

Abstract
Genetic and functional studies have revealed that both common and rare variants of several nicotinic acetylcholine receptor (nAChR) subunits are associated with nicotine dependence (ND). In this study, we identified variants in 30 candidate genes including nicotinic receptors in 200 sib pairs selected from the Mid-South Tobacco Family (MSTF) population with equal numbers of African Americans (AAs) and European Americans (EAs). We selected 135 of the rare and common variants and genotyped them in the Mid-South Tobacco Case-Control (MSTCC) population, which consists of 3088 AAs and 1430 EAs. None of the genotyped common variants showed significant association with smoking status (smokers vs. non-smokers), Fagerström Test for Nicotine Dependence (FTND) scores, or indexed cigarettes per day (CPD) after Bonferroni correction. Rare variants in NRXN1, CHRNA9, CHRNA2, NTRK2, GABBR2, GRIN3A, DNM1, NRXN2, NRXN3, and ARRB2 were significantly associated with smoking status in the MSTCC AA sample, with Weighted Sum Statistic (WSS) P values ranging from 2.42 × 10−3 to 1.31 × 10−4 after 106 phenotype rearrangements. We also observed a significant excess of rare nonsynonymous variants exclusive to EA smokers in NRXN1, CHRNA9, TAS2R38, GRIN3A, DBH, ANKK1/DRD2, NRXN3, and CDH13 with WSS P values between 3.5 × 10−5 and 1 × 10−6. Variants rs142807401 (A432T) and rs139982841 (A452V) in CHRNA9 and variants V132L, V389L, rs34755188 (R480H), and rs75981117 (N549S) in GRIN3A are of particular interest because they are found in both the AA and EA samples. A significant aggregate contribution of rare and common coding variants in CHRNA9 to the risk for ND (SKAT-C: P= 0.0012) was detected by applying the combined sum test in MSTCC EAs. Together, our results indicate that rare variants alone or combined with common variants in a subset of 30 biological candidate genes contribute substantially to the risk of ND.
Keywords: CHRNA9, common variants, GRIN3A, nicotinic acetylcholine receptor, nicotine dependence, rare genetic variants
INTRODUCTION
In recent years, candidate gene and genome-wide association studies (GWAS) have identified several common genetic variants associated with the risk of nicotine dependence (ND). These genes include the nicotinic acetylcholine receptor (nAChR) subunit genes CHRNA5, CHRNA3, and CHRNB4 (clustered on human chromosome 15q) and the CHRNA6 and CHRNB3 genes (clustered on chromosome 8p).1–3 Examples of findings involving genes other than nicotinic receptors are the nicotine metabolism gene CYP2A6,2 the dopamine receptor gene DRD2 and its closely linked gene ANKK1,4, 5 the dopamine hydroxylase gene DBH,6 the brain-derived neurotrophic factor gene BDNF,6, 7 and the synaptic maintenance gene NRXN1.8, 9 However, the variants of these susceptibility genes can explain only a small to modest part of the estimated heritability for ND; e.g., alleles of the CHRNA5-CHRNA3-CHRNB4 nAChR gene cluster explain < 1% of the variance in the amount smoked.10 On the other hand, there is increasing evidence that both common and rare or low-frequency genetic variants are playing a significant role in the involvement of each susceptibility gene for ND and other complex human diseases.11–13
Several studies have revealed that rare variants of nAChR subunits are associated with ND both genetically and functionally. Wessel et al.14 investigated the contribution of common and rare variants in 11 nAChR genes to Fagerström Test for Nicotine Dependence (FTND) scores in 448 European-American (EA) smokers who participated in a smoking cessation trial. Significant association was found for common and rare variants of CHRNA5 and CHRNB2, as well as for rare variants of CHRNA4. Xie et al.15 followed up on the CHRNA4 finding by sequencing exon 5, where most of the rare nonsynonymous variants were detected, in 1,000 ND cases and 1,000 non-ND control subjects with equal numbers of EAs and African Americans (AAs), and reported that functional rare variants within CHRNA4 might reduce ND risk. Recently, Haller et al.16 detected protective effects of rare missense variants at conserved residues in CHRNB4 and examined functional effects of the three major association signal contributors (T375I and T91I in CHRNB4 and R37H in CHRNA3) in vitro, the minor alleles of which increased cellular response to nicotine. However, like the other two studies, Haller et al.16 limited their sequencing targets to nAChR subunits.
To address whether genes other than nAChR subunit genes having common variants associated with ND also contain rare ND susceptibility variants, this study was conducted with the goal of determining both the individual and the cumulative effects of rare and common variants in genes/regions implicated in ND candidate gene studies and/or GWAS through pooled sequencing of a subset of our Mid-South Tobacco Family (MSTF) samples followed by conducting validation in an independent case-control sample. Additionally, we implemented a three-step strategy to identify association signals of rare and common variants within the same genomic region. First, we evaluated each common variant individually with a univariate statistic; i.e., logistic and linear regression models. Second, rare variants were grouped by genomic regions and analysed using burden tests, i.e., the Weighted Sum Statistic (WSS);17 third, we tested for combined effects of rare and common variants with a unified statistical test that allows both types of variants to contribute fully to the overall test statistic.18
MATERIALS AND METHODS
Subjects
Four hundred subjects (200 sib pairs) were selected for variant discovery from the MSTF population based on ethnic group (AAs or EAs), smoking status (smokers or non-smokers), and FTND scores (light smokers: FTND < 4 or heavy smokers: FTND 4). The reasons for us to choose participants from our family study as discovery samples for deep-sequencing analysis were based on the following two main factors. First, recent studies have shown that rare variants are enriched in family data. If one family member has a rare allele, half of the siblings are expected to carry it, and hence, variants that are rare in the general population could be very commonly present in certain families.19 Second, family-based designs are advantageous for their robustness to population stratification. Participants in this family-based study were recruited between 1999 and 2004 primarily from the Mid-South states within the USA. More detailed descriptions of demographic and clinical data for these participants can be found in Supplementary Table 1 and previous publications from our group.9, 20–22
Subjects used for variant validation and analysis were recruited from the same geographical area during 2005–2011 as part of the Mid-South Tobacco Case-Control (MSTCC) study under the same recruitment criteria used for the MSTF sample except the subjects were required to be biologically unrelated to each other. Written informed consent was obtained from all participants under the aegis of a human research protocol approved by the IRB of the University of Virginia and University of Mississippi Medical Center. Questionnaires assessing various smoking-related behaviours and other characteristics of interest were administered to participants. Individuals exhibiting substance dependence or abuse other than for alcohol were excluded. The MSTCC sample included 3,088 unrelated AAs (1,454 smokers and 1,634 non-smokers) and 1,430 unrelated EAs (758 smokers and 672 non-smokers). All smokers had smoked at least 100 cigarettes in their lifetimes, while non-smokers were required to have smoked 1–99 cigarettes in their lifetimes, but had no tobacco use in the past year. The ND of each smoker was assessed by the FTND, a commonly used measure, as well as indexed cigarettes per day (CPD) based on a 0 to 3 scale (0: 1–10 CPD, 1: 11–20 CPD, 2: 21–30 CPD and 3: > 30 CPD). Detailed characteristics of the MSTCC AA and EA samples are summarized in Table 1.
Table 1.
Demographic and Phenotypic Characteristics of MSTCC AA and EA Samples
| Characteristic | AA (N = 3,088) | EA (N = 1,430) | ||
|---|---|---|---|---|
| Smokers | Non-smokers | Smokers | Non-smokers | |
| Sample size | 1,454 | 1,634 | 758 | 672 |
| Female (%) | 681 (46.8) | 962 (58.9) | 380 (50.1) | 451 (67.1) |
| Age, years (SD) | 43.6 (12.5) | 42.1 (14.2) | 41.6 (12.2) | 45.1 (14.9) |
| Indexed CPD (SD) | 1.9 (0.4) | N/A | 1.9 (0.5) | N/A |
| FTND Score (SD) | 8.6 (1.2) | N/A | 8.0 (1.9) | N/A |
Notes:
1) SD = standard deviation; N/A = not applicable.
2) Indexed CPD and FTND scores are for smokers only.
3) Indexed CPD: 0 (1–10 CPD), 1 (11–20 CPD), 2 (21–30 CPD), 3 (>30 CPD).
4) FTND Score: possible range 0–10.
Sequencing and Genotyping
We used a customized capture panel of 30 targeted genes, which included nAChR subunit genes and several neurotransmitter receptor and metabolism genes. Almost all of these genes have been reported by our or other research groups to be associated with at least one ND measure in either AA or EA samples. Please refer to Table 2 for the detailed gene list and related references. The coding regions, UTR regions, and flanking sequences of these genes were covered by the Agilent Sure Select Capture panel (250 kb). We divided the 400 samples from the MSTF study into eight pools based on ethnic group, smoking status, and FTND scores to conduct high-throughput sequencing (50 samples/pool).23 The concentration of each DNA sample was first measured using the QuantiT™ dsDNA assay kit (Life Technologies, Carlsbad, CA) and then 50 DNA samples were pooled in equimolar amounts, as suggested by the manufacturers. Each pooled DNA sample was subjected to library preparation, targeted capture, and high-throughput sequencing (72 bp paired-end) according to the protocols suggested by the manufacturers. Base quality recalibration and alignment were performed using the Burrows-Wheeler Aligner (BWA)24 referencing hg19. We used Syzygy11 to call variants from the pooled targeted resequencing data.
Table 2.
Biological Information on Rare and Common Variants of 30 Candidate Genes
| Gene | (SNP type)/amino acid change | dbSNP ID | Chr. | Hg19 position | Allele 1/Allele 2 | Allele 1 freq. in AA (%) | Allele 1 freq. in EA (%) | PhyloP score | SIFT prediction | Polyphen prediction | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHRNB2 | E34G | rs200223952 | 1 | 154541974 | G/A | 0 | 0.07 | 4.33 | Tolerated | Benign | 20 |
| Y178* | – | 154543833 | A/C | 0.02 | 0 | 0.08 | Premature stop codon | ||||
| NRXN1 | T274P | rs77665267 | 2 | 50280522 | G/T | 0.03 | 0.07 | 5.01 | Damaging | Benign | 9 |
| R206L | – | 50280725 | A/C | 0.11 | 0.04 | 4.46 | Damaging | Probably damaging | |||
| (Intron) | rs10208208 | 50593914 | T/G | 14.82 | 2.32 | 0.05 | |||||
| (Intron) | rs10490227 | 50659515 | T/C | 23.63 | 13.73 | −0.43 | |||||
| (Intron) | rs6721498 | 50713012 | G/A | 49.37 | 52.02 | −1.17 | |||||
| Y367* | – | 50765614 | T/G | 0.02 | 0 | −0.12 | Premature stop codon | ||||
| S62* | – | 50850606 | T/G | 0.02 | 0 | 6.04 | Premature stop codon | ||||
| (Intron) | rs2193225 | 51079482 | C/T | 21.54 | 50.36 | −1.19 | |||||
| CHRNA1† | E436D | rs61737716 | 2 | 175613317 | A/C | 0 | 0.04 | 0.65 | Tolerated | Probably damaging | |
| DRD3† | G9S | rs6280 | 3 | 113890815 | T/C | 26.30 | 63.00 | 0.11 | Tolerated | Benign | 60 |
| (Intron) | rs7638876 | 113894300 | T/C | 19.20 | 62.20 | −2.06 | |||||
| (Intergenic) | rs9825563 | 113900220 | G/A | 48.98 | 32.71 | −0.30 | |||||
| CHRNA9 | A312T | rs56210055 | 4 | 40356031 | A/G | 7.19 | 0.85 | 6.20 | Damaging | Possibly damaging | |
| A315V | rs55633891 | 40356041 | T/C | 15.07 | 12.55 | 4.48 | Damaging | Benign | |||
| A432T | rs142807401 | 40356391 | A/G | 0.06 | 0.07 | 4.45 | Tolerated | Benign | |||
| A452V | rs139982841 | 40356452 | T/C | 0.14 | 0.04 | 6.02 | Damaging | Probably damaging | |||
| DRD1 | (Intergenic) | rs265975 | 5 | 174862195 | C/T | 35.36 | 60.71 | −0.31 | 61 | ||
| (3′ UTR) | rs686 | 174868700 | A/G | 43.08 | 63.57 | −0.26 | |||||
| R226W | – | 174869427 | A/G | 0.02 | 0 | 2.92 | Damaging | Probably damaging | |||
| (5′ UTR) | rs4532 | 174870150 | C/T | 11.44 | 33.39 | −0.80 | |||||
| DDC | (Intron) | rs1451371 | 7 | 50553051 | C/T | 30.62 | 47.20 | 0.72 | 21 | ||
| (Intron) | rs3735273 | 50596864 | T/C | 36.10 | 20.96 | −0.41 | |||||
| E61D | rs11575292 | 50611601 | A/C | 1.38 | 0.18 | 0.39 | Tolerated | Probably damaging | |||
| (Intron) | rs921451 | 50623285 | C/T | 22.22 | 30.50 | −0.14 | |||||
| TAS2R38 | R274C | rs114288846 | 7 | 141673087 | A/G | 1.93 | 0.11 | 0.48 | Damaging | Probably damaging | 62 |
| V262A | rs1726866 | 141672705 | A/G | 32.88 | 49.82 | 0.88 | Tolerated | Benign | |||
| W135G | rs139843932 | 141672670 | C/A | 0.78 | 0.04 | 2.68 | Damaging | Probably damaging | |||
| CHRNA2 | S488* | – | 8 | 27320497 | T/G | 0.02 | 0.07 | 1.74 | Premature stop codon | ||
| R121L | – | 27324833 | A/C | 0.02 | 0 | 1.50 | Damaging | Possibly damaging | |||
| T22I | rs2472553 | 27328511 | A/G | 16.62 | 13.32 | −0.33 | Tolerated | Benign | |||
| CHRNB3 | (Intergenic) | rs10958725 | 8 | 42524584 | G/T | 30.63 | 74.85 | 0.35 | 63 | ||
| (Intergenic) | rs10958726 | 42535909 | T/G | 39.79 | 75.02 | −0.13 | |||||
| (Intergenic) | rs4736835 | 42547033 | C/T | 34.85 | 74.75 | −1.27 | |||||
| (Intergenic) | rs6474412 | 42550498 | T/C | 34.78 | 74.54 | −1.64 | |||||
| (5′ UTR) | rs4950 | 42552633 | A/G | 27.16 | 73.95 | −0.40 | |||||
| (Intron) | rs13280604 | 42559586 | A/G | 27.40 | 73.94 | 0.43 | |||||
| (Intron) | rs6474415 | 42562938 | A/G | 23.03 | 73.66 | −0.69 | |||||
| H410Y | – | 42587678 | T/C | 0.05 | 0 | 2.31 | Damaging | Possibly damaging | |||
| K451E | rs35327613 | 42591735 | G/A | 4.91 | 0.25 | 1.60 | Tolerated | Benign | |||
| NTRK2 | L140F | rs150692457 | 9 | 87322819 | C/G | 0.35 | 0 | 0.64 | Damaging | Probably damaging | 64 |
| (Intron) | rs1187272 | 87404086 | A/G | 37.19 | 66.53 | 2.11 | |||||
| C623* | – | 87563481 | A/C | 0.02 | 0 | −0.14 | Premature stop codon | ||||
| GABBR2 | P742Q | – | 9 | 101068407 | T/G | 0.03 | 0 | 5.15 | Damaging | Probably damaging | 65 |
| G671C | - | 101068621 | A/C | 0.02 | 0 | 5.07 | Damaging | Probably damaging | |||
| (Intron) | rs2491397 | 101205162 | T/C | 44.61 | 51.63 | 0.70 | |||||
| (Intron) | rs2184026 | 101304348 | T/C | 6.31 | 22.78 | −0.78 | |||||
| A120A | rs3750344 | 101340316 | C/T | 26.07 | 18.20 | 0.33 | |||||
| GRIN3A | (Intron) | rs11788456 | 9 | 104348150 | G/A | 45.09 | 44.93 | 0.20 | 53 | ||
| (Intron) | rs17189632 | 104368002 | A/T | 36.72 | 43.59 | 0.11 | |||||
| N549S | rs75981117 | 104433048 | C/T | 0.11 | 0.51 | 3.22 | Damaging | Possibly damaging | |||
| R480H | rs34755188 | 104433255 | T/C | 0.33 | 1.88 | 4.16 | Damaging | Probably damaging | |||
| V389L | – | 104449017 | A/C | 0.02 | 0.04 | 4.27 | Damaging | Possibly damaging | |||
| V132L | – | 104499868 | A/C | 0.02 | 0.04 | 3.84 | Damaging | Probably damaging | |||
| DNM1 | L16M | rs61757224 | 9 | 130965795 | A/C | 0.05 | 0.19 | 1.07 | Damaging | Probably damaging | 66 |
| S126* | – | 130981002 | A/C | 0.05 | 0 | 5.99 | Premature stop codon | ||||
| R228L | – | 130982360 | T/G | 0.03 | 0 | 6.05 | Damaging | Probably damaging | |||
| Y231* | – | 130982464 | A/C | 0.02 | 0 | 2.36 | Premature stop codon | ||||
| F336F | rs3003609 | 130984755 | T/C | 11.29 | 54.62 | −0.03 | |||||
| DBH | (Intergenic) | rs3025343 | 9 | 136478355 | A/G | 2.03 | 10.37 | 0.63 | 2 | ||
| I340T | rs182974707 | 136509437 | C/T | 0.06 | 0.04 | 1.84 | Tolerated | Possibly damaging | |||
| A362V | rs75215331 | 136513028 | T/C | 0.06 | 0.07 | 5.39 | Damaging | Probably damaging | |||
| Y389* | – | 136513110 | A/C | 0.06 | 0 | 1.76 | Premature stop codon | ||||
| T395P | – | 136513126 | C/A | 0.02 | 0 | 4.42 | Damaging | Probably damaging | |||
| G482R | rs41316996 | 136521654 | A/G | 0.06 | 0.32 | 1.89 | Damaging | Probably damaging | |||
| R549C | rs6271 | 136522274 | T/C | 1.58 | 6.39 | 1.68 | Damaging | Probably damaging | |||
| CHAT | (5′ UTR) | rs1880676 | 10 | 50824117 | A/G | 4.91 | 23.21 | 2.03 | 67 | ||
| A120T | rs3810950 | 50824619 | A/G | 4.89 | 23.19 | 0.88 | Tolerated | Benign | |||
| E188G | rs75011234 | 50827946 | G/A | 0.33 | 0.36 | 1.64 | Damaging | Probably damaging | |||
| L243F | rs8178990 | 50830171 | T/C | 1.14 | 4.98 | 2.26 | Damaging | Probably damaging | |||
| G284S | rs146236256 | 50833616 | A/G | 0 | 0.04 | 5.92 | Damaging | Probably damaging | |||
| P299L | rs868749 | 50833662 | T/C | 0.02 | 0.04 | 6.01 | Damaging | Probably damaging | |||
| LOC100188947† | (Intron) | rs1329650 | 10 | 93348120 | T/G | 9.50 | 26.86 | −2.00 | 1 | ||
| (Intron) | rs1028936 | 93349797 | C/A | 8.10 | 18.32 | −0.33 | |||||
| CHRNA10 | R421C | rs2231548 | 11 | 3687429 | A/G | 1.22 | 0.07 | 2.36 | Damaging | Probably damaging | |
| R351W | rs139793380 | 3687639 | A/G | 0.10 | 0.04 | 1.65 | Damaging | Probably damaging | |||
| L348R | rs147150654 | 3687647 | C/A | 0.21 | 0 | 3.01 | Damaging | Probably damaging | |||
| V248L | rs2231542 | 3688615 | A/C | 0.08 | 0 | 1.19 | Damaging | Possibly damaging | |||
| W86G | – | 3690532 | C/A | 0.02 | 0.04 | 4.55 | Damaging | Probably damaging | |||
| E85G | rs77958837 | 3690534 | C/T | 0.02 | 0 | 2.80 | Tolerated | Benign | |||
| T77N | rs55719530 | 3690558 | T/G | 1.67 | 1.92 | 3.86 | Damaging | Probably damaging | |||
| BDNF | V74M | rs6265 | 11 | 27679916 | T/C | 3.13 | 14.36 | 3.63 | Damaging | Possibly damaging | 7 |
| (Intron) | rs6484320 | 27703188 | T/A | 7.48 | 18.11 | 1.01 | |||||
| E6K | rs66866077 | 27720937 | T/C | 1.27 | 5.84 | 0 | Damaging | Benign | |||
| (Intron) | rs2030324 | 27726915 | G/A | 47.29 | 49.27 | 0.12 | |||||
| (Intron) | rs7934165 | 27731983 | A/G | 47.22 | 49.12 | 1.07 | |||||
| NRXN2 | T1371P | – | 11 | 64390287 | G/T | 0.51 | 0.76 | 4.09 | Damaging | Probably damaging | |
| V53G | – | 64410118 | C/A | 0.03 | 0 | 2.81 | Damaging | Possibly damaging | |||
| E267G | – | 64457927 | C/T | 0.02 | 0 | 2.37 | Damaging | Possibly damaging | |||
| ARRB1† | H198P | – | 11 | 74989678 | G/T | 0.02 | 0.04 | 4.33 | Damaging | Possibly damaging | 68 |
| ANKK1 | C52W | rs111789052 | 11 | 113258762 | G/C | 6.09 | 0.40 | 2.25 | Damaging | Probably damaging | 5 |
| R122H | rs35877321 | 113264382 | A/G | 0.22 | 1.27 | 0.70 | Damaging | Possibly damaging | |||
| R185Q | rs115800217 | 113265724 | A/G | 10.26 | 0.94 | 0.62 | Tolerated | Probably damaging | |||
| R237* | rs56047699 | 113266815 | T/C | 0.02 | 0.04 | 1.36 | Premature stop codon | ||||
| S313* | - | 113268045 | A/C | 0.19 | 0.11 | −0.43 | Premature stop codon | ||||
| G318R | rs11604671 | 113268059 | A/G | 10.92 | 42.70 | −0.01 | Tolerated | Benign | |||
| P351S | rs186633697 | 113269742 | T/C | 0.21 | 0 | 0.03 | Tolerated | Benign | |||
| E376K | rs56299709 | 113269817 | A/G | 1.24 | 0.04 | 3.29 | Tolerated | Probably damaging | |||
| R445C | rs78229381 | 113270024 | T/C | 6.16 | 0.58 | 0.66 | Damaging | Probably damaging | |||
| E458G | rs184645039 | 113270064 | G/A | 0.54 | 0.11 | 4.23 | Damaging | Probably damaging | |||
| H490R | rs2734849 | 113270160 | G/A | 16.71 | 43.24 | −1.12 | Tolerated | Benign | |||
| E587* | rs113005509 | 113270450 | T/G | 2.28 | 0.14 | 0.84 | Premature stop codon | ||||
| Q657* | rs202222056 | 113270660 | T/C | 0.49 | 0 | 1.08 | Premature stop codon | ||||
| R734C | – | 113270891 | T/C | 0.03 | 0.11 | 0.06 | Damaging | Probably damaging | |||
| DRD2 | E181* | – | 11 | 113286325 | A/C | 0.02 | 0.04 | 3.68 | Premature stop codon | 5 | |
| (Intron) | rs2075654 | 113289066 | T/C | 4.25 | 19.73 | 0.49 | |||||
| (Intron) | rs2075652 | 113294898 | A/G | 4.87 | 1.12 | −0.01 | |||||
| (Intron) | rs4586205 | 113307129 | T/G | 35.61 | 71.86 | −0.88 | |||||
| NRXN3 | Y234* | rs199840331 | 14 | 79181259 | A/C | 0.02 | 0 | −0.33 | Premature stop codon | ||
| G696* | – | 79433576 | T/G | 0.16 | 0.04 | 6.33 | Premature stop codon | ||||
| T99P | – | 79933611 | C/A | 0.05 | 0.04 | 5.18 | Damaging | Possibly damaging | |||
| CHRNA5 | (Intron) | rs588765 | 15 | 78865425 | T/C | 29.46 | 38.84 | −0.27 | 69 | ||
| V134I | rs2229961 | 78880752 | A/G | 0.40 | 0.95 | 5.99 | Damaging | Probably damaging | |||
| K167R | rs80087508 | 78882233 | G/A | 1.87 | 0.11 | 5.01 | Damaging | Probably damaging | |||
| D398N | rs16969968 | 78882925 | A/G | 6.01 | 29.51 | 3.19 | Tolerated | Benign | |||
| CHRNA3 | (3′ UTR) | rs578776 | 15 | 78888400 | G/A | 46.33 | 65.19 | 0.09 | 69 | ||
| H217Y | rs72650603 | 78894335 | A/G | 0.05 | 0.22 | 6.42 | Damaging | Probably damaging | |||
| Y215Y | rs1051730 | 78894339 | A/G | 12.81 | 30.20 | 2.54 | |||||
| (Intron) | rs6495308 | 78907656 | C/T | 29.74 | 29.26 | −1.56 | |||||
| R37H | rs8192475 | 78911230 | T/C | 1.04 | 4.40 | 3.28 | Damaging | Possibly damaging | |||
| CHRNB4 | R497C | – | 15 | 78917483 | A/G | 0.05 | 0 | −1.82 | Damaging | Probably damaging | 69 |
| F462V | – | 78917588 | C/A | 0.02 | 0 | 4.75 | Damaging | Probably damaging | |||
| R349C | rs56235003 | 78921602 | A/G | 0.10 | 0.61 | 1.40 | Damaging | Probably damaging | |||
| P145A | – | 78922214 | C/G | 0.02 | 0 | 5.76 | Damaging | Probably damaging | |||
| S140G | rs56218866 | 78922229 | C/T | 4.25 | 0.83 | 2.22 | Tolerated | Possibly damaging | |||
| T91I | rs12914008 | 78923505 | A/G | 0.73 | 3.58 | 1.72 | Tolerated | Benign | |||
| N41S | rs75495090 | 78927863 | C/T | 1.40 | 0.22 | 4.40 | Damaging | Probably damaging | |||
| CDH13 | N39S | rs72807847 | 16 | 82892037 | G/A | 3.18 | 0.83 | 1.25 | Tolerated | Benign | |
| V464I | rs200591230 | 83711918 | A/G | 0.02 | 0.07 | 3.39 | Damaging | Probably damaging | |||
| ARRB2 | T84P | – | 17 | 4619841 | C/A | 0.02 | 0 | 4.29 | Damaging | Probably damaging | 68 |
| H281Q | – | 4622686 | A/C | 0.03 | 0 | −0.47 | Damaging | Possibly damaging | |||
| CHRNA4† | (3′ UTR) | rs2236196 | 20 | 61977556 | A/G | 35.26 | 73.67 | −0.24 | 20 | ||
| P457L | rs201739273 | 61981180 | A/G | 0.03 | 0 | 0.49 | Damaging | Possibly damaging | |||
| (Intron) | rs2273504 | 61988061 | A/G | 15.84 | 17.85 | −0.53 | |||||
Notes:
1)† = none or only one rare variant validated in this gene, so burden rare variant analysis was not applicable; - = not reported in dbSNP database by 2/17/2014; Chr. = chromosome; Freq. = frequency; Ref. = reference.
2) SNP positions are based on human genome reference assembly build 37.1 (hg19).
3) PhyloP score is basewise vertebrate conservation score.
Together, about 62 GB (868 million reads) of raw sequencing data was obtained from deep-sequencing analysis of the eight pooled DNA samples, with an average of 108 million reads per pooled sample. After appropriate quality control and data filtering, more than 80% of the raw sequencing data was mapped to hg19. A total of 147 million reads were mapped to the targeted regions, which were 100% covered with a median coverage of 106× for each individual DNA sample. Minor allele frequencies (MAF) were calculated for 25 common variants within coding regions and compared with our previous genotyping results based on the TaqMan® assay for individual DNA samples, which revealed that the MAF correlations between the results of the two methods are 0.97 for AA samples and 0.90 for EA samples.23
After removing intronic and synonymous variants, we identified 430 putative functional variants with a minimum read of more than 500 and an MAF of more than 0.75% from our deep-sequencing analysis of pooled DNA samples. Next, based on their SIFT25 and PolyPhen26 scores and MAF rankings, we selected 130 variants, which included 118 rare and 12 common variants, for further validation using independent MSTCC samples. An additional 62 common variants were chosen from the literature on association studies of the 30 genes for validation, based on the fact that they had been reported to be nominally or significantly associated with different ND measures (for a detailed list of these reports, please see Table 2). Selection of the 130 rare and common variants was based on the SIFT25 and PolyPhen26 predictions with the following criteria: 1) all premature stop codons; 2) damaging variants presented in either smoker or non-smoker samples; and 3) damaging and benign variants with an MAF ratio > 1.5 between the smoker and non-smoker samples with the goal of increasing the likelihood of detecting significant single nucleotide polymorphisms (SNPs) from the two groups. These SNPs were genotyped on the TaqMan® OpenArray® genotyping system (Life Technologies, Carlsbad, CA) for the case control samples. All experiments related to deep sequencing and genotyping validation were performed in the Laboratory of Neurogenetics at the NIAAA, NIH.
Data analysis
We arbitrarily used a 5% MAF threshold to define rare and common variants for all samples. Conservation status was determined by the basewise vertebrate conservation PhyloP score.27 A site was defined as conserved when its PhyloP score was ≥ 2, corresponding to a P value of 0.01. Both SIFT25 and PolyPhen26 were used to predict the effect of nonsynonymous variants on protein structure and function. SIFT yields two predictions: tolerated and damaging, and PolyPhen offers three: benign, possibly damaging, and probably damaging. Because all samples were recruited from the same geographical region of Mississippi following exactly the same inclusion and exclusion criteria, significant population stratification was not detected in smokers vs. non-smokers in either AAs or EAs based on principal component analysis of 49 and 51 common variants included in this study, respectively, for each ethnic group (Supplementary Figure 2) and other genotyping results on the same samples (data not shown).
For common variants, we performed individual SNP-based association analysis with smoking status using logistic regression models and with FTND and indexed CPD using linear regression models as implemented in PLINK.28 Additive, dominant, and recessive genetic models were tested for each SNP, adjusted for sex and age in the AA and EA samples separately. All common variants were in Hardy-Weinberg equilibrium within population.
As reported that grouping rare variants together would increase statistical power for association analysis, we used the WSS pooling method 17 to test for association of rare variants with smoking status. This method is applicable to genomic regions with at least two rare nonsynonymous variants. In most cases, one genomic region contained a single gene, the exceptions being the ANKK1/DRD2 and CHRNA5/A3/B4 gene clusters. The WSS method can accommodate only binary response variables because of its intrinsic characteristics.17 In WSS, rare variant counts within the same genomic region for each individual are accumulated rather than collapsed, as implemented in the Cohort Allelic Sums Test (CAST).29 This method puts greater weight on alleles with lower frequencies in controls, which have a higher tendency to be functional both biologically and statistically. Scores for all subjects are then ordered, and the WSS is computed as the sum of ranks for all cases. Variants over-represented in cases will have larger WSS values. Then 106 permutations were performed to determine P values for each genomic region. Limited by computational burden, 108 permutations were implemented only when 106 phenotype rearrangements were insufficient to acquire an exact P value.
After obtaining association results for common and rare variants separately, we evaluated the cumulative effects of both rare and common variants on smoking status using the combined sum tests (i.e., SKAT-C and Burden-C) and adaptive sum tests (i.e., SKAT-A and Burden-A) with age and sex controlled.18 Smoking status was used as the sole response variable for the following two reasons: 1) to keep analysis results consistent with rare variant analysis; and 2) the other two phenotypes (FTND and indexed CPD) are available for smokers only, use of which means excluding around half of the samples and rare variants presented only in non-smoker samples. The combined sum tests choose the weight parameter in such a manner that rare and common variants contribute equally to the overall test statistic. In contrast, the adaptive sum tests are more powerful if the overall effect sizes of rare and common variants are very different, for example, when a trait is associated only with rare or common variants in the region. Because the relative contribution of rare and common variants to ND risk is unknown, we used both tests to estimate their combined effects. Burden and variance-component (e.g., SKAT) tests are two major types of group-wise association tests proposed for rare variant analysis, which in this case were extended to accommodate combined analysis of rare and common variants by adjusting the weighting scheme. Only genomic regions with at least one rare and one common variant can be analysed by this approach.
To determine the effect directions of significant results obtained from the above group-wise tests, we performed case control-based association analysis for each rare variant using PLINK.28 Then rare variants were separated into two groups based on their estimated odds ratios (OR): if OR > 1, the rare variant was predicted to increase smoking risk; if OR < 1, the rare variant was considered to be protective. However, limited by low frequencies of the rare variants and our moderate sample size used in this study, the OR was not available for every rare variant, which happened mostly for rare variants with fewer copies of the minor allele. In this case, we assigned the variant to the risk group if more minor alleles were present in smokers; otherwise, to the protective group. For collapsing methods, such as the WSS test, the statistical power decreases dramatically as the proportion of functional variants excluded from the analysis increases.30 Also, because most of the genes or genomic regions investigated in this study have only 2 to 4 rare variants, splitting them on the basis of their effect directions would provide little information about association with the phenotype of interest given our sample sizes.31
As a result, we only performed effect direction specific combined and adaptive sum tests, not WSS, as described above to further characterize cumulative variant effect directions. Even though we put rare and common variants with the same effect direction together, some of the groups still had limited number of variants. For groups with one rare variant and one common variant, SKAT-C and Burden-C tests are equivalent, so do SKAT-A and Burden-A tests; if only rare or common variants exist in a group, SKAT-C will provide the same results as SKAT-A, which also applies to Burden-C and Burden-A; in cases of only one rare or common variant, all four tests are equivalent to logistic regression analysis.
Bonferroni corrections were used to select significant association results for all analyses. Uncorrected P values are presented throughout the manuscript.
RESULTS
Description of variants and their functionality prediction
There existed 135 out of the 192 variants selected for validation in the MSTCC samples based on genotyping results, which include 33 novel variants (25%; without rs numbers in the dbSNP database as searched on 2/17/2014) in 30 candidate genes (Table 2). As shown in Figure 1A, 58% of these variants (n = 78) are missense; 11% (n = 15) are nonsense–premature stop codons; and 2% (n = 3) are synonymous; the remaining 29% (n = 39) are from intronic, intergenic, or untranslated regions. Of the 93 non synonymous variants, 79 (85%) were predicted to be damaging by PolyPhen, SIFT, or both. The prediction concordance rate between SIFT and PolyPhen programs was 51% (69/135); 14 of 69 were predicted as tolerated by SIFT and benign by PolyPhen; the remaining 55 were predicted to be damaging by SIFT and possibly or probably damaging by PolyPhen. All 33 novel variants were non synonymous; they will be mentioned as amino acid change throughout the manuscript. Additionally, 55%of the coding variants were located at conserved sites (53/96; PhyloP score ≥ 2)27 compared with only 5% of non-coding variants (2/39). The proportion of conserved sites is significantly different among the coding and non-coding variants (Fisher’s Exact p = 1.59×10−8).
Figure 1.
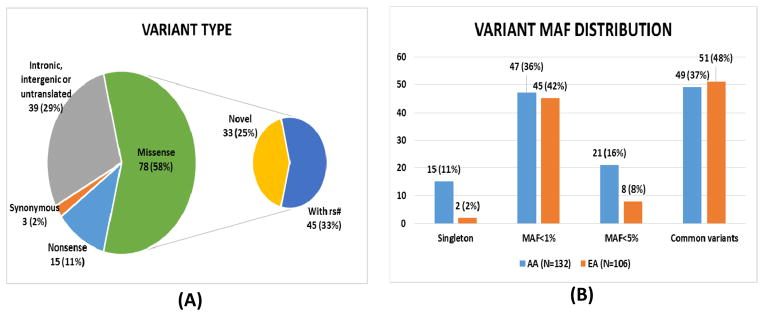
Descriptive statistics of the 135 validated variants. (A) Proportions of different variant types. Almost 70% of the validated variants lead to amino acid changes. All novel-identified variants (without rs# in dbSNP database by 2/17/2014) are missense. (B) The MAF distribution of variants for the AA and EA samples. The four categories are singleton-only one copy of a rare allele identified in the AA and EA samples, MAF < 1%, MAF < 5%, and common variants. The AA sample has more singletons and low-frequency variants (1% < MAF < 5%) and fewer common variants than the EA sample.
Of the validated variants, 67% are rare (91/135; MAF < 5%) in AAs, EAs, or both (Table 2), many appearing only once in all individuals (17/91 = 19% are singletons) and 7 appearing once only in both the AA and EA samples. Among the 44 common variants, 77% (34/44) belong to non-coding regions compared with 5% (5/91) of the 91 rare variants (Fisher’s exact test P = 8.82 × 10−18), which is consistent with data from exome sequencing studies that non-synonymous coding variants are significantly skewed toward low frequencies.32 Figure 1B compares the MAF distributions in the AA and EA samples for different MAF groups, revealing a higher percentage of singletons and rare variants with MAF between 1% and 5%, and a lower proportion of common variants in the AA sample relative to the EA sample.
Association analysis results for common variants
There are 24 SNPs across 12 genes (DRD3, CHRNA9, DRD1, DDC, CHRNB3, NTRK2, GABBR2, BDNF, ANKK1, DRD2, CHRNA3, and CHRNA4) and one genomic region (LOC100188947) that show nominally significant association (P< 0.05) with smoking status, FTND, or indexed CPD in the AA sample (Supplementary Table 2). Of them, rs1051730 in CHRNA3 has the lowest P value, 0.0016 (OR = 2.45; 95% confidence interval [CI] = 1.41, 4.26), which is nominally associated with smoking statusunder the recessive model. Twenty-one SNPs of 8 genes (NRXN1, CHRNA9, TAS2R38, CHRNB3, NTRK2, DBH, CHAT, BDNF, and CHRNA3) and one genomic region (LOC100188947) are nominally associated with the three phenotypes in the EA sample. Both rs1726866 of TAS2R38 and rs2030324 of BDNF have the smallest P value, 0.0017, in the EA sample. The SNP rs1726866 shows nominal damaging effects toward FTND (beta= 0.30; 95% CI = 0.11, 0.49) under the additive model, while rs2030324 nominally protects against FTND (beta= −0.51; 95% CI = −0.83, 0.19) under the recessive model.
The SNPs rs55633891 in CHRNA9, 5 SNPs (rs10958725, rs10958726, rs4736835, rs6474412, and rs13280604) in CHRNB3, rs1187272 in NTRK2, rs1329650 in LOC100188947, and rs6484320 in BDNF show nominally significant associations in both the AA and EA samples (Supplementary Table 2). However, none of these SNPs survives Bonferroni correction (threshold of significance for AAs = 1.13 × 10−4 for 49 variants, 3 genetic models, and 3 phenotypes; for EAs = 1.09 × 10−4for 51 variants, 3 genetic models, and 3 phenotypes). Of note, some variants have MAF > 5% in only one sample, which were not called common variants based on our definition, but we performed individual variant analysis for these SNPs.
Association analysis results for rare variants
By using the WSS method, 10 genes (NRXN1, CHRNA9, CHRNA2, NTRK2, GABBR2, GRIN3A, DNM1, NRXN2, NRXN3, and ARRB2) are significantly associated with smoking status in the AA sample (Table 3), with P values ranging from 1.31 × 10−4 for CHRNA2 to 2.42 × 10−3 for GRIN3A based on 106 permutations. The family-wise error rate (FWER) for 19 genomic regions or genes tested in AAs, which contain at least two nonsynonymous rare variants, is 2.63 × 10−3 (0.05/19). There are 7 genes (NRXN1, CHRNA9, TAS2R38, GRIN3A, DBH, NRXN3, and CDH13) and 1 gene cluster (ANKK1/DRD2) showing significant associations, at P values between 1 × 10−6 (DBH and NRXN3) and 3.5 × 10−5 (CDH13) in the EA sample based on 106 or 108 permutations (i.e., permuting subjects’ smoker/non-smoker status for 106 or 108 times; see Table 3). With 11 genes tested for EAs, the FWER threshold is 4.55 × 10−3 (0.05/11). TAS2R38 (P= 2 × 10−6), NRXN3 (P = 1 × 10−6), and CDH13 (P= 3.5 × 10−5) are the three genes that required 108 permutations in order to obtain a reliable P value.
Table 3.
Significant Rare Variant Association Results Using Weighted Sum Statistic (WSS) in AA and EA Samples
| Gene | AA Sample | EA Sample | ||
|---|---|---|---|---|
| SNPs | Permuted p value | SNPs | Permuted p value | |
| NRXN1 |
rs77665267 (p.T274P) - (p.R206L) - (p.Y367*) - (p.S62*) |
2.28×10−4 |
rs77665267 (p.T274P) - (p.R206L) |
2×10−6 |
| CHRNA9 |
rs142807401 (p.A432T) rs139982841 (p.A452V) |
3.81×10−4 | rs56210055 (p.A312T) rs142807401 (p.A432T) rs139982841 (p.A452V) |
8×10−6 |
| TAS2R38 |
rs139843932 (p.W135G) rs114288846 (p.R274C) |
0.5346 |
rs139843932 (p.W135G) rs114288846 (p.R274C) |
2×10−6† |
| CHRNA2 | - (p.S488*) - (p.R121L) |
1.31×10−4 | N/A | N/A |
| NTRK2 | rs150692457 (p.L140F) - (p.C623*) |
4.25×10−4 | N/A | N/A |
| GABBR2 | - (p.P742Q) - (p.G671C) |
1.58×10−4 | N/A | N/A |
| GRIN3A |
rs75981117 (p.N549S) rs34755188 (p.R480H) - (p.V389L) - (p.V132L) |
2.42×10−3 |
rs75981117 (p.N549S) rs34755188 (p.R480H) - (p.V389L) - (p.V132L) |
8×10−6 |
| DNM1 | rs61757224 (p.L16M) - (p.S126*) - (p.R228L) - (p.Y231*) |
3.53×10−4 | N/A | N/A |
| DBH |
rs182974707 (p.I340T) rs75215331 (p.A362V) - (p.Y389*) - (p.T395P) rs41316996 (p.G482R) rs6271 (p.R549C) |
0.2427 |
rs182974707 (p.I340T) rs75215331 (p.A362V) rs41316996 (p.G482R) |
1×10−6 |
| NRXN2 | - (p.T1371P) - (p.V53G) - (p.E267G) |
1.49×10−3 | N/A | N/A |
| ANKK1/DRD2 |
rs35877321 (p.R122H) rs56047699 (p.R237*) - (p.S313*) rs186633697 (p.P351S) rs56299709 (p.E376K) rs184645039 (p.E458G) rs113005509 (p.E587*) rs202222056 (p.Q657*) - (p.R734C) - (p.E181*) |
0.8114 | rs111789052 (p.C52W) rs35877321 (p.R122H) rs115800217 (p.R185Q) rs56047699 (p.R237*) - (p.S313*) rs56299709 (p.E376K) rs78229381 (p.R445C) rs184645039 (p.E458G) rs113005509 (p.E587*) - (p.R734C) - (p.E181*) |
6×10−6 |
| NRXN3 | rs199840331 (p.Y234*) - (p.G696*) - (p.T99P) |
2.17×10−4 |
- (p.G696*) - (p.T99P) |
1×10−6† |
| CDH13 |
rs72807847 (p.N39S) rs200591230 (p.V464I) |
0.5231 |
rs72807847 (p.N39S) rs200591230 (p.V464I) |
3.5×10−5† |
| ARRB2 | - (p.T84P) - (p.H281Q) |
1.32×10−4 | N/A | N/A |
P value based on 108 permutations.
Notes:
1) Permuted p value = value based on 106 permutations; - = not reported in dbSNP database by 2/17/2014; N/A = not applicable; i.e., without two rare nonsynonymous variants in gene or region.
2) SNPs included in both AA and EA rare variant analysis are underlined.
3) Significant association p values after correction for multiple testing(p< 2.63×10−3 for AA sample and p< 4.55×10−3 for EA sample) are given in bold. See “Materials and Methods” for details.
The genes NRXN1, CHRNA9, GRIN3A, and NRXN3 have significantly larger WSS values in both AAs and EAs. NRXN1 has two nonsynonymous substitutions (R206L andrs77665267) and two premature stop codons (S62* andY367*) in the AA sample (P= 2.28 × 10−4), while only R206L and rs77665267 were detected in the EA sample (P= 2 × 10−6). The two nonsynonymous variants (rs142807401 and rs139982841) of CHRNA9 are found in both the AA (P= 3.81 × 10−4) and EA (P= 8 × 10−6) samples, as are the four SNPs (V132L, V389L, rs34755188, andrs75981117) of GRIN3A (P = 2.42 × 10−4 in AAs; P= 8 × 10−6 in EAs). For NRXN3, there are two premature stop codons (rs199840331 and G696*) and one nonsynonymous variant (T99P) included in the analysis for AA subjects (P = 2.17 × 10−4) and one premature stop codon (G696*) and one nonsynonymous variant (T99P) included in the analysis for EA subjects (P = 1 × 10−6).
Association analysis results for rare and common variants
CHRNA9, with two rare variants (rs142807401 and rs139982841) and two common variants (rs56210055 and rs55633891), and DRD1, with one rare variant (R226W) and three common variants (rs265975, rs686, and rs4532), are nominally associated with smoking status after correcting for sex and age in the AA sample (Table 4). The P values are 0.0495 for CHRNA9 using Burden-A method and 0.0458 using Burden-C, and 0.0430 using Burden-A for DRD1. All four variants of CHRNA9 result in amino acid changes, among which rs56210055 has an MAF of 7.19% in AAs, but only 0.85% in EAs. So in the EA sample, with three rare variants (rs56210055, rs142807401, and rs139982841) and one common variant (rs55633891), CHRNA9 shows significant association, with P values of 0.0012, 0.0032, 0.0036, and 0.0080 using SKAT-C, Burden-C, SKAT-A, and Burden-A, respectively (Table 4). The first three P values survive multiple testing correction for 12 genes, which have at least one rare and one common variant and were eligible to be included in this analysis in the EA sample (0.05/12 = 0.0042). Both rare and common variants of CHRNA9 contribute to the risk for ND in EAs and possibly in AAs.
Table 4.
Significant Combined and Adaptive Sum Test Results of Cumulative Rare- and Common-Variant Effects on Smoking Status in AA and EA Samples
| Gene | AA Sample | EA Sample | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rare variant(s) | Common variant(s) | Effect direction | P value† Separated | Pooled | Rare variant(s) | Common variant(s) | Effect direction | P-value† | ||
| Separated | Pooled | |||||||||
| NRXN1 |
rs77665267 (p.T274P) -(p.S62*) |
rs10208208 rs6721498 |
↑ | 0.5553 0.3023 0.5354 0.2996 |
0.5537 0.9368 0.5398 0.9275 |
rs77665267 (p.T274P) rs10208208 |
rs10490227 | ↑ | 0.1016 0.0527 0.0738 0.0362 |
0.1062 0.0772 0.0683 0.0505 |
|
- (p.R206L) - (p.Y367*) |
rs10490227 rs2193225 |
↓ | 0.4244 0.1991 0.5232 0.3547 |
- (p.R206L) |
rs6721498 rs2193225 |
↓ | 0.4648 0.2849 0.4506 0.4596 |
|||
| CHRNA9 |
rs139982841 (p.A452V) |
↑ | 0.2381 0.2381 0.2381 0.2381 |
0.0706 0.0766 0.1448 0.0495 |
rs142807401 (p.A432T) |
rs55633891 (p.A315V) |
↑ |
0.0143 0.0143 0.0246 0.0246 |
0.0012 0.0032 0.0036 0.0080 |
|
|
rs142807401 (p.A432T) |
rs56210055 (p.A312T) rs55633891 (p.A315V) |
↓ |
0.0381 0.0143 0.0942 0.0353 |
rs56210055 (p.A312T) rs139982841 (p.A452V) |
↓ |
0.0119 0.0072 0.0119 0.0072 |
||||
| DRD1 | - (p.R226W) |
rs265975 rs4532 |
↑ | 0.0572 0.0393 0.0549 0.0372 |
0.1121 0.0458 0.1224 0.0430 |
N/A | ||||
| rs686 | ↓ | 0.8101 0.8101 0.8101 0.8101 |
||||||||
| ANKK1/DRD2 |
rs56299709 (p.E376K) - (p.R734C) - (p.E181*) rs2075654 |
rs111789052 (p.C52W) rs115800217 (p.R185Q) rs11604671 (p.G328R) rs78229381 (p.R445C) rs2734849 (p.H490R) |
↑ | 0.4445 0.1179 0.3915 0.1370 |
0.3915 0.0950 0.4850 0.1361 |
rs111789052 (p.C52W) rs35877321 (p.R122H) rs56299709 (p.E376K) rs78229381 (p.R445C) rs184645039 (p.E458G) - (p.R734C) |
rs2075654 rs4586205 |
↑ | 0.3828 0.1708 0.2649 0.1303 |
0.5566 0.8627 0.4306 0.8619 |
|
rs35877321 (p.R122H) rs56047699 (p.R237*) - (p.S313*) rs186633697 (p.P351S) rs184645039 (p.E458G) rs113005509 (p.E587*) rs202222056 (p.Q657*) rs2075652 |
rs4586205 | ↓ | 0.4965 0.0371 0.5029 0.0273 |
rs115800217 (p.R185Q) rs56047699 (p.R237*) - (p.S313*) rs113005509 (p.E587*) - (p.E181*) rs2075652 |
rs11604671 (p.G328R) rs2734849 (p.H490R) |
↓ | 0.7714 0.3605 0.8413 0.2840 |
|||
| CHRNA5/ A3/B4 |
rs72650603 (p.H217Y) rs8192475 (p.R37H) - (p.R497C) - (p.F462V) rs56235003 (p.R349C) - (p.P145A) rs12914008 (p.T91I) rs75495090 (p.N41S) |
rs16969968 (p.D398N) rs1051730 (p.Y215Y) |
↑ | 0.0901 0.0323 0.0701 0.0596 |
0.2406 0.4546 0.1398 0.4909 |
rs2229961 (p.V134I) rs72650603 (p.H217Y) rs8192475 (p.R37H) |
rs588765 rs16969968 (p.D398N) rs1051730 (p.Y215Y) |
↑ | 0.7389 0.2600 0.6989 0.3322 |
0.7570 0.9566 0.7185 0.9078 |
|
rs2229961 (p.V134I) rs80087508 (p.K167R) rs56218866 (p.S140G) |
rs588765 rs578776 rs6495308 |
↓ | 0.5737 0.4017 0.4497 0.4983 |
rs80087508 (p.K167R) rs56235003 (p.R349C) rs56218866 (p.S140G) rs12914008 (p.T91I) rs75495090 (p.N41S) |
rs578776 rs6495308 |
↓ | 0.6270 0.3729 0.5879 0.5716 |
|||
Notes:
1) P values for each gene or region were obtained by four statistical methods; i.e., SKAT-C, Burden-C, SKAT-A, and Burden-A;
= p values from top to bottom for each gene or region were obtained in the abovementioned order.
2) Only genes or regions with at least one rare and one common variant were eligible for the pooled analysis; N/A = not applicable.
3) “↑” = variants increase smoking risk estimated from individual variant-based odds ratios (if available) or minor allele counts in Cases and Controls, “↓” = variants decrease smoking risk; effect direction specific tests were applied with p values listed under “Separated”.
4) SNP rs numbers are based on dbSNP database (accessed on 2/17/2014).
5) SNPs included in both AA and EA samples for this analysis are underlined.
6) Nominal significant associations (p< 0.05) for both “Pooled” and “Separated” analyses are given in bold, including p values, SNP, and gene names. See the section of “Materials and Methods” for details.
Nominally significant associations were also detected in effect-direction separated analysis for NRXN1, CHRNA9, DRD1, ANKK1/DRD2, and CHRNA5/A3/B4 (Table 4). Two rare variants (rs77665267 and rs10208208) and one common variant (rs10490227) of NRXN1 in EAs show a P value of 0.0362 using the Burden-A method, indicating a possible combined risk effect of the three variants. The common variant, rs10490227 did not show any significant association with smoking status in individual SNP-based analysis; however, it is nominally associated with FTND (Supplementary Table 2). For CHRNA9, its nominal association in AAs seems to be driven mainly by one rare variant (rs142807401) and two common variants (rs56210055 and rs55633891) with decreased probability of smoking. SNPs rs142807401 and rs55633891 have opposite effects in the EA sample, which suggests population-specific effects or is simply caused by the rough assignment of effect directions as described in Materials and Methods. Three of the four variants in DRD1, which increase smoking risk, result in a nominal association in the AA sample (Burden-C P= 0.0393 and Burden-A P= 0.0372).
Burden-C and Burden-A methods worked as expected for the effect-direction separated analysis according to their theoretical designs and assumptions. BesidesNRXN1, CHRNA9, and DRD1, these two methods discovered nominal associations between the two genomic regions (ANKK1/DRD2 and CHRNA5/A3/B4) that contain the most variants in this study and smoking status in the AA samples as well. Eight rare variants and one common variant in ANKK1/DRD2 together decrease smoking risk, while eight rare variants and two common variants in CHRNA5/A3/B4 display the opposite effect (Table 4).
For groups with rare variants only, the combined and adaptive sum tests revealed nominal associations between TAS2R38, GRIN3A, DNM1, DBH, and smoking status, respectively, in either AAs or EAs (Supplementary Table 4). This can be seen as a confirmation of the association signals detected by the WSS method. Non-significant association results for rare variant analysis and rare and common variant combined analysis are presented in Supplementary Tables 3 and 4.
DISCUSSION
Although none of the 44 common variants showed significant association with any of the three nicotine phenotypes (smoking status, FTND, and indexed CPD) after Bonferroni correction in this study, rare variants in 10 genes (NRXN1, CHRNA9, CHRNA2, NTRK2, GABBR2, GRIN3A, DNM1, NRXN2, NRXN3, and ARRB2) in the AA sample and 7 genes (NRXN1, CHRNA9, TAS2R38, GRIN3A, DBH, NRXN3, and CDH13) plus 1 gene cluster (ANKK1/DRD2) in the EA sample are significantly associated with smoking status using the WSS method. Further, we also detected a significant cumulative effect of both rare and common variants in CHRNA9 that contribute to smoking status with age and sex controlled in the EA sample when applying both the combined and the adaptive sum test.
Among the common variants that are nominally associated with any of the three ND measures, SNP rs1051730 is of great interest. This SNP has the smallest common variant association P value in the AA sample, which has been reported as the most significant genome-wide association in meta-analyses of subjects of European ancestry (P= 2.75 × 10−73).2, 3, 6, 33 Another wasrs16969968, the most robust genetic finding on chromosome 15q25 in subjects of European ancestry, with a P value of 5.57 × 10−72.2, 3, 6, 33 Although we did not find significant associations for these two SNPs in our EA sample, which is likely attributable to the small sample size (758 smokers vs. 672 non-smokers), the nominally significant association presented for the AA sample is of interest, providing an independent replication of the association of this SNP with smoking in our independent samples.
HapMap data show that rs1051730 and rs16969968 are in strong linkage disequilibrium in European and Asian populations but not in AAs (r2= 0.40).34 In a meta-analysis of AA samples, Chen et al.34 found that rs16969968 is more strongly associated with heavy smoking (P= 0.0011) than is rs1051730 (P= 0.011). In our AA sample, however, only rs1051730 is nominally associated with smoking status (P= 0.0016; OR= 2.45; 95% CI = 1.41, 4.26) under the recessive model even though the correlation coefficient between rs1051730 and rs16969968 is 0.42; this is consistent with the HapMap data. As a coding synonymous variant, rs1051730 is expected to have less functional significance than rs16969968, a missense mutation (aspartate to asparagine). So while the functional significance of rs16969968 has been demonstrated in vitro35 and to some extent via α5 knockout mouse models that show a role for the gene,36 the functional relevance of rs1051730 is undetermined. Based on our study result, we suspect that rs1051730 is in linkage disequilibrium (LD) with another functional missense variant with a large effect but low MAF, other than rs16969968, in our AA sample; or it changes CHRNA3 expression in a significant way.
For rare variants, although we have 10 and 8 genomic regions significantly associated with smoking status in the AA and EA samples, respectively, the two ethnic samples provide replication for each other only for four genes that overlapped across the samples: NRXN1, CHRNA9, GRIN3A, and NRXN3. Among the four genes, CHRNA9 and GRIN3A have rare nonsynonymous variants that are seen in both populations, which could be of importance in an evolutionary functional context because of the implication that they are ancient. Because CHRNA9 is also significantly and nominally associated with smoking status for rare and common variant combined analysis in both the EA and AA sample, it will be discussed first.
CHRNA9, which codes for nAChR α9, is located on chromosome 4p15.1-p14 and contains five exons and four introns.37 The protein is composed of 479 amino acids (UniProtKB/Swiss-Prot ID: Q9UGM1; RefSeq ID: NP_060051) and contains two highly conserved domains, which are the neurotransmitter-gated ion-channel ligand binding domain (aa 31–236) and the neurotransmitter-gated ion-channel transmembrane region (aa 244–457).38 The nAChR α9 can form homo- or hetero-oligomericcation-selective channels in conjunction with nAChR α1039 and is usually expressed in the cochlea, keratinocytes, pituitary gland, B-cells, and T-cells.39–41 Both α9 and α10 nAChR subunits also are coexpressed in dorsal root ganglion neurons.42
The four variants in CHRNA9 that contribute to the association signals are rs56210055 (p.A312T), rs55633891 (p.A315V), rs142807401 (p.A432T), and rs139982841 (p.A452V). All have PhyloP Scores >4 (Table 2). Both ala312 and ala315 lie within a transmembrane region composed of 22 amino acids (aa 302–323), whereas ala432 and ala452 are located within the cytoplasmic region (aa 324–457). The rs139982841 variant has also been identified in lung cancer tissues in the catalogue of somatic mutations in cancer (COSM587183).
Other researchers have reported nominally significant association of CHRNA9 (rs4861065) with ND in a female Israeli sample43 and of CHRNA9 (rs766988 and rs4861065) with response inhibition, as well as of CHRNA9 (rs4861065) with selective attention in a subset of the same sample, in which neurocognitive functions are putatively implicated in ND susceptibility.44 Chikova et al.45 revealed that rs56159866 and rs6819385 in CHRNA9 are associated with an increased risk of lung cancer, while three SNPs, rs55998310, rs56291234, and rs182073550 (single nucleotide deletion) protect against lung cancer.
All these SNPs are either synonymous variations or within intronic or UTR regions, and therefore lack any obvious direct functional effect but may affect protein production at the transcriptional and/or translational levels or simply manifest association through linkage disequilibrium with other functional variants. In contrast, the four variants we reported in this study all cause amino acid changes, among which rs56210055 (p.A312T) and rs55633891 (p.A315V) may affect nAChR stability or the permeability of the ion channel, while rs142807401 (p.A432T) and rs139982841 (p.A452V) may influence downstream signalling characteristics based on the amino acid locations they affect. Based on the effect direction specific analysis results shown in Table 4, these four variants may have a mixture of risk and protective effects in affecting smoking risk. Thus, future functional studies are warranted for these four SNPs inCHRNA9.
GRIN3A is localized on chromosome 9q34 and consists of nine exons,46 which code for glutamate receptor ionotropic NMDA 3A (GluN3A). The deduced protein contains 1115 amino acids (UniProtKB/Swiss-Prot ID: Q8TCU5; RefSeq ID: NP_597702.2) and shows 92.7% identity to rat NMDA receptor 3A.46 Functional NMDA receptors are heterotetramers composed of two ζ subunits (GluN1) and two ε subunits (GluN2A, GluN2B, GluN2C, or GluN2D) or third subunits (GluN3A or GluN3B), which serve critical functions in neuronal development, functioning, and degeneration of the mammalian central nervous system.47 GluN3A suppresses NMDA receptor functions in a dominant-negative way.48, 49 GluN3A-containing NMDA receptors display reduced Ca2+ permeability and low sensitivity to Mg2+ blockade.50, 51 The transcript of GRIN3A was detected by in situ hybridization in human fetal spinal cord and forebrain.52
All four substituted amino acids, val132, val389, arg480, andasn549, are located in the extracellular region of GluN3A and are conserved, with PhyloP scores > 3 (Table 2). We have previously reported common variants of GRIN3A significantly associated with different ND measures in the MSTF population.53 Different variants within GRIN3A have also been associated with Alzheimer’s disease54 and schizophrenia.55 The recent work by Takata et al.55 identified disease association of a missense variant in GRIN3A (p.R480G, rs149729514; P = 0.00042; OR = 1.58) in a Japanese schizophrenia case-control cohort. This association was supported by their meta-analysis with independent Han-Chinese case-control and family samples (combined P= 3.3 × 10−5). However, as the authors suggested, the GRIN3A R480G variant was not detected in AA and EA populations, and thus it seems to be Asian specific.
In this study, instead of finding the glycine substitution at residue 480, we identified a histidine substitution at the same position of GluN3A in both AAs and EAs. The ingenious connection between the two studies confers great functional importance for this residue not only in ND, but also in other psychiatric disorders. Another variant, rs75981117 (p.N549S), is an N-linked glycosylation site on GluN3A, which could be important for both the structure and function of the protein. SNPs rs75981117 (p.N549S), rs34755188 (p.R480H), and V389L together show a nominal protective effect against smoking risk in AAs (Supplementary Table 4). The functional importance of the four variants may show in ND-related mouse models, as Marco et al.56 recently discovered that overexpression of GluN3A in mouse striatum mimicked the synapse loss observed in Huntington’s disease mouse models, whereas genetic deletion of GluN3A prevented synapse degeneration, ameliorated motor and cognitive decline, and reduced striatal atrophy and neuronal loss in the YAC128 Huntington’s disease mouse model.
Because of space limitations, we cannot elaborate on the potential functional importance of the rare variants we identified in NRXN1, CHRNA2, TAS2R38, NTRK2, GABBR2, DNM1, DBH, NRXN2, ANKK1/DRD2, NRXN3, and CDH13 here. To interpret the results of this study more appropriately, five main limitations need to be considered. First, rare variants are usually population specific, or even sample specific, which, on one hand, makes replication very difficult and on the other hand, reveals that the rare variants identified in this study are just a starting point. Association studies of these biological candidate genes in other populations and samples are thus warranted. Second, we limited our search to biological candidate genes, which makes these findings not surprising at the gene level. If we are to uncover new genes, more comprehensive and hypothesis-free analyses, particularly genome-wide sequencing analyses of rare variants, are needed. Third, although none of the 44 common variants showed significant association with any of the three nicotine phenotypes after Bonferroni correction, this does not mean common variants in general are not important in affecting smoking risk. The primary reason for our failure to identify significant association of these common variants with ND measures is more likely related to our sample size, especially for EAs, with a sample size of only 1430. Another reason may be the selection of these common variants from our previous studies, 30 and 7 of which showed nominal or significant associations in preceding analysis of MSTF and MSTCC samples, respectively (see Supplementary Table 2). Nineteen out of the 30 common variants chosen based on previous MSTF study results were found nominally associated with at least one of the three ND measures (i.e., smoking status, FTND, and indexed CPD) in either AA or EA case control samples; however, all 7 common variants selected from one meta-analysis study on CHRNB3 including MSTCC samples showed nominal significance in this study composed solely of MSTCC subjects. Such analysis result difference is likely caused by sample difference – family and case control samples. Although both samples were recruited from the same geographical locations, they were recruited at different time periods with the family samples recruited from 1999–2004 and the case control samples recruited from 2005–2011. This difference is also consistent with regression to the mean for two samples. Fourth, it is hard to dissect the contribution of each rare variant and the relative contributions of rare vs. common variants, hampered by our sample size and the statistical methods we applied. Five, although our subjects were recruited from the same geographical area and the two ancestry-based groups; i.e., AA and EA, are well separated according to our previous reports using common variants,57, 58 we still could not completely rule out the possibility of some hidden distributional differentiation of rare and low-frequency variants in our samples, considering the insights provided by the 1000 Genomes Project analyses59 and currently lack of the genome-wide profiles of these variants.
We used one type of burden test; i.e., WSS,17 to accumulate counts of rare variants in separate genomic regions and then examined their overrepresentation in cases vs. controls. The burden test is a compromise between extremely low allele frequency and limited statistical power, which enables detection of pooled rare variant effects but is incapable of disentangling individual effects of rare variants. For combined analysis of rare and common variants, we implemented the combined and adaptive sum tests;18 the former assumes equal contribution of rare and common variants, and the latter presumes rare variants have different effects than common variants. Without knowing the relative contribution of rare and common variants to any trait of interest, we highly encourage applying both tests to analyze the same dataset as used in this study. We also performed effect direction-specific analyses to examine the combined effect directions of rare and common variants. Because of the limited number of rare variants available for each gene or genomic region and the expected substantial power loss of burden tests when functional variants are excluded, this analytical strategy was applied only to the combined and adaptive sum tests. Nominal association results provided evidence for combined-effect direction speculation of the variant groups; however, no significant association was discovered. This strategy will be more effective with a larger number and more accurate classification of rare variants.
This study demonstrates for the first time the contribution of common and, particularly, rare variants within a subset of biological candidate genes besides nAChR subunit genes, to the risk for ND. Our findings about these variants, especially rs56210055 (p.A312T), rs55633891 (p.A315V), rs142807401 (p.A432T), and rs139982841 (p.A452V) in CHRNA9 and V132L, V389L, rs34755188 (p.R480H), and rs75981117 (p.N549S) in GRIN3A are interesting and encouraging and deserve further study using both in vitro and in vivo approaches.
Supplementary Material
Figure S1. Density plot of estimated minor allele frequency (MAF) distributions from pooled-sequencing analysis of the 200 sib pairs. Many more rare variants were discovered in AAs than in EAs.
Figure S2. Scatter plots of principal components 1 and 2 for smokers and non-smokers based on principal component analysis of 49 and 51 common variants available for AAs and EAs, respectively. Smokers and non-smokers are uniformly mixed within the two ethnic groups. Significant population stratification was not detected between smokers and non-smokers.
Acknowledgments
We acknowledge the invaluable contributions of personal information and blood samples by all participants in the study. This project was supported by National Institutes of Health grant R01 DA012844 to MDL.
References
- 1.TAG. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42(5):448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42(5):436–440. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gelernter J, Yu Y, Weiss R, Brady K, Panhuysen C, Yang BZ, et al. Haplotype spanning TTC12 and ANKK1, flanked by the DRD2 and NCAM1 loci, is strongly associated to nicotine dependence in two distinct American populations. Hum Mol Genet. 2006;15(24):3498–3507. doi: 10.1093/hmg/ddl426. [DOI] [PubMed] [Google Scholar]
- 5.Huang W, Payne TJ, Ma JZ, Beuten J, Dupont RT, Inohara N, et al. Significant association of ANKK1 and detection of a functional polymorphism with nicotine dependence in an African-American sample. Neuropsychopharmacology. 2009;34(2):319–330. doi: 10.1038/npp.2008.37. [DOI] [PubMed] [Google Scholar]
- 6.Tobacco-and-Genetics-Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beuten J, Ma JZ, Payne TJ, Dupont RT, Quezada P, Huang W, et al. Significant association of BDNF haplotypes in European-American male smokers but not in European-American female or African-American smokers. Am J Med Genet B Neuropsychiatr Genet. 2005;139(1):73–80. doi: 10.1002/ajmg.b.30231. [DOI] [PubMed] [Google Scholar]
- 8.Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16(1):24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nussbaum J, Xu Q, Payne TJ, Ma JZ, Huang W, Gelernter J, et al. Significant association of the neurexin-1 gene (NRXN1) with nicotine dependence in European- and African-American smokers. Hum Mol Genet. 2008;17(11):1569–1577. doi: 10.1093/hmg/ddn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43(11):1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asselbergs FW, Guo Y, van Iperen EP, Sivapalaratnam S, Tragante V, Lanktree MB, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet. 2012;91(5):823–838. doi: 10.1016/j.ajhg.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diogo D, Kurreeman F, Stahl EA, Liao KP, Gupta N, Greenberg JD, et al. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet. 2013;92(1):15–27. doi: 10.1016/j.ajhg.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wessel J, McDonald SM, Hinds DA, Stokowski RP, Javitz HS, Kennemer M, et al. Resequencing of nicotinic acetylcholine receptor genes and association of common and rare variants with the Fagerstrom test for nicotine dependence. Neuropsychopharmacology. 2010;35(12):2392–2402. doi: 10.1038/npp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie P, Kranzler HR, Krauthammer M, Cosgrove KP, Oslin D, Anton RF, et al. Rare nonsynonymous variants in alpha-4 nicotinic acetylcholine receptor gene protect against nicotine dependence. Biological psychiatry. 2011;70(6):528–536. doi: 10.1016/j.biopsych.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haller G, Druley T, Vallania FL, Mitra RD, Li P, Akk G, et al. Rare missense variants in CHRNB4 are associated with reduced risk of nicotine dependence. Hum Mol Genet. 2012;21(3):647–655. doi: 10.1093/hmg/ddr498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5(2):e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92 (6):841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi G, Rao DC. Optimum designs for next-generation sequencing to discover rare variants for common complex disease. Genet Epidemiol. 2011;35(6):572–579. doi: 10.1002/gepi.20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li MD, Beuten J, Ma JZ, Payne TJ, Lou XY, Garcia V, et al. Ethnic- and gender-specific association of the nicotinic acetylcholine receptor alpha4 subunit gene (CHRNA4) with nicotine dependence. Hum Mol Genet. 2005;14(9):1211–1219. doi: 10.1093/hmg/ddi132. [DOI] [PubMed] [Google Scholar]
- 21.Ma JZ, Beuten J, Payne TJ, Dupont RT, Elston RC, Li MD. Haplotype analysis indicates an association between the DOPA decarboxylase (DDC) gene and nicotine dependence. Hum Mol Genet. 2005;14(12):1691–1698. doi: 10.1093/hmg/ddi177. [DOI] [PubMed] [Google Scholar]
- 22.Beuten J, Ma JZ, Payne TJ, Dupont RT, Crews KM, Somes G, et al. Single- and Multilocus Allelic Variants within the GABAB Receptor Subunit 2 (GABAB2) Gene Are Significantly Associated with Nicotine Dependence. Am J Hum Genet. 2005;76(5):859–864. doi: 10.1086/429839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Yang Z, Ma JZ, Payne TJ, Li MD. Introduction to deep sequencing and its application to drug addiction research with a focus on rare variants. Molecular neurobiology. 2014;49(1):601–614. doi: 10.1007/s12035-013-8541-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20(1):110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutation research. 2007;615(1–2):28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83(3):311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91(2):224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42(11):969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 33.Saccone NL, Schwantes-An TH, Wang JC, Grucza RA, Breslau N, Hatsukami D, et al. Multiple cholinergic nicotinic receptor genes affect nicotine dependence risk in African and European Americans. Genes Brain Behav. 2010;9(7):741–750. doi: 10.1111/j.1601-183X.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen LS, Saccone NL, Culverhouse RC, Bracci PM, Chen CH, Dueker N, et al. Smoking and genetic risk variation across populations of European, Asian, and African American ancestry--a meta-analysis of chromosome 15q25. Genet Epidemiol. 2012;36(4):340–351. doi: 10.1002/gepi.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, et al. Variants in Nicotinic Receptors and Risk for Nicotine Dependence. Am J Psychiatry. 2008;165(9):1163–1171. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471(7340):597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lustig LR, Peng H. Chromosome location and characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) gene. Cytogenetic and genome research. 2002;98(2–3):154–159. doi: 10.1159/000069804. [DOI] [PubMed] [Google Scholar]
- 38.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic acids research. 2011;39:D225–229. doi: 10.1093/nar/gkq1189. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, et al. A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Molecular pharmacology. 2002;61(1):150–159. doi: 10.1124/mol.61.1.150. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen VT, Ndoye A, Grando SA. Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. The American journal of pathology. 2000;157(4):1377–1391. doi: 10.1016/s0002-9440(10)64651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng H, Ferris RL, Matthews T, Hiel H, Lopez-Albaitero A, Lustig LR. Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNA10) in lymphocytes. Life sciences. 2004;76 (3):263–280. doi: 10.1016/j.lfs.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 42.Lips KS, Pfeil U, Kummer W. Coexpression of alpha 9 and alpha 10 nicotinic acetylcholine receptors in rat dorsal root ganglion neurons. Neuroscience. 2002;115(1):1–5. doi: 10.1016/s0306-4522(02)00274-9. [DOI] [PubMed] [Google Scholar]
- 43.Greenbaum L, Kanyas K, Karni O, Merbl Y, Olender T, Horowitz A, et al. Why do young women smoke? I. Direct and interactive effects of environment, psychological characteristics and nicotinic cholinergic receptor genes. Mol Psychiatry. 2006;11(3):312–322. 223. doi: 10.1038/sj.mp.4001774. [DOI] [PubMed] [Google Scholar]
- 44.Rigbi A, Kanyas K, Yakir A, Greenbaum L, Pollak Y, Ben-Asher E, et al. Why do young women smoke? V. Role of direct and interactive effects of nicotinic cholinergic receptor gene variation on neurocognitive function. Genes Brain Behav. 2008;7(2):164–172. doi: 10.1111/j.1601-183X.2007.00329.x. [DOI] [PubMed] [Google Scholar]
- 45.Chikova A, Bernard HU, Shchepotin IB, Grando SA. New associations of the genetic polymorphisms in nicotinic receptor genes with the risk of lung cancer. Life Sci. 2012;91 (21–22):1103–1108. doi: 10.1016/j.lfs.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersson O, Stenqvist A, Attersand A, von Euler G. Nucleotide sequence, genomic organization, and chromosomal localization of genes encoding the human NMDA receptor subunits NR3A and NR3B. Genomics. 2001;78(3):178–184. doi: 10.1006/geno.2001.6666. [DOI] [PubMed] [Google Scholar]
- 47.Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, et al. Cysteine regulation of protein function--as exemplified by NMDA-receptor modulation. Trends in neurosciences. 2002;25(9):474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 48.Ciabarra AM, Sullivan JM, Gahn LG, Pecht G, Heinemann S, Sevarino KA. Cloning and characterization of chi-1: a developmentally regulated member of a novel class of the ionotropic glutamate receptor family. J Neurosci. 1995;15(10):6498–6508. doi: 10.1523/JNEUROSCI.15-10-06498.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sucher NJ, Akbarian S, Chi CL, Leclerc CL, Awobuluyi M, Deitcher DL, et al. Developmental and regional expression pattern of a novel NMDA receptor-like subunit (NMDAR-L) in the rodent brain. J Neurosci. 1995;15(10):6509–6520. doi: 10.1523/JNEUROSCI.15-10-06509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Otano I, Schulteis CT, Contractor A, Lipton SA, Trimmer JS, Sucher NJ, et al. Assembly with the NR1 subunit is required for surface expression of NR3A-containing NMDA receptors. J Neurosci. 2001;21(4):1228–1237. doi: 10.1523/JNEUROSCI.21-04-01228.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sasaki YF, Rothe T, Premkumar LS, Das S, Cui J, Talantova MV, et al. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. Journal of neurophysiology. 2002;87 (4):2052–2063. doi: 10.1152/jn.00531.2001. [DOI] [PubMed] [Google Scholar]
- 52.Eriksson M, Nilsson A, Froelich-Fabre S, Akesson E, Dunker J, Seiger A, et al. Cloning and expression of the human N-methyl-D-aspartate receptor subunit NR3A. Neuroscience letters. 2002;321(3):177–181. doi: 10.1016/s0304-3940(01)02524-1. [DOI] [PubMed] [Google Scholar]
- 53.Ma JZ, Payne TJ, Nussbaum J, Li MD. Significant association of glutamate receptor, ionotropic N-methyl-D-aspartate 3A (GRIN3A), with nicotine dependence in European- and African-American smokers. Hum Genet. 2010;127(5):503–512. doi: 10.1007/s00439-010-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu HP, Lin WY, Liu SH, Wang WF, Tsai CH, Wu BT, et al. Genetic variation in N-methyl-D-aspartate receptor subunit NR3A but not NR3B influences susceptibility to Alzheimer’s disease. Dementia and geriatric cognitive disorders. 2009;28(6):521–527. doi: 10.1159/000254757. [DOI] [PubMed] [Google Scholar]
- 55.Takata A, Iwayama Y, Fukuo Y, Ikeda M, Okochi T, Maekawa M, et al. A population-specific uncommon variant in GRIN3A associated with schizophrenia. Biol Psychiatry. 2013;73(6):532–539. doi: 10.1016/j.biopsych.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 56.Marco S, Giralt A, Petrovic MM, Pouladi MA, Martinez-Turrillas R, Martinez-Hernandez J, et al. Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington’s disease models. Nature medicine. 2013;19(8):1030–1038. doi: 10.1038/nm.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li MD, Ma JZ, Payne TJ, Lou XY, Zhang D, Dupont RT, et al. Genome-wide linkage scan for nicotine dependence in European Americans and its converging results with African Americans in the Mid-South Tobacco Family sample. Mol Psychiatry. 2008;3 (4):407–416. doi: 10.1038/sj.mp.4002038. [DOI] [PubMed] [Google Scholar]
- 58.Li MD, Payne TJ, Ma JZ, Lou XY, Zhang D, Dupont RT, et al. A genomewide search finds major susceptibility Loci for nicotine dependence on chromosome 10 in african americans. Am J Hum Genet. 2006;79(4):745–751. doi: 10.1086/508208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang W, Payne TJ, Ma JZ, Li MD. A functional polymorphism, rs6280, in DRD3 is significantly associated with nicotine dependence in European-American smokers. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1109–1115. doi: 10.1002/ajmg.b.30731. [DOI] [PubMed] [Google Scholar]
- 61.Huang W, Ma JZ, Payne TJ, Beuten J, Dupont RT, Li MD. Significant association of DRD1 with nicotine dependence. Hum Genet. 2008;123(2):133–140. doi: 10.1007/s00439-007-0453-9. [DOI] [PubMed] [Google Scholar]
- 62.Mangold JE, Payne TJ, Ma JZ, Chen G, Li MD. Bitter taste receptor gene polymorphisms are an important factor in the development of nicotine dependence in African Americans. J Med Genet. 2008;45(9):578–582. doi: 10.1136/jmg.2008.057844. [DOI] [PubMed] [Google Scholar]
- 63.Cui WY, Wang S, Yang J, Yi SG, Yoon D, Kim YJ, et al. Significant association of CHRNB3 variants with nicotine dependence in multiple ethnic populations. Mol Psychiatry. 2013;18(11):1149–1151. doi: 10.1038/mp.2012.190. [DOI] [PubMed] [Google Scholar]
- 64.Beuten J, Ma JZ, Payne TJ, Dupont RT, Lou XY, Crews KM, et al. Association of Specific Haplotypes of Neurotrophic Tyrosine Kinase Receptor 2 Gene (NTRK2) with Vulnerability to Nicotine Dependence in African-Americans and European-Americans. Biological psychiatry. 2007;61(1):48–55. doi: 10.1016/j.biopsych.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 65.Beuten J, Ma JZ, Payne TJ, Dupont RT, Crews KM, Somes G, et al. Single- and multilocus allelic variants within the GABA(B) receptor subunit 2 (GABAB2) gene are significantly associated with nicotine dependence. Am J Hum Genet. 2005;76(5):859–864. doi: 10.1086/429839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Q, Huang W, Payne TJ, Ma JZ, Li MD. Detection of genetic association and a functional polymorphism of dynamin 1 gene with nicotine dependence in European and African Americans. Neuropsychopharmacology. 2009;34(5):1351–1359. doi: 10.1038/npp.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei J, Ma JZ, Payne TJ, Cui W, Ray R, Mitra N, et al. Replication and extension of association of choline acetyltransferase with nicotine dependence in European and African American smokers. Hum Genet. 2010;127(6):691–698. doi: 10.1007/s00439-010-0818-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun D, Ma JZ, Payne TJ, Li MD. Beta-arrestins 1 and 2 are associated with nicotine dependence in European American smokers. Mol Psychiatry. 2008;13(4):398–406. doi: 10.1038/sj.mp.4002036. [DOI] [PubMed] [Google Scholar]
- 69.Li MD, Xu Q, Lou XY, Payne TJ, Niu T, Ma JZ. Association and interaction analysis of variants in CHRNA5/CHRNA3/CHRNB4 gene cluster with nicotine dependence in African and European Americans. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(3):745–756. doi: 10.1002/ajmg.b.31043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Density plot of estimated minor allele frequency (MAF) distributions from pooled-sequencing analysis of the 200 sib pairs. Many more rare variants were discovered in AAs than in EAs.
Figure S2. Scatter plots of principal components 1 and 2 for smokers and non-smokers based on principal component analysis of 49 and 51 common variants available for AAs and EAs, respectively. Smokers and non-smokers are uniformly mixed within the two ethnic groups. Significant population stratification was not detected between smokers and non-smokers.