach with acquisition of 12 images/well and scoring for Grb2 positive pit formation. Comparison of untreated and EGF-treated cells illustrates a robust dynamic range (Fig. 2b). For screening, SCC61 cells were pre-treated with compounds for 5 min, and then treated with EGF (10 ng/ml) or vehicle for 50 min. Compounds that alter Grb2 positive pit formation could fall into one of 4 possible classes. Class 1-reduces pits in the presence of EGF; Class 2-increases pits in the presence of EGF; Class 3-reduces pits in the absence of EGF and Class 4-increases pits in the absence of EGF. Pit count phenotypes were grouped based on the normalized ratio of pit count with and without EGF and identified as belonging to one of the four Classes. Fourteen preliminary hit compounds were selected for further analysis, based on phenotypic robustness as well as the combination of phenotype and whether or not the docked location predicted interaction with EGF.
As anticipated, Grb2-positive pits induced by EGF treatment were reduced by the control EGFR inhibitors cetuximab and erlotinib (Fig. 2c). Among compounds screened, F0922-0900 represents a Class 1 hit compound, F2738-2186 represents a Class 2 hit compound, and F3109-0096 represents a Class 4 hit compound. No hit compounds were identified in Class 3. Typical screen results for the 14 preliminary hit compounds are shown in Fig. 2d, with results normalized to EGF-treated cells, or with results normalized to untreated baseline conditions to emphasize Class 4. The most effective Class I and Class 4 hit compounds had contact sites that were dispersed across the entire dimer-dimer interface, while the two robust Class 2 hit compounds were derived from a flanking box that largely avoids EGF.
Compound effect on cell viability
The 14 preliminary hit compounds were independently assessed for their influence on cell viability, with effects benchmarked to the EGFR-inhibiting agents erlotinib and cetuximab. We also assessed whether these compounds could potentiate the effects of erlotinib or cetuximab on cell viability. For these analyses, we employed both the EGFR-expressing SCC61 cells, which were used for initial candidate compound screening (Fig. 3a), and a second highly EGFR-dependent cell line, A431 human squamous carcinoma cells (Fig. 3b).
Fig. 3.
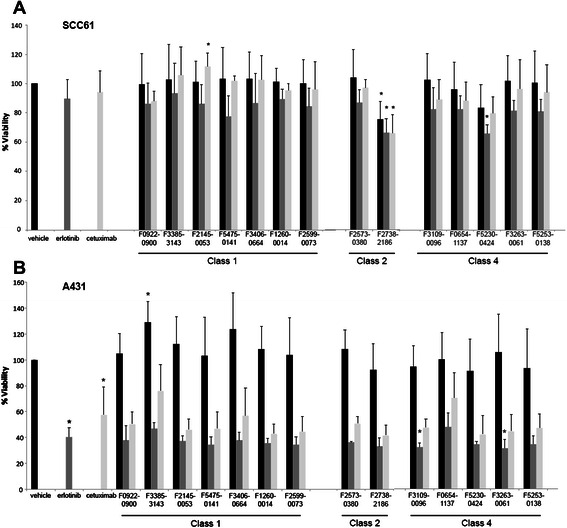
Viability of SCC61 and A431 cells treated with test compounds. a SCC61 or b A431 cells were treated for 72 h with vehicle (black bars) or indicated agents. Erlotinib (grey bars) was used at 2 μM, and cetuximab (white bars) at 4 μM, while test compounds were used at 10 μM. *, P <0.05 in reference to vehicle treated cells
In SCC61 cells, neither 2 μM erlotinib nor 4 μM cetuximab significantly affected cell viability after 72 h of incubation. Under these conditions, 10 μM of the Class 2 compound F2738-2186 reduced cell viability by 25 % relevant to vehicle treated cells (p = 0.0064) (Fig. 3a). Combination of F2738-2186 with erlotinib or cetuximab did not further decrease viability in reference to F2738-2186 used alone. Class 4 compound F5230-0424 modestly reduced viability in combination with erlotinib (p value 0.001).
In contrast, the same concentrations of erlotinib (p = 0.0001) and cetuximab (p = 0.0001) both significantly reduced cell viability in A431 cells. Interestingly, no test compound significantly reduced the viability of A431 cells, although one Class I compound, F3385-3143, significantly increased cell viability (p = 0.0171) (Fig. 3b). However, in combination with erlotinib, two Class 4 compounds modestly but significantly enhanced the erlotinib-dependent decrease in cell viability. Further, in combination with cetuximab, a number of compounds showed a trend towards decreased viability, although results did not rise to statistical significance.
EGFR-Y1068, EGFR-Y1173, and ERK1/2-T202/Y204 phosphorylation in response to compound treatment
To assess whether the preliminary hit compounds influenced the activity state of EGFR or its downstream effector ERK1/2, SCC61 cells were treated with vehicle, cetuximab, erlotinib, or 1 of 6 of the preliminary hits for 1 h, and then treated with EGF or vehicle for 45 min. Whole cell lysates were then analyzed for levels of total, Y1068-phosphorylated, and Y1173-phosphorylated EGFR, and total or T202/Y204-phosphorylated ERK1/2 (Fig. 4a, b and c). In vehicle-treated cells, in the absence of EGF, very low basal phosphorylation of EGFR and ERK1/2 was observed, while EGF treatment induced EGFR-Y1068-phosphorylation 5.4-fold, EGFR-Y1173-phosphorylation 8.2-fold, and ERK1/2-T202/Y204 phosphorylation 3.6-fold.
Fig. 4.
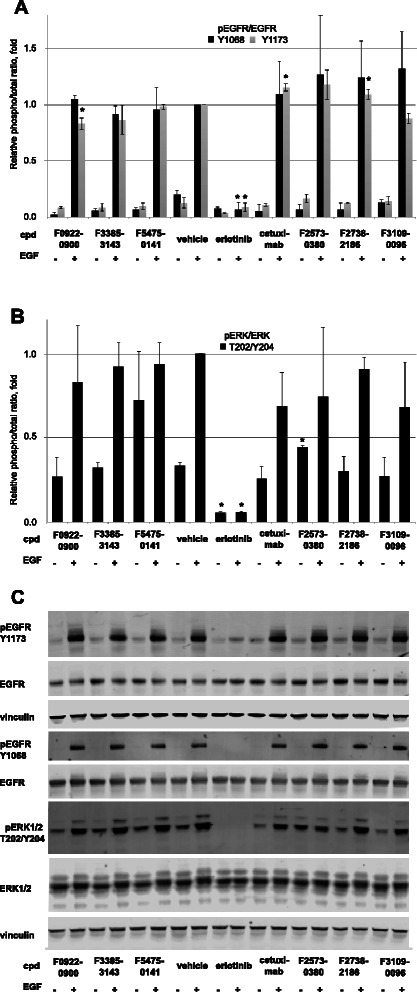
EGFR and ERK1/2 phosphorylation following treatment of SCC61 cells with test compounds. a Averaged quantification of Western blots analyzing Y1068- or Y1173-phosphorylated EGFR following 1 h pre-treatment with compounds, vehicle, cetuximab, or erlotinib, followed by 45 min treatment with vehicle or EGF +/− EGF. Values shown are normalized to total EGFR. b Averaged quantification of Western blots analyzing T202/Y204-phosphorylated ERK1/2 following 1 h pre-treatment with compounds, vehicle, cetuximab, or erlotinib, followed by 45 min treatment with vehicle or EGF +/− EGF. Values shown are normalized to total ERK1/2. c Representative Western blots for quantified data shown in parts a and b
Each of these EGF-induced phosphorylation events was entirely blocked by treatment with the EGFR kinase inhibitor erlotinib. In contrast, cetuximab treatment slightly elevated EGF-induced Y1173-EGFR phosphorylation. Among the compounds assessed, the two Class 2 compounds, F2738-2186 and F2573-0380, each also slightly elevated EGF-dependent Y1173-EGFR phosphorylation, while the class I compound F0922-0900 slightly reduced this phosphorylation. Neither cetuximab nor any of the six preliminary hit compounds tested significantly affected Y1068-EGFR phosphorylation. Finally, F2573-0380 slightly increased basal levels of T202/Y204-ERK1/2, in the absence of EGF treatment.
Analysis of hit compounds
The data obtained from screening and biological assays are summarized in Additional file 7: Table S2. Among the 14 compound hit set, 4 of the 14 were derived from the subset (28 %) of the total library that is predicted to interact with EGF. Of the six compounds evaluated for their effects on phosphorylation (Fig. 5), 2 of the 6 (33 %) are predicted to interact with EGF. The predicted binding pockets have some distinct commonalities. With regard to molecular interactions, Fig. 5 shows that 4 of the 6 hits dock to Arg-470, and the 2 that are not predicted to interact with this residue, are predicted to interact with neighboring residues (e.g., Ser-468, Asn-469, Glu-472, Asn-473). For the compounds that are predicted to interact with EGF, the putative EGF interactions all involve EGF residues in the region of Lys48-Leu52.
Fig. 5.
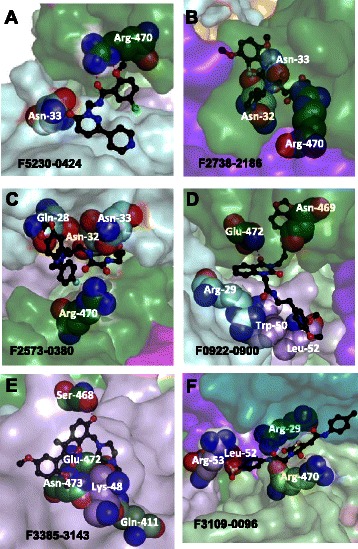
Identified hits in their docked poses highlighting key intermolecular interactions. Panels a-c show ligands that dock in Box 2, panels d and e show ligands that dock in Box 3, panel f shows ligands that dock in Box 1. Each ligand is shown in ball-and-stick representation with the carbons in black and other atoms in CPK. The surrounding binding pockets formed by EGFR and EGF are shown as transparent surfaces and are colored by domain as in Fig. 1. In each panel, residues from EGFR or EGF that form hydrogen bonds or ring-stacking hydrophobic interactions with the ligand are shown as spheres with the carbons colored by domain and other atoms in CPK. Hydrogen bonds are shown as dashed red lines. The orientation varies among the panels to best show the structure of each ligand
Discussion
Based on the data presented above, 14 of 109 compounds predicted to stabilize a recently defined homo-tetramer of EGFR influenced intracellular trafficking of the EGFR effector Grb2. As with the EGFR-binding antibody cetuximab, these compounds had little effect on EGFR auto-phosphorylation, or activation of the EGFR effector ERK1/2. Encouragingly, several compounds potentiated the activity of clinical EGFR inhibitors. While the identified compounds are implied to bind to tetrameric EGFR, where domain III is a component of the tetramer interface, cetuximab binds to domain III of monomeric EGFR in a conformation that is not competent for multimerization (PDB id 1YY9 [40]). Thus both are targeted to modulate the EGFR quaternary structure equilibrium, but with different stabilization targets, allowing a combination therapy to target multiple EGFR structural isoforms to limit the quaternary structure dynamics essential for EGFR function. These findings in sum suggest that targeting the extracellular EGFR prone tetramer is a strategy worthy of further evaluation in identification of clinical leads.
Homo-multimer stabilization as an approach to allosteric drug discovery is gaining momentum as more drug targets are appreciated to participate in quaternary structure equilibria among functionally distinct homomeric assemblies [41–43]. In prior work, we have used a carefully constructed homology model of a homo-multimeric enzyme as a target for the identification of multimer-specific allosteric inhibitor molecules [44]. In that case the quaternary structure equilibrium includes high activity octamers and low activity hexamers whose interconversion requires dissociation, hinge motion, and re-association events [45]. Using the unique prone tetramer of EGFR as a target, we deduced that stabilization of the head-to-head interface could potentially alter EGFR function by shifting the equilibrium toward this tetramer. The high hit rate for bioactive compounds observed in this study of regulation of Grb2 internalization is similar to our prior studies that used in silico docking followed by in vitro validation to identify multimer-specific allosteric inhibitors [44, 46], and is presumed to be far above that expected for 100 randomly selected drug-like compounds, providing support to the tetramer model.
Our use of Grb2 internalization as a readout for stabilization of the EGFR tetramer was recently validated by an elegant biophysical analysis of EGFR multimerization and its relationship to Grb2 association [23]. That study, which employed fluorescence energy transfer methods, a C-terminal GFP-labeled EGFR, and an RFP-labeled Grb2, concluded that, for these labeled constructs, 2 % of EGFR is monomeric, 5 % is dimeric, <1 % is trimeric, and 94 % is tetrameric. Cluster distribution at 10 nM EGF shows nearly 95 % of the EGFR tetramers containing at least one bound Grb2, while monomers, dimers, and trimers are predominantly (~98 %) free of Grb2. However, identification of compounds in the Grb2 screening assay is not diagnostic of the mechanism of action of the compound; it is formally possible that the compounds are stabilizing, destabilizing, or not affecting stability, but rather altering EGFR oligomeric structure in a manner that impacts the magnitude or the kinetics of internalization and recycling. These points all bear further investigation. It will be important to perform classic medicinal chemical optimization, and establishment of structure-activity relationships in relation to the docking boxes to validate the mode of action of compounds emerging as hits. Finally, further investigation of the compounds identified in this screen should expand to explore their effect on the mobilization of total and phosphorylated (active) EGFR, and other EGFR-dependent effector pathways.
Conclusion
Our current study demonstrates that focusing on EGFR tetramer-specific surface cavities as targets for virtual screening, combined with a high content cell-based screening approach, can identify novel compounds that modulate EGFR function, presumably via a novel mechanism. While the current studies do not establish whether the active compounds bind to the EGFR tetramer as predicted by docking, nevertheless, the cited demonstration that Grb2 specifically interacts with a tetrameric form of EGFR provides an exciting validation to hypotheses that drove our experimental design.
Acknowledgments
The authors were supported by pilot funding from the Fox Chase Cancer Center Head and Neck Keystone Program; NIH R01 ES003654-27 (to EKJ); R21 CA181287 and R01 CA063366 (to EAG); by a subsidy of the Russian Government to support the Program of competitive growth of Kazan Federal University (to IS); and by the NIH Cancer Center Core Grant P30 CA006927 (to Fox Chase Cancer Center). We acknowledge Cynthia Myers of the FCCC Organic Synthesis Facility for confirming the molecular structures of the hit compounds. We graciously thank Dr. Martyn D. Winn of the Scientific Computing Department, STFC Daresbury Laboratory, Warrington, UK, for providing coordinates for a head-to-head EGFR tetramer model and allowing our inclusion of the coordinates as Additional file 1: Data S1.
Abbreviations
- EGFR
Epidermal growth factor receptor
- EGF
Epidermal growth factor
- F0654-1137
3-({[2-(4-fluorophenyl)-6,8-dimethyl-5,7-dioxo-5H,6H,7H,8H-[1,3]diazino[4,5-days]pyrimidin-4-yl] sylfanyl}methyl)pyridine-1-ium
- F0922-0900
N-(2H-1,3-benzodioxol-5-ylmethyl)-3-[1-({[2-(3,4-dimethoxyphenyl)ethyl]carbamoyl}methyl)-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]propanamide
- F1260-0014
4-[(2S,3Z)-3-[hydroxy9thiophen-2-yl)methylidene]-1-[3-(1H-imidazol-3-ium-1-yl)propyl]-4,5-dioxopyrrolidin-2-yl]pyridine-1-ium
- F2145-0053
4-(2-azaniumylethanesulfonyl)-1-(4-methoxyphenyl)piperazin-1-ium
- F2573-0380
1-[(1R)-1-(2H-1,3-benzodioxol-5-yl)-2-{[(3-hydroxypropyl)carbamoyl]formamideo}ethyl]-4-(4-fluorophenyl)piperazin-1-ium
- F2599-0073
(9R)-1,2,9-trimethyl-8-({[(4-nitrophenyl)carbamoyl]methyl}sulfanyl)-2,6-dioxo-2,3,6,9-tetrahydro-1H-purin-9-ium
- F2738-2186
2-{[N-(4,7-dimethoxy-1,3-benzothiazol-2-yl)-2-(2,5-dioxopyrrolidin-1-yl)acetamido]methyl}pyridine-1-ium
- F3109-0096
2-[(5Z)-5-{[(4-methylphenyl)carbamoyl]methylidene}-2,4-dioxo-1,3-thiazolidin-3-yl]-N-(4-sulfamoylphenyl)acetamide
- F3263-0061
(2R,6R)-1-[(2R)-3-(4-acetamidophenoxy0-2-hydroxypropyl]-2,6-dimethylpiperidin-1-ium
- F3385-3143
4-{[3-(2,5-dimethoxyphenyl)-7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl]methyl}-1-(2-hydroxyethyl)piperazin-1ium
- F3406-0664
1-(3-{[(2-fluorophenyl)methyl]carbamoyl}phenyl)-2-({[(2-methylphenyl)carbamoyl]methyl}sulfanyl)-1H-imidazol-3-ium
- F5230-0424
4-(1-{2-[(5-chloro-2-methoxyphenyl)formamido]ethyl}-6-oxo-1,6-dihydropyridazin-3-yl)pyridine-1-ium
- F5253-0138
4-[3-[(2-oxo-1,2-dihydropyridin-3-yl)formamido]propanesulfonyl}-1-phenylpiperazin-1-ium
- F5475-0141
1-(2H-1,3-benzodioxol-5-ylmethyl)-4-[1-(4-fluorophenyl)cyclopropanecarbonyl]piperazin-1-ium
Additional files
Coordinates for the prone EGFR tetramer.
Coordinates for docking box 1.
Coordinates for docking box 2.
Coordinates for docking box 3.
Flow Chart describing computational docking of the Life Chemicals, Inc. small molecule library to the head-to-head prone model of tetrameric EGFR.
Life Chemicals, Inc. compounds selected for in vitro characterization. Presented are the chemical structures, manufacturer’s ID, molecular weight, and the Glide XP score.
Summary table of compounds selected from primary screen and activity in secondary assays. Life Chemicals, Inc. compounds selected for in vitro characterization. Presented are the, manufacturer’s ID, chemical structures, hit class determined in the primary screen (see Fig. 2), activity in EGFR pY1173 western analysis (see Fig. 4), activity in phospho-ERK western analysis (see Fig. 4), activity in viability assays of SCC61 cells, and activity in viability assays of A431 cells. S.S. denotes statistically significant, N.S. denotes not significant, and - - denotes not determined.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UDR performed the computational docking studies through selection of the compounds for in vitro characterization. ASN and HL performed Western analysis of signaling consequences of compound administration. AP performed the screen and image acquisition and image analysis. SHL designed and executed the preparation of the molecular illustrations. IGS provided extensive input on data analysis for the in vitro studies. YZ supervised the image-based data and statistical analysis. MKR assisted in designing the cell-based Grb2 internalization screen. MBE designed the cell-based Grb2 internalization screen and supervised its execution. EAG and EKJ collaboratively developed the overall rationale for the described studies and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ursula D. Ramirez, Email: Ursula.Ramirez@fccc.edu
Anna S. Nikonova, Email: Anna.Nikonova@fccc.edu
Hanqing Liu, Email: hanqing@ujs.edu.cn.
Anna Pecherskaya, Email: anna_pech@yahoo.com.
Sarah H. Lawrence, Email: Sarah.Lawrence@fccc.edu
Ilya G. Serebriiskii, Email: Ilya.Serebriiskii@fccc.edu
Yan Zhou, Email: Yan.Zhou@fccc.edu.
Matthew K. Robinson, Email: Matthew.Robinson@fccc.edu
Margret B. Einarson, Email: Margret.Einarson@fccc.edu
Erica A. Golemis, Email: Erica.Golemis@fccc.edu
Eileen K. Jaffe, Email: Eileen.Jaffe@fccc.edu
References
- 1.Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell. 2000;102(2):211–20. doi: 10.1016/S0092-8674(00)00026-X. [DOI] [PubMed] [Google Scholar]
- 2.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–30. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89(7):2965–9. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopper-Borge EA, Nasto RE, Ratushny V, Weiner LM, Golemis EA, Astsaturov I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin Ther Targets. 2009;13(3):339–62. doi: 10.1517/14712590902735795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gan HK, Burgess AW, Clayton AH, Scott AM. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012;72(12):2924–30. doi: 10.1158/0008-5472.CAN-11-3898. [DOI] [PubMed] [Google Scholar]
- 6.Janjigian YY, Azzoli CG, Krug LM, Pereira LK, Rizvi NA, Pietanza MC, et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin Cancer Res. 2011;17(8):2521–7. doi: 10.1158/1078-0432.CCR-10-2662. [DOI] [PubMed] [Google Scholar]
- 7.Weickhardt AJ, Price TJ, Chong G, Gebski V, Pavlakis N, Johns TG, et al. Dual targeting of the epidermal growth factor receptor using the combination of cetuximab and erlotinib: preclinical evaluation and results of the phase II DUX study in chemotherapy-refractory, advanced colorectal cancer. J Clin Oncol. 2012;30(13):1505–12. doi: 10.1200/JCO.2011.38.6599. [DOI] [PubMed] [Google Scholar]
- 8.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor perspectives in biology. 2014;6(4):a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Needham SR, Zanetti-Domingues LC, Hirsch M, Rolfe DJ, Tynan CJ, Roberts SK, et al. Structure-function relationships and supramolecular organization of the EGFR (epidermal growth factor receptor) on the cell surface. Biochem Soc Trans. 2014;42(1):114–9. doi: 10.1042/BST20130236. [DOI] [PubMed] [Google Scholar]
- 10.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541–52. doi: 10.1016/S1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11(2):507–17. doi: 10.1016/S1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 12.Alvarado D, Klein DE, Lemmon MA. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell. 2010;142(4):568–79. doi: 10.1016/j.cell.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Villeneuve G, Wang Z. Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005;6(10):942–8. doi: 10.1038/sj.embor.7400491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawano A, Takayama S, Matsuda M, Miyawaki A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev Cell. 2002;3(2):245–57. doi: 10.1016/S1534-5807(02)00224-1. [DOI] [PubMed] [Google Scholar]
- 15.Ariotti N, Liang H, Xu Y, Zhang Y, Yonekubo Y, Inder K, et al. Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Mol Cell Biol. 2010;30(15):3795–804. doi: 10.1128/MCB.01615-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clayton AH, Tavarnesi ML, Johns TG. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry. 2007;46(15):4589–97. doi: 10.1021/bi700002b. [DOI] [PubMed] [Google Scholar]
- 17.Clayton AH, Walker F, Orchard SG, Henderson C, Fuchs D, Rothacker J, et al. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J Biol Chem. 2005;280(34):30392–9. doi: 10.1074/jbc.M504770200. [DOI] [PubMed] [Google Scholar]
- 18.Kozer N, Barua D, Orchard S, Nice EC, Burgess AW, Hlavacek WS, et al. Exploring higher-order EGFR oligomerisation and phosphorylation–a combined experimental and theoretical approach. Mol Biosyst. 2013;9(7):1849–63. doi: 10.1039/c3mb70073a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehra R, Serebriiskii IG, Dunbrack RL, Jr, Robinson MK, Burtness B, Golemis EA. Protein-intrinsic and signaling network-based sources of resistance to EGFR- and ErbB family-targeted therapies in head and neck cancer. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2011;14(6):260–79. doi: 10.1016/j.drup.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kastner J, Loeffler HH, Roberts SK, Martin-Fernandez ML, Winn MD. Ectodomain orientation, conformational plasticity and oligomerization of ErbB1 receptors investigated by molecular dynamics. J Struct Biol. 2009;167(2):117–28. doi: 10.1016/j.jsb.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Tynan CJ, Roberts SK, Rolfe DJ, Clarke DT, Loeffler HH, Kastner J, et al. Human epidermal growth factor receptor (EGFR) aligned on the plasma membrane adopts key features of Drosophila EGFR asymmetry. Mol Cell Biol. 2011;31(11):2241–52. doi: 10.1128/MCB.01431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murali R, Brennan PJ, Kieber-Emmons T, Greene MI. Structural analysis of p185c-neu and epidermal growth factor receptor tyrosine kinases: oligomerization of kinase domains. Proc Natl Acad Sci U S A. 1996;93(13):6252–7. doi: 10.1073/pnas.93.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozer N, Barua D, Henderson C, Nice EC, Burgess AW, Hlavacek WS, et al. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry. 2014;53(16):2594–604. doi: 10.1021/bi500182x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen CH, Chernis GA, Hoang VQ, Landgraf R. Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc Natl Acad Sci U S A. 2003;100(16):9226–31. doi: 10.1073/pnas.1332660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Park E, Kani K, Landgraf R. Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc Natl Acad Sci U S A. 2012;109(33):13237–42. doi: 10.1073/pnas.1200105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landgraf R, Eisenberg D. Heregulin reverses the oligomerization of HER3. Biochemistry. 2000;39(29):8503–11. doi: 10.1021/bi000953+. [DOI] [PubMed] [Google Scholar]
- 27.Park E, Baron R, Landgraf R. Higher-order association states of cellular ERBB3 probed with photo-cross-linkable aptamers. Biochemistry. 2008;47(46):11992–2005. doi: 10.1021/bi8004208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 30.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 31.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12(2):104–17. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 32.Sukhanova A, Gorin A, Serebriiskii IG, Gabitova L, Zheng H, Restifo D, et al. Targeting C4-demethylating genes in the cholesterol pathway sensitizes cancer cells to EGF receptor inhibitors via increased EGF receptor degradation. Cancer discovery. 2013;3(1):96–111. doi: 10.1158/2159-8290.CD-12-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499(7457):166–71. doi: 10.1038/nature12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aguilar H, Urruticoechea A, Halonen P, Kiyotani K, Mushiroda T, Barril X, et al. VAV3 mediates resistance to breast cancer endocrine therapy. Breast cancer research: BCR. 2014;16(3):R53. doi: 10.1186/bcr3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dayekh K, Johnson-Obaseki S, Corsten M, Villeneuve PJ, Sekhon HS, Weberpals JI, et al. Monensin inhibits epidermal growth factor receptor trafficking and activation: synergistic cytotoxicity in combination with EGFR inhibitors. Mol Cancer Ther. 2014;13(11):2559–71. doi: 10.1158/1535-7163.MCT-13-1086. [DOI] [PubMed] [Google Scholar]
- 36.Prewett M, Rockwell P, Rockwell RF, Giorgio NA, Mendelsohn J, Scher HI, et al. The biologic effects of C225, a chimeric monoclonal antibody to the EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor Immunol. 1996;19(6):419–27. doi: 10.1097/00002371-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Prewett M, Rockwell P, Rose C, Goldstein N. Anti-tumor and cell cycle responses in KB cells treated with a chimeric anti-EGFR monoclonal antibody in combination with cisplatin. Int J Oncol. 1996;9(2):217–24. doi: 10.3892/ijo.9.2.217. [DOI] [PubMed] [Google Scholar]
- 38.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102(21):7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3(140):ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Jaffe EK. Impact of quaternary structure dynamics on allosteric drug discovery. Curr Top Med Chem. 2013;13(1):55–63. doi: 10.2174/1568026611313010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabizon R, Friedler A. Allosteric modulation of protein oligomerization: an emerging approach to drug design. Frontiers in chemistry. 2014;2:9. doi: 10.3389/fchem.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, et al. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell. 2013;154(4):763–74. doi: 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence SH, Ramirez UD, Tang L, Fazliyez F, Kundrat L, Markham GD, et al. Shape shifting leads to small-molecule allosteric drug discovery. Chem Biol. 2008;15(6):586–96. doi: 10.1016/j.chembiol.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaffe EK, Lawrence SH. Allostery and the dynamic oligomerization of porphobilinogen synthase. Arch Biochemi Biophys. 2012;519(2):144–53. doi: 10.1016/j.abb.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawrence SH, Ramirez UD, Selwood T, Stith L, Jaffe EK. Allosteric inhibition of human porphobilinogen synthase. J Biol Chem. 2009;284(51):35807–17. doi: 10.1074/jbc.M109.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]