

ABSTRACT
Skin is the most common site of Staphylococcus aureus infection. While most of these infections are self-limited, recurrent infections are common. Keratinocytes and recruited immune cells participate in skin defense against infection. We postulated that S. aureus is able to adapt to the milieu within human keratinocytes to avoid keratinocyte-mediated clearance. From a collection of S. aureus isolated from chronically infected patients with atopic dermatitis, we noted 22% had an agr mutant-like phenotype. Using several models of human skin infection, we demonstrate that toxin-deficient, agr mutants of methicillin-resistant S. aureus (MRSA) USA300 are able to persist within keratinocytes by stimulating autophagy and evading caspase-1 and inflammasome activation. MRSA infection induced keratinocyte autophagy, as evidenced by galectin-8 and LC3 accumulation. Autophagy promoted the degradation of inflammasome components and facilitated staphylococcal survival. The recovery of more than 58% agr or RNAIII mutants (P < 0.0001) of an inoculum of wild-type (WT) MRSA from within wortmannin-treated keratinocytes compared to control keratinocytes reflected the survival advantage for mutants no longer expressing agr-dependent toxins. Our results illustrate the dynamic interplay between S. aureus and keratinocytes that can result in the selection of mutants that have adapted specifically to evade keratinocyte-mediated clearance mechanisms.
IMPORTANCE
Human skin is a major site of staphylococcal infection, and keratinocytes actively participate in eradication of these pathogens. We demonstrate that methicillin-resistant Staphylococcus aureus (MRSA) is ingested by keratinocytes and activates caspase-1-mediated clearance through pyroptosis. Toxin-deficient MRSA mutants are selected within keratinocytes that fail to induce caspase-1 activity and keratinocyte-mediated clearance. These intracellular staphylococci induce autophagy that enhances their intracellular survival by diminishing inflammasome components. These findings suggest that S. aureus mutants, by exploiting autophagy, can persist within human keratinocytes.
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) USA300 is the major cause of skin and soft tissue infections in the United States (1, 2), usually infecting patients with no underlying immune defects (3). While these skin infections are typically local, they provide a source of organisms for recurrent/persistent colonization and a reservoir for systemic dissemination. The host response to skin infection is shared by local keratinocytes and immune cells that are recruited to the disruption in the epithelial barrier (4, 5). Despite the recruitment of neutrophils once infection is established at a cutaneous site, it can be difficult to clear and may require surgical drainage, even if appropriate antibiotics are employed (6).
Human skin is a complex immune and physical barrier (4) composed of multiple layers of proliferating and differentiating keratinocytes linked by tight junctions (7). Keratinocyte production of antimicrobial peptides to kill bacteria is well established, whereas exactly how keratinocytes kill ingested bacteria, including staphylococci, is not fully understood. S. aureus-induced keratinocyte death may result in death of the bacteria or release of organisms to be cleared by recruited phagocytes. Compensation for keratinocyte loss induced by infection is ongoing, as human keratinocytes are continually in the process of proliferation, maturation, and cell death (7). Keratinocytes undergo pyroptosis, a caspase-1-dependent activation of the NLRP3 inflammasome resulting in cell death (8–10). This is triggered by α-hemolysin (Hla), additional staphylococcal toxins, including the Panton-Valentine leukocidin (PVL) (11) and other two-component toxins (12) which are under control of the agr locus (13). Caspase-1-dependent pyroptosis results in the production of interleukin 1β (IL-1β) to recruit neutrophils, a process facilitated by the constitutive expression of pro-IL-1β in keratinocytes (8). Activation of the inflammasome functions to eradicate infecting organisms and to recruit neutrophils to eliminate extracellular bacteria (14, 15).
S. aureus has evolved multiple mechanisms to promote survival within the context of human skin (16, 17). Differentiating keratinocytes are actively undergoing autophagy (18), a process that is often important in the clearance of intracellular pathogens and provides a source of nutrients through catabolism (19, 20). Autophagy can also serve to limit the availability of inflammasome components and decrease proinflammatory signaling (20, 21). If keratinocytes contribute significantly to S. aureus defense, it is likely that these organisms have acquired mechanisms to evade keratinocyte-mediated clearance (16). We postulated that MRSA USA300 evades keratinocyte-mediated clearance through the selection of toxin-deficient mutants that can persist intracellularly. Using both laboratory-derived mutants of the epidemic MRSA USA300 strain LAC and S. aureus isolated from atopic dermatitis (AD) patients, we demonstrate selection of toxin-deficient agr mutants within autophagic keratinocytes that have increased ability to persist within human skin.
RESULTS
Characterization of S. aureus from atopic dermatitis patients.
Atopic dermatitis (AD) is a common inflammatory skin condition affecting 20% of the population. AD patients typically have chronic or recurrent S. aureus infection (22); thus, they are a likely source for S. aureus strains that have adapted to human keratinocytes. As part of an ongoing study to correlate S. aureus genotypes, phenotypes, and clinical outcomes in AD patients, 85 S. aureus isolates from superficial skin cultures of 133 patients were characterized, and 22% were noted to be toxin deficient, as defined by lack of hemolysis on sheep blood agar plates, likely indicating mutation in the agr or hla locus (23). We postulated that strains lacking toxin production would be less likely to activate keratinocyte clearance mechanisms and might have increased intracellular persistence. The first 10 of these isolates for which whole-genome sequences were available were screened for survival within keratinocytes using a gentamicin protection assay and for induction of IL-1β as a marker of inflammasome activation compared with wild-type (WT) MRSA USA300 LAC and an agr null mutant control (Fig. 1A). The results of four typical AD strains are shown; both the agr control strain and strain AD4 had significantly increased intracellular persistence at 24 h compared to the WT USA300. Strain AD7 had an intermediate phenotype, whereas strains AD5 and AD10 were more similar to the USA300 control and did not accumulate to any appreciable extent within the keratinocytes. The strains with increased intracellular persistence (AD4 and AD7), as well as the agr control, induced less IL-1β, a marker of inflammasome activation (Fig. 1B). This correlated with lack of detectable Hla (α-hemolysin) (Fig. 1C) and significantly decreased expression of RNAIII by AD4 (Fig. 1D). AD7 was found to have a stop codon mutation in the hla locus consistent with its lack of Hla production (Fig. 1E). Comparison of whole-genome sequences of these strains to the appropriate agr reference strains revealed no additional mutations in the agr loci. While AD5 behaved much like the prototypic USA300 reference strain, AD10, which was efficiently cleared from the keratinocytes, expressed relatively less Hla and induced modest amounts of IL-1β, suggesting additional mechanisms of clearance from within the keratinocyte.
FIG 1 .

Characterization of S. aureus isolated from atopic dermatitis (AD) patients. (A) Intracellular persistence within HaCaT keratinocytes detected by gentamicin protection assays of S. aureus isolates from AD patients compared with WT MRSA USA300 LAC and agr null mutant controls. (B and C) IL-1β in HaCaT cell supernatant at 24 h postinfection (B) and the corresponding immunoblot for Hla production (C). Values that are significantly different (P < 0.05) from the WT value in panel B are indicated by three asterisks. (D) RNAIII expression. (E) The Hla locus of strain AD7 contains a stop codon mutation consistent with lack of Hla production. (F) Hld presence and expression, as detected by synergistic hemolysis with Hlβ in the collection of AD clinical isolates (MSSA, methicillin-sensitive S. aureus), suggesting that most are likely to express Hld. Gentamicin protection assays were performed at least three separate times; the results of a representative experiment are shown. The graphs in panels A, B and D show the means plus standard deviations (SD) (error bars) of sextuplicate samples.
To ascertain whether additional hemolysins contribute to clearance from keratinocytes, we screened for expression of delta-hemolysin (Hld) (phenol-soluble modulin gamma [Psm-γ]). The S. aureus delta-hemolysin has been suggested to be important in the pathogenesis of AD through its targeting of mast cells (24), and Hld mutants have been linked to chronic bone infection (25). Accordingly, the AD isolates were all screened for the presence and expression of the hld locus. The clinical isolates, including AD10, contained the hld locus (Fig. 1F). Expression of Hld in the total collection of AD isolates was assessed by enhancement of hemolysis in the presence of the β-hemolysin (Hlb) (strain RN4220) determined by cross-streaking (with the caveat that this is not completely specific for Hld) (26). Hld production in the characterized AD strains did correlate with intracellular persistence. To better characterize how S. aureus induced keratinocyte-mediated clearance, we more critically analyzed the intracellular uptake and persistence of the common WT USA300 LAC and defined agr and toxin-deficient mutants.
Increased intracellular persistence of toxin-deficient mutants.
The relative ability of the WT and USA300 mutants to persist intracellularly was quantified using a gentamicin protection assay in the HaCaT cell line and in primary keratinocytes (HEKn) (Fig. 2). The agr locus regulates expression of several toxins, including Hla and Psmα, and affects PVL production (lukS and lukF) as well (27, 28). Uptake of WT and agr null mutant strains into the HaCaT keratinocytes was equivalent and mediated by the fibronectin binding proteins (29), primarily FnBPA (fibronectin binding protein A) (Fig. 2A and B). USA300 mutants lacking hla or agr accumulated to a significantly greater extent within keratinocytes than WT organisms did, and a small but significant fraction persisted intracellularly for more than 48 h (Fig. 2C); lack of PVL expression did not enhance intracellular persistence under these conditions. Psmα mutants, but not Psmβ or Psmδ mutants, had increased intracellular persistence at 24 h (Fig. 2C). agr mutants with other genetic backgrounds had significantly increased intracellular survival followed for up to 96 h (Fig. 2D). Controls for keratinocyte viability using trypan blue exclusion (Fig. 2E) or lactate dehydrogenase (LDH) release (Fig. 2F) after a 24-h incubation with the various S. aureus strains were not significantly different, suggesting that only a small fraction of the cells in culture are infected and killed by staphylococci.
FIG 2 .

Uptake and intracellular survival of S. aureus USA300 mutants. (A) Uptake of WT S. aureus and agr mutant by HaCaT cells determined by flow cytometry. (B to D) Gentamicin protection assays were performed to quantify the ability of WT and mutant strains to persist within human keratinocytes (B), toxin mutants (C), and agr mutants (D). (B) FnBP mutants (fnbA or fnbB) in the presence of fibronectin (+ Fn) or with no Fn. (C) Toxin mutants hla (α-hemolysin), agr, Panton-Valentine leukocidin-negative (PVL−) (lukS and lukF), and phenol-soluble modulins (psm) α, β, and ε (hld) and triple mutant mutants in HaCaT and HEKn cells (human keratinocytes in primary culture). (D) agr mutant (strain RN7206) in strain RN6374 and agr mutant strain UAMS-1 in UAMS-155 strain. (E) HaCaT viability following 24 h of incubation with the strains listed by trypan blue exclusion. (F) Cytotoxicity as detected by LDH assay. Values that are significantly different from the value for the WT strain under the same condition by one-way ANOVA and Dunnett’s posttest are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. Each experiment was performed at least three times, and the results of a representative assay are shown. Data graphed display the means of sextuplicate samples plus standard deviations (SD) (error bars).
MRSA USA300 bacteria escape from the endosome to cause keratinocyte pyroptosis.
For S. aureus to gain access to the keratinocyte cytosol, the staphylococci must first escape from the endosome, a process associated with toxin and Psmα expression (30). S. aureus can be cleared by the phagolysosome (31) or escape to the cytosol where organisms expressing toxin could activate caspase-1-mediated pyroptosis through the NLRP3 inflammasome (9, 10), resulting in keratinocyte death (32). Alternative mechanisms of cell death induced by staphylococci include apoptosis, a caspase-3-dependent process, and necroptosis, a RIP1-RIP3-MLKL (RIP1 stands for receptor-interacting protein 1 and MLKL stands for mixed-lineage kinase domain-like) mode of cell death (33). Using keratinocytes in primary culture as well as the HaCaT cell line, we found that caspase-1 inhibition, but not caspase-3 inhibition, resulted in significantly increased intracellular survival of S. aureus USA300 at 24 h (Fig. 3A). Caspase-1 inhibition also increased the intracellular persistence of the agr mutant (Fig. 3B), suggesting that additional non-agr-dependent gene products may contribute to inflammasome activation and that agr mutants can escape from the keratinocyte endosome. The effect of caspase-1 inhibition on staphylococcal persistence suggested that many of the endocytosed staphylococci readily gain access to the cytosol. Treating HaCaT cells with chloroquine or balflinocycin to limit endosomal acidification had no significant effect upon the numbers of staphylococci retained (Fig. 3C). Imaging studies were performed using organotypic cultures; human skin grafts were grown on a feeder layer of fibroblasts to determine the location of S. aureus USA300 within the multiple strata of normal human skin as well their distribution within individual skin cells (Fig. 3D). S. aureus was observed within the stratum granulosum. In electron micrographs, both WT and agr mutant staphylococci appeared to be proliferating within the keratinocytes, not necessarily within membrane-bound compartments. Confocal imaging using either EEA-1, a fluorescent marker for the early endosome, or monodansyl cadaverine (MDC), a marker for the late autophagosome (34), demonstrated colocalization of WT and agr mutants within autophagosomal compartments; however, some organisms were apparently free in the cytosol, indicating that the agr null mutants are not limited to the endosome, and the WT USA300 bacteria are not exclusively cytosolic in human keratinocytes (Fig. 3F).
FIG 3 .

Intracellular persistence of S. aureus USA300 in keratinocytes treated with inhibitors of caspases and endosomal acidification. (A and B) Intracellular persistence of WTS. aureus USA300 (A) and USA300 agr null mutant (B) in the presence of caspase-1 or caspase-3 inhibitors in HaCaT and HEKn cells. (C) Effects of chloroquine and bafilomycin pretreatment on intracellular persistence. Values in panels A to C are the means plus standard deviations (SD) (error bars) of sextup licate wells. (D and E) Light micrographs (D) and electron micrographs (E) of WT and agr mutant-infected organotypic cultures (skin grafts maintained on fibroblast feeder cells), showing accumulation of staphylococci in the stratum granulosum (black arrows) and organisms that do not appear to be within a membrane-bound compartment. The photomicrographs in panels D and E are representative images of infections using at least three separate grafts and multiple sections of each graft. (F) Confocal images of infected HaCaT cells with fluorescent labeling of EEA-1 (early endosome [green]) and MDC (autophagosome [green]); demonstrating some colocalization (yellow) of both WT S. aureus and S. aureus agr mutant (red) with the autophagosomal MDC marker, but not with the early endosome EEA-1 at 24 h postinfection. Intracellular persistence assays were performed at least three times, and the results of a representative experiment are shown. Confocal imaging was done at least twice on separate samples, and representative images are shown.
WT MRSA USA300 and agr null mutant induce autophagy in human skin grafts.
The ingestion of staphylococci by keratinocytes is a likely stimulus for autophagy, a process which generates energy in response to metabolic or other types of stress by inducing degradation of cellular organelles, including those containing pathogens (35, 36). To characterize the interplay between S. aureus and autophagy in human keratinocytes, we used human skin grafts maintained on SCID mice to follow the consequences of S. aureus infection (see Fig. S1 in the supplemental material). These grafts reflect the expected architecture of healthy skin with a basal proliferative layer, the differentiating keratinocytes of the stratum spinosum and stratum granulosum, and the terminally differentiated stratum corneum at the surface (37) (Fig. 4A). Staphylococci were visualized intercalating in between adjacent corneocytes, indicating that they could infect the epidermis without major disruption of the epithelial barrier (Fig. 4B). Pathology was apparent 72 h following the application of 5 × 108 CFU of strain USA300 in 10 µl of phosphate-buffered saline (PBS) onto the surface of the intact graft (Fig. 4C). Infection with the WT organisms resulted in hyperkeratosis of the stratum corneum with small mounds of serum and neutrophilic crust, with very few bacterial colonies apparent. Many more clusters of bacteria are seen in the agr mutant-infected graft (Fig. 4D). Neutrophil infiltration is apparent in WT MRSA-infected grafts (Fig. 4E); hyperkeratosis is observed, but bacteria are not visualized. The stratum corneum of the agr mutant-infected grafts also shows hyperkeratosis and parakeratosis (Fig. 4F). Significantly, there is broad erosion of the epidermis, covered by fibrin and numerous neutrophils seen at higher magnification, and bacteria are observed within vacuoles (Fig. 4G). Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining revealed the fragmented DNA of dead cells in the infected grafts, but not in PBS controls, consistent with keratinocyte death as a mechanism of staphylococcal clearance (Fig. 4H).
FIG 4 .

Histology of WT S. aureus USA300 and agr mutant infection (108 CFU applied in 10 μl of PBS) of human skin grafts maintained on SCID mice at 72 h postinfection. (A to D) Gram staining with trichrome counterstaining of PBS control (A) or S. aureus infection (B to D). (B) WT S. aureus infection at 72 h, showing WT USA300 intercalation through corneocytes. (C and D) WT S. aureus (C) and agr mutant (D) infection demonstrating clusters of staphylococci (black arrows) in the agr mutant-infected graft. (E to G) Hematoxylin and eosin staining of WT and agr mutant-infected grafts demonstrating significant corneal erosion and clusters of neutrophils (yellow arrows) in the WT infection. (F and G) agr mutant infection is associated with more significant corneal erosion and neutrophilic accumulation (yellow arrows), but clusters of staphylococci are also noted (black arrows). (D) TUNEL staining (brown) of PBS- and WT USA300-exposed sections, demonstrating focal cell death in the stratum granulosum. At least three different mice were grafted and infected with WT or agr mutant or functioned as a PBS control. Numerous sections were obtained from each mouse, and representative images are shown.
S. aureus infection induces autophagy in human skin.
Confocal imaging of the infected grafts was done to assess accumulation of the markers of autophagy. Galectin-8 which is recruited to damaged endosomes, especially those damaged by pathogens and targets them for autophagosomal clearance (38), colocalized with the organisms in the infected skin grafts (Fig. 5A) and was more abundant in the agr null mutant infections. LC3, a marker for autophagosome formation (34), was detected in grafts infected with WT S. aureus and more prominently with agr null mutant S. aureus, but it did not colocalize with the organisms at this time point, 3 days postinoculation. Images of spa null mutant, agr mutant, and WT staphylococci are shown (see Fig. S2 in the supplemental material) to demonstrate that protein A-associated binding of the antistaphylococcus antibody is not responsible for apparent differences in the numbers of fluorescence-labeled WT and agr mutant staphylococci. Control images using the secondary antibody alone were entirely negative.
FIG 5 .

S. aureus induces autophagy in keratinocytes. (A) Confocal images of galectin-8 (red)- or LC3 (red)-labeled sections of SCID-hu grafts from mice infected with WT S. aureus (SA) or agr mutant (green). Representative images from two separate experiments are shown. (B) Electron micrograph of cells infected with agr null mutant demonstrating the double membrane surrounding staphylococci and adjacent mitochondria. (C) Intracellular recovery of WT S. aureus USA300 and agr mutant from HaCaT and HEKn cells treated with wortmannin (24-h infection) or 3-MA (2-h infection). Data shown represent the means plus standard errors of sextuplicate samples; at least two independent experiments were done, and the results of a representative experiment are shown. The P values in the graphs compare the value for the strain shown to the value for no inhibitor by Student’s t test. (D) Immunoblot of WT S. aureus- or agr mutant-infected HaCaT cells compared to a PBS control detecting galectin-8 and LC3II. (E) Flow cytometric analysis of autophagy in HaCaT cells exposed to WT USA300 or agr null mutant by Cyto-ID (LC3) staining in the presence of wortmannin, 3-MA (negative control), and chloroquine (positive control) to increase autophagosome formation.
Electron micrographs of infected human keratinocytes in organotypic cultures revealed S. aureus USA300 agr mutant within a characteristic double-membrane-bound compartment, seen in apposition to mitochondria, typical of the autophagosome (20) (Fig. 5B). To determine how induction of autophagy affects the intracellular persistence of S. aureus, we treated keratinocytes with the autophagy inhibitor wortmannin or 3-methyladenine (3-MA), which when used for a limited time, has a similar effect (39), and quantified USA300 staphylococci by gentamicin protection assay (Fig. 5C). For both the WT and agr mutant, there was significantly decreased intracellular survival in the HaCaT and primary keratinocytes treated with inhibitors of autophagy, suggesting that these organisms may be exploiting autophagy to persist within the keratinocytes. Immunoblots performed with HaCaT cells at early time points after S. aureus infection indicated induction of galectin-8 by 2 h postinfection, but no consistent changes in LC3II (Fig. 5D).
The relative induction of autophagy by WT and agr null mutant staphylococci was evaluated using a fluorescence microscopy and cytometric analysis of LC3 (Cyto-ID [Enzo Life Sciences]) (40) (Fig. 5E). Infection with WT USA300 induced Cyto-ID staining in 4.9% of the cells compared with 34% of the cells exposed to the agr null mutant. Treatment of the keratinocytes with wortmannin to inhibit autophagy decreased the numbers of either WT or agr null mutant staphylococci that were Cyto-ID positive as expected, and 3-MA essentially blocked autophagy altogether in this assay system. Chloroquine, by decreasing acidification of the endosome, resulted in increased Cyto-ID-positive cells, consistent with the interference of autophagosomal clearance (41). These data suggest that S. aureus that can persist within keratinocytes may exploit autophagy to evade clearance.
Induction of autophagy enhances MRSA persistence through effects on the inflammasome.
In immune cells, induction of autophagy results in consumption of nonessential cellular components, including the inflammasome (42). Autophagic degradation of the NLRP3 component ASC (apoptosis-associated speck-like protein containing CARD [caspase activation and recruitment domain]), an adaptor protein that functions in inflammasome assembly, limits pyroptosis, IL-1β production (21), and inflammatory signaling (43) important in neutrophil recruitment (44) and critical for S. aureus clearance in vivo. S. aureus-infected HaCaT cells were pretreated with wortmannin to block autophagy, and relative amounts of ASC were assessed by immunoblotting. The wortmannin-treated keratinocytes were found to have increased amounts of ASC in S. aureus-infected cells compared to medium controls (Fig. 6A). To confirm that inhibition of autophagy increases inflammasome function, small interfering RNA (siRNA) knockdown of two components of the autophagy pathway, Nod2, a cytosolic receptor for S. aureus cell wall components (45), and ATG5 (35) was performed, and the effects on IL-1β production were measured (Fig. 6B to E). Inhibition of autophagy resulted in increased induction of IL-1β, consistent with the proposed effect of autophagy on inflammasome-mediated signaling. To determine whether the clinical AD isolates of S. aureus similarly induce autophagy in human keratinocytes, we monitored infected SCID mouse-human (SCID-hu) grafts for the relative induction of galectin-8 and LC3 in response to S. aureus AD4 or AD10 infection (Fig. 6F and G). The Hla-deficient strain AD4, which had increased intracellular persistence in HaCaT cells (Fig. 1), was not effectively cleared from the skin graft at 72 h postinoculation and was readily detected within the stratum granulosum. AD4 colocalized with the autophagosomal marker galcetin-8 but not with LC3. This was in contrast to AD10; the AD10 strain that was efficiently cleared from HaCaT cells was also cleared from the human grafts at 72 h.
FIG 6 .

Induction of autophagy decreases inflammasome activation. (A) Immunoblot of HaCaT cells detecting ASC in HaCaT cells in the presence (+) or absence (−) of wortmannin (Wm) after 2 h of infection with WT S. aureus USA300 or agr mutant and corresponding densitometry standardized to the GAPDH control. (B and C) Immunoblots of HaCaT cells detecting Nod2 (B) and ATG5 (C) knockdown by siRNA compared to nontargeting siRNA pool control in HaCaT cells exposed to media (med) or to WT S. aureus or agr mutant. (D and E) IL-1β production determined by ELISA in the corresponding Nod2 and ATG5 knockdowns and nontargeting siRNA-transfected controls all at 24 h postinfection. (F and G) Light microscopy of Gram-positive trichrome-stained (F) and confocal images (G) of sections obtained from S. aureus AD4- and AD10-infected human skin grafts on SCID mice after 72 h of infection using anti-S. aureus antibody (SA) (green), anti-galectin-8 (red), or anti-LC3 (red). Colocalization (yellow) of galectin-8 and the AD4 isolate is observed, but not with the AD10 strain. siRNA knockdowns were done twice, and the results of a representative experiment are shown.
Selection of agr mutants from within infected keratinocytes.
S. aureus bacteria readily adapt to environmental immune pressure through the selection and proliferation of specific mutants (16). We postulated that WT organisms cleared by caspase-1-dependent pyroptosis in the cytosol would be at a selective disadvantage within keratinocytes compared to the agr mutant bacteria, which lacking toxin secretion, fail to trigger inflammasome-mediated clearance. Moreover, under autophagic conditions and diminished inflammasome activity, intracellular accumulation of sufficient numbers of WT organisms would enable selection of spontaneous agr-like mutants. To determine whether this selective process occurs in infected keratinocytes, we screened the phenotypes of S. aureus recovered from within keratinocytes infected with WT S. aureus alone or with inhibitors or with a mixed WT-agr mutant (1:1) infection using hemolysis on blood agar plates as a marker for WT (Hla toxin-producing) colonies (Fig. 7A). Following WT staphylococcal infection, 6.5% of the colonies recovered from within the keratinocytes at 24 h were nonhemolytic. In a mixed WT-agr mutant (1:1) infection, the staphylococci recovered were predominantly (83.3%) nonhemolytic (P < 0.0001 compared to the value for the WT). In wortmannin-treated cells, only 41.4% of the recovered staphylococci retained the WT phenotype, in contrast to 93.5% of the colonies infecting keratinocytes under control conditions. Control experiments were performed to verify that wortmannin or small amounts of gentamicin that might gain access to the cytosol of S. aureus (in the absence of the keratinocytes) did not induce the generation of agr mutants (see Fig. S3 in the supplemental material). The increased recovery of the toxin-deficient mutants from within keratinocytes is consistent with their ability to avoid inflammasome-mediated clearance, which would be enhanced in the absence of autophagy. Analysis of 10 nonhemolytic colonies isolated from keratinocytes originally infected with WT USA300 in wortmannin-treated cells revealed loss of Hla production, a major toxin associated with activation of the inflammasome (Fig. 7B) and/or lack of RNAIII, a mediator of agr signaling and Hla production (13) (Fig. 7C). Thus, there is active selection of toxin-deficient S. aureus within keratinocytes that is influenced by the effects of autophagy in enhancing intracellular survival.
FIG 7 .
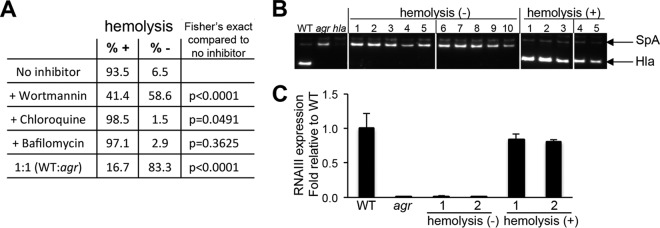
Selection of S. aureus mutants from within infected HaCaT cells. (A) Phenotypes of colonies recovered from HaCaT cells infected with WT S. aureus USA300 alone or with inhibitors of autophagy or endosomal acidification or with a mixed WT-agr mutant (1:1) infection. The percentages of colonies that are hemolysis positive (+) or negative (−) are shown. (B) Hla expression detected by immunoblotting in colonies recovered in the presence of wortmannin (the increased expression of surface protein A [SpA] in the agr or hla mutant is noted). (C) RNAIII expression in representative colonies. At least four separate experiments were performed with recovery of approximately 100 to 150 WT colonies. The data shown represent a compilation of the results of four experiments at the 24-h time point.
DISCUSSION
Intact human skin is remarkably resistant to S. aureus infection, even by the highly virulent USA300 strains. Nonetheless, staphylococcal skin infection usually initiated through autoinoculation of inapparent breaks in the integrity of the epidermis or at sites of trauma, is a major clinical problem. Our data suggest that human keratinocytes participate in USA300 clearance through many of the same mechanisms as have been well described for immune cells. These mechanisms include uptake by α5β1 integrins, escape from the endosome that is not dependent upon expression of agr-associated toxins, and induction of pyroptosis, suggested by caspase-1-associated cytotoxicity and production of IL-1β. S. aureus infection stimulates keratinocyte autophagy, which does not appear to contribute to eradication, but instead facilitates intracellular persistence through suppression of inflammasome signaling, a mechanism of immune evasion shared by many human pathogens (46).
S. aureus clearance through endosomal acidification appears less important in keratinocytes than has been reported for immune cells and other types of epithelial and endothelial cells (30, 31, 47, 48). Endosomal escape and the ability to survive within the cytosol are likely dependent on both the strain and cell type. For example, in studies using S. aureus RN4220 and HeLa cells, endosomal escape was mediated by the agr-dependent delta toxin synergizing with theβ-toxin (30), whereas escape from the RHEK-1 keratinocyte cell line required PVL production in a community-acquired MRSA (CA-MRSA) strain from Taiwan (11). Studies with a number of more-toxigenic strains (MW2, LAC, and USA400) indicated that Psmα expression is critical for endosomal escape and cytosolic replication in 293 (human embryonic kidney), THP-1, and endothelial cell lines (30). By using human keratinocyte lines, cells in primary culture, and human skin grafts in situ, we attempted to model more closely what occurs in differentiated human skin. Our data suggest that keratinocytes rapidly clear even a high inoculum of staphylococciand that this occurs at least partially through staphylococcal escape from the endosome and activation of the inflammasome and pyroptosis. As human keratinocytes have constitutive expression of pro-IL-1β, they may be primed for inflammasome activation (8), and ongoing proliferation and differentiation compensate for loss of infected cells. TUNEL staining of the USA300-infected human grafts but not PBS controls delineated occasional TUNEL-positive dead keratinocytes, consistent with the induction of pyroptosis by the WT S. aureus infection, and consequent death of the host cell.
Keratinocyte ingestion of S. aureus, by either WT or agr mutants, stimulates autophagy. This was detected in the differentiated human skin grafts as well as in the keratinocyte lines. Keratinocytes are continually being replaced, thus there is ongoing autophagy as part of their self-replacement program (18). By immunoblotting and immunofluorescence imaging (Cyto-ID), we noted a background level of LC3-positive cells that was increased by WT and especially by agr mutant S. aureus infection. The agr mutant staphylococci that do not activate the inflammasome were occasionally observed within the classic double-membrane-bound compartment typical of the autophagosome. However, evidence of the classic autophagic pathway and staphylococcal removal through LC3-associated phagocytosis was not observed (35). A substantial number of ingested staphylococci colocalized with galetin-8, which labels the bacterially damaged endosome/autophagosome, as well as with MDC, a marker for the late autophagosome (34). As inhibitors of autophagy increased staphylococcal clearance from within the keratinocyte, it appears that this pathway is activated but ineffective in S. aureus eradication.
S. aureus adaptation to the intracellular milieu of human keratinocytes was enhanced by autophagy. Autophagic keratinocytes did not eradicate the infecting organisms; instead, they provided a milieu more conducive to intracellular persistence. S. aureus-induced autophagy targeted inflammasome components, specifically ASC, resulting in decreased generation of IL-1β and inhibition of pyroptosis, a consequence of autophagy that has been well described for immune cells (21). Consistent with our observations that caspase-1-dependent pyroptosis is a major mechanism of keratinocyte clearance of S. aureus infection (8), consumption of inflammasome components as a consequence of autophagy served to increase intracellular survival. When autophagy was limited pharmacologically, there was greater selective pressure for recovery of agr-like mutants from within the keratinocytes, mutants that could not activate pyroptosis and clearance.
Enhanced recovery of agr mutant staphylococci was also observed in infected human skin grafts. These grafts were resistant to superficial exposure to S. aureus USA300, and the vast majority of the inoculum was readily cleared. However, consistent with the data obtained using cell lines, there were substantially more agr null mutant organisms associated with the human skin grafts than the WT organisms, and activation of autophagy was clearly demonstrated by galectin-8 and LC3 immunofluorescence. The burden of infection served to stimulate autophagy that would enhance staphylococcal persistence by consuming ASC and diminishing pyroptosis. These skin graft models did not directly assess the role of recruited phagocytes in S. aureus clearance, an important factor in in vivo infection (14), but demonstrate the contribution of the secreted toxins and their effects in promoting staphylococcal clearance by keratinocytes (12, 49).
Multiple S. aureus USA300 toxins appear to contribute to keratinocyte-mediated clearance mechanisms, as would be expected due to the ability of the pore-forming toxins to mediate escape from the endosome and to activate inflammasome signaling (10, 30, 48). Keratinocyte uptake of the WT and various toxin-deficient strains was roughly equivalent, and increased intracellular survival was most consistently observed with the agr, hla, and psmα null mutants. Although the PVL leukotoxin can activate inflammasome signaling, we did not observe intracellular persistence of the PVL-negative mutant, which still expresses other agr-dependent toxins. The agr locus, through sarA and RNAIII, regulates expression of hla and psmα (13, 50). Hla-associated pathology has been clearly documented in murine models of skin infection (51), and keratinocyte pyroptosis likely contributes to this process. Nonetheless, it is difficult to tease out the contributions of individual toxins, as there are multiple interactions between the different agr, psmα, and hla loci (50), and the abundance of the various human toxin-specific receptors on keratinocytes has not been rigorously established.
The ability of USA300 MRSA to actively adapt to conditions within human keratinocytes is likely an important factor in its success as a skin pathogen. S. aureus survival within an intracellular niche has been well described at many sites, including neutrophils (52), osteoblasts, macrophages (53), sinus cells (54), mammary and pulmonary epithelial and endothelial cell lines (30, 48). Thus, finding a small population of S. aureus within human keratinocytes is not unexpected. The evolution of S. aureus during asymptomatic carriage documents ongoing adaptation of these pathogens to the host (55) and includes mutation at the agr locus (16). Clinical studies indicate that human nasal carriage of the same S. aureus strains can persist over weeks if not longer (56). The appearance of agr mutants, with predominantly single point mutations from clinical infections has been interpreted to confer a short-term advantage for survival even if these mutations are only transient (57). The pathogenicity and clinical relevance of toxin-deficient strains have been recognized with the recovery of agr mutants from systemic human S. aureus infections (23). The source of agr defective strains in patients with bloodstream infection was often nasal carriage (58), which is likely the source of autoinoculation for skin infection as well (59). While we found that only a small fraction of the infecting organisms appear to persist within keratinocytes, the increased recovery of the agr mutants from within skin cells, both from chronically infected patients and in a laboratory setting, suggests that these strains may be relevant to human skin colonization. Of note, decreased neutrophil-mediated killing of USA300 agr null mutant has been described, suggesting that these strains may be less likely to be eradicated in an in vivo setting (60).
The adaptation of MRSA to human skin and occasional selection of agr defective S. aureus that persist intracellularly may be sufficient to maintain infection within human keratinocytes in areas such as the nares, as well as in diseased conditions such as atopic dermatitis. This small intracellular population may be especially refractory to eradication, as they are protected from many antibiotics, antibodies, and complement as well from the activity of the inflammasome and neutrophils. Therapeutic strategies to target the toxins associated with inflammasome activation and skin pathology (61), such as Hla, while effective at eradicating the WT organisms, may also provide additional selective pressure for the intracellular persistence of these mutants.
MATERIALS AND METHODS
Bacterial strains, human cells, and reagents.
MRSA USA300 FPR3757 (LAC) strains, specifically WT, spa null mutant (62), and mutant strains, were grown in LB broth overnight at 37°C, diluted 1:100, and grown to an optical density at 600 nm (OD600) of 1.0 for keratinocyte infections. WT LAC and the α-hemolysin (Hla) mutant were provided by Juliane Bubeck-Wardenburg (University of Chicago) (63). WT and Panton-Valentine leukocidin (PVL) mutants were provided by Frank DeLeo (64) (National Institute of Allergy and Infectious Diseases, Rocky Mountain Laboratories, MT). Phenol-soluble modulin (Psm) (psmα, psmβ, or hld) single and triple mutants (65) and Δagr mutants (13) were provided by Michael Otto (National Institute of Allergy and Infectious Diseases, MD). WT and agr-matched pairs in the RN and UAMS background were provided by Bo Shopsin (New York University Langone Medical Center, NY). The human keratinocyte HaCaT cell line was obtained from Angela Christiano (Columbia University, NY) and grown in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum with penicillin and streptomycin at 37°C and 5% CO2 in a humidified environment. HEKn primary human keratinocytes were purchased from Life Technologies and grown in DermaLife K complete medium (Lifeline Cell Technology). The antibodies used were as follows: mouse anti-human integrin α5β1 (Chemicon International); rabbit anti-LC3B (recognizes LC3I and LC3II) (Sigma); rabbit anti-galectin-8 (H-80), mouse anti-Staphylococcus aureus (704) (which recognizes both WT and spa null mutant strains), and rabbit anti-GAPDH(anti-glyceraldehyde-3-phosphate dehydrogenase antibody) (FL-335) (Santa Cruz Biotechnology); rabbit anti-ASC [TMS1] (Millipore); mouse anti-ATG5 and mouse anti-EEA-1 (MBL International). Fibronectin (Life Technologies) at 10 µg/ml was used to pretreat keratinocytes and organotypic cultures prio r to MRSA infection. Cyto-ID autophagy detection reagent was purchased from Enzo Life Sciences and used per the manufacturer’s instructions, and cells were analyzed on a Tali image cytometer (Life Technologies). Monodansyl cadaverine (MDC) (5 mM) was from Santa Cruz Biotechnology. The inhibitors and concentrations used were caspase-1 inhibitor (cell permeable) (catalog no. 40011; Calbiochem) (20 µM) and caspase-3 inhibitor (catalog no. 235420; Calbiochem) (20 µM) from EMD Millipore; wortmannin (2 μM), chloroquine (100 μM), and bafilomycin (200 nM) from Sigma; 3-methyladenine (3-MA) (5 mM) from Selleckchem. Controls contained the media and 5% dimethyl sulfoxide (DMSO).
Organotypic cultures.
Organotypic cultures of human keratinocytes in primary culture were obtained from the Cell and Tissue Kinetics Core of the Columbia University Department of Dermatology Skin Disease Research Center. Human keratinocytes were grown at an air-liquid interface supported by a dermal substitute matrix as a three-dimensional model system composed of the dermal and epidermal compartments. Following 24-h stimulation with strain USA300 or PBS, human organotypic skin equivalents were fixed for 24 h in 4% paraformaldehyde prior to embedding in paraffin blocks.
Infection of keratinocytes and gentamicin protection assay.
HaCaT or HEKn cells grown to confluence and without antibiotics for 18 h prior to infection were pretreated for 1 h with 10 µg/ml fibronectin. Bacterial cultures grown to log phase (to an OD600 of 1.0) were pelleted and resuspended in media without antibiotics. HaCaT cells were infected with MRSA (WT or mutants) at a multiplicity of infection (MOI) of 10 and incubated at 37°C for 2 h. Extracellular bacteria were killed by the addition of gentamicin to a final concentration of 500 μg/ml. Intracellular infections were then allowed to continue in the presence of gentamicin for up to 96 h. To quantify intracellular bacteria, cells were disassociated with TrypLE express cell dissociation enzyme (Life Technologies), and dilutions were plated on LB agar plates for CFU enumeration. For assays with inhibitors (caspase-1, caspase-3, wortmannin, and 3-MA), controls containing the inhibitor in 5% DMSO in the absence of bacteria were included, along with controls containing just bacteria, to verify a lack of toxicity. Intracellular accumulation of staphylococci was quantified 24 h after the initial 2-h infection; additional time points are noted in the figures. Keratinocyte culture supernatants were collected for IL-1β enzyme-linked immunosorbent assay (ELISA) (R&D Systems). Cytotoxicity was determined using the LDH assay (Roche) using multiply frozen and thawed keratinocytes as a positive control. Alternatively, cell viability was determined using the Countess automated cell counter and trypan blue (Life Technologies).
RNA analysis and siRNA.
RNA was isolated using the eZNA total RNA kit (Omega Bio-Tek) followed by DNase treatment using a DNA-free DNA removal kit (Life Technologies). cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative reverse transcription-PCR (qRT-PCR) was performed using Power SYBR green PCR master mix (Applied Biosystems) in a StepOne Plus thermal cycler (Applied Biosystems). The primers for RNAIII were 5′ GGGATGGCTTAATAACTCATAC 3′ and 5′ GGAAGGAGTGATTTCAATGG 3′. ON-TARGETplus SMARTpool Human ATG5 siRNA, Nod2 and ON-TARGETplus nontargeting control pool siRNAs were purchased from Dharmacon and transfected into HaCaT cells using Lipofectamine RNAiMAX (Life Technologies) per the manufacturer’s instructions. After 72 h, the cells were stimulated with S. aureus USA300 (MOI of 10) in media containing fibronectin or PBS plus fibronectin for 2 h, treated with gentamicin as described above, and allowed to incubate for 24 h. Culture supernatants were collected and analyzed for IL-1β, and cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-Cl, 50 mM NaCl, 0.1% SDS, 1% Triton X-100, 10% glycerol, 2 mM EDTA, 0.5% sodium deoxycholate) containing 1× Halt protease and phosphatase single-use inhibitor cocktail (Thermo Scientific). Proteins were separated on Bolt 4% to 12% bis-Tris Plus gels (Life Technologies), transferred using iBLot dry blotting system (Invitrogen), and blocked with 5% milk in TBST (Tris-buffered saline plus Tween) (50 mM Tris [pH 7. 5], 150 mM NaCl, 0.05% Tween) or 5% bovine serum albumin (BSA) in TBST for 1 h at room temperature. Immunodetection was performed using anti-Nod2 (Santa Cruz), anti-ATG5 (Cell Signaling), anti-ASC (EMD-Millipore), and β-actin or GAPDH (Sigma-Aldrich). Densitometry was performed using ImageJ.
SCID-hu mouse model.
Skin grafts were performed as follows. NOD-scid IL2Rγ null (NSG) mice (Jackson Laboratories) (66) were grafted with human neonatal foreskin skin per Columbia University institutional review board (IRB) protocol AAAN6306. Freshly obtained skin was placed in DMEM containing penicillin and streptomycin, trimmed of dermal tissue, and cut to 1 cm by 1 cm. Mice were anesthetized with isofluorane at 4% at an O2 flow rate of 1 liter/min for 2 to 3 min for induction and maintained at 2% isofluorane with an O2 flow rate of 1 liter/min. Local anesthesia at the surgical site was achieved by a subcutaneous injection of 0.5% marcaine at 1 mg/kg of body weight and 5 mg/kg carprofen intraperitoneal (i.p.) injection. After the mice were shaved, the surgical site was locally cleaned with Betadine and alcohol. Incisions were made with strabismus scissors, and a flap of mouse skin (1 cm by 1 cm) was removed and replaced with processed foreskin that was secured with single interrupted sutures at all the corners of the graft. The surgical site was protected with petroleum jelly-soaked nonadhering gauze secured with a circumferential bandage. The mice were given a daily dose of carprofen by i.p. at 5 mg/kg for the next 2 days. Mouse bandages were inspected every day for the first 2 days after surgery and then weekly for the following 3 to 6 weeks. For infections, mice were anesthetized with ketamine and xylazine, and S. aureus was applied dropwise in a 10-µl aliquot of PBS containing 108 CFU of S. aureus. Skin grafts were excised after 72 h and fixed in 4% paraformaldehyde for 24 h for paraffin sectioning and staining.
Animal work in this study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, the Animal Welfare Act, and U.S. federal law. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University (protocol AAAG7408).
Hla and Hld expression in atopic dermatitis strains.
S. aureus strains, including clinical isolates (previously obtained from superficial skin swabs of atopic dermatitis patients and stored at −80°C), were grown in LB to stationary phase, and culture supernatants were harvested for Western immunoblots. Primary anti-α-hemolysin (anti-Hla) rabbit antibody was a gift from Juliane Bubeck-Wardenburg, University of Chicago, and used at 1:10,000 dilution. The presence of Hld in the clinical isolates was assessed by sequencing. Hemolysis was detected by growth on sheep blood agar plates (Becton, Dickinson) (5% in TSB). Synergistic hemolysis between Hld and Hlb was evaluated using the cross-streak method with S. aureus RN4220 by the method of Cheung et al. (26).
RNAIII expression in atopic dermatitis strains.
Overnight cultures of S. aureus laboratory strains and 10 clinical isolates from atopic dermatitis patients were diluted 1:100 in LB broth and grown at 37°C to an OD600 of 1.0, and bacteria were pelleted, resuspended, and incubated for 2 h at 37°C in cell wall lysis mixture (6 µg/ml lysostaphin, 2.7 µg/ml mutanolysin, and 16.7 µg/ml lysozyme in 50 µM Tris-HCl−10 μM EDTA [pH 8]). RNA was extracted from bacteria, cDNA was synthesized, and qPCR was performed as described above to assess for S. aureus RNAIII expression.
Whole-genome sequencing.
Clinical isolates of S. aureus were collected as part of an ongoing clinical study (approved by IRB AAAI5956) and subjected to whole-genome sequencing. Genomic DNA was prepared from overnight cultures using the DNeasy blood and tissue kit (Qiagen) after a 2-h incubation at 37°C in cell wall lysis mixture (described above). Genome libraries were prepped using the Nextera XT kit and sequenced using the Illumina MiSeq instrument (paired-end reads of 250 bp). Genome assembly was done using ABySS60 at multiple kmer sizes (kmer = 23, 27, 31, 35, 39, and 63), and we selected the assembly with the highest N50 value (kmer = 39) for further analysis. The RAST tool (http://rast.nmpdr.org/rast.cgi) was used for genome annotation.
Statistics.
Samples with normal distribution were analyzed using the Student’s t test. Multiple comparisons were analyzed using one-way analysis of variance (ANOVA) with Dunnett’s posttest. Differences in groups were considered significant if P was <0.05. Statistics were performed with GraphPad Prism version 4.00.
SUPPLEMENTAL MATERIAL
SCID-hu skin grafts in situ. Image of SCID mouse with intact human graft. Visual inspection of the infected graft at 72 h postinoculation. No obvious differences were observed in the gross appearance of the skin grafts exposed to PBS, WT S. aureus USA300, or the agr null mutant strain. Download
Visualization of S. aureus USA300 and mutants with anti-S. aureus antibody (Ab). The anti-S. aureus staining achieved in HaCaT cells exposed to WT USA300 or to the spa null mutant or agr null mutant are compared. The cells were stained for MDC (green), S. aureus (red), or To-Pro-3 (DNA) (blue), demonstrating roughly equivalent detection of S. aureus mutants that have very different amounts of surface protein A expression. Download
Effects of wortmannin and gentamicin on the recovery of mutants like the agr mutant (nonhemolytic mutants). S. aureus USA300 was grown in the presence of 10 µg/ml of wortmannin and/or 0.1 μg/ml of gentamicin for 24 or 72 h. Colonies were plated onto LB medium, and individual clones were screened for hemolysis on sheep blood agar plates. Download
Footnotes
Citation Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, Brown A, Lauren C, Dowd M, West E, Horst B, Planet P, Prince A. 2015. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. mBio 6(2):e00289-15. doi:10.1128/mBio.00289-15.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Talan DA, Krishnadasan A, Gorwitz RJ, Fosheim GE, Limbago B, Albrecht V, Moran GJ, EMERGEncy ID Net Study Group . 2011. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin Infect Dis 53:144–149. doi: 10.1093/cid/cir308. [DOI] [PubMed] [Google Scholar]
- 3.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heath W, Carbone F. 2013. The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 14:978–985. doi: 10.1038/ni.2680. [DOI] [PubMed] [Google Scholar]
- 5.Brown ML, O’Hara FP, Close NM, Mera RM, Miller LA, Suaya JA, Amrine-Madsen H. 2012. Prevalence and sequence variation of Panton-Valentine leukocidin in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains in the United States. J Clin Microbiol 50:86–90. doi: 10.1128/JCM.05564-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singer AJ, Talan DA. 2014. Management of skin abscesses in the era of methicillin-resistant Staphylococcus aureus. N Engl J Med 370:1039–1047. doi: 10.1056/NEJMra1212788. [DOI] [PubMed] [Google Scholar]
- 7.Lippens S, Denecker G, Ovaere P, Vandenabeele P, Declercq W. 2005. Death penalty for keratinocytes: apoptosis versus cornification. Cell Death Differ 12(Suppl 2):1497–1508. doi: 10.1038/sj.cdd.4401722. [DOI] [PubMed] [Google Scholar]
- 8.Soong G, Chun J, Parker D, Prince A. 2012. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis 205:1571–1579. doi: 10.1093/infdis/jis244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA. 2009. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PloS One 4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munoz-Planillo R, Franchi L, Miller LS, Nunez G. 2009. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol 183:3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi CY, Lin CC, Liao IC, Yao YC, Shen FC, Liu CC, Lin CF. 2014. Panton-Valentine leukocidin facilitates the escape of Staphylococcus aureus from human keratinocyte endosomes and induces apoptosis. J Infect Dis 209:224–235. doi: 10.1093/infdis/jit445. [DOI] [PubMed] [Google Scholar]
- 12.DuMont AL, Torres VJ. 2014. Cell targeting by the Staphylococcus aureus pore-forming toxins: it’s not just about lipids. Trends Microbiol 22:21–27. doi: 10.1016/j.tim.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. 2008. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 32:150–158. doi: 10.1016/j.molcel.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishna S, Miller LS. 2012. Innate and adaptive immune responses against Staphylococcus aureus skin infections. Semin Immunopathol 34:261–280. doi: 10.1007/s00281-011-0292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JS, Guo Y, Ramos RI, Hebroni F, Plaisier SB, Xuan C, Granick JL, Matsushima H, Takashima A, Iwakura Y, Cheung AL, Cheng G, Lee DJ, Simon SI, Miller LS. 2012. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog 8:e1003047. doi: 10.1371/journal.ppat.1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitzgerald JR. 2014. Evolution of Staphylococcus aureus during human colonization and infection. Infect Genet Evol 21:542–547. doi: 10.1016/j.meegid.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 17.Planet PJ, LaRussa SJ, Dana A, Smith H, Xu A, Ryan C, Uhlemann AC, Boundy S, Goldberg J, Narechania A, Kulkarni R, Ratner AJ, Geoghegan JA, Kolokotronis SO, Prince A. 2013. Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. mBio 4(6):e00889-13. doi: 10.1128/mBio.00889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aymard E, Barruche V, Naves T, Bordes S, Closs B, Verdier M, Ratinaud M-H. 2011. Autophagy in human keratinocytes: an early step of the differentiation? Exp Dermatol 20:263–268. doi: 10.1111/j.1600-0625.2010.01157.x. [DOI] [PubMed] [Google Scholar]
- 19.Deretic V. 2012. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol 24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. 2012. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boguniewicz M, Leung DY. 2011. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev 242:233–246. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shopsin B, Drlica-Wagner A, Mathema B, Adhikari R, Kreiswirth B, Novick R. 2008. Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J Infect Dis 198:1171–1175. doi: 10.1086/592051. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, Villaruz AE, Cheung GY, McGavin MJ, Travers JB, Otto M, Inohara N, Nunez G. 2013. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature 503:397–401. doi: 10.1038/nature12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valour F, Rasigade JP, Trouillet-Assant S, Gagnaire J, Bouaziz A, Karsenty J, Lacour C, Bes M, Lustig S, Benet T, Chidiac C, Etienne J, Vandenesch F, Ferry T, Laurent F, Lyon BJI Study Group . 10 February 2015. Delta-toxin production deficiency in Staphylococcus aureus: a diagnostic marker of bone and joint infection chronicity linked with osteoblast invasion and biofilm formation. Clin Microbiol Infect doi: 10.1016/j.cmi.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 26.Cheung GY, Duong AC, Otto M. 2012. Direct and synergistic hemolysis caused by Staphylococcus phenol-soluble modulins: implications for diagnosis and pathogenesis. Microbes Infect 14:380–386. doi: 10.1016/j.micinf.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto M. 2014. Staphylococcus aureus toxins. Curr Opin Microbiol 17:32–37. doi: 10.1016/j.mib.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Date SV, Modrusan Z, Lawrence M, Morisaki JH, Toy K, Shah IM, Kim J, Park S, Xu M, Basuino L, Chan L, Zeitschel D, Chambers HF, Tan MW, Brown EJ, Diep BA, Hazenbos WL. 2014. Global gene expression of methicillin-resistant Staphylococcus aureus USA300 during human and mouse infection. J Infect Dis 209:1542–1550. doi: 10.1093/infdis/jit668. [DOI] [PubMed] [Google Scholar]
- 29.Ingham KC, Brew S, Vaz D, Sauder DN, McGavin MJ. 2004. Interaction of Staphylococcus aureus fibronectin-binding protein with fibronectin: affinity, stoichiometry, and modular requirements. J Biol Chem 279:42945–42953. doi: 10.1074/jbc.M406984200. [DOI] [PubMed] [Google Scholar]
- 30.Grosz M, Kolter J, Paprotka K, Winkler AC, Schafer D, Chatterjee SS, Geiger T, Wolz C, Ohlsen K, Otto M, Rudel T, Sinha B, Fraunholz M. 2014. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol 16:451–465. doi: 10.1111/cmi.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole J, Aberdein J, Jubrail J, Dockrell DH. 2014. The role of macrophages in the innate immune response to Streptococcus pneumoniae and Staphylococcus aureus: mechanisms and contrasts. Adv Microb Physiol 65:125–202. doi: 10.1016/bs.ampbs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. 2014. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 34.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deretic V. 2012. Autophagy: an emerging immunological paradigm. J Immunol 189:15–35. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, von Hoven G, Boller K, Husmann M. 2010. Pro-autophagic signal induction by bacterial pore-forming toxins. Med Microbiol Immunol 199:299–608. doi: 10.1007/s00430-010-0163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ku CC, Besser J, Abendroth A, Grose C, Arvin AM. 2005. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J Virol 79:2651–2658. doi: 10.1128/JVI.79.5.2651-2658.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. 2010. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warnes G. 2014. Measurement of autophagy by flow cytometry. Curr Protoc Cytom 68:9.45.1–9.45.10. doi: 10.1002/0471142956.cy0945s68. [DOI] [PubMed] [Google Scholar]
- 41.Amano A, Nakagawa I, Yoshimori T. 2006. Autophagy in innate immunity against intracellular bacteria. J Biochem 140:161–167. doi: 10.1093/jb/mvj162. [DOI] [PubMed] [Google Scholar]
- 42.Byrne BG, Dubuisson JF, Joshi AD, Persson JJ, Swanson MS. 2013. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. mBio 4(1):e00620-12. doi: 10.1128/mBio.00620-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee H-M, Shin D-M, Yuk J-M, Shi G, Choi D-K, Lee S-H, Huang S, Kim J-M, Kim C, Lee J-H, Jo E-K. 2011. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol 186:1248–1306. doi: 10.4049/jimmunol.1001954. [DOI] [PubMed] [Google Scholar]
- 44.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deshmukh H, Hamburger J, Ahn S, McCafferty D, Yang S, Fowler V. 2009. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect Immun 77:1376–1458. doi: 10.1128/IAI.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulland TK, Ferguson PJ, Sutterwala FS. 2015. Evasion of inflammasome activation by microbial pathogens. J Clin Invest 125:469–477. doi: 10.1172/JCI75254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nauseef WM. 2007. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 48.Jarry TM, Memmi G, Cheung AL. 2008. The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell Microbiol 10:1801–1814. doi: 10.1111/j.1462-5822.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 49.Pishchany G, McCoy AL, Torres VJ, Krause JC, Crowe JE Jr, Fabry ME, Skaar EP. 2010. Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 8:544–550. doi: 10.1016/j.chom.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berube BJ, Sampedro GR, Otto M, Bubeck Wardenburg J. 2014. The psmα locus regulates production of Staphylococcus aureus alpha-toxin during infection. Infect Immun 82:3350–3358. doi: 10.1128/IAI.00089-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, Bubeck Wardenburg J. 2014. Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J Infect Dis 210:1012–1018. doi: 10.1093/infdis/jiu223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol 164:3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 53.Hamza T, Li B. 2014. Differential responses of osteoblasts and macrophages upon Staphylococcus aureus infection. BMC Microbiol 14:207. doi: 10.1186/s12866-014-0207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svider PF, Husain Q, Mauro KM, Folbe AJ, Baredes S, Eloy JA. 2014. Impact of mentoring medical students on scholarly productivity. Int Forum Allergy Rhinol 4:138–142. doi: 10.1002/alr.21247. [DOI] [PubMed] [Google Scholar]
- 55.Golubchik T, Batty EM, Miller RR, Farr H, Young BC, Larner-Svensson H, Fung R, Godwin H, Knox K, Votintseva A, Everitt RG, Street T, Cule M, Ip CL, Didelot X, Peto TE, Harding RM, Wilson DJ, Crook DW, Bowden R. 2013. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PloS One 8:e61319. doi: 10.1371/journal.pone.0061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slingerland BC, Tavakol M, McCarthy AJ, Lindsay JA, Snijders SV, Wagenaar JA, van Belkum A, Vos MC, Verbrugh HA, van Wamel WJ. 2012. Survival of Staphylococcus aureus ST398 in the human nose after artificial inoculation. PloS One 7:e48896. doi: 10.1371/journal.pone.0048896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shopsin B, Eaton C, Wasserman GA, Mathema B, Adhikari RP, Agolory S, Altman DR, Holzman RS, Kreiswirth BN, Novick RP. 2010. Mutations in agr do not persist in natural populations of methicillin-resistant Staphylococcus aureus. J Infect Dis 202:1593–1599. doi: 10.1086/656915. [DOI] [PubMed] [Google Scholar]
- 58.Smyth DS, Kafer JM, Wasserman GA, Velickovic L, Mathema B, Holzman RS, Knipe TA, Becker K, von Eiff C, Peters G, Chen L, Kreiswirth BN, Novick RP, Shopsin B. 2012. Nasal carriage as a source of agr-defective Staphylococcus aureus bacteremia. J Infect Dis 206:1168–1177. doi: 10.1093/infdis/jis483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garbacz K, Piechowicz L, Baranska-Rybak W, Dabrowska-Szponar M. 2011. Staphylococcus aureus isolated from patients with recurrent furunculosis carrying Panton-Valentine leukocidin genes represent agr specificity group IV. Eur J Dermatol 21:43–46. doi: 10.1684/ejd.2010.1151. [DOI] [PubMed] [Google Scholar]
- 60.Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. 2010. agr-dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun 2:546–559. doi: 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, Gresham HD. 2014. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PloS Pathog 10:e1004174. doi: 10.1371/journal.ppat.1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin FJ, Gomez MI, Wetzel DM, Memmi G, O’Seaghdha M, Soong G, Schindler C, Prince A. 2009. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest 119:1931–1939. doi: 10.1172/JCI35879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, Wack A. 2013. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog 9:e1003773. doi: 10.1371/journal.ppat.1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, Kreiswirth BN, DeLeo FR. 2006. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis 194:1761–1770. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 65.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 66.Pearson T, Greiner DL, Shultz LD. 2008. Humanized SCID mouse models for biomedical research. Curr Top Microbiol Immunol 324:25–51. doi: 10.1007/978-3-540-75647-7_2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SCID-hu skin grafts in situ. Image of SCID mouse with intact human graft. Visual inspection of the infected graft at 72 h postinoculation. No obvious differences were observed in the gross appearance of the skin grafts exposed to PBS, WT S. aureus USA300, or the agr null mutant strain. Download
Visualization of S. aureus USA300 and mutants with anti-S. aureus antibody (Ab). The anti-S. aureus staining achieved in HaCaT cells exposed to WT USA300 or to the spa null mutant or agr null mutant are compared. The cells were stained for MDC (green), S. aureus (red), or To-Pro-3 (DNA) (blue), demonstrating roughly equivalent detection of S. aureus mutants that have very different amounts of surface protein A expression. Download
Effects of wortmannin and gentamicin on the recovery of mutants like the agr mutant (nonhemolytic mutants). S. aureus USA300 was grown in the presence of 10 µg/ml of wortmannin and/or 0.1 μg/ml of gentamicin for 24 or 72 h. Colonies were plated onto LB medium, and individual clones were screened for hemolysis on sheep blood agar plates. Download